DOI:
10.1039/D3RA03940D
(Paper)
RSC Adv., 2023,
13, 25002-25006
Preparation of spiroborate supramolecular and peapod polymers containing a photoluminescent ruthenium(II) complex†
Received
13th June 2023
, Accepted 16th August 2023
First published on 23rd August 2023
Abstract
The immobilization of functional metal complexes onto polymer supports remains one of the most important research areas. In this study, we prepared spiroborate supramolecular and peapod polymers containing a cationic photoluminescent ruthenium(II) complex. The supramolecular polymer was obtained by mixing spiroborate cyclic trimer bearing homoallyl group and a ruthenium(II) tris(bipyridyl) complex, and was further converted into the corresponding peapod polymer by olefin metathesis polymerization. The structure of these polymers was determined by 1H NMR, dynamic light scattering, inductively coupled plasma-atomic emission spectroscopy, energy dispersive X-ray analyses, and atomic force microscopy. The absorption and emission behaviors of the ruthenium(II) complex were almost the same for the free form and the supramolecular polymer in the mixed solvent of N,N-dimethylformamide and chloroform, although the emission intensity decreased when the chloroform portion was increased. On the other hand, the hypsochromism was observed upon the emission of the ruthenium(II) complex in the peapod polymer, probably due to the rigidochromic effect of the tight encapsulation by the peapod structure.
Introduction
The development of polypyridyl ruthenium(II) (Ru(II)) complexes bound to polymer supports is one of the most important research topics because of their wide applications in such areas as photocatalysts or photoelectrodes for solar cells.1 The preparation of these polymer materials typically involves embedding the complex into the polymer main chain or side chain via a covalent bond.2,3 In both cases, introducing a functionality to the target complex is inevitable to fix the complex onto the polymer chain via a covalent bond.
We have reported the preparation of a supramolecular polymer via the iterative molecular recognition of twin-bowl-shaped spiroborate cyclic trimer with a tricationic iridium(III) (Ir(III)) complex.4 We have also demonstrated that the covalent bond formation between the adjacent spiroborate twin bowls in the supramolecular polymer led to the formation of a peapod-type nanotube structure in which the Ir(III) complexes were encapsulated in the peapod cavity formed along with the polymer and topologically fixed onto the polymer main chain.5 In this system, no covalent bond formation is required for the fixation of the guest complex onto the polymer chain. The fact that the guest complex can be fixed without the need for any chemical transformation or introduction of functional groups used for bond formation would be quite advantageous for the development of functional organic materials. In this manuscript, we report the preparation of a spiroborate peapod polymer containing the tris(2,2′-bipyridine)ruthenium(II) complex [Ru(bpy)3]2+. The Ru(II) complex was topologically fixed onto the polymer chain in the peapod structure without any functionalization of the complex. The optical properties of the Ru(II) complex inside the peapod polymer were also elucidated.
Results and discussion
Preparation of spiroborate supramolecular polymer M-SP and peapod polymer M-PP
The preparation of the supramolecular polymer and the peapod polymer was carried out according to the previously reported procedures (Scheme 1).4b,5 The supramolecular polymer (Ru-SP) was obtained by mixing equimolar amounts of spiroborate twin bowl (TB·(Me2NH2)3), [Ru(bpy)3](PF6)2, and potassium trifluoromethanesulfonate (KOTf) in N,N-dimethylformamide (DMF), followed by precipitation with methanol. Potassium ion was employed to achieve a high degree of polymerization by adjusting the counter charges. Potassium ion was incorporated into the central cavity of TB3− to form the stable complex [TB·K]2−, that could iteratively associate with [Ru(bpy)3]2+ without accumulating negative charges on the polymer chain. Ru-SP was then subjected to olefin metathesis polymerization using Grubbs 2nd generation catalyst, followed by precipitation with methanol and washing with acetonitrile to obtain the peapod polymer (Ru-PP). Fe-SP and Fe-PP, containing the iron(II) (Fe(II)) complex [Fe(bpy)3]2+ instead of [Ru(bpy)3]2+, were also prepared by the same procedure.
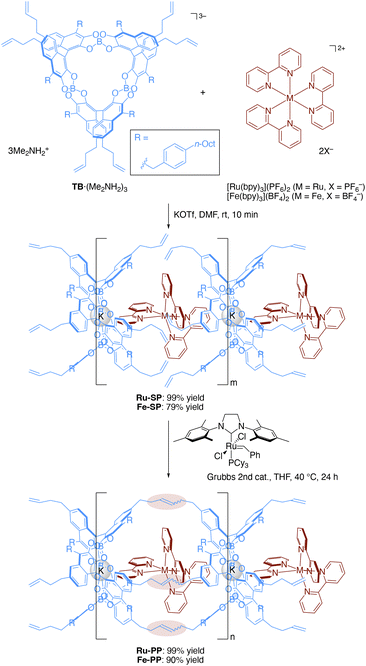 |
| | Scheme 1 Preparation of supramolecular polymer M-SP and peapod polymer M-PP (M = Ru or Fe). The chemical yield was determined on the basis of the theoretical molecular weight, that was calculated for [TB·K·M(bpy)3] (for M-SP) or [TB·K·M(bpy)3–3C2H4] (for M-PP). | |
Structural evaluation of Ru-SP and Ru-PP
Structural evaluation of Ru-SP and Ru-PP commenced with 1H NMR measurement. The proton signals of [Ru(bpy)3]2+ were shifted upfield and slightly broadened in the 1H NMR spectrum of Ru-SP, indicating the association of [Ru(bpy)3]2+ with [TB·K]2− (Fig. 1c). In the 1H NMR spectrum of Ru-PP, all signals were markedly broadened, making it difficult to assign [Ru(bpy)3]2+ proton signals (Fig. 1d).5 In the peapod structure, the motion of the components would be restricted, resulting in the marked broadening of the proton signals.
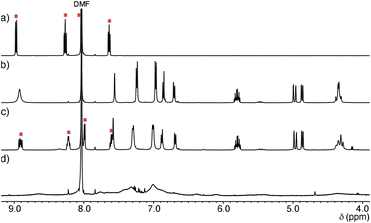 |
| | Fig. 1 Partial 1H NMR spectra (500 MHz, 25 °C in DMF-d7) of (a) [Ru(bpy)3](PF6)2 (2 mM), (b) TB·(Me2NH2)3 (2 mM), (c) Ru-SP (3 mg mL−1), and (d) Ru-PP (3 mg mL−1). Signals marked by red asterisks are assigned to [Ru(bpy)3]2+. | |
The formation of the peapod structure in Ru-PP was further confirmed by the dynamic light scattering (DLS) measurement. The average hydrodynamic diameter (Dh) of Ru-SP in DMF could only be detected at 2.2 nm owing to the dissociation of the polymer structure (Fig. 2). In the case of Ru-PP, the main peak was detected at Dh = 13.4 nm, together with a small amount of a larger component (Dh = 66 nm). This indicates that the covalent bond formation occurred between the adjacent TB3−s in the supramolecular polymer by the olefin metathesis polymerization.
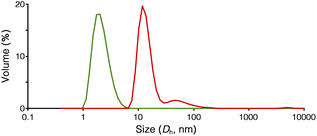 |
| | Fig. 2 Size characterization of Ru-SP (green) and Ru-PP (red) using DLS (0.5 g L−1 in DMF, 20 °C). | |
The molar ratio of TB3− to [Ru(bpy)3]2+ in the obtained supramolecular and peapod polymers was estimated by inductively coupled plasma-atomic emission spectroscopy (ICP-AES). The molar amounts were 0.97 and 0.24 mmol in 1 g of Ru-SP for boron (B) and Ru, respectively (Table 1, Fig. S1†), which meant that the molar ratio of TB3− to [Ru(bpy)3]2+ was approximately 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 in Ru-SP, and around 25% of [Ru(bpy)3]2+ was lost during the purification process. On the other hand, ICP-AES analysis of Ru-PP gave the molar amounts of 0.83 and 0.27 mmol g−1 for B and Ru, respectively. In this case, the ratio of TB3− to Ru was 1:1, which indicated an increase in the ratio of Ru compared with that in Ru-SP. To elucidate the cause of this increase in Ru, ICP-AES analysis was carried out for Fe-SP and Fe-PP. For Fe-SP, the molar amounts were 0.93 and 0.24 mmol g−1 for B and Fe, respectively, and the host/guest ratio was the same as that of Ru-SP. In the case of Fe-PP, the molar amounts were 0.82 and 0.21 mmol g−1 for B and Fe, respectively, and in addition, 0.077 mmol g−1 of Ru was detected. It was thought that Fe-PP contained a Ru center derived from the Grubbs 2nd generation catalyst used in the polymerization step. In this case, the atomic ratio of B, Fe, and Ru was estimated to be approximately 12:3:1, which meant that a single Ru center was bound to the peapod polymer per heptameric repeating unit, which was expected to contain four TB3− units and three [Ru(bpy)3]2+ units. From these results, it was concluded that Ru-PP would also contain the catalyst-derived Ru center in a similar ratio. Energy dispersive X-ray (EDX) analysis of Ru-PP revealed that the atomic ratio of Ru to potassium (K) was approximately 55:35 (Fig. S2†). This indicates that around 35% of potassium ion was lost during the preparation of Ru-PP. In addition, phosphorus (P), probably derived from the Grubbs 2nd generation catalyst, was detected, although the molar ratio (Ru:P = 10:55) was smaller than expected.
:3 in Ru-SP, and around 25% of [Ru(bpy)3]2+ was lost during the purification process. On the other hand, ICP-AES analysis of Ru-PP gave the molar amounts of 0.83 and 0.27 mmol g−1 for B and Ru, respectively. In this case, the ratio of TB3− to Ru was 1:1, which indicated an increase in the ratio of Ru compared with that in Ru-SP. To elucidate the cause of this increase in Ru, ICP-AES analysis was carried out for Fe-SP and Fe-PP. For Fe-SP, the molar amounts were 0.93 and 0.24 mmol g−1 for B and Fe, respectively, and the host/guest ratio was the same as that of Ru-SP. In the case of Fe-PP, the molar amounts were 0.82 and 0.21 mmol g−1 for B and Fe, respectively, and in addition, 0.077 mmol g−1 of Ru was detected. It was thought that Fe-PP contained a Ru center derived from the Grubbs 2nd generation catalyst used in the polymerization step. In this case, the atomic ratio of B, Fe, and Ru was estimated to be approximately 12:3:1, which meant that a single Ru center was bound to the peapod polymer per heptameric repeating unit, which was expected to contain four TB3− units and three [Ru(bpy)3]2+ units. From these results, it was concluded that Ru-PP would also contain the catalyst-derived Ru center in a similar ratio. Energy dispersive X-ray (EDX) analysis of Ru-PP revealed that the atomic ratio of Ru to potassium (K) was approximately 55:35 (Fig. S2†). This indicates that around 35% of potassium ion was lost during the preparation of Ru-PP. In addition, phosphorus (P), probably derived from the Grubbs 2nd generation catalyst, was detected, although the molar ratio (Ru:P = 10:55) was smaller than expected.
Table 1 ICP-AES analysis of M-SP and M-PP (M = Ru or Fe)a
| |
Ru-SP |
Ru-PP |
Fe-SP |
Fe-PP |
| Each sample was subjected to wet ashing using the mixed acid of HNO3 aq/HClO4 aq/H2SO4 and diluted with deionized water prior to analysis. Molar amount of atom per 1 g of polymer sample. Atomic ratio. |
| B (mmol g−1)b |
0.97 |
0.83 |
0.93 |
0.82 |
| Ru (mmol g−1) |
0.24 |
0.27 |
— |
0.077 |
| Fe (mmol g−1) |
— |
— |
0.24 |
0.21 |
| B/3:Mc |
4:3 |
1:1 |
4:3 |
1:1 |
Optical properties of Ru-SP and Ru-PP
The optical properties of [Ru(bpy)3]2+ in the forms of supramolecular polymer and peapod polymer were evaluated. From ICP-AES analysis, it was estimated that 40 mg of Ru-SP and Ru-PP incorporated approximately 9.6 and 8.4 μmol of [Ru(bpy)3]2+, respectively. In DMF solution, [Ru(bpy)3](PF6)2, Ru-SP, and Ru-PP showed similar absorption behavior (εmax = 453 nm), and almost the same emission peaks were given by [Ru(bpy)3](PF6)2 and Ru-SP, as the metal to ligand charge transfer (MLCT) band at λmax = 602 nm (Fig. 3a).6 As shown in the DLS analysis, Ru-SP was almost completely dissociated in DMF solution, and [Ru(bpy)3]2+ existed almost in the free form. Their emission maxima are the same even in DMF/chloroform mixed solvent (λmax = 596 and 593 nm in DMF/chloroform = 5:5 and 1:9 (v/v), respectively, Fig. 3b and c). It was noteworthy that the emission intensity of Ru-SP decreased when the ratio of chloroform was increased. In the less polar solvent, the electrostatic interaction of TB3− and [Ru(bpy)3]2+ became stronger, resulting in the quenching of the emission of [Ru(bpy)3]2+. On the other hand, a small blue shift was found for the emission spectrum of Ru-PP in comparison with that of [Ru(bpy)3](PF6)2. In DMF, the emission maximum of Ru-PP was found at λmax = 598 nm, and the width of the emission shift from [Ru(bpy)3](PF6)2 (Δλmax) was 4 nm. This hypsochromic effect became more pronounced when the ratio of chloroform in the solvent was increased, and Δλmax finally reached up to 16 nm in DMF/chloroform = 1:9 (Fig. 3c). We anticipated that this blue shift was caused by the rigidochromic behavior of [Ru(bpy)3]2+ in Ru-PP.7–9 The guest complex in the peapod polymer was tightly encapsulated in the cavity formed by the two adjacent spiroborate twin bowls and lost its motional freedom. In this case, the excited state of [Ru(bpy)3]2+ would have an asymmetric charge distribution, and reorientation of the distribution took place very slowly inside the cavity. This led to the long lifetime of the Franck–Condon excited state, and the successive emission occurred directly from this unrelaxed state.
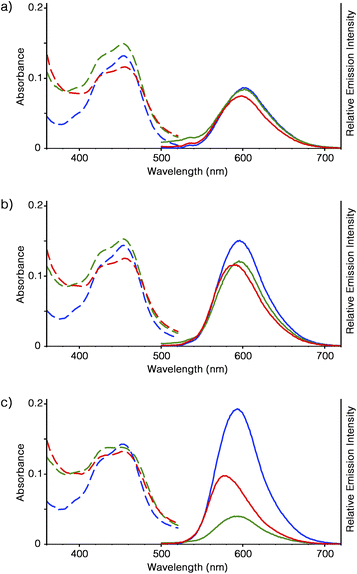 |
| | Fig. 3 Absorption (left, dashed lines) and emission (right, solid lines, λex = 460 nm) spectra of [Ru(bpy)3](PF6)2 (10 mM, blue), Ru-SP (40 mg L−1, green), and Ru-PP (40 mg L−1, red) in DMF/chloroform (v/v) = (a) 10:0, (b) 5:5, and (c) 1:9 at 25 °C. | |
AFM observation of Ru-SP and Ru-PP
The structure of Ru-SP and Ru-PP was evaluated by atomic force microscopy (AFM). On a highly oriented pyrolytic graphite (HOPG) substrate, no specific aggregation morphology could be found for Ru-SP (Fig. S3†). On the other hand, Ru-PP gave some island-shaped aggregates, and the thickness were estimated to be about 1.5 nm from the cross-sectional height profile (Fig. 4). Since this value shows good agreement with the thickness of the spiroborate peapod polymer reported previously, those aggregates could be regarded as the monolayers of the peapod polymer.5
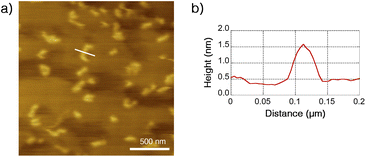 |
| | Fig. 4 (a) Dynamic-mode AFM image (2.0 × 2.0 μm2 on a HOPG substrate) of Ru-PP and (b) height profile on a cross-section of the white line. | |
Conclusions
Spiroborate supramolecular and peapod polymers were prepared by using photoluminescent Ru(II) complex [Ru(bpy)3]2+ as the cationic monomer component. The spiroborate supramolecular polymer was easily obtained by mixing homoallylated spiroborate twin bowl and [Ru(bpy)3](PF6)2 in DMF, followed by precipitation with methanol. This supramolecular polymer was then converted into the spiroborate peapod polymer via olefin metathesis polymerization by using Grubbs 2nd generation catalyst. The formation of these polymers was confirmed by 1H NMR, ICP-AES, and DLS analyses. ICP-AES analysis revealed that these polymers were composed of spiroborate twin bowl and [Ru(bpy)3]2+ in the ratio of approximately 4:3, and residual Grubbs catalyst-derived Ru component was also detected in the peapod polymer. The optical properties of [Ru(bpy)3]2+ in the forms of supramolecular and peapod polymers, and as free form [Ru(bpy)3](PF6)2 were evaluated. In the supramolecular polymer, [Ru(bpy)3]2+ showed almost the same absorption and emission behavior as the free form in DMF, although the emission intensity was decreased when the ratio of chloroform in the solvent was increased because the enhanced interaction between Ru complex and the twin bowl resulted in emission quenching. On the other hand, a significant blue shift of the emission of [Ru(bpy)3]2+ in the form of the peapod polymer was observed, probably because of the rigidochromic effect caused by the tight encapsulation inside the peapod cavity. In AFM observation, some island-shaped monolayer aggregates were observed for the peapod polymer.
Experimental section
General
1H NMR spectra were recorded on an Agilent Unity INOVA 500 at 25 °C. Chemical shifts were reported in δ ppm referenced to an internal tetramethylsilane standard.
Materials
Spiroborate twin bowl (TB·(Me2NH2)3) was prepared according to the reported procedure.5 All commercially available reagents were used without further purification. The chemical yield was determined based on the theoretical molecular weight, that was calculated for [TB·K·M(bpy)3] for M-SP or [TB·K·M(bpy)3–3C2H4] for M-PP.
Synthetic procedures and characterization
Preparation of Ru-SP: a typical procedure. In a 20 mL flask, TB·(Me2NH2)3 (424.4 mg, 160.0 μmol), [Ru(bpy)3](PF6)2 (137.7 mg, 160.0 μmol), and potassium trifluoromethanesulfonate (36.2 mg, 192.0 μmol) were dissolved in 4.0 mL of DMF at room temperature, and the mixture was stirred at intact temperature for 10 min. Methanol (50 mL) was then added to the solution, and the resulting brown precipitate was collected by centrifugation. The precipitate was washed with methanol and dried under reduced pressure to give Ru-SP as a brown solid (480.6 mg, 99% yield); 1H NMR (500 MHz, DMF-d7) δ (ppm) 8.92 (d, J = 8 Hz, 3H), 8.90 (d, J = 8 Hz, 3H), 8.23 (d, J = 8 Hz, 3H), 8.21 (d, J = 8 Hz, 3H), 7.99 (d, J = 6 Hz, 6H), 7.61 (t, J = 8 Hz, 6H), 7.58 (s, 6H), 7.29 (d, J = 6 Hz, 12H), 7.00 (d, J = 6 Hz, 12H), 6.88 (d, J = 9 Hz, 6H), 6.69 (d, J = 9 Hz, 6H), 5.79 (ddt, J = 17, 11, 7 Hz, 6H), 4.97 (dd, J = 17, 2 Hz, 6H), 4.86 (dt, J = 11, 1 Hz, 6H), 4.40–4.25 (m, 12H), 2.62 (d, J = 8 Hz, 12H), 2.49 (d, J = 8 Hz, 12H), 2.26 (q, J = 8 Hz, 12H), 1.52 (br s, 12H), 1.32–1.18 (m, 60H), 0.86 (t, J = 7 Hz, 18H).
Fe-SP. 79% yield as a purple solid; 1H NMR (500 MHz, DMF-d7) δ (ppm) 8.92 (d, J = 6 Hz, 3H), 8.88 (d, J = 6 Hz, 3H), 8.30–8.22 (m, 6H), 7.68–7.52 (m, 18H), 7.34–7.22 (m, 12H), 7.01 (d, J = 7 Hz, 12H), 6.88 (d, J = 9 Hz, 6H), 6.69 (d, J = 9 Hz, 6H), 5.88–5.72 (m, 6H), 4.96 (d, J = 17 Hz, 6H), 4.86 (d, J = 10 Hz, 6H), 4.50–4.10 (m, 12H), 2.65–2.55 (m, 12H), 2.54–2.42 (m, 12H), 2.30–2.20 (m, 12H), 1.57–1.46 (m, 12H), 1.35–1.15 (m, 60H), 0.86 (t, J = 7 Hz, 18H).
Preparation of Ru-PP: a typical procedure. In a 20 mL 2-necked round-bottomed flask, Ru-SP (237.4 mg, 76.0 μmol) and Grubbs 2nd catalyst (32.2 mg, 38.0 μmol) was dissolved in THF, and stirred at 40 °C for 24 h. After allowed to room temperature, methanol (50 mL) was added to the solution, and the brown precipitate was collected by centrifugation. The precipitate was washed with acetonitrile and dried under reduced pressure to give Ru-PP as a dark brown solid (231.7 mg, 99% yield); 1H NMR (500 MHz, DMF-d7) δ (ppm) 8.64 (br s), 7.77 (br s), 7.32 (br s), 7.01 (br s), 6.38 (br s), 6.10 (br s), 5.48 (br s), 4.36 (br s), 2.49 (br s), 1.51 (br s), 1.25 (br s), 0.86 (br s).
Fe-PP. 90% yield as a dark purple solid; 1H NMR (500 MHz, DMF-d7) δ (ppm) 7.59 (br s), 7.32 (br s), 6.99 (br s), 6.36 (br s), 5.78 (br s), 5.38 (br s), 4.95 (br s), 2.50 (br s), 1.52 (br s), 1.25 (br s), 0.86 (br s).
Author contributions
WM performed syntheses. WM and HD performed solution studies. MN performed EDX and AFM studies. All authors contributed to experiment design and interpretation. HD and MN wrote the manuscript. All authors proofread and improved the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 26410103.
References
-
(a) B. Happ, A. Winter, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 2222–2255 RSC
 ;
(b) Z. A. Morseth, L. Wang, E. Puodziukynaite, G. Leem, A. T. Gilligan, T. J. Meyer, K. S. Schanze, J. R. Reynolds and J. M. Papanikolas, Acc. Chem. Res., 2015, 48, 818–827 CrossRef CAS PubMed ;
(c) X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 CrossRef CAS PubMed ;
(d) G. Leem, B. D. Sherman and K. S. Schanze, Nano Convergence, 2017, 4, 37 CrossRef PubMed .
;
(b) Z. A. Morseth, L. Wang, E. Puodziukynaite, G. Leem, A. T. Gilligan, T. J. Meyer, K. S. Schanze, J. R. Reynolds and J. M. Papanikolas, Acc. Chem. Res., 2015, 48, 818–827 CrossRef CAS PubMed ;
(c) X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 CrossRef CAS PubMed ;
(d) G. Leem, B. D. Sherman and K. S. Schanze, Nano Convergence, 2017, 4, 37 CrossRef PubMed . -
(a) U. S. Schubert and C. Eschbaumer, Angew. Chem., Int. Ed., 2002, 41, 2892–2926 CrossRef CAS ;
(b) R. Dobrawa and F. Würthner, J Polym Sci A Polym Chem, 2005, 43, 4981–4995 CrossRef CAS ;
(c) W. K. Chan, Coord. Chem. Rev., 2007, 251, 2104–2118 CrossRef CAS ;
(d) G. R. Whittell and I. Manners, Adv. Matter., 2007, 19, 3439–3468 CrossRef CAS ;
(e) A. Wild, A. Winter, F. Schlütter and U. S. Schubert, Chem. Soc. Rev., 2011, 40, 1459–1511 RSC ;
(f) A. Breivogel, C. Kreitner and K. Heinze, Eur. J. Inorg. Chem., 2014, 5468–5490 CrossRef CAS ;
(g) A. Winter and U. S. Schubert, Chem. Soc. Rev., 2016, 45, 5311–5357 RSC ;
(h) T. Mede, M. Jäger and U. S. Schubert, Chem. Soc. Rev., 2018, 47, 7577–7627 RSC .
-
(a) J. N. Younathan, S. F. McClanahan and T. J. Meyer, Macromolecules, 1989, 22, 1048–1054 CrossRef CAS ;
(b) G. F. Strouse, L. A. Worl, J. N. Younathan and T. J. Meyer, J. Am. Chem. Soc., 1989, 111, 9101–9102 CrossRef CAS ;
(c) L. M. Dupray and T. J. Meyer, Inorg. Chem., 1996, 35, 6299–6307 CrossRef CAS ;
(d) C. N. Fleming, K. A. Maxwell, J. M. DeSimone, T. J. Meyer and J. M. Papanikolas, J. Am. Chem. Soc., 2001, 123, 10336–10347 CrossRef CAS PubMed ;
(e) Y. Sun, Z. Chen, E. Puodziukynaite, D. M. Jenkins, J. R. Reynolds and K. S. Schanze, Macromolecules, 2012, 45, 2632–2642 CrossRef CAS ;
(f) Z. Fang, A. Ito, S. Keinan, Z. Chen, Z. Watson, J. Rochette, Y. Kanai, D. Taylor, K. S. Schanze and T. J. Meyer, Inorg. Chem., 2013, 52, 8511–8520 CrossRef CAS PubMed ;
(g) G. Leem, S. Keinan, J. Jiang, Z. Chen, T. Pho, Z. A. Morseth, Z. Hu, E. Puodziukynaite, Z. Fang, J. M. Papanikolas, J. R. Reynolds and K. S. Schanze, Polym. Chem., 2015, 6, 8184–9193 RSC ;
(h) G. Leem, B. D. Sherman, A. J. Burnett, Z. A. Morseth, K.-R. Wee, J. M. Papanikolas, T. J. Meyer and K. S. Schanze, ACS Energy Lett., 2016, 1, 339–343 CrossRef CAS ;
(i) Y. Zhao, S. Kim, Y. K. Eom, S. R. Valandro and K. S. Schanze, ACS Appl. Energy Mater., 2021, 4, 7030–7039 CrossRef CAS .
-
(a) H. Danjo, K. Hirata, S. Yoshigai, I. Azumaya and K. Yamaguchi, J. Am. Chem. Soc., 2009, 131, 1638–1639 CrossRef CAS PubMed ;
(b) H. Danjo, K. Hirata, M. Noda, S. Uchiyama, K. Fukui, M. Kawahata, I. Azumaya, K. Yamaguchi and T. Miyazawa, J. Am. Chem. Soc., 2010, 132, 15556–15558 CrossRef CAS PubMed .
- H. Danjo, T. Nakagawa, A. Morii, Y. Muraki and K. Sudoh, ACS Macro Lett., 2017, 6, 62–65 CrossRef CAS PubMed .
- D. W. Thompson, A. Ito and T. J. Meyer, Pure Appl. Chem., 2013, 85, 1257–1513 CrossRef CAS .
- A. J. Lee, Comments Inorg. Chem., 1995, 16, 319–346 Search PubMed .
-
(a) T. G. Kotch and A. J. Lee, Inorg. Chem., 1993, 32, 2570–2575 CrossRef CAS ;
(b) J. Wu, M. M. Abu-Omar and S. H. Tolbert, Nano Lett., 2001, 1, 27–31 CrossRef CAS ;
(c) Y.-J. Pu, R. E. Harding, S. G. Stevenson, E. B. Namdas, C. Tedeschi, J. P. J. Markham, R. J. Rummings, P. L. Burn and I. D. W. Samuel, J. Mater. Chem., 2007, 17, 4255–4262 RSC ;
(d) I. Roppolo, E. Celasco, M. Sangermano, A. Garcia, T. Gacoin, J.-P. Boilot and S. Perruchas, J. Mater. Chem. C, 2013, 1, 5725–5732 RSC ;
(e) M. Xue and J. I. Zink, J. Phys. Chem. Lett., 2014, 5, 839–842 CrossRef CAS PubMed ;
(f) A. E. Norton, K. P. S. Zanoni, M.-A. Dourges, L. P. Ravaro, M. K. Abdolmakeki, A. S. S. de Camargo, T. Toupance, W. B. Connick and S. Chatterjee, J. Mater. Chem. C, 2021, 9, 6193–6207 RSC .
-
(a) K. Matsui, K. Sasaki and N. Takahashi, Langmuir, 1991, 7, 2866–2868 CrossRef CAS ;
(b) F. N. Castellano, T. A. Heimer, M. T. Tandhasetti and G. J. Meyer, Chem. Mater., 1994, 6, 1041–1048 CrossRef CAS ;
(c) P. Innocenzi, H. Kozuka and T. Yoko, J. Phys. Chem. B, 1997, 101, 2285–2291 CrossRef CAS ;
(d) W. R. Glomm, S. Volden, J. Sjöblom and M. Lindgren, Chem. Mater., 2005, 17, 5512–5520 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b and
Hiroshi Danjo
b and
Hiroshi Danjo



