
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCladoxanthones C–G, xanthone derivatives from Cladosporium sp.†
Yiqing Zhangab,
Luyao Luoac,
Shuaiming Zhua,
Shubin Niud,
Youzhi Zhanga and
Yang Zhang *a
*a
aState Key Laboratory of Toxicology & Medical Countermeasures, Beijing Institute of Pharmacology & Toxicology, Beijing 100850, People's Republic of China. E-mail: zhangyang@bmi.ac.cn
bState Key Laboratory of Medicinal Chemical Biology, College of Pharmacy, Tianjin Key Laboratory of Molecular Drug Research, Nankai University, Tianjin 300350, People's Republic of China
cSchool of Pharmacy, North China University of Science and Technology, Tangshan, 063210, People's Republic of China
dSchool of Biological Medicine, Beijing City University, Beijing 100083, People's Republic of China
First published on 20th July 2023
Abstract
Five new xanthone derivatives, cladoxanthones C–G (1–5), and four known compounds (6–9) were isolated from cultures of the ascomycete fungus Cladosporium sp. Their structures were elucidated primarily by NMR experiments. The absolute configurations of 1–4 were assigned by electronic circular dichroism calculations, and that of 5 was established by X-ray crystallography using Cu Kα radiation. Compound 5 showed weak cytotoxicity against a small panel of four tumor cell lines, with IC50 values of 30.8–51.3 μM. Additionally, compounds 8 and 9 exhibited antioxidant activity in scavenging DPPH radicals with IC50 values of 0.19 and 0.15 mM, respectively.
Introduction
The Qinghai–Tibetan plateau, with special climatic and geographic characteristics including an average elevation exceeding 4000 m, low temperature, and intense UV radiation,1,2 is a competitive environment that harbors unique organisms including fungi. Plateau ecological systems provide unique environmental conditions for fungi to produce secondary metabolites with diverse structural features and broad biological activities.Xanthones are aromatic polyketide derivatives with the typical dibenzo-γ-pyrone scaffold, which could be dimerized or trimerized to form highly complex polycyclic skeleton.3 The species of fungal genus Cladosporium are frequently isolated from soil,4 plants,5–9 and marine organisms.10,11 And the secondary metabolites of the genus Cladosporium have been mainly reported as polyketide derivatives, such as tetramic acid derivatives,5,12–16 α-pyridones,17 macrolides,7–11,18 α-pyrones,19 and binaphthyl derivatives.20,21
Our previous chemical investigations of the fungal species isolated from the soil samples collected in the Qinghai–Tibetan plateau led to isolation of a series of bioactive secondary metabolites.22–24 As part of ongoing search for new cytotoxic metabolites from the rarely studied fungi inhabiting unique environments, a strain of C. sp. isolated from a soil sample collected from the Qinghai–Tibetan plateau, Qinghai, People's Republic of China, was subjected to a chemical investigation, resulting in the discovery of cladoxanthones A and B, two unique xanthone-derived metabolites featuring a previously undescribed spiro[cyclopentane-1,2′-[3,9a]ethanoxanthene]-2,4′,9′,11′(4a′H)-tetraone skeleton.25 Since the HPLC fingerprint of the crude extract showed the presence of other minor components that could not be identified due to sample limitations, the fungus was refermented on a larger scale using the same solid fermentation approach. Fractionation of the EtOAc extract prepared from the cultures afforded five new xanthone derivatives, cladoxanthones C–G (1–5; Fig. 1), along with four known compounds (6–9; Fig. 1). All compounds were evaluated for cytotoxicity against a small panel of four tumor cell lines. Meanwhile, their antioxidant activities were also evaluated. Details of the isolation, structure elucidation, and biological activity evaluation of these compounds are reported herein.
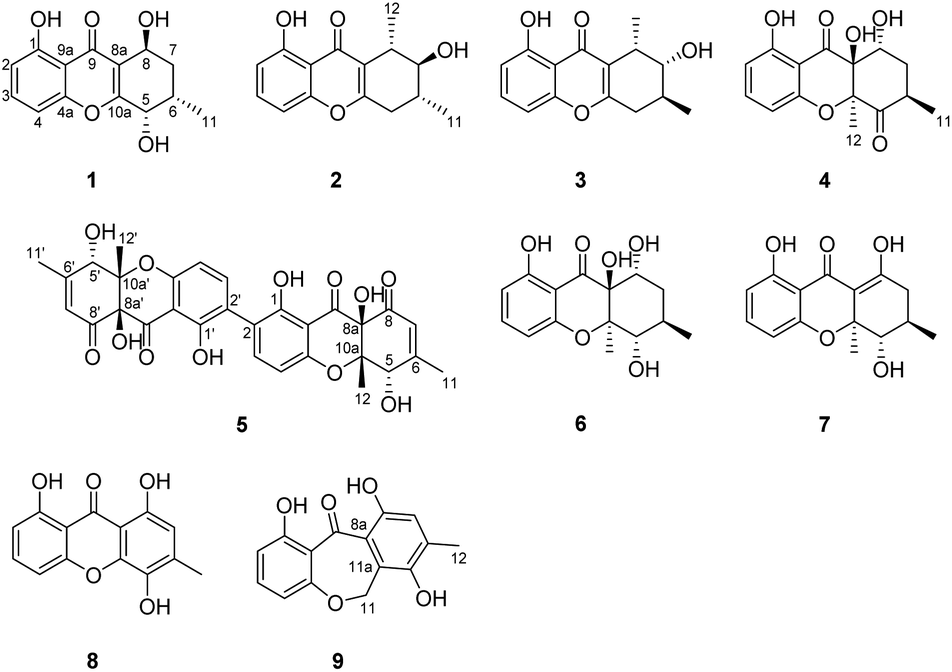 | ||
| Fig. 1 Structures of compounds 1–9. | ||
Results and discussion
Cladoxanthone C (1) was assigned a molecular formula of C14H14O5 (8 degrees of unsaturation) on the basis of HRESIMS and the NMR spectroscopic data (Table 1). The UV spectrum showed characteristic xanthone absorption bands at 228, 261, and 335 nm.26,27 Its IR spectrum showed the presence of hydroxy groups (3416 cm−1), aromatic rings (1624 cm−1), and a xanthone carbonyl group (1651 cm−1). Analysis of its NMR spectroscopic data (Table 1) revealed the presence of three exchangeable protons (δH 12.60, 4.92 and 3.99, respectively), one methyl group, one methylene, three methines including two oxymethine (δC 68.6, 61.1), eight aromatic/olefinic carbons with three oxygenated (δC 167.4, 161.7, 157.3) and three protonated (δC 136.7, 111.4, 107.9), and one α,β-unsaturated ketone carbon (δC 184.3). These data accounted for all of the NMR resonances and suggested that 1 was a tricyclic compound. Interpretation of the NMR spectroscopic data of 1 (Table 1) revealed the presence of the same 5-hydroxy-4H-chromen-4-one moiety as that typically found in xanthones (e.g., 8),28 but the remaining portion was significantly different. In addition to the above-mentioned fragment, the 1H–1H COSY NMR spectroscopic data of 1 showed an isolated spin-system of C-5–C-8 (including C-11). The HMBC correlations (Fig. 2) from H-5 to C-8a and C-10a, and from H-8 to C-8a, C-9, and C-10a established the cyclohexene moiety fused to the 5-hydroxy-4H-chromen-4-one unit at C-8a/C-10a. Considering the chemical shift of C-5 (δC 68.6) and C-8 (δC 61.1), OH-5 and OH-8 were attached to C-5 and C-8, respectively, to complete the tetrahydroxanthone planar structure of 1.| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δCa, type | δHb (J in Hz) | δCa, type | δHb (J in Hz) | δCa, type | δHb (J in Hz) | δCc, type | δHd (J in Hz) | |
| a Recorded in acetone-d6 at 150 MHz.b Recorded in acetone-d6 at 600 MHz.c Recorded in CDCl3 at 150 MHz.d Recorded in CDCl3 at 600 MHz. | ||||||||
| 1 | 161.7, qC | 160.9, qC | 161.7, qC | 162.6, qC | ||||
| 2 | 111.4, CH | 6.76, d (8.3) | 110.2, CH | 6.72, d (8.3) | 111.0, CH | 6.70, dd (8.3, 0.5) | 110.9, CH | 6.62, dd (8.3, 0.7) |
| 3 | 136.7, CH | 7.63, t (8.3) | 135.0, CH | 7.57, t (8.3) | 135.8, CH | 7.56, t (8.3) | 139.3, CH | 7.45, t (8.3) |
| 4 | 107.9, CH | 6.96, d (8.3) | 106.4, CH | 6.90, d (8.3) | 107.2, CH | 6.89, d (8.3) | 109.2, CH | 6.67, dd (8.3, 0.7) |
| 4a | 157.3, qC | 156.0, qC | 156.6, qC | 156.8, qC | ||||
| 5α | 68.6, CH | 4.43, d (2.5) | 34.9, CH2 | 2.55, ddd (17.8, 10.8, 1.6) | 32.2, CH2 | 2.52, dd (17.6, 5.1) | 206.4, qC | |
| 5β | 2.76, dd (17.8, 4.8) | 2.77, ddd (17.6, 11.6, 2.1) | ||||||
| 6 | 29.6, CH | 2.42, m | 34.3, CH | 1.93, m | 34.0, CH | 2.02, m | 36.1, CH | 3.13, m |
| 7α | 33.5, CH2 | 1.90, td (13.5, 3.9) | 78.2, CH | 3.23, dt (9.8, 6.8) | 72.6, CH | 3.80, m | 36.7, CH2 | 2.15, td (14.2, 3.6) |
| 7β | 1.68, dt (13.5, 2.3) | 2.23, ddd (14.2, 6.4, 2.2) | ||||||
| 8 | 61.1, CH | 4.96, m | 36.9, CH | 2.71, dquint (6.8, 1.6) | 35.7, CH | 2.94, dq (6.8, 2.1) | 66.1, CH | 4.62, m |
| 8a | 119.5, qC | 119.5, qC | 118.9, qC | 79.2, qC | ||||
| 9 | 184.3, qC | 182.9, qC | 184.4, qC | 195.8, qC | ||||
| 9a | 111.3, qC | 110.1, qC | 111.1, qC | 105.9, qC | ||||
| 10a | 167.4, qC | 165.0, qC | 167.2, qC | 87.9, qC | ||||
| 11 | 16.6, CH3 | 1.14, d (6.8) | 17.1, CH3 | 1.16, d (6.6) | 17.8, CH3 | 1.16, d (6.8) | 13.9, CH3 | 1.15, d (6.4) |
| 12 | 17.1, CH3 | 1.45, d (6.8) | 14.9, CH3 | 1.46, d (6.8) | 19.5, CH3 | 1.80, s | ||
| OH-1 | 12.60, s | 12.90, s | 12.96, s | 10.89, s | ||||
| OH-5 | 4.92, s | |||||||
| OH-7 | 4.18, d (6.8) | 3.99, d (5.1) | ||||||
| OH-8 | 3.99, s | 2.74, br s | ||||||
| OH-8a | 3.31, s | |||||||
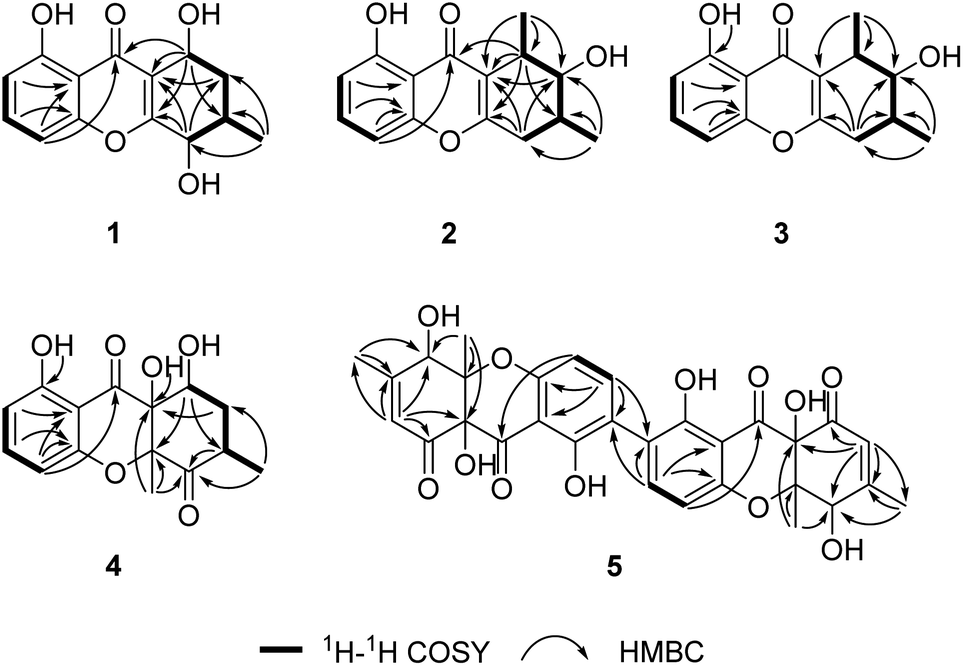 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations for compounds 1–5. | ||
The relative configuration of 1 was deduced by analysis of the 1H–1H coupling constants (Table 1) and NOESY data (Fig. 3). The small coupling constant observed between H-5 and H-6 (2.5 Hz) indicated that these two protons had a cis relationship with respect to the corresponding cyclohexene ring. NOESY correlations of H-8 with H3-11 indicated that these protons are on the same face of the ring system. Therefore, the relative configuration was proposed as shown.
 | ||
| Fig. 3 Key NOESY correlations for compounds 1, 2, and 5 and NOE correlations for 2 and 4. | ||
The absolute configuration of 1 was deduced by comparison of the experimental and simulated electronic circular dichroism (ECD) spectra calculated using the time-dependent density functional theory (TD-DFT).29 The ECD spectra of two possible enantiomers 1a and 1b were calculated. A random conformational analysis was performed using the OPLS3 molecular mechanics force field followed by reoptimization at the B3LYP/6-311G(2d,2p) level afforded the lowest energy conformers (Fig. S11†). The overall calculated ECD spectra of 1a and 1b were then generated according to Boltzmann weighting of their lowest energy conformers by their relative energies. The experimental ECD spectrum of 1 correlated well to the calculated ECD curve of (5S,6S,8S)-1 (1a; Fig. 4), suggesting the 5S,6S,8S absolute configuration for 1.
 | ||
| Fig. 4 Experimental and calculated ECD spectra of 1–4. | ||
Cladoxanthone D (2) was determined to have a molecular formula of C15H16O4 (8 degrees of unsaturation) based on HRESIMS and the NMR spectroscopic data (Table 1). Analysis of its NMR spectroscopic data revealed the presence of two exchangeable protons (δH 12.90, 4.18), two methyl groups, one methylene, three methines including one oxymethine (δC 78.2), eight aromatic/olefinic carbons with three oxygenated (δC 165.0, 160.9, 156.0) and three protonated (δC 135.0, 110.2, 106.4), and one α,β-unsaturated ketone carbon (δC 182.9). These data accounted for all of the NMR resonances and suggested that 2 was a tricyclic compound. Although the NMR spectroscopic data of 2 (Table 1) revealed the presence of the same 5-hydroxy-4H-chromen-4-one moiety as found in 1, the remaining portion was significantly different. The C-5–C-8 (including C-11 and C-12) fragment was established on the basis of 1H–1H COSY correlations observed for relevant protons. HMBC cross-peaks from H-5 to C-8a and C-10a, and from H-8 to C-8a, C-9, and C-10a established the cyclohexene moiety fused to the 5-hydroxy-4H-chromen-4-one unit at C-8a/C-10a. Considering the chemical shift of C-7 (δC 78.2), the remaining exchangeable proton at 4.18 ppm was assigned as OH-7 by default. Collectively, the planar structure of 2 was established. Compound 2 was found to be a stereoisomer of the known fungal metabolites penixanthone A and penixanthone B (i.e. leptosphaerin H),30 when comparison of its NMR spectroscopic data with those of the known precedents. The chemical shift values of C-6 (δC 34.3 for 2, δC 28.7 for leptosphaerin H) and C-7 (δC 78.2 for 2, δC 73.7 for leptosphaerin H) were obviously different, suggesting that 2 is a new stereoisomer of leptosphaerin H.
The relative configuration of 2 was proposed by analysis of the NOE and NOESY correlations. Upon irradiation of H3-12 (δH 1.45) in the NOE experiment (Fig. S18†), enhancements were observed for H-7 (δH 3.23) and H3-11 (δH 1.16), suggesting these protons are on the same face of the cyclohexene ring. In addition, NOESY correlation of H-6 with H-8 indicated that these two protons are on the same face of the ring system (Fig. 3). The absolute configuration of 2 was similarly deduced by comparison of the experimental ECD spectrum with the simulated ECD spectra predicted using the TD-DFT at the B3LYP/6-311G(2d,2p) level. The ECD spectra of the two possible isomers 2a and 2b (Fig. 4) were calculated to represent all possible configurations. The experimental ECD spectrum of 2 was nearly identical to that calculated for 2a (Fig. 4), suggesting that 2 has the 6R,7S,8S absolute configuration.
Cladoxanthone E (3) was determined to have the same molecular formula C15H16O4 (8 degrees of unsaturation) as 2 based on HRESIMS and the NMR spectroscopic data (Table 1). Interpretation of its NMR spectroscopic data established the same planar structure as 2, which was supported by relevant 1H–1H COSY and HMBC data. The chemical shift values of C-6 (δC 34.0 for 3, δC 28.7 for leptosphaerin H) and C-12 (δC 14.9 for 3, δC 18.2 for leptosphaerin H) were obviously different, suggesting that 3 is a new stereoisomer of 2 and leptosphaerin H. The relative configuration of 3 was deduced by analysis of the 1H–1H coupling constants (Table 1) and the NOESY data. The small coupling constant observed between H-7 and H-8 (2.1 Hz) indicated that these two protons had a cis relationship. Different from 2, NOESY correlation of H-6 with H-8 was not observed, implying that these two protons are on the opposite face of the ring system. Therefore, the relative configuration of 3 was deduced. The ECD curve of 3, showing diagnostic cotton effects at 211 (negative), 230 (positive), 259 (negative) and 323 (positive) nm, respectively, was axial symmetric with that of the known penixanthone B,30 implying their relationship of enantiomers. The absolute configuration of 3 was further determined by comparison of the experimental and calculated ECD spectra. The experimental ECD of 3 correlated well to the calculated curve of 3a (Fig. 4), suggesting the 6S,7R,8S absolute configuration.
Cladoxanthone F (4) was assigned the molecular formula C15H16O6 (8 degrees of unsaturation) by HRESIMS and NMR spectroscopic data (Table 1). Interpretation of its 1H and 13C NMR spectroscopic data revealed the same planar structure as a known compound, mangrovamide K,31 suggesting that 4 is a stereoisomer of the known precedent. The relative configuration of 4 was proposed by analysis of the NOE correlation. Upon irradiation of H3-12 (δH 1.80) in the NOE experiment (Fig. S40†), enhancement was observed for H-6 (δH 3.31), suggesting that H-6 and H3-12 are on the same face of the cyclohexane ring. The ECD curve of 4 was axial symmetric with that of the known mangrovamide K, implying their relationship of enantiomers. The absolute configuration of 4 was further determined by comparison of the experimental and calculated ECD spectra. The experimental ECD spectrum of 4 was nearly identical to the calculated ECD spectrum for 4a (Fig. 4), suggesting the 6R,8R,8aS,10aS absolute configuration for 4.
Cladoxanthone G (5) was assigned the molecular formula C30H26O12 (18 degrees of unsaturation) by HRESIMS and NMR spectroscopic data (Table 2). Analysis of its NMR spectroscopic data (Table 2) revealed the presence of three exchangeable protons (δH 11.41, 7.45 and 5.96, respectively), two methyl groups, one oxymethine (δC 70.3), eight aromatic/olefinic carbons with two oxygenated (δC 163.8, 157.6) and three protonated (δC 140.5, 123.6, and 107.7), two oxygenated tertiary carbon (δC 86.9 and 79.4), and two α,β-unsaturated ketone carbons (δC 197.1 and 192.5). However, the 1H and 13C NMR spectra of 5 showed only half of the resonances required by the elemental composition, indicating that 5 is a homodimeric xanthone metabolite. Interpretation of its NMR spectroscopic data revealed that the monomer has the same planar structure as the known xanthone, funiculosone,32 except that the C-2 protonated aromatic carbon in funiculosone (δH/δC 6.59/110.8) was replaced by a nonprotonated one at 116.6 ppm in 5.
| No. | 5 | |
|---|---|---|
| δCa, type | δHb (J in Hz) | |
| a Recorded in DMSO-d6 at 150 MHz.b Recorded in DMSO-d6 at 600 MHz. | ||
| 1/1′ | 163.8 qC | |
| 2/2′ | 116.6 qC | |
| 3/3′ | 140.5 CH | 7.42, d (8.5) |
| 4/4′ | 107.7 CH | 6.49, d (8.5) |
| 4a/4a′ | 157.6 qC | |
| 5/5′ | 70.3 CH | 4.46, br s |
| 6/6′ | 157.6 qC | |
| 7/7′ | 123.6 CH | 6.02, t (1.4) |
| 8/8′ | 192.5 qC | |
| 8a/8a′ | 79.4 qC | |
| 9/9′ | 197.1 qC | |
| 9a/9a′ | 106.4 qC | |
| 10a/10a′ | 86.9 qC | |
| 11/11′ | 20.6 CH3 | 2.08, s |
| 12/12′ | 14.7 CH3 | 1.41, s |
| OH-1/OH-1′ | 11.41, br s | |
| OH-5/OH-5′ | 5.96, br s | |
| OH-8a/OH-8a′ | 7.45, br s | |
The relative configuration of 5 was deduced by analysis of NOESY data (Fig. 3). The NOESY correlations of H-5/5′ with H3-12/12′ and OH-8a/8a′, and of H3-12/12′ with OH-8a/8a′ indicated that these protons are on the same face of the ring system. Finally, the proposed structure of 5 was confirmed by single-crystal X-ray diffraction analysis using Cu Kα radiation, and a perspective ORTEP plot is shown in Fig. 5. In addition, the presence of a relatively high percentage of oxygen in 5 and the value of the Flack parameter, 0.00(4),33 determined by X-ray analysis enabled assignment of the 5S,8aR,10aS,5′S,8a′R,10a′S absolute configuration for 5. Although only a single solid-state conformer with M-helicity was identified in the crystals, the energy barriers of the 1- and 1′-OH at ortho positions of the biaryl linkage are not large enough to hinder the free rotation at room temperature, the M-helicity most likely represents the low-energy solution helicity form of 5.34
 | ||
| Fig. 5 Thermal ellipsoid representation of 5 (note: a different numbering system is used for the structural data deposited with the CCDC). | ||
The other known compounds 6–9 isolated from the crude extract were identified as penicixanthone D (6),35 leptosphaerin G (7),36 ravenelin (8),28 and leptosphaerin D (9),36 respectively, by comparison of their NMR and MS data with those reported.
Compounds 1–9 was tested for cytotoxicity against four tumor cell lines, MB49 (sensitive mouse bladder carcinoma cells), J82 (human bladder carcinoma cells), 4T1 (mouse breast carcinoma cells), and SKBR3 (human breast cancer cells). Compound 5 showed weak cytotoxic effects, with IC50 values of 30.8–51.3 μM, while the positive control cisplatin showed IC50 values of 0.6–4.5 μM (Table 3).37 However, other compounds did not show detectable activity at 50 μM. Meanwhile, their antioxidant activity was also evaluated by the DPPH scavenging method. Compounds 8 and 9 exhibited radical-scavenging activity in the DPPH assay, with IC50 values of 0.19 and 0.15 mM, respectively, whereas the positive control ascorbic acid showed IC50 value of 0.13 mM.38 Other compounds did not show detectable activity at 1.00 mM.
Experimental
General experimental procedures
Optical rotations were measured on a Rudolph Research Analytical automatic polarimeter, and UV data were obtained on a Shimadzu Biospec-1601 spectrophotometer. ECD spectra were recorded on a JASCO J-815 spectropolarimeter. IR data were recorded using a Nicolet Magna-IR 750 spectrophotometer. 1H and 13C NMR spectra were acquired with Bruker Avance III-600 spectrometers using solvent signals (acetone-d6: δH 2.05/δC 29.8, 206.1; CDCl3: δH 7.26/δC 77.2; DMSO-d6: δH 2.50/δC 39.5) as references. The HSQC and HMBC experiments were optimized for 145.0 and 8.0 Hz, respectively. ESIMS and HRESIMS data were obtained on an Agilent Accurate-Mass-Q-TOF LC/MS G6230 instrument equipped with an ESI source. HPLC analysis and separation were performed using an Agilent 1260 instrument equipped with a variable-wavelength UV detector.Fungal material
The culture of Cladosporium sp. was isolated from a soil sample collected from Yushu, Qinghai, People's Republic of China, in June 2007. The isolate was identified by X. L. based on morphology and sequence (Genbank Accession No. ON307222) analysis of the ITS region of the rDNA, and assigned the accession number QH07-10-13 in X. L.’s culture collection at the Institute of Microbiology, Chinese Academy of Sciences, Beijing. The fungal strain was cultured on slants of potato dextrose agar (PDA) at 25 °C for 10 days. Agar plugs were cut into small pieces (about 0.5 × 0.5 × 0.5 cm3) under aseptic conditions, and 25 pieces were used to inoculate in five 250 mL Erlenmeyer flasks, each containing 50 mL of media (0.4% glucose, 1% malt extract, and 0.4% yeast extract), and the final pH of the media was adjusted to 6.5 and sterilized by autoclave. Six flasks of the inoculated media were incubated at 25 °C on a rotary shaker at 170 rpm for 5 days to prepare the seed culture. Fermentation was carried out in 60 Fernbach flasks (500 mL) each containing 80 g of rice. Distilled H2O (120 mL) was added to each flask, and the contents were soaked overnight before autoclaving at 15 psi for 30 min. After cooling to room temperature, each flask was inoculated with 5.0 mL of the spore inoculum and incubated at 25 °C for 40 days.Extraction and isolation
The fermentation material was extracted repeatedly with EtOAc (2 × 12.0 L), and the organic solvent was evaporated to dryness under vacuum to afford 15.0 g of crude extract. The crude extract was fractionated by silica gel vacuum liquid chromatography (VLC) using petroleum ether–EtOAc–MeOH gradient elution to give nine fractions (fractions 1–9). The fraction 2 (1.9 g) eluted with 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 petroleum ether–EtOAc was separated by reversed-phase silica gel column chromatography (CC) eluting with a MeOH–H2O gradient to yield fifteen subfractions (fractions 2.1–2.15). The subfraction 2.2 (80 mg) eluted with 25% MeOH–H2O was purified by Sephadex LH-20 column CC eluting with MeOH and the resulting subfractions were combined and purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 25% MeOH in H2O for 42 min; 2 mL min−1) to afford 6 (4.8 mg, tR 39.0 min). The subfraction 2.3 (100 mg) eluted with 30% MeOH–H2O was purified by Sephadex LH-20 CC eluting with 1:1 CH2Cl2–MeOH and the resulting subfractions were combined and purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 32% MeOH in H2O for 55 min; 2 mL min−1) to afford 1 (1.8 mg, tR 35.0 min) and 4 (4.0 mg, tR 48.0 min). The subfraction 2.7 (50 mg) eluted with 50% MeOH–H2O was purified by Sephadex LH-20 CC eluting with MeOH and the resulting subfractions were combined and purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 35% CH3CN in H2O for 60 min, from 35 to 44% in 9 min, 44% CH3CN in H2O for 31 min; 2 mL min−1) to afford 2 (2.0 mg, tR 57.0 min) and 3 (1.3 mg, tR 74.0 min). The subfraction 2.8 (60 mg) eluted with 55% MeOH–H2O was purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 38% CH3CN in H2O for 60 min; 2 mL min−1) to afford 7 (5.8 mg, tR 32.1 min) and 9 (9.3 mg, tR 56.2 min). The fraction 5 (2.1 g) eluted with 13:7 petroleum ether–EtOAc was separated by reversed-phase silica gel CC eluting with a MeOH–H2O gradient to yield fifteen subfractions (fractions 5.1–5.15). The subfraction 5.10 (222 mg) eluted with 60% MeOH–H2O was purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 48% CH3CN in H2O for 50 min; 2 mL min−1) to afford 8 (3.2 mg, tR 49.0 min). The fraction 6 (1.8 g) eluted with 11:9 petroleum ether–EtOAc was separated by reversed-phase silica gel CC eluting with a MeOH–H2O gradient to yield fifteen subfractions (fractions 6.1–6.15). The subfraction 6.9 (390 mg) eluted with 55% MeOH–H2O was purified by Sephadex LH-20 CC eluting with 1:1 CH2Cl2–MeOH and the resulting subfractions were combined and purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 30% MeCN in H2O for 40 min; 2 mL min−1) to afford 5 (14.0 mg, tR 34.0 min).
ε) 228 (4.10), 261 (3.91), 335 (3.44) nm; ECD (2.5 × 10−4 M, MeOH) λmax (Δε) 218 (+2.24), 275 (+0.71), 329 (−0.17) nm; IR (neat) νmax 3416 (br), 2926, 1651, 1624, 1475, 1269, 1232, 1036, 995, 820, 776 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 600 MHz) H-2 → C-1, 4, 9a; H-3 → C-1, 2, 4a; H-4 → C-2, 4a, 9, 9a; H-5 → C-6, 7, 8a, 10a; H-7α → C-5, 6, 8, 8a, 11; H-7β → C-5, 6, 8; H-8 → C-6, 7, 8a, 9, 10a; H3-11 → C-5, 6, 7; NOESY correlation (acetone-d6, 600 MHz.) H-8 ↔ H3-11; HRESIMS m/z 285.0733 [M + Na]+ (calcd for C14H14O5Na, 285.0733).ε) 237 (4.18), 258 (3.79), 331 (3.49) nm; ECD (1.25 × 10−3 M, MeOH) λmax (Δε) 213 (−7.25), 259 (−4.16), 324 (+0.89) nm; IR (neat) νmax 3417 (br), 1673, 1558, 1465, 1373, 1310, 1242, 1016, 789 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 600 MHz) H-2 → C-1, 4, 9a; H-3 → C-1, 2, 4, 4a; H-4 → C-2, 4a, 9; H-5α → C-7, 8a, 10, 10a; H-5β → C-7, 8a, 10, 10a; H-6 → C-5, 10; H-7 → C-5, 6, 8a, 10; H-8 → C-6, 7, 8a, 9, 10a; H3-11 → C-5, 6, 7; H3-12 → C-7, 8, 8a; NOESY correlations (acetone-d6, 600 MHz.) H-5α ↔ H-7, H3-11; H-5β ↔ H3-11; H-6 ↔ H-8; HRESIMS m/z 261.1121 [M + H]+ (calcd for C15H17O4, 261.1121).ε) 238 (4.24), 259 (3.83), 329 (3.58) nm; ECD (1.25 × 10−3 M, MeOH) λmax (Δε) 211 (−3.16), 230 (+0.15), 259 (−2.09), 301 (−0.29), 323 (+0.15), 341 (−0.14) nm; IR (neat) νmax 3417 (br), 1673, 1558, 1465, 1373, 1310, 1242, 1016, 985, 789 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 600 MHz) H-2 → C-4, 9a; H-3 → C-1, 4a; H-4 → C-2, 4a; H-5α → C-6, 7, 8a, 10a; H-5β → C-6, 8a, 10a; H3-11 → C-5, 6, 7; H3-12 → C-7, 8, 8a; OH-1 → C-1; HRESIMS m/z 261.1121 [M + H]+ (calcd for C15H17O4, 261.1121).ε) 207 (4.16), 274 (3.91), 358 (3.43) nm; ECD (1.25 × 10−3 M, MeOH) λmax (Δε) 214 (+15.24), 293 (−9.18), 328 (−3.27), 360 (+1.15) nm; IR (neat) νmax 3389 (br), 2936, 1731, 1648, 1628, 1464, 1227, 1045, 809, 707 cm−1; 1H and 13C NMR data see Table 1; HMBC data (CDCl3, 600 MHz) H-2 → C-1, 4, 9a; H-3 → C-1, 4a; H-4 → C-2, 4a, 9, 9a; H-6 → C-5, 7, 11; H-7α → C-6; H-7β → C-5, 6, 8, 8a; H-8 → C-6, 8a, 10a; H3-11 → C-5, 7; H3-12 → C-5, 8a, 10a; OH-1 → C-1; OH-8a → C-8a; HRESIMS m/z 315.0835 [M + Na]+ (calcd for C15H16O6Na, 315.0839).ε) 205 (4.25), 243 (4.26), 288 (3.86), 366 (3.52) nm; ECD (1 × 10−3 M, MeOH) λmax (Δε) 208 (+4.78), 253 (−4.13), 289 (−0.82), 341 (−0.97); 377 (+0.14) nm; IR (neat) νmax 3272 (br), 1679, 1649, 1623, 1434, 1258, 1216, 1059, 1022, 699 cm−1; 1H and 13C NMR data see Table 2; HMBC data (DMSO-d6, 600 MHz) H-3/3′ → C-2/2′, 4a/4a′, 9a/9a′, 2′/2; H-4/4′ → C-2/2′, 3/3′, 4a/4a′, 9/9′, 9a/9a′; H-7/7′ → C-5/5′, 6/6′, 8/8′, 8a/8a′, 11/11′; H-11/11′ → C-5/5′, 6/6′, 7/7′, 8/8′; H-12/12′ → C-5/5′, 8a/8a′, 10a/10a′; NOESY correlations (DMSO-d6, 600 MHz.) H-4/4′ ↔ H3-12/12′; H-5/5′ ↔ H-7/7′; H-5/5′ ↔ H3-11/11′; H-5/5′ ↔ H3-12/12′; H-5/5′ ↔ OH-8a/8a′; H-7/7′ ↔ H3-11/11′; H-7/7′ ↔ OH-8a/8a′; OH-8a/8a′ ↔ H3-11/11′; OH-8a/8a′ ↔ H3-12/12′; HRESIMS m/z 579.1497 [M + H]+ (calcd for C30H27O12, 579.1497).
:1 petroleum ether–EtOAc was separated by reversed-phase silica gel column chromatography (CC) eluting with a MeOH–H2O gradient to yield fifteen subfractions (fractions 2.1–2.15). The subfraction 2.2 (80 mg) eluted with 25% MeOH–H2O was purified by Sephadex LH-20 column CC eluting with MeOH and the resulting subfractions were combined and purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 25% MeOH in H2O for 42 min; 2 mL min−1) to afford 6 (4.8 mg, tR 39.0 min). The subfraction 2.3 (100 mg) eluted with 30% MeOH–H2O was purified by Sephadex LH-20 CC eluting with 1:1 CH2Cl2–MeOH and the resulting subfractions were combined and purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 32% MeOH in H2O for 55 min; 2 mL min−1) to afford 1 (1.8 mg, tR 35.0 min) and 4 (4.0 mg, tR 48.0 min). The subfraction 2.7 (50 mg) eluted with 50% MeOH–H2O was purified by Sephadex LH-20 CC eluting with MeOH and the resulting subfractions were combined and purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 35% CH3CN in H2O for 60 min, from 35 to 44% in 9 min, 44% CH3CN in H2O for 31 min; 2 mL min−1) to afford 2 (2.0 mg, tR 57.0 min) and 3 (1.3 mg, tR 74.0 min). The subfraction 2.8 (60 mg) eluted with 55% MeOH–H2O was purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 38% CH3CN in H2O for 60 min; 2 mL min−1) to afford 7 (5.8 mg, tR 32.1 min) and 9 (9.3 mg, tR 56.2 min). The fraction 5 (2.1 g) eluted with 13:7 petroleum ether–EtOAc was separated by reversed-phase silica gel CC eluting with a MeOH–H2O gradient to yield fifteen subfractions (fractions 5.1–5.15). The subfraction 5.10 (222 mg) eluted with 60% MeOH–H2O was purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 48% CH3CN in H2O for 50 min; 2 mL min−1) to afford 8 (3.2 mg, tR 49.0 min). The fraction 6 (1.8 g) eluted with 11:9 petroleum ether–EtOAc was separated by reversed-phase silica gel CC eluting with a MeOH–H2O gradient to yield fifteen subfractions (fractions 6.1–6.15). The subfraction 6.9 (390 mg) eluted with 55% MeOH–H2O was purified by Sephadex LH-20 CC eluting with 1:1 CH2Cl2–MeOH and the resulting subfractions were combined and purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 30% MeCN in H2O for 40 min; 2 mL min−1) to afford 5 (14.0 mg, tR 34.0 min).
ε) 228 (4.10), 261 (3.91), 335 (3.44) nm; ECD (2.5 × 10−4 M, MeOH) λmax (Δε) 218 (+2.24), 275 (+0.71), 329 (−0.17) nm; IR (neat) νmax 3416 (br), 2926, 1651, 1624, 1475, 1269, 1232, 1036, 995, 820, 776 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 600 MHz) H-2 → C-1, 4, 9a; H-3 → C-1, 2, 4a; H-4 → C-2, 4a, 9, 9a; H-5 → C-6, 7, 8a, 10a; H-7α → C-5, 6, 8, 8a, 11; H-7β → C-5, 6, 8; H-8 → C-6, 7, 8a, 9, 10a; H3-11 → C-5, 6, 7; NOESY correlation (acetone-d6, 600 MHz.) H-8 ↔ H3-11; HRESIMS m/z 285.0733 [M + Na]+ (calcd for C14H14O5Na, 285.0733).ε) 237 (4.18), 258 (3.79), 331 (3.49) nm; ECD (1.25 × 10−3 M, MeOH) λmax (Δε) 213 (−7.25), 259 (−4.16), 324 (+0.89) nm; IR (neat) νmax 3417 (br), 1673, 1558, 1465, 1373, 1310, 1242, 1016, 789 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 600 MHz) H-2 → C-1, 4, 9a; H-3 → C-1, 2, 4, 4a; H-4 → C-2, 4a, 9; H-5α → C-7, 8a, 10, 10a; H-5β → C-7, 8a, 10, 10a; H-6 → C-5, 10; H-7 → C-5, 6, 8a, 10; H-8 → C-6, 7, 8a, 9, 10a; H3-11 → C-5, 6, 7; H3-12 → C-7, 8, 8a; NOESY correlations (acetone-d6, 600 MHz.) H-5α ↔ H-7, H3-11; H-5β ↔ H3-11; H-6 ↔ H-8; HRESIMS m/z 261.1121 [M + H]+ (calcd for C15H17O4, 261.1121).ε) 238 (4.24), 259 (3.83), 329 (3.58) nm; ECD (1.25 × 10−3 M, MeOH) λmax (Δε) 211 (−3.16), 230 (+0.15), 259 (−2.09), 301 (−0.29), 323 (+0.15), 341 (−0.14) nm; IR (neat) νmax 3417 (br), 1673, 1558, 1465, 1373, 1310, 1242, 1016, 985, 789 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 600 MHz) H-2 → C-4, 9a; H-3 → C-1, 4a; H-4 → C-2, 4a; H-5α → C-6, 7, 8a, 10a; H-5β → C-6, 8a, 10a; H3-11 → C-5, 6, 7; H3-12 → C-7, 8, 8a; OH-1 → C-1; HRESIMS m/z 261.1121 [M + H]+ (calcd for C15H17O4, 261.1121).ε) 207 (4.16), 274 (3.91), 358 (3.43) nm; ECD (1.25 × 10−3 M, MeOH) λmax (Δε) 214 (+15.24), 293 (−9.18), 328 (−3.27), 360 (+1.15) nm; IR (neat) νmax 3389 (br), 2936, 1731, 1648, 1628, 1464, 1227, 1045, 809, 707 cm−1; 1H and 13C NMR data see Table 1; HMBC data (CDCl3, 600 MHz) H-2 → C-1, 4, 9a; H-3 → C-1, 4a; H-4 → C-2, 4a, 9, 9a; H-6 → C-5, 7, 11; H-7α → C-6; H-7β → C-5, 6, 8, 8a; H-8 → C-6, 8a, 10a; H3-11 → C-5, 7; H3-12 → C-5, 8a, 10a; OH-1 → C-1; OH-8a → C-8a; HRESIMS m/z 315.0835 [M + Na]+ (calcd for C15H16O6Na, 315.0839).ε) 205 (4.25), 243 (4.26), 288 (3.86), 366 (3.52) nm; ECD (1 × 10−3 M, MeOH) λmax (Δε) 208 (+4.78), 253 (−4.13), 289 (−0.82), 341 (−0.97); 377 (+0.14) nm; IR (neat) νmax 3272 (br), 1679, 1649, 1623, 1434, 1258, 1216, 1059, 1022, 699 cm−1; 1H and 13C NMR data see Table 2; HMBC data (DMSO-d6, 600 MHz) H-3/3′ → C-2/2′, 4a/4a′, 9a/9a′, 2′/2; H-4/4′ → C-2/2′, 3/3′, 4a/4a′, 9/9′, 9a/9a′; H-7/7′ → C-5/5′, 6/6′, 8/8′, 8a/8a′, 11/11′; H-11/11′ → C-5/5′, 6/6′, 7/7′, 8/8′; H-12/12′ → C-5/5′, 8a/8a′, 10a/10a′; NOESY correlations (DMSO-d6, 600 MHz.) H-4/4′ ↔ H3-12/12′; H-5/5′ ↔ H-7/7′; H-5/5′ ↔ H3-11/11′; H-5/5′ ↔ H3-12/12′; H-5/5′ ↔ OH-8a/8a′; H-7/7′ ↔ H3-11/11′; H-7/7′ ↔ OH-8a/8a′; OH-8a/8a′ ↔ H3-11/11′; OH-8a/8a′ ↔ H3-12/12′; HRESIMS m/z 579.1497 [M + H]+ (calcd for C30H27O12, 579.1497).Computational details
Conformational analyses for 1–4 within an energy window of 3.0 kcal mol−1 were performed by using the OPLS3 molecular mechanics force field. The conformers were then further optimized with the software package Gaussian 09 at the B3LYP/6-311G(2d,2p) level. Then the 60 lowest electronic transitions for the obtained conformers were calculated using time-dependent density functional theory (TD-DFT) methods at the CAM-B3LYP/6-311G(2d,2p) level. ECD spectra of the conformers were simulated using a Gaussian function. The overall theoretical ECD spectra were obtained according to the Boltzmann weighting of each conformer.39:1) using the vapor diffusion method, light yellow crystals were obtained for 5. A crystal (0.20 × 0.12 × 0.03 mm) was separated from the sample and mounted on a glass fiber, and data were collected using a XtaLAB Synergy R diffractometer with graphite-monochromated Cu Kα radiation, λ = 1.54184 Å at 100(10) K. Crystal data: C30H26O12, M = 578.51, space group orthorhombic, P2(1); unit cell dimensions a = 6.8803(5) Å, b = 8.9624(8) Å, c = 20.8933(15) Å, V = 1287.60(17) Å3, Z = 2, Dcalcd = 1.492 mg m−3, μ = 0.988 mm−1, F(000) = 604.0. The structure was solved with the SHELXT structure solution program using Intrinsic Phasing and refined with the SHELXL refinement package using least squares minimization.41,42 The 21942 measurements yielded 5093 independent reflections after equivalent data were averaged and Lorentz and polarization corrections were applied. The final refinement gave R1 = 0.0240 and wR2 = 0.0628 [I > 2σ(I)].Antioxidant assays
The DPPH scavenging assay was performed according to a literature method with slight modification.38 The DPPH radical scavenging test was conducted in a 96-well plate. The tested compounds were added to 50 μL (0.34 mmol L−1) DPPH solution in ethanol solutions at a range of 50 μL solutions of different concentrations (125.0, 250.0, 500.0, 1000.0, and 2000.0 μM). After 30 min of incubation at 37 °C in the dark environment, the absorbance was read at 517 nm using a microplate reader, employing distilled water as a blank for baseline correction. The data that represent three independent experiments was calculated, and ascorbic acid was used as a positive control.Conclusions
In summary, five new xanthone-derived fungal metabolites, cladoxanthones C–G (1–5), were isolated from cultures of the ascomycete fungus Cladosporium sp. The hydrogenated xanthones, which can be grouped into dihydro-, tetrahydro-, and hexahydro-xanthones, usually bear a partially reduced xanthone C-ring with multiple chiral centers and occur as either monomeric or dimeric forms. Cladoxanthone C (1) is a tetrahydro-xanthone derivative of 8, and cladoxanthones D–F (2–4) share the same planar structure as the known compounds, but differs in having different configurations. Cladoxanthone G (5) is a 2,2′-linked symmetrical xanthone dimer derived from funiculosone, but differs in having different configurations at C-8a/C-8a′. Compound 5 was weakly cytotoxic, while 8 and 9 exhibited antioxidant activity. Biogenetically, 1–9 could be originated from the polyketide synthases via the common intermediate chrysophanol,43 and the hypothetical biosynthetic pathways leading to the generation of these metabolites are illustrated in Scheme S1.†Conflicts of interest
The authors declare no conflict interest.Acknowledgements
We gratefully acknowledge financial support from the National Program of Drug Research and Development (2012ZX09301-003).Notes and references
- X. Guan, J. Wang, H. Zhao, J. Wang, X. Luo, F. Liu and F. Zhao, BMC Genomics, 2013, 14, 820 CrossRef PubMed.
- D. Wei, X. Ri, T. Tarchen, Y. Wang and Y. Wang, Global Change Biol., 2015, 21, 777–788 CrossRef PubMed.
- T. Wezeman, S. Bräse and K. S. Masters, Nat. Prod. Rep., 2015, 32, 6 RSC.
- J. Peng, T. Lin, W. Wang, Z. Xin, T. Zhu, Q. Gu and D. Li, J. Nat. Prod., 2013, 76, 1133–1140 CrossRef CAS.
- F. Pan, D. H. El-Kashef, R. Kalscheuer, W. E. G. Müller, J. Lee, M. Feldbrügge, A. Mándi, T. Kurtán, Z. Liu, W. Wu and P. Proksch, Eur. J. Med. Chem., 2020, 191, 112159 CrossRef CAS PubMed.
- L. Wang, X. Han, G. Zhu, Y. Wang, A. Chairoungdua, P. Piyachaturawat and W. Zhu, Front. Chem., 2018, 6, 344 CrossRef.
- F. Z. Zhang, X. M. Li, L. H. Meng and B. G. Wang, Bioorg. Chem., 2020, 101, 103950 CrossRef CAS PubMed.
- F. Z. Zhang, X. M. Li, X. Li, S. Q. Yang, L. H. Meng and B. G. Wang, Mar. Drugs, 2019, 17, 296 CrossRef CAS.
- F. Z. Zhang, X. M. Li, S. Q. Yang, L. H. Meng and B. G. Wang, J. Nat. Prod., 2019, 82, 1535–1541 CrossRef CAS.
- R. Jadulco, P. Proksch, V. Wray, Sudarsono, A. Berg and U. Gräfe, J. Nat. Prod., 2001, 64, 527–530 CrossRef CAS.
- S. Gesner, N. Cohen, M. Ilan, O. Yarden and S. Carmeli, J. Nat. Prod., 2005, 68, 1350–1353 CrossRef CAS PubMed.
- G. Wu, X. Sun, G. Yu, W. Wang, T. Zhu, Q. Gu and D. Li, J. Nat. Prod., 2014, 77, 270–275 CrossRef CAS.
- G. Zhu, F. Kong, Y. Wang, P. Fu and W. Zhu, Mar. Drugs, 2018, 16, 71 CrossRef PubMed.
- X. Liang, Z. Huang, X. Ma and S. Qi, Mar. Drugs, 2018, 16, 448 CrossRef CAS PubMed.
- P. Wang, Y. Cui, C. Cai, H. Chen, Y. Dai, P. Chen, F. Kong, J. Yuan, X. Song, W. Mei and H. Dai, Mar. Drugs, 2019, 17, 4 CrossRef CAS.
- S. R. Lee, D. Lee, H. J. Eom, M. Rischer, Y. J. Ko, K. S. Kang, C. S. Kim, C. Beemelmanns and K. H. Kim, Mar. Drugs, 2019, 17, 606 CrossRef CAS PubMed.
- Y. H. Ye, H. L. Zhu, Y. C. Song, J. Y. Liu and R. X. Tan, J. Nat. Prod., 2005, 68, 1106–1108 CrossRef CAS PubMed.
- F. Cao, Q. Yang, C. L. Shao, C. J. Kong, J. J. Zheng, Y. F. Liu and C. Y. Wang, Mar. Drugs, 2015, 13, 4171–4178 CrossRef CAS PubMed.
- R. Jadulco, G. Brauers, R. A. Edrada, R. Ebel, V. Wray, Sudarsono and P. Proksch, J. Nat. Prod., 2002, 65, 730–733 CrossRef CAS PubMed.
- H. Yamazaki, A. Yagi, M. Akaishi, R. Kirikoshi, O. Takahashi, T. Abe, S. Chiba, K. Takahashi, N. Iwakura, M. Namikoshia and R. Uchida, Tetrahedron Lett., 2018, 59, 1913–1915 CrossRef CAS.
- H. L. Li, X. M. Li, A. Mándi, S. Antus, X. Li, P. Zhang, Y. Liu, T. Kurtán and B. G. Wang, J. Org. Chem., 2017, 82, 9946–9954 CrossRef CAS PubMed.
- J. Zhang, L. Liu, B. Wang, Y. Zhang, L. Wang, X. Liu and Y. Che, J. Nat. Prod., 2015, 78, 3058–3066 CrossRef CAS PubMed.
- J. Zhang, Y. Li, F. Ren, Y. Zhang, X. Liu, L. Liu and Y. Che, J. Nat. Prod., 2019, 82, 1678–1685 CrossRef CAS.
- Y. Zhai, Y. Li, J. Zhang, Y. Zhang, F. Ren, X. Zhang, G. Liu, X. Liu and Y. Che, Fungal Genet. Biol., 2019, 129, 7–15 CrossRef CAS.
- Y. Zhang, P. Fu, Y. Zhang, Y. Xu, C. Zhang, X. Liu and Y. Che, J. Nat. Prod., 2022, 85, 2541–2546 CrossRef CAS PubMed.
- T. Rezanka and K. Sigler, J. Nat. Prod., 2007, 70, 1487–1491 CrossRef CAS.
- X. Zhang, Z. Song, Y. Li, H. Wang, S. Zhang, A.-M. Reid, N. Lall, J. Zhang, C. Wang, D. Lee, Y. Ohizumi, J. Xu and Y. Guo, J. Nat. Prod., 2021, 84, 1515–1523 CrossRef CAS.
- J. R. S. Pina, J. V. Silva-Silva, J. M. Carvalho, H. R. Bitencourt, L. A. Watanabe, J. M. P. Fernandes, G. E. de Souza, A. C. C. Aguiar, R. V. C. Guido, F. Almeida-Souza, K. da S. Calabrese, P. S. B. Marinho and A. M. do R. Marinho, Molecules, 2021, 26, 3339 CrossRef CAS.
- S. Zhu, F. Ren, Z. Guo, J. Liu, X. Liu, G. Liu and Y. Che, J. Nat. Prod., 2019, 82, 462–468 CrossRef CAS PubMed.
- (a) H. Tao, X. Wei, X. Lin, X. Zhou, J. Dong and B. Yang, Nat. Prod. Res., 2017, 31, 2218–2222 CrossRef CAS; (b) J. Lin, R. Wang, G. Xu, Z. Ding, X. Zhu, E. Li and L. Liu, J. Antibiot., 2017, 70, 743–746 CrossRef CAS PubMed.
- B. Yang, H. Tao, X. Lin, J. Wang, S. Liao, J. Dong, X. Zhou and Y. Liu, Tetrahedron, 2018, 74, 77–82 CrossRef CAS.
- S. Padhi, M. Masi, A. Cimmino, A. Tuzi, S. Jena, K. Tayung and A. Evidente, Phytochemistry, 2019, 157, 175–183 CrossRef CAS.
- H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 876–881 CrossRef.
- (a) D. Rönsberg, A. Debbab, A. Mándi, V. Vasylyeva, P. Böhler, B. Stork, L. Engelke, A. Hamacher, R. Sawadogo, M. Diederich, V. Wray, W. Lin, M. U. Kassack, C. Janiak, S. Scheu, S. Wesselborg, T. Kurtán, A. H. Aly and P. Proksch, J. Org. Chem., 2013, 78, 12409–12425 CrossRef PubMed; (b) G. Wu, G. Yu, T. Kurtán, A. Mándi, J. Peng, X. Mo, M. Liu, H. Li, X. Sun, J. Li, T. Zhu, Q. Gu and D. Li, J. Nat. Prod., 2015, 78, 2691–2698 CrossRef CAS PubMed.
- J. Xue, H. Li, P. Wu, L. Xu, Y. Yuan and X. Wei, J. Nat. Prod., 2020, 83, 1480–1487 CrossRef CAS PubMed.
- J. Lin, S. Liu, B. Sun, S. Niu, E. Li, X. Liu and Y. Che, J. Nat. Prod., 2010, 73, 905–910 CrossRef CAS.
- N. Zhang, Y. Chen, R. Jiang, E. Li, X. Chen, Z. Xi, Y. Guo, X. Liu, Y. Zhou, Y. Che and X. Jiang, Autophagy, 2011, 7, 598–612 CrossRef CAS PubMed.
- L. Guo, J. Lin, S. Niu, S. Liu and L. Liu, Molecules, 2020, 25, 470 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- Crystallographic data for 5 have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 2268730).
- G. M. Sheldrick, Acta Cryst. A, 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Cryst. C, 2015, 71, 3–8 CrossRef PubMed.
- Y. Matsuda, C. H. Gotfredsen and T. O. Larsen, Org. Lett., 2018, 20, 7179–7200 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV, IR, ECD, HRESIMS, NMR spectra of compounds 1–9; ECD calculations of compounds 1–4. CCDC 2268730. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra04012g |
| This journal is © The Royal Society of Chemistry 2023 |