
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polythioethers bearing side groups for efficient degradation by E1cB reaction: reaction design for polymerization and main-chain scission†
Ryo Kawatani a,
Keito Hagiwaraa,
Anri Tanakaa and
Yasuhiro Kohsaka*ab
a,
Keito Hagiwaraa,
Anri Tanakaa and
Yasuhiro Kohsaka*ab
aFaculty of Textile Science and Technology, Shinshu University, 3-15-1 Tokida, Ueda, Nagano 386-8567, Japan. E-mail: kohsaka@shinshu-u.ac.jp
bResearch Initiative for Supra-Materials (RISM), Interdisciplinary Cluster for Cutting Edge Research (ICCER), Shinshu University, 4-17-1 Wakasato, Nagano City, Nagano 380-8553, Japan
First published on 10th July 2023
Abstract
We have previously reported the polycondensation by the tandem reactions of dithiols and α-(bromomethyl)acrylates, consisting of conjugate substitution (SN2′ reaction) and conjugate addition (Michael addition) reactions. The resulting polythioethers underwent a main-chain scission (MCS) by E1cB reaction, which is the reverse reaction of conjugate addition, although it was not quantitative due to the equilibrium. Herein, the modification of the structures of polythioethers led to irreversible MCS, whereby the β-positions of ester moieties were substituted with a phenyl group. This slight modification in the polymer structure influenced the monomer structures and polymerization mechanisms. The understanding of reaction mechanisms by model reactions was required to obtain high molecular weights of polythioethers. It was clarified that the consequent additions of 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), and PBu3 were effective to achieve high molecular weight. The resulting polythioethers decomposed by irreversible MCS via E1cB reaction with DBU.
Introduction
The main-chain scission (MCS) of polymers leads to a high decrease in molecular weight through a small number of reactions. As a result, the thermal properties, such as glass transition temperature (Tg) and melting temperature (Tm), mechanical properties, such as Young modulus and elasticity, and the solubility were drastically changed. The changes in physical properties by MCS have been applied to photoresists,1 dismantling adhesions,2 degradable crosslinked polymers,3 and a prospective strategy for controlling and supporting biodegradation.4–6 Therefore, the developments of polymers accepting MCS by specific stimuli are important issues.Although the method to break a carbon–carbon (C–C) covalent bond is limited,7–11 those of carbon–heteroatom (C–X) are often performed.12–16 Recently, Hoye et al. applied the E1cB reaction (retro-oxa-Michael addition) to the MCS of polyester.17 The polyester was prepared by ring-opening polymerization of a δ-lactone derivative bearing a carbonyl pendant at the γ-position, and the E1cB reaction by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) resulted in MCS producing an acrylate monomer at a high yield (95%). This study suggests that the E1cB reaction is effective for quantitative and selective MCS.
Polythioethers are typically synthesized by the polyaddition of thiols with alkenes18 or alkynes,19 whereas the radical ring-opening polymerization of cyclic α-(thiomethyl)acrylates is another practical route.20 The former, polyaddition, are based on thiol–ene and thiol–yne click reaction that progresses quantitatively even under ambient condition. Thiol–ene reactions include both radical and ionic mechanisms.21 The latter is the so-called Michael addition reaction between thiols and α,β-unsaturated carbonyl compounds, such as acrylates, which is reverse of the E1cB reaction. Therefore, optimization of molecular and reaction designs would lead to both polymerizations by Michael addition and efficient MCS by E1cB reaction under suitable conditions.
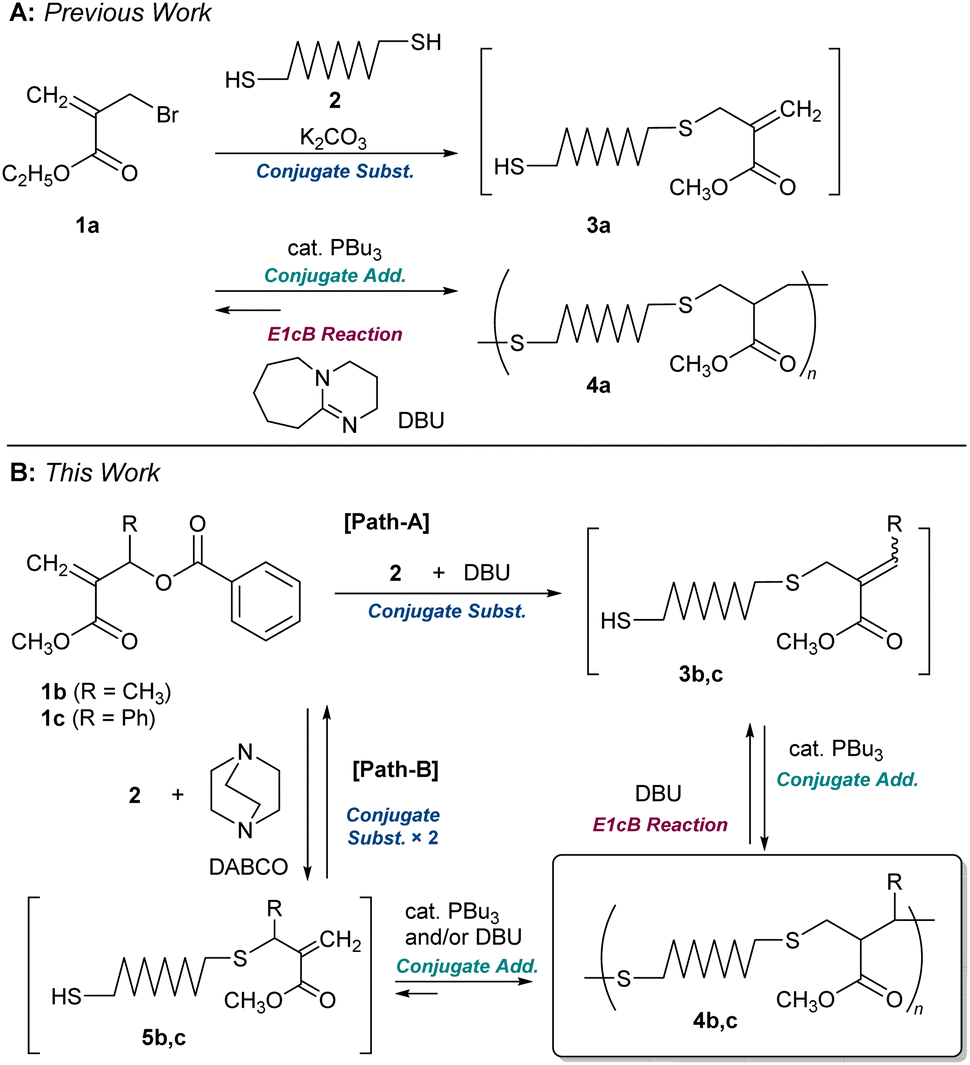
We recently reported the polycondensation via a tandem reaction of conjugate substitution and addition reactions of α-(bromomethyl)acrylate 1a and dithiol 2 (Scheme 1A).16,22 The product, 4a, underwent MCS via E1cB reaction in the presence of DBU.15 However, since the E1cB reactions were not irreversible, the end-capping of formed thiol-end with monothiol was required to promote MCS. However, the MCS was not quantitative; for example, 4a (Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 and Đ = 1.89) decomposed to an oligomer (Mn = 2100 and Đ = 1.83) even in the presence of 5.0 equimolar monothiol.
700 and Đ = 1.89) decomposed to an oligomer (Mn = 2100 and Đ = 1.83) even in the presence of 5.0 equimolar monothiol.
 | ||
| Scheme 1 Synthesis and MCS of polythioethers. (A) Our previous report.22 (B) This report. | ||
Herein, new polythioethers 4b and 4c, bearing methyl and phenyl substituents next to the sulphur atom, respectively, were designed to achieve more efficient MCS (Scheme 1B). The allylic substituents were found as the key leading to the MCS by the irreversible E1cB reaction. On the other hand, substitution at the allylic position complicated the polymerization. An understanding of the reaction mechanism and careful examination of the conditions were required to achieve a high degree of polymerization.
Results and discussion
Molecular design and model reactions
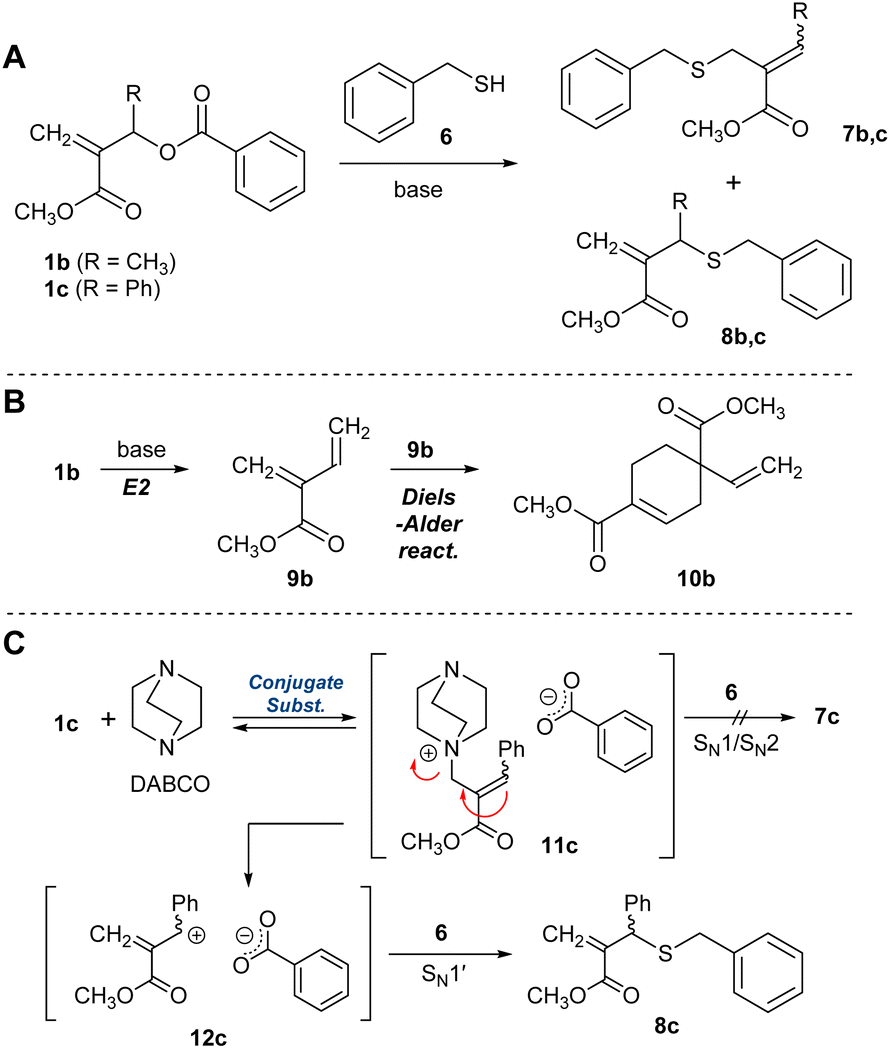
For the difficulty in synthesis, 1b and 1c, which have no halogen atom but a benzoyloxy group as a leaving group for conjugate substitution, were prepared. Two routes are possible to access 4b:23 one is a direct conjugate substitution with 2, and the subsequent conjugate addition (Scheme 1B, path A). As this is a reversible reaction, the reaction condition is expected to be the key to achieving a high degree of polymerization. The other is the conjugate addition reaction of 5b, which seems more reactive than 3b due to the exo-olefin structure (path B). Thus, we initially planned the polymerization via path B. For this strategy, the issue of how 5b can be prepared from 1b must be addressed.Yu et al. have reported the synthesis of an exo-methylene product by the nucleophilic substitution reaction of the analogue of 1b and a phenol catalysed by Et3N,24 which was a hint to prepare 5b from 1b, although the product yield was not quantitative. As is well known, quantitative and selective conversion is necessary for polycondensation. Therefore, a model experiment with benzyl mercaptan (6) was conducted in CDCl3 to optimize the reaction condition for the selective and quantitative synthesis of an exo-olefin 8b (Scheme 2A). Without a base, no reaction occurred (Table 1, entry 1). Then, 6 was added to a mixture of DABCO and 1b, but an E2 reaction and the subsequent Diels–Alder reaction to yield 9b and 10b, respectively, were observed (Scheme 2B, entry 2, Fig. S3†). To avoid these side reactions, 1b and 6 were mixed in advance, and DABCO was added (entry 3, Fig. S4†). The main product was exo-olefin 8b, with some slight side reactions. To reduce the side reactions, the feed of DABCO was decreased to a catalytic amount, but selectivity was unchanged, thus slowing down the process (entry 4, Fig. S5†). Weak bases, such as Et3N (entry 5) and iPr2NEt (entry 6), resulted in low conversion and selectivity, while a stronger base, DBU, yielded 7b as the main product (entry 7). The side reactions are not desirable for polycondensation as they lead to a low degree of polymerization. Thus, 1c, which has a phenyl group instead of methyl group at the allylic substituent, was prepared as a substrate free from the possibility of an E2 reaction.24 As expected, the selective formation of 8c was achieved with DABCO (entry 10, Fig. S6†). Thus, the combination of 1c and DABCO seemed suitable for path B in Scheme 1B. Notably, bases with low nucleophilicity, such as iPr2NEt (entry 12) and DBU (entry 13), were effective in preparing 7c. Since iPrNEt2 resulted in decreased conversion, a combination of 1c and DBU seemed suitable for the preparation of 7c and path A in Scheme 1B.
 | ||
| Scheme 2 A model reaction of 1b and c with 6 (A) and the proposed reaction mechanisms (B) and (C). | ||
| Entrya | 1 | Baseb (equimol.) | Time [h] | Conv.c [%] | Compositionc [%] | |||
|---|---|---|---|---|---|---|---|---|
| 7 | 8 | 9 | 10 | |||||
| a 1: 60 μmol, [1]/[6] = 1/1.2, CDCl3: 0.70 mL, 25 °C. A base was added after mixing 1 and 6.b Equimolar to 1.c Determined by 1H NMR spectra.d 1b and DABCO were mixed before adding 6. | ||||||||
| 1 | 1b | — | 1 | 0 | ||||
| 2d | 1b | DABCO (1.2) | 1 | 86 | 85 | 15 | ||
| 3 | 1b | DABCO (1.2) | 1 | >99 | 1 | 95 | 1 | 2 |
| 4 | 1b | DABCO (0.33) | 15 | 81 | 1 | 94 | 3 | 2 |
| 5 | 1b | Et3N (1.2) | 24 | 26 | 58 | 38 | 0 | 4 |
| 6 | 1b | iPr2NEt (1.2) | 24 | Trace | ||||
| 7 | 1b | DBU (1.2) | 3 | >99 | 96 | 2 | 1 | 1 |
| 8 | 1b | DBU (0.33) | 36 | 27 | 95 | 1 | 0 | 4 |
| 9 | 1c | — | 1 | 0 | ||||
| 10 | 1c | DABCO (1.2) | 1 | 97 | <1 | >99 | 0 | 0 |
| 11 | 1c | Et3N (1.2) | 24 | 35 | 61 | 31 | 0 | 0 |
| 12 | 1c | iPr2NEt (1.2) | 24 | 18 | >99 | <1 | 0 | 0 |
| 13 | 1c | DBU (1.2) | 1 | 97 | 97 | 3 | 0 | 0 |
The experiments suggested the following reaction mechanism. A poor nucleophilic base, e.g. DBU, led to the deprotonation of 6 and the subsequent conjugate substitution reaction to yield 7c. In contrast, a nucleophilic base, such as DABCO, preferred the conjugate substitution to 1c than the deprotonation of 6 (Scheme 2C). Then, the endo-olefin intermediate, 11c was formed. Since the product 8c involved an SN1′ mechanism, i.e., the elimination–substitution mechanism, was considered reasonable rather than SN1 and SN2 reaction25 toward 7c. In this context, the phenyl substituent might have a decisive effect on promoting the formation of 8c, as the resonance effect stabilizes the intermediate 12c.
Polymerization
Since DBU was expected to yield the intermediate 3c selectively and quantitatively (Scheme 1B), the polycondensations of 1c and 2 through path A were conducted in the presence of DBU (Table 2, entries 1–3). Polymerization conditions were referred from our previous papers22 to compare polymerization behaviors with 4a (Mn = 10700 and Đ = 1.89). However, lower molecular weight polymers were obtained (Table 2, entry 1, Mn = 5600 and Đ = 2.10). The polymerization was further monitored by size-exclusion chromatograms (SECs, Fig. S9†). The molecular weight increased after 7 h but decreased after 24 h, suggesting MCS by E1cB reaction after the propagating reaction. Therefore, the reaction system seemed to have reached equilibrium. The addition of PBu3 to promote the conjugate addition (propagating reaction) resulted in a slight increase in molecular weight (entries 4 and 5, Fig. S10†).
| Entrya | Base (equimol.) | Solvent | Temp. [°C] | Time [h] | Yield [%] | Mnb | Đb |
|---|---|---|---|---|---|---|---|
| a 1c: 0.750 mmol, [1c]/[2]/[base]/[catalyst] = 1/1.0/1.2/0.2. Base: DBU, catalyst: PBu3, solvent: 0.75 mL.b Determined by SEC (THF, 40 °C, polystyrene standards).c Polycondensation for 24 h with DBU, and then, PBu3 was added. | |||||||
| 1 | 1.2 | CHCl3 | 25 | 24 | >99 | 5600 | 2.10 |
| 2 | 1.2 | CH3CN | 25 | 24 | >99 | 4900 | 2.32 |
| 3 | 1.2 | CH3CN | 50 | 24 | >99 | 4700 | 1.91 |
| 4 | 1.2 | CH3CN | 25 | 24 + 5c | 37 | 6600 | 1.60 |
| 5 | 2.2 | CH3CN | 25 | 24 + 5c | 28 | 7300 | 1.74 |
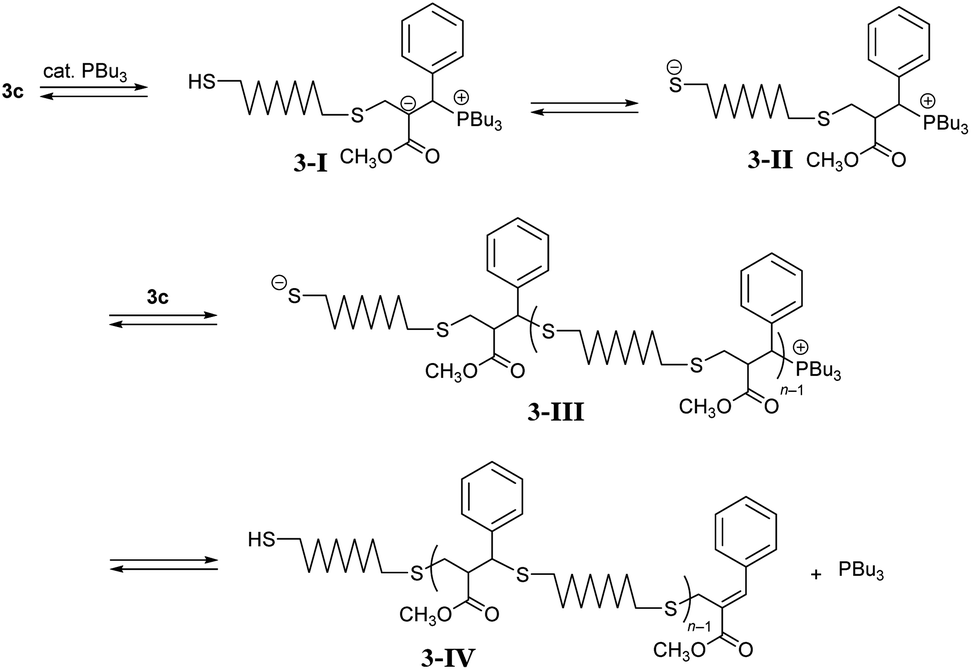
Next, polymerizations through path B were investigated using DABCO and PBu3 in CHCl3 (Table 3, entry 1). However, the resulting product was a polymer with a low degree of polymerization (Mn = 1000 and Đ = 2.25). Similar results were obtained in CH3CN (entries 2 and 3). To investigate the reason of unsuccessful polycondensation, the reaction was monitored in CD3CN by 1H NMR spectra (Fig. S11†). After 1 h, 5c was observed as the main product, indicating reaction proceeding through path B as expected. However, signal X assigned to the endo-olefin proton was observed around 7.7 ppm, which became more pronounced after 16 h, suggesting an unexpected reaction that inhibited further propagation. A possible mechanism of the side reaction is described in Scheme 3. The conjugate addition proceeds through the addition of PBu3 to an acrylate skeleton to form enolate intermediate 3-I.21 The subsequent proton transfer forms thiolate anion 3-II, and the conjugate addition follows. Herein, the elimination from the phosphonium end of 3-III to the chain end 3-IV is possible. A basic catalyst, such as Et3N, that directly deprotonates thiols was also effective in promoting conjugate substitution,25 and the weak base was expected to decrease the side reaction. Thus, Et3N (entry 4) and iPr2NEt (entry 5) were analysed but found ineffective in increasing the molecular weight, probably due to the low activity. A stronger base, DBU, was more effective (entry 6, Mn = 5400, and Đ = 2.16), although the molecular weight was still lower than polymers obtained in entry 5 in Table 2. Stronger bases than DBU were expected to be ineffective in increasing the molecular weight because they promoted to MCS by E1cB reaction. So in the place of such stronger bases, PBu3, a catalyst which promote the Michael addition, was used with DBU to enhance the propagation. However, the polymerization, initiated with DABCO and promoted by adding DBU and PBu3 simultaneously, resulted in a similar molecular weight (entry 7). In entry 8, the reaction of 1c and 2, initiated with DABCO, was monitored by SEC (Fig. S12†). After 1 h, DBU was added to promote conjugate addition. As a result, Mn increased from 510 to 7100 after a further 1 h. Then, PBu3 was added and the reaction was allowed to proceed for 22 h at which point the Mn had increased to 13000. Thus, the addition of PBu3 at the early stage of reaction was ineffective because of the side reaction of elimination from the phosphonium intermediate 3-III. However, the addition of PBu3 after the almost complete consumption of the acrylate chain end promoted further propagation. In this stage, the elimination from the chain end to prevent further propagation was not a fatal problem.
| Entrya | Catalyst | Solvent | Temp. [°C] | Yield [%] | Mnb | Đb |
|---|---|---|---|---|---|---|
| a 1c: 0.750 mmol, [1c]/[2]/[base]/[catalyst] = 1/1.0/1.2/0.2. Solvent: 0.75 mL.b Determined by SEC (THF, 40 °C, polystyrene standards).c 1c: 0.500 mmol, [1c]/[2]/[base]/[catalyst] = 1/1.0/1.2/0.2. Solvent: 0.50 mL. Catalysts were added after 1 h and the reaction was conducted for more than 22 h.d 1c: 0.500 mmol, [1c]/[2]/[base]/[catalyst] = 1/1.0/1.2/0.2. Solvent: 0.50 mL DBU and PBu3 were added after 1 h, at the same time, and the reaction was conducted for more than 22 h.e 1c: 0.500 mmol, [1c]/[2]/[base]/[catalyst] = 1/1.0/1.2/0.2. Solvent: 0.50 mL DBU and PBu3 were added after 1 h and 2 h, respectively, and the reaction was conducted for more than 22 h. | ||||||
| 1 | PBu3 | CHCl3 | 25 | 10 | 1000 | 2.25 |
| 2 | PBu3 | CH3CN | 25 | 27 | 1600 | 2.09 |
| 3 | PBu3 | CH3CN | 50 | 66 | 1100 | 2.02 |
| 4 | Et3Nc | CH3CN | 25 | 51 | 1900 | 1.78 |
| 5 | iPr2NEtc | CH3CN | 25 | 49 | 1800 | 2.31 |
| 6 | DBUc | CH3CN | 25 | 83 | 5400 | 2.16 |
| 7d | DBU + PBu3 | CH3CN | 25 | 29 | 5600 | 2.05 |
| 8e | DBU/PBu3 | CH3CN | 25 | 57 | 13000 |
1.63 |
 | ||
| Scheme 3 A proposed side reaction in the polycondensation of 3c catalyzed by PBu3. | ||
Main-chain scission
The polythioether 4c obtained in Table 3 (entry 6) was treated with DBU (1.2 equimolar to the repeating unit) in various solvents for 17 h (Fig. 1A). It was noted that the isolated 4c used in the MCS experiments were not completely soluble in these solvents even though the polymerization reactions in CH3CN reported above proceeded in a homogeneous system. As the polymer was not completely dissolved in CH3CN and DMSO, the MCS in these solvents resulted in incomplete degradation (Fig. 1B). On the other hand, the reaction in DMF proceeded in a homogeneous system, leading to efficient MCS to small molecules. Fig. 1C shows the 1H NMR spectra before and after the reaction. The signals k–m assigned to the main-chain structure were scarcely observed after the reaction, while the signals x–z specific to the chain-end structure were observed at a high intensity. These changes suggest the MCS by E1cB reaction. In this reaction, not only the change from exo-olefin to endo-olefin but also the extension of a conjugated system to cover the cinnamate-like moieties is the driving forces to shift the equilibrium from polymerization to MCS. Furthermore, the amount of effective DBU in the polymerization system should be lower than that of MCS experiment, even if equal amount of DBU was used; in polymerization system, the acetic acid was formed as a byproduct in the conjugate substitution reaction, which played to deactivate the DBU. In addition, PBu3 contributed equilibrium shift to the propagation side. In other words, the MCS reaction of the obtained polymers, employed using higher effective concentration of DBU and in the absence of PBu3, resulted in effective degradation.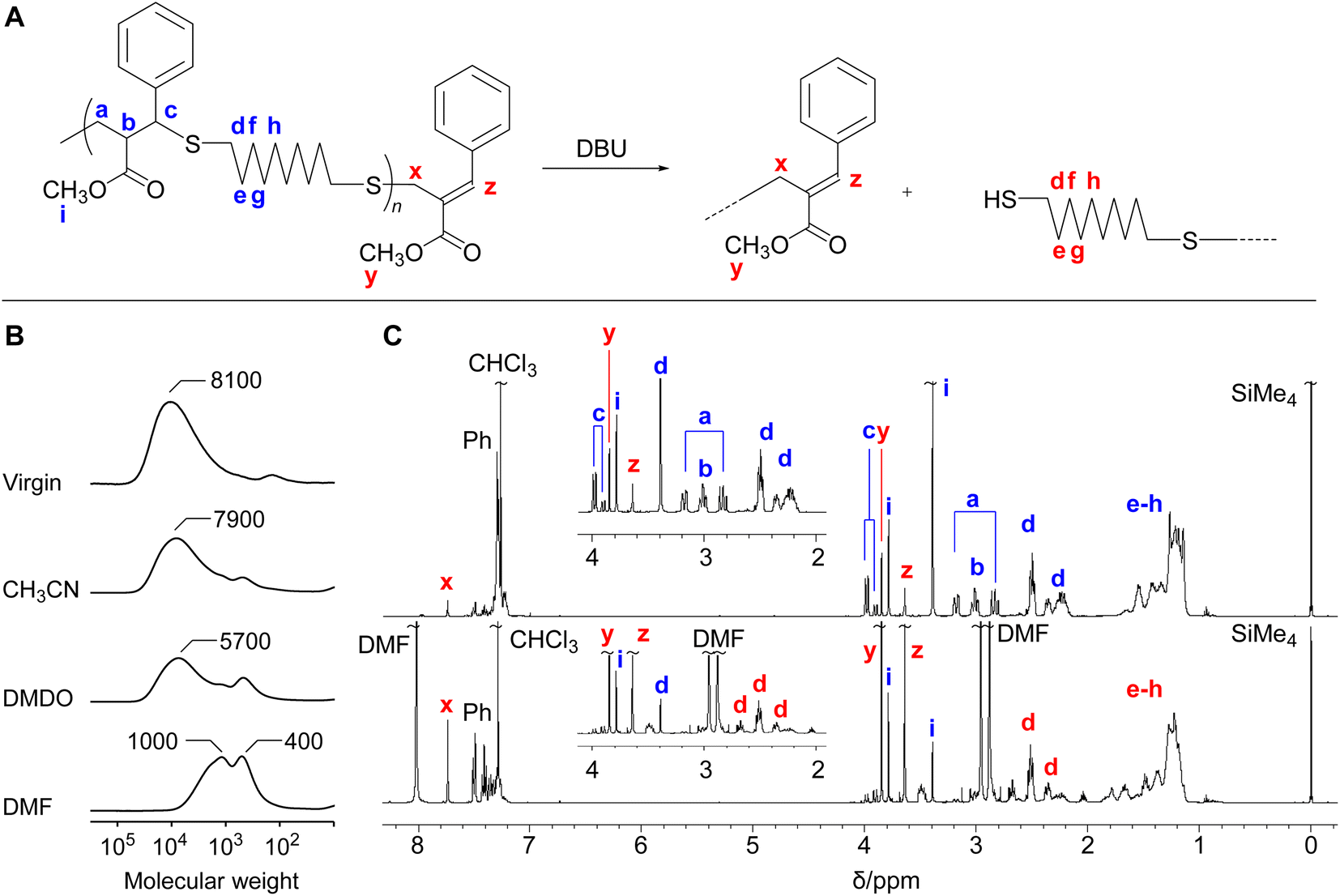 | ||
| Fig. 1 (A) MCS of 4c by E1cB reaction. (B) SECs before and after MCS in various solvents. The labels associated with peaks indicate the peak-top molecular weight (Mp). (C) 1H NMR spectra of 4c (entry 6) and after MCS in DMF. | ||
Conclusions
In short, the designed polythioether, 4c, underwent efficient MCS by E1cB reaction. The incorporation of a phenyl substituent was significant both in the polymerization and MCS: in polymerization, the phenyl substituent led to the formation of an inactive endo-olefin chain-end, while it was a key to achieving efficient MCS. In conventional polymer chemistry, the modification of the backbone structure has been the typical strategy to realize MCS, and the introduction of a ‘weak’ or ‘dynamic’ covalent bond is always in discussion.26,27 In contrast, the above results cast a spotlight on the design of the side group. In this study, polymerization (path A) and MCS were based on the same equilibrium system. However, the polymerization through a different route (path B) enabled a high molecular weight (Mn > 104). Therefore, the molecular weight increased compared to a complete equilibrium system. From the above points discussed, our conclusion is that ‘side-group design’ and ‘backbone design’ are important for both polymerization and MCS.Author contributions
K. H. proposed the plan and employed experiments except those for Fig. S10–S12,† which were conducted by A. T., R. K. repeated the model experiments and proposed the reaction mechanism by SN1′ reaction. The draft of this article was written by R. K. and Y. K. Y. K. supervised the research project.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was financially supported by MEXT KAKENHI No. 19H02763. Benzoyl chloride for the preparation of 1c was a kind gift from Iharanikkei Chemical Industry Co., Ltd.Notes and references
- K. J. Lawrie, I. Blakey, J. P. Blinco, H. H. Cheng, R. Gronheid, K. S. Jack, I. Pollentier, M. J. Leeson, T. R. Younkind and A. K. Whittaker, J. Mater. Chem., 2011, 21, 5629 RSC.
- E. Sato, C. Omori, M. Yuri, Y. Koda and H. Horibe, ACS Appl. Polym. Mater., 2019, 1, 2140 CrossRef CAS.
- N. Hakuto, K. Saito, M. Kirihara and Y. Kotsuchibashi, Polym. Chem., 2020, 11, 2469 RSC.
- Y. Tachibana, T. Baba and K. Kasuya, Polym. Degrad. Stab., 2017, 137, 67 CrossRef CAS.
- S. Pal, A. Das, S. Maitiand and P. De, J. Biomater. Sci., Polym. Ed., 2012, 23, 2105 CrossRef CAS PubMed.
- Y. Kohsaka, M. Yamashita, Y. Matsuhashi and S. Yamashita, Eur. Polym. J., 2019, 120, 109185 CrossRef CAS.
- D. Messmer, O. Bertran, R. Kissner, C. Alemán and A. D. Schlüter, Chem. Sci., 2019, 10, 6125 RSC.
- J. Steinkoenig, M. M. Zieger, H. Mutlu and C. Baner-Kowollik, Macromolecules, 2017, 50, 5385 CrossRef CAS.
- A. Kazama and Y. Kohsaka, Polym. Chem., 2019, 10, 2764 RSC.
- A. Kazama and Y. Kohsaka, Polym. Chem., 2022, 13, 6484 RSC.
- X. Y. Oh, Y. Ge and A. Goto, Chem. Sci., 2021, 12, 13546–13556 RSC.
- S. Mete, P. Mukherjee, B. Maiti, S. Pal, K. P. Ghorai and P. De, Macromolecules, 2018, 51, 8912 CrossRef CAS.
- A. Kanazawa and S. Aoshima, ACS Macro Lett., 2015, 4, 783 CrossRef CAS PubMed.
- Y. Kohsaka and K. Nagai, Eur. Polym. J., 2020, 141, 110049 CrossRef CAS.
- Y. Kohsaka and K. Nagai, Macromol. Rapid Commun., 2021, 42, 2000570 CrossRef CAS PubMed.
- Y. Kohsaka, T. Miyazaki and T. Hagiwara, Polym. Chem., 2018, 9, 1610 RSC.
- G. W. Fahnhorst and T. R. Hoye, ACS Macro Lett., 2018, 7, 143 CrossRef CAS PubMed.
- (a) Y. Song, J. He and Y. Zhang, Macromol. Rapid Commun., 2020, 41, 2000456 CrossRef CAS PubMed; (b) G. Herwig and A. P. Dove, ACS Macro Lett., 2019, 8, 1268 CrossRef CAS PubMed; (c) O. Daglar, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2019, 52, 2258 CrossRef; (d) O. Daglar, B. Alkan, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Eur. Polym. J, 2022, 162, 110931 CrossRef CAS; (e) B. Pektas, G. Sagdic, O. Daglar, S. Luleburgaz, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Polymer, 2022, 253, 124909 CrossRef.
- (a) Y. B. Cheng, S. J. Zhi, A. Qin and B. Z. Tang, Chin. Sci. Bull., 2013, 58, 2711 CrossRef; (b) B. Li, J. Wang, B. He, A. Qin and B. Z. Tang, Chin. J. Chem., 2022, 40, 2001 CrossRef CAS.
- (a) R. A. Evans, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1994, 27, 7935 CrossRef CAS; (b) J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805 CrossRef CAS PubMed.
- (a) A. Lowe, Polym. Chem., 2010, 1, 17 RSC; (b) A. Lowe, Polym. Chem., 2014, 5, 4820 RSC.
- Y. Kohsaka, K. Hagiwara and K. Ito, Polym. Chem., 2017, 8, 976 RSC.
- K. Hagiwara and Y. Kohsaka, Polym. Chem., 2020, 11, 5128 RSC.
- C. Yu, L. Xu, S. Tu, Z. Li and B. Li, J. Fluorine Chem., 2006, 127, 1540 CrossRef CAS.
- R. K. Sankar, R. S. Kumbhare, A. T. Dharmaraja and H. Chakrapani, Chem. Commun., 2014, 50, 15323 RSC.
- F. García and M. M. J. Smulders, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3551 CrossRef PubMed.
- H. Otsuka, Polym. J., 2013, 45, 879 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03751g |
| This journal is © The Royal Society of Chemistry 2023 |