
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn insight into the hydrogen bonding, halogen bonding and chalcogen bonding interactions in manganese(III) complexes with N2O2donor salicylidine Schiff base ligands†
Mridul Karmakara,
Wahedur Ska,
Rosa M. Gomila b,
Michael. G. B. Drewc,
Antonio Frontera*b and
Shouvik Chattopadhyay*a
b,
Michael. G. B. Drewc,
Antonio Frontera*b and
Shouvik Chattopadhyay*a
aDepartment of Chemistry, Jadavpur University, Kolkata-700032, West Bengal, India. E-mail: shouvik.chattopadhyay@jadavpuruniversity.in
bDepartamento de Química, Universitat de les IllesBalears, Crta.deValldemossakm 7.5, 07122 Palma, Baleares, Spain. E-mail: toni.frontera@uib.es
cSchool of Chemistry, The University of Reading, P. O. Box 224, Whiteknights, Reading RG6 6AD, UK
First published on 13th July 2023
Abstract
Four manganese(III) complexes, [MnL1(H2O)2]ClO4·H2O (1), [MnL2(H2O)2]ClO4 (2), [MnL3(DMSO)(H2O)]ClO4 (3) and [MnL4(DMSO)(H2O)]ClO4 (4), where H2L1 = N,N′-bis(5-bromosalicylidene)-1,3-diaminopropane, H2L2 = 2,2-dimethyl-N,N-bis(3-methyloxysalicylidene)-1,3-diaminopropane, H2L3 = N,N′-bis(5-chlorosalicylidene)-2,2-dimethyl-1,3-diaminopropane and H2L4 = 2-hydroxy-N,N′-bis(3-ethyloxysalicylidene)-1,3-diaminopropane are tetradentate N2O2-donor ligands and DMSO = dimethyl sulfoxide, have been synthesized and characterised by elemental analysis, IR and UV-vis spectroscopy and single-crystal X-ray diffraction studies. All are monomeric complexes. Complex 1 crystallises in orthorhombic space group P212121, complex 3 crystallises in triclinic space group P-1, whereas complexes 2 and 4 crystallize in monoclinic space groups, C2/c and C2/m respectively. In all the complexes, manganese(III) has a six-coordinated pseudo-octahedral geometry in which imine nitrogen atoms and phenolate oxygen atoms of the deprotonated di-Schiff base constitute the equatorial plane. In complexes 1 and 2, water molecules are present in the fifth and sixth coordination sites in the axial positions while in complexes 3 and 4 they are occupied by one water and one DMSO. The coordinated water molecules initiate hydrogen-bonded networks in all complexes. DFT calculations have been carried out to analyze two aspects of these complexes viz. the formation of halogen (HaB) and chalcogen bonding (ChB) interactions in complexes 1 and 3 where the electron donor is the perchlorate anion and the acceptor either bromine or chlorine atoms for the HaBs and the sulfur atom of the coordinated DMSO for the ChB. In addition, other intermolecular effects are discussed in the solid state for complexes 1, 2 and 4, where the hydrogen atoms of the coordinated water molecules interact with the electron rich cavities formed by the phenolate and alkyloxy oxygen atoms of the Schiff-base ligand.
Introduction
Synthetic inorganic chemists have used several salen type N2O2-donor Schiff base ligands to form varieties of manganese(III) complexes.1–6 The complexes are usually octahedral, where the tetradentate H2 salen-type ligand occupies four coordination sites and other appropriate co-ligands occupy the axial sites. It is well established that the four donor atoms (two nitrogens and two oxygens) of the salen ligand around the metal centre can assume two different arrangements: (i) all imine and phenolate donor atoms occupy the equatorial positions (favoured by the presence of two monodentate ligands),7–17 and (ii) one oxygen atom is displaced from the equatorial plane, occupying an axial position of the coordination polyhedron (favoured by the presence of a bidentate chelating ligand).7,18–23 In this work, we shall concentrate on the first type arrangement, where [MnIII(Schiff base)]+ cations (with a high spin state) may have a strong uni-axial magnetic anisotropy created by the Jahn–Teller effect in an octahedral ligand field.24 In some complexes, the axial sites are filled by water molecules and these complexes may be utilized for the construction of several water bridged complexes. These complexes may also be important in understanding the mechanism of ligand replacement reactions. In these complexes, the hydrogen atoms of the coordinated water molecules will also be involved in hydrogen bonding.25Obviously, the most commonly used approach for engineering the supramolecular structure of these complexes is to utilize hydrogen bonds. However, many other non-covalent interactions, such as, π-stacking, cation–π, C–H⋯π, lone-pair⋯π, anion⋯π, hydrophobic interactions, etc., are also important for the synthesis and the stabilization of the different supramolecular architectures of the complexes. Recently, σ-hole interaction involving a positive electrostatic potential region (called an σ-hole, created by the anisotropic distribution of the electron density) and an electron rich center of the molecule is widely studied and has been found relevance in stabilizing the supramolecular assemblies. σ-Hole interactions involving group 6, 7 and 8 metals are called osme bonds, wolfium bonds and matere bonds respectively.26 Such interactions are called spodium bonds and tetrel bonds for group 12 and group 14 elements respectively.27 The non-covalent interactions involving the elements of halogen and chalcogen groups are called halogen bonding (HaB) and chalcogen bonding (ChB) interactions respectively.28–30
In the present work, we have used 1,3-diaminopropane moiety to prepare some salen-type Schiff base ligands, which have, in turn, been used to synthesize a series of mononuclear manganese(III) complexes. In each of the complexes, the imine nitrogen atoms of the Schiff bases form six-membered chelate rings with the manganese(III) centers, see Scheme 1.
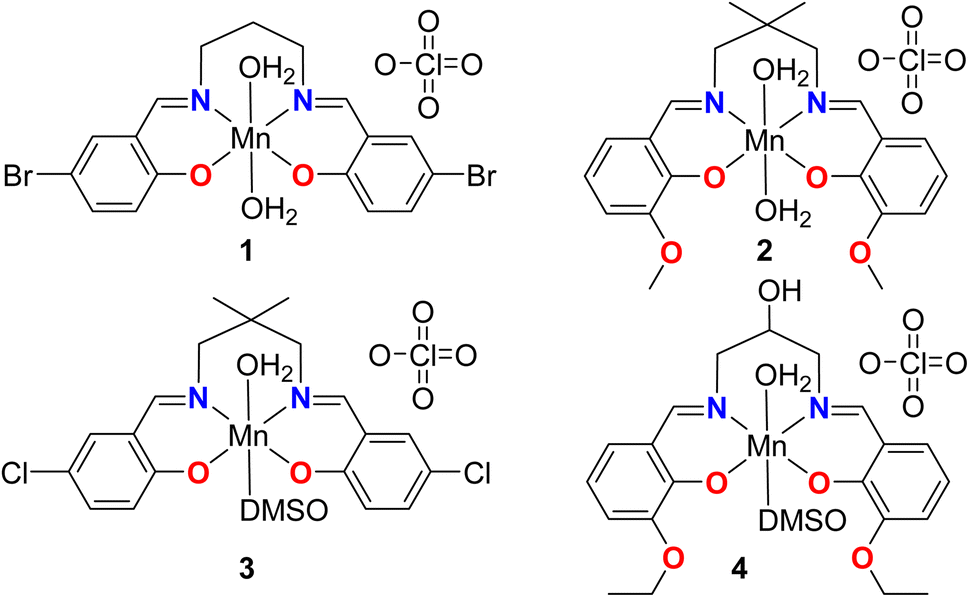 | ||
| Scheme 1 Manganese complexes 1–4 reported in this work. | ||
The DFT study is utilized to analyze the formation of recurrent motifs in the solid state of complexes consisting of supramolecular dimers where the hydrogen-atoms of the coordinated water molecules interact with the electron rich cavity formed by the phenolic and alkyloxy oxygen atoms of the Schiff-base ligand, as detailed in the following sections. We have also rationalized the formation of halogen bonding and chalcogen bonding interactions in complexes, where the electron donor is the perchlorate anion and the acceptor either bromine or chlorine atoms for the HaBs and the sulfur-atom of the coordinated DMSO for the ChB, see Scheme 2. In particular compound 1 forms infinite chains of perchlorate anions bridges by lattice water molecules with appended Mn(III) complexes where bifurcated halogen bonds are establishes (see Scheme 2A). Moreover, compound 3 forms infinite 1D chains where the cationic Mn(III) complexes are interconnected by the perchlorate anions that forms concurrent halogen and chalcogen bonds (see Scheme 2B). As all four complexes are cationic with perchlorate counter anions, the potential energy density at the bond CP has also been calculated to estimate the strength of the bifurcated HaB free from the pure coulombic attraction between the counter ions.
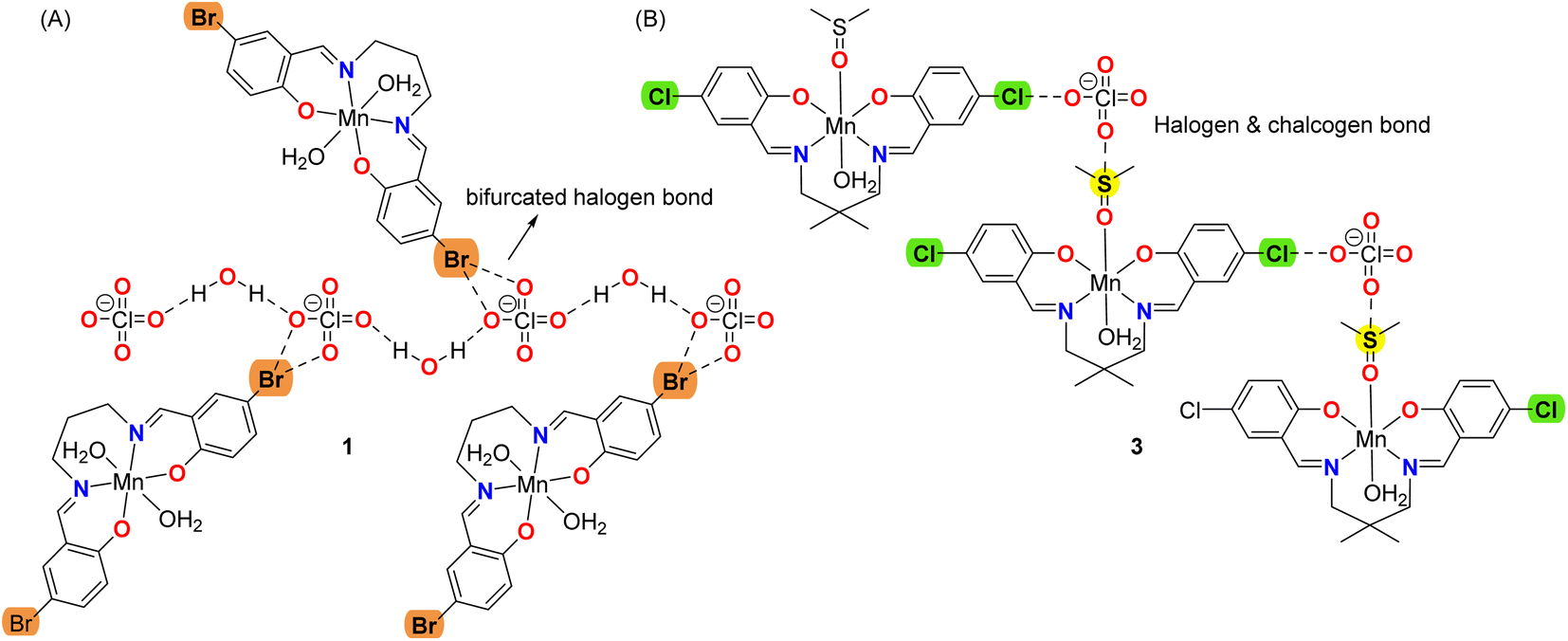 | ||
| Scheme 2 Representation of the halogen bonding interactions in 1 and concurrent halogen and chalcogen bonds in 3. | ||
Experimental
The starting ingredients and solvents utilized in the experiment were all of reagent-grade quality, readily available, and procured from Sigma-Aldrich. Without any further purification, all of them were used for experiments.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in 10 mL methanol for around 4 hours. Then the solution was cooled down to room temperature and a methanol/water (4:1) solution (5 mL) of manganese(II) perchlorate hexahydrate (0.362 g, ∼1 mmol) was added to it with constant stirring. The yellow colour of the solution turned into dark brown. After 1 h, the resulting solution was filtered and the solution was kept undisturbed. Deep brown, blocked-shaped, single crystals of 1, suitable for X-ray diffraction, were obtained after a few days by slow evaporation of the resulting solution in an open atmosphere. It was re-crystallized from methanol solution.
:1 molar ratio in 10 mL methanol for around 4 hours. Then the solution was cooled down to room temperature and a methanol/water (4:1) solution (5 mL) of manganese(II) perchlorate hexahydrate (0.362 g, ∼1 mmol) was added to it with constant stirring. The yellow colour of the solution turned into dark brown. After 1 h, the resulting solution was filtered and the solution was kept undisturbed. Deep brown, blocked-shaped, single crystals of 1, suitable for X-ray diffraction, were obtained after a few days by slow evaporation of the resulting solution in an open atmosphere. It was re-crystallized from methanol solution.Complex 1: yield: 401 mg (∼62%). Calc. for C17H20Br2ClMnN2O9 (FW 646.55): C, 31.58; H, 3.12; N, 4.33%. Found: C, 31.57; H, 3.14; N, 4.35%. FT-IR (KBr, cm−1): 1610 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N); 2948 (νC−H); 3364 (νO−H). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN), 221 (3.4 × 104), 275 (1.8 × 104), 377 (5.8 × 103), 579 (1.5 × 103). Magnetic moment = 5.02 B. M. HRMS (ESI, positive ion mode, acetonitrile) m/z: 492.7210 (cald. 493.06) for [Mn(L1)]+.
N); 2948 (νC−H); 3364 (νO−H). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN), 221 (3.4 × 104), 275 (1.8 × 104), 377 (5.8 × 103), 579 (1.5 × 103). Magnetic moment = 5.02 B. M. HRMS (ESI, positive ion mode, acetonitrile) m/z: 492.7210 (cald. 493.06) for [Mn(L1)]+.
Complex 2: yield: 364 mg (∼65%). Calc. for C21H28ClMnN2O10 (FW 558.84): C, 45.13; H, 5.05; N, 5.01%. Found: C, 45.15; H, 5.08; N, 5.04%. FT-IR (KBr, cm−1): 1607 (νCN); 2938 (νC–H); 3363 (νO–H). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN), 230 (4.9 × 104), 288 (2.2 × 104), 393 (1.03 × 104), 570 (4.6 × 103). Magnetic moment = 5.03 B. M. HRMS (ESI, positive ion mode, acetonitrile) m/z: 423.0227 (cald. 423.37) for [Mn(L2)]+.
:1 molar ratio. The solution mixture was then cooled to room temperature. A methanol/water (4:1) solution (5 mL) of manganese(II) perchlorate hexahydrate (0.362 g, ∼1 mmol) was added to it with continuous stirring. The colour of the solution changes from yellow to dark brown. A few drops of DMSO were added to the reaction mixture. The reaction mixture was then kept undisturbed for slow evaporation in an open atmosphere. Deep brown block-shaped single crystals of 3, suitable for X-ray diffraction, were obtained after 10–11 days. It was re-crystallized from DMSO.Complex 3: yield: 434 mg (∼69%). Calc. for C21H26Cl3MnN2O8S (FW 627.79): C, 40.18; H, 4.17; N, 4.46%. Found: C, 40.24; H, 4.32; N, 4.53%. FT-IR (KBr, cm−1): 1605–1632 (νCN); 2949 (νC–H); 3370 (νO–H). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN), 224 (2.86 × 104), 277 (1.41 × 104), 388 (5.2 × 103), 520 (9.6 × 102). Magnetic moment = 5.01 B. M. HRMS (ESI, positive ion mode, acetonitrile) m/z: 430.9119 (cald. 432.21) for [Mn(L3)]+.
Complex 4: yield: 405 mg (∼64%). Calc. for C23H30ClMnN2O11S (FW 632.95): C, 43.64; H, 4.78; N, 4.43%. Found: C, 43.67; H, 4.79; N, 4.44%. FT-IR (KBr, cm−1): 1603 (νCN); 2936 (νC–H); 3403 (νO–H). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN), 228 (3.93 × 104), 287 (1.7 × 104), 391 (6.5 × 103), 567 (3.9 × 103). Magnetic moment = 5.03 B. M. HRMS (ESI, positive ion mode, acetonitrile) m/z: 439.0023 (cald. 437.35) for [Mn(L4)]+.
X-Ray crystallography
Suitable single crystals of all complexes were collected and mounted on glass fibers. A ‘Bruker D8 QUEST area detector’, equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å), was used to measure the diffraction intensities of the complexes. The molecular structures were solved by direct methods and refined by full-matrix least squares on F2, using the SHELX-16/3 program.31 Anisotropic thermal parameters were used for the refinement of non-hydrogen atoms. Hydrogen atoms attached to oxygen atoms were located by different Fourier maps and were refined with distance constraints where possible else kept at fixed positions. All other hydrogen atoms were placed in their geometrically idealized positions and constrained to ride on their parent atoms. In 4, disorder was found in the DMSO and the CH–OH moiety of the ligand. Multi-scan empirical absorption corrections were applied to the data using the program SADABS.32 The details of crystallographic data and refinements of all complexes are listed in Table S1.† Important bond lengths and bond angles are given in Table 1.| Complex | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| a Symmetry transformation, a = x, 1 − y, z. | ||||
| Mn(1)–O(1) | 1.881(2) | 1.898(2) | 1.865(4) | — |
| Mn(1)–O(2) | 1.903(2) | 1.894(2) | 1.892(4) | 1.890(2) |
| Mn(1)–N(1) | 2.026(3) | 2.023(3) | 2.013(5) | 2.040(3) |
| Mn(1)–N(2) | 2.030(3) | 2.036(3) | 1.997(5) | — |
| Mn(1)–O(3) | 2.206(2) | 2.233(3) | 2.193(5) | 2.251(3) |
| Mn(1)–O(4) | 2.221(2) | 2.225(3) | 2.326(5) | 2.248(4) |
| O(1)–Mn(1)–O(2) | 85.04(10) | 87.37(10) | 88.9(2) | —– |
| O(1)–Mn(1)–O(3) | 89.64(11) | 90.99(10) | 98.1(2) | —– |
| O(1)–Mn(1)–O(4) | 94.23(11) | 92.37(11) | 89.7(2) | —– |
| O(1)–Mn(1)–N(1) | 90.43(11) | 89.19(12) | 91.3(2) | — |
| O(1)–Mn(1)–N(2) | 174.44(12) | 177.14(11) | 175.6(2) | — |
| O(2)–Mn(1)–O(3) | 92.86(11) | 91.91(10) | 90.1(2) | 92.41(9) |
| O(2)–Mn(1)–O(4) | 91.79(11) | 94.33(11) | 94.5(2) | 91.40(10) |
| O(2)–Mn(1)–N(1) | 175.45(10) | 176.49(15) | 178.6(2) | 90.45(10) |
| O(2)–Mn(1)–N(2) | 90.53(11) | 89.82(12) | 90.6(2) | — |
| O(3)–Mn(1)–O(4) | 174.20(10) | 173.04(10) | 171.0(2) | 174.81(11) |
| O(3)–Mn(1)–N(1) | 87.52(12) | 88.82(11) | 88.5(2) | 87.63(9) |
| O(3)–Mn(1)–N(2) | 87.21(11) | 88.59(11) | 86.2(2) | — |
| O(4)–Mn(1)–N(1) | 88.13(11) | 85.13(11) | 87.0(2) | 88.82(10) |
| O(4)–Mn(1)–N(2) | 89.27(11) | 88.35(12) | 86.0(2) | — |
| N(1)–Mn(1)–N(2) | 94.01(11) | 93.63(13) | 89.3(2) | — |
| O(2)–Mn(1)–O(2)a | — | — | — | 85.44(12) |
| O(2)–Mn(1)–N(1)a | — | — | — | 175.89(9) |
| N(1)–Mn(1)–N(1)a | — | — | — | 93.66(14) |
Computational details
The calculations reported herein were performed using the Turbomole 7.2 program.33 The crystallographic coordinates were used for the calculations of the supramolecular assemblies. We used the crystallographic coordinates for the assemblies because we are interested in evaluating the interactions as they stand in the solid state. The level of theory used for the calculations was RI-BP86-D3/def2-TZVP.34–38 The MEP surface plots were generated using the wavefunction obtained at the same level of theory and the 0.001 a.u. isosurface to simulate the van der Waals envelope. The topological analysis of the electron density was carried out according to the quantum theory of atoms in molecules (QTAIM) method proposed by Bader39 and the reduced density gradient (RDG) isosurfaces (NCIplot)40 and represented using the VMD program.41 They were computed using the MultiWFN program42 at the PB86-D3/def2-TZVP level of theory. The NBO analysis43 was performed using the same level of theory and the NBO 7.0 program.44Hirshfeld surface analysis
The structure input files in CIF format was used to calculate the Hirshfeld surfaces45–47 and the associated 2D-fingerprint plots48–50 using Crystal Explorer software.51 The Hirshfeld surface is a crucial and useful tool for studying intermolecular interactions while preserving a whole-molecule perspective. The proximity of intermolecular contacts in a molecule is well defined by parameters de and di, where de refers to the distances to the nearest atoms outside, and di refers to distances to the nearest atoms inside respectively. Another parameter, dnorm, has been introduced to account for the relative sizes of atoms. A distinctive red-white-blue colour scheme has been used to display intermolecular contacts. The red spots on the Hirshfeld map indicate shorter contacts, white spots denote the contacts around the van der Waals separation and blue spots are for larger contacts. The interactions in the crystal are succinctly summarised by 2D-fingerprint displays.Results and discussion
Synthesis
The Schiff bases (H2L1–H2L4) were prepared by refluxing the appropriate diamine and salicylaldehyde derivatives in 1:2 molar ratio in methanol following the literature method.52–56 These Schiff bases were not isolated and purified, but the methanol solution of the Schiff bases were used in situ for the synthesis of manganese(III) complexes, [MnL1(H2O)2]ClO4·H2O (1), [MnL2(H2O)2]ClO4 (2), [MnL3(DMSO)(H2O)]ClO4 (3), [MnL4(DMSO)(H2O)]ClO4 (4) by adding manganese(II) perchlorate hexahydrate with constant stirring. The formation of the complexes is shown in Scheme 3. It is to be mentioned here that all the ligands have already been well characterized and used by different research groups for the synthesis of different transition and non-transition metal complexes for many years.57–66
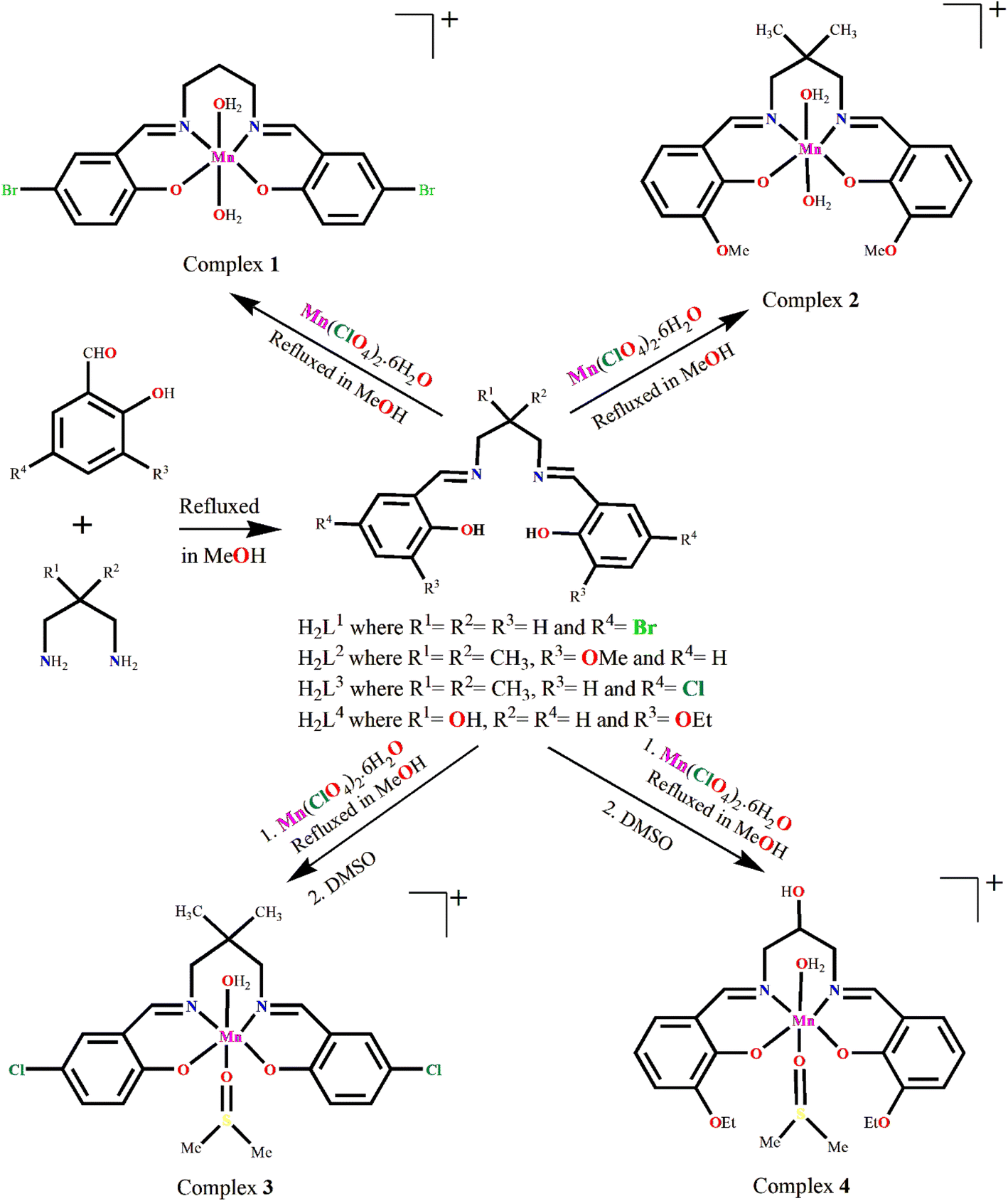 | ||
| Scheme 3 Synthetic route to complexes 1–4. Counter perchlorate anions and lattice solvent molecules, if any, have been omitted for clarity. | ||
In the present case, mononuclear octahedral complexes containing the tetradentate Schiff base ligands (dianions of H2L1, H2L2, H2L3, and H2L4 respectively) are formed, with one/two coordinating water molecule(s). In each case manganese(II) is converted to manganese(III) species by aerial oxidation, as has also been observed previously.67–69 Thus, the efforts to prepare complexes in N2 or Ar atmosphere were not successful. X-Ray-quality single crystals of complexes 3 and 4 were grown in DMSO medium. We have tried to obtain single crystals of 3 and 4 from other solvents also, but the efforts were unsuccessful. X-Ray diffraction data shows that one DMSO molecule is as co-ligand in the coordination sphere of manganese(III) in each of complexes 3 and 4 (vide infra) and this explains the necessity of using DMSO in their synthesis.
It is to be mentioned here that complexes with coordinated water molecules may open up the possibilities of preparing several water-bridged dimeric complexes. To understand the mechanism of ligand replacement reactions, complexes with coordinated water molecules are important. In each complex, the hydrogen atoms of the water molecules are involved in H-bonding.
Description of structures
H2O for complexes [MnL1(H2O)2]ClO4·H2O (1) and [MnL2(H2O)2]ClO4 (2), whereas X = H2O and Y = DMSO for complexes [MnL3(DMSO)(H2O)]ClO4 (3) and [MnL4(DMSO)(H2O)]ClO4 (4); L = L1 (in 1), L2 (in 2), L3 (in 3), and L4 (in 4). A lattice water molecule is also present in the asymmetric unit of complex 1. Complex 1 crystallizes in the orthorhombic space group P212121. Complex 2 crystallizes in the monoclinic space group C2/c. Single-crystal X-ray analysis reveals that complex 3 crystallizes in the orthorhombic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Single crystal X-ray diffraction analysis reveals that complex 4 crystallizes in the monoclinic space group C2/m and has mirror symmetry.
. Single crystal X-ray diffraction analysis reveals that complex 4 crystallizes in the monoclinic space group C2/m and has mirror symmetry.Each mononuclear complex contains a manganese(III) center with six-coordination pseudo-octahedral geometry. In each complex, two imine nitrogen atoms {N(1) and N(2) for complexes 1–3; and N(1), N(1)a for complex 4}, and two phenolate oxygen atoms, {O(1) and O(2) for complexes 1–3; and O(2), O(2)a for complex 4}, of the deprotonated di-Schiff base (L2−) constitute the equatorial plane {symmetry transformation, a = x, 1 − y, z}. Two oxygen atoms {O(3) and O(4)} from two coordinated water molecules (in 1 and 2) or one from water and another from DMSO (in 3 and 4), are attached to the manganese(II) centers in axial sites. Manganese(III)–oxygen and manganese(III)–nitrogen distances fall within the range observed for structurally characterized manganese(III) complexes.1–17 The axial manganese(III)–oxygen distances are much longer compared to the equatorial manganese(III)–oxygen and manganese(III)–nitrogen distances (Table 1), as were also observed in similar complexes.1–17 The elongation of axial bonds indicates clear evidence of Jahn–Teller distortion, as expected for high-spin manganese(III) with d4 (t32ge1g) electronic configuration. The basal bond angles are all close to 90° (Table 1). The donor atoms, {O(1), O(2), N(1), N(2) in complexes 1–3; and O(2), O(2)a, N(1), N(1)a in 4} in the equatorial plane are approximately co-planar with no atoms deviating from the plane by more than 0.050 Å in any complex. The deviations of all the coordinating atoms in the basal plane from the mean plane passing through them and that of manganese(III) from the same plane are gathered in Table 2. The perspective views of complexes along with the selected atom numbering scheme is shown in Fig. 1 and 2.
| Complex | N(1) | N(2) | O(1) | O(2) | Mn(III) |
|---|---|---|---|---|---|
| 1 | 0.029(2) | −0.028(2) | −0.033(2) | 0.033(2) | −0.026(2) |
| 2 | 0.009(2) | −0.009(2) | −0.010(2) | 0.010(2) | 0.001(2) |
| 3 | 0.046(2) | −0.047(2) | −0.050(3) | 0.050(3) | 0.024(2) |
| 4 | 0.000 | — | 0.000 | — | 0.051(2) |
 | ||
| Fig. 1 Perspective views of complexes 1 (a) and 2 (b) with the selective atom numbering scheme. All the hydrogen atoms (except those of coordinating water molecules), the non-coordinated perchlorate ions, and a lattice water molecule have been omitted for clarity. | ||
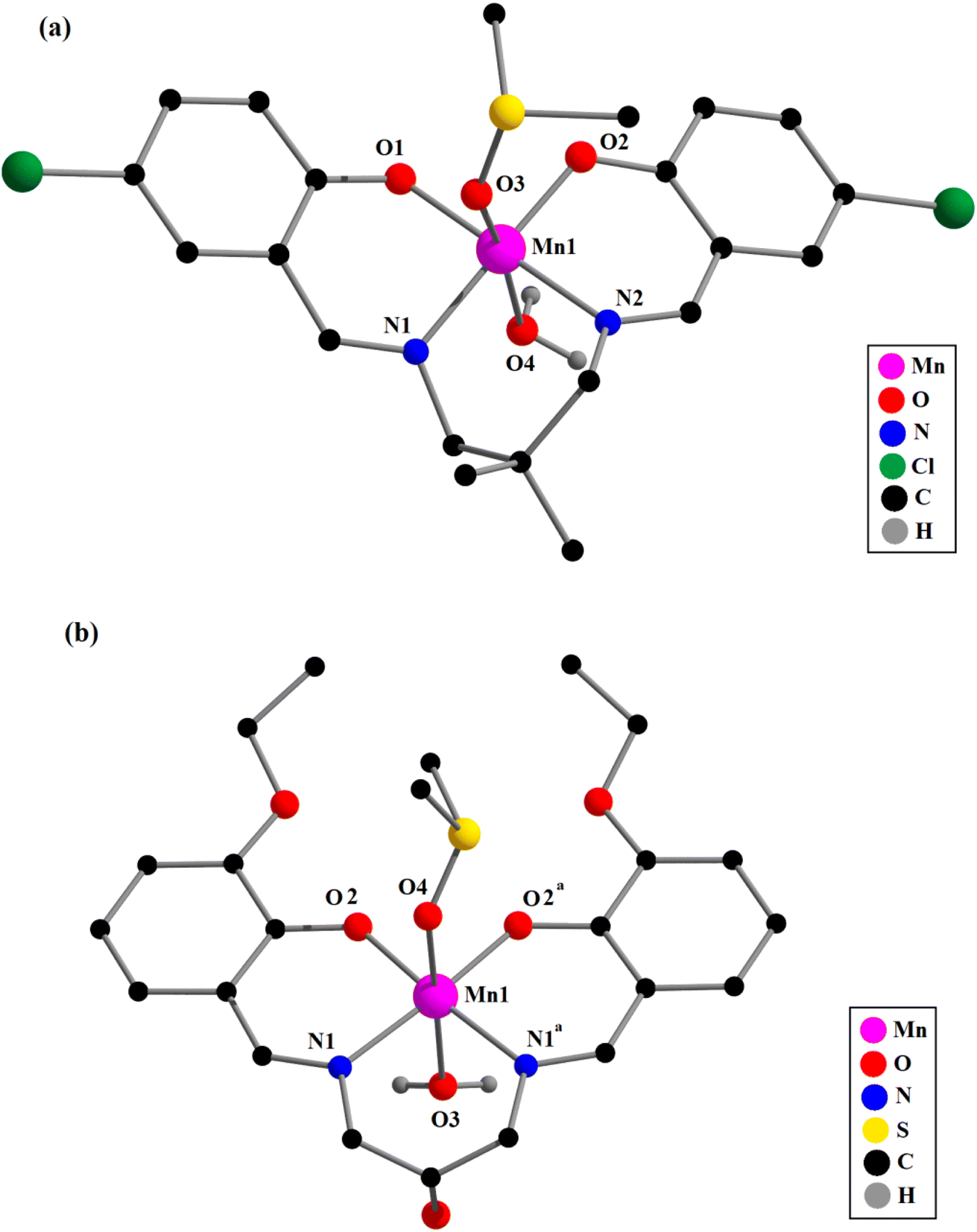 | ||
| Fig. 2 Perspective views of complexes 3 (a) and 4 (b) with the selective atom numbering schemes. All the hydrogen atoms (except hydrogen-atoms of coordinating water molecules) and a non-coordinated perchlorate ion are omitted for clarity. In 4, which has mirror symmetry, only the major component of the disordered CH–OH group and the DMSO ligand is shown. Symmetry transformation, a = x, 1 − y, z. | ||
The saturated six-membered chelate rings [Mn(1)–N(1)–C(8)–C(9)–C(10)–N(2) in 1, Mn(1)–N(1)–C(9)–C(10)–C(13)–N(2) in 2, Mn(1)–N(1)–C(8)–C(9)–C(12)–N(2) in 3, and Mn(1)–N(1)–C(10)–C(11)–C(10)a–N(1)a in 4] have different conformations, as confirmed by puckering analysis.70–72 The conformation of the rings are shown in Fig. 3 while their puckering parameters are given in Table 3.
 | ||
| Fig. 3 Half-chair conformations of six-membered chelate rings in complexes 1 and 2. Twist-boat conformation of six-membered chelation ring of complex 3. Envelope like conformation of six-membered chelation ring of complex 4. Symmetry transformation, a = x, 1 − y, z. | ||
| Puckering amplitude, q (Å) | Torsion angle, θ (°) | Phase angle, ϕ (°) | Conformation | ||
|---|---|---|---|---|---|
| a Symmetry transformation, a = x, 1 − y, z. | |||||
| 1 | Mn(1)–N(1)–C(8)–C(9)–C(10)–N(2) | 0.526(4) | 32.4(3) | 173.8(7) | Approximate half-chair |
| 2 | Mn(1)–N(1)–C(9)–C(10)–C(13)–N(2) | 0.540(5) | 146.7(4) | 351.2(8) | Approximate half-chair |
| 3 | Mn(1)–N(1)–C(8)–C(9)–C(12)–N(2) | 0.745(7) | 89.1(5) | 261.9(5) | Twist boat |
| 4 | Mn(1)–N(1)–C(10)–C(11)–C(10)a–N(1)a | 0.498(7) | 115.5(6) | 360.0(6) | Approximate envelope |
DFT study on supramolecular interaction
The DFT study is focused on two main aspects of the solid-state structures of complexes 1–4. One is the formation of halogen (HaB) and chalcogen bonding (ChB) interactions in complexes 1 and 3 where the electron donor is the perchlorate anion and the acceptor either bromine or chlorine atoms for the HaBs and the sulfur atom of the coordinated DMSO for the ChB. The other one is the analysis of the formation of recurrent motifs in the solid state of complexes 1, 2 and 4. They consist of supramolecular dimers where the hydrogen atoms of the coordinated water molecules interact with the electron rich cavity formed by the phenolic and alkyloxy oxygen atoms of the Schiff-base ligand. Dimensions of the hydrogen bonds in the structures are given in Table 4.| Complex | Atoms involved (D–H⋯A) | Distance D–H (Å) | Distance H⋯A (Å) | Distance D⋯A (Å) | Angle ∠D−H⋯A (°) | Symmetry |
|---|---|---|---|---|---|---|
| 1 | O4–H3W⋯O1 | 0.82 | 2.30 | 3.079(4) | 158.3(2) | x + 1/2, 3/2 − y, 1 − z |
| O4–H3W⋯O2 | 0.82 | 2.44 | 2.982(3) | 124.6(2) | x + 1/2, 3/2 − y, 1 − z | |
| O3–H1W⋯O1 | 0.82(3) | 2.64(6) | 3.108(4) | 117(5) | x − 1/2, 3/2 − y, 1 − z | |
| O3–H1W⋯O2 | 0.82(3) | 2.06(2) | 2.879(3) | 175(6) | x − 1/2, 3/2 − y, 1 − z | |
| O3–H2W⋯O10 | 0.85(2) | 1.99(3) | 2.799(4) | 159(6) | −1 + x, y, z | |
| O4–H4W⋯O10 | 0.85(2) | 1.95(3) | 2.794(4) | 173(6) | ||
| O10–H10W⋯O6 | 0.83(2) | 1.96(2) | 2.789(4) | 173(6) | 2 − x, y − 1/2,3/2 − z | |
| O10–H11W⋯O8 | 0.82(2) | 2.09(3) | 2.894(6) | 169(7) | ||
| C5–H5⋯Br2 | 0.93 | 3.03 | 3.949(4) | 170.5 | x, −1 + y, z | |
| C11–H11⋯O8 | 0.93 | 2.64 | 3.273(5) | 125.8 | −1 + x, y, z | |
| C13–H13⋯O6 | 0.93 | 2.61 | 3.428(5) | 146.5 | −1 + x, y, z | |
| 2 | O4–H3W⋯O5 | 0.85(2) | 1.93(3) | 2.761(4) | 164(10) | −x + 3/2, y + 1/2, −z + 3/2 |
| O4–H4W⋯O6 | 0.85(2) | 2.03(4) | 2.840(4) | 160(11) | −x + 3/2, y + 1/2, −z + 3/2 | |
| O3–H2W⋯O2 | 0.85(2) | 2.05(5) | 2.811(4) | 149(9) | − x + 3/2, y − 1/2, −z + 3/2 | |
| O3–H2W⋯O6 | 0.85(2) | 2.68(9) | 3.107(4) | 113(8) | − x + 3/2, y − 1/2, −z + 3/2 | |
| O3–H1W⋯O1 | 0.85(2) | 2.10(6) | 2.778(3) | 137(7) | −x + 3/2, y − 1/2, −z + 3/2 | |
| O3–H1W⋯O5 | 0.85(2) | 2.41(7) | 3.092(4) | 138(8) | −x + 3/2, y − 1/2, −z + 3/2 | |
| C8–H8⋯O8 | 0.93 | 2.59 | 3.433(8) | 151.4 | ||
| 3 | O4–H1W⋯O2 | 0.85(2) | 1.92(3) | 2.762(6) | 171(12) | 2 − x, 1 − y, 1 − z |
| O4–H2W⋯O5 | 0.86(2) | 2.23(6) | 3.044(9) | 158(13) | ||
| C5–H5⋯O8 | 0.93 | 2.60 | 3.227(11) | 124.8 | ||
| C7–H7⋯O8 | 0.93 | 2.57 | 3.365(11) | 144.1 | 1 − x, 2 − y, 1 − z | |
| C8–H8B⋯O3 | 0.97 | 2.56 | 3.148(9) | 119.3 | ||
| C12–H12B⋯O4 | 0.97 | 2.56 | 3.199(8) | 123.0 | ||
| C13–H13⋯O7 | 0.93 | 2.40 | 3.188(11) | 141.8 | 1 − x, 1 − y, 1 − z | |
| C20–H20C⋯O1 | 0.96 | 2.57 | 3.399(12) | 144.7 | ||
| C21–H21B⋯Cl2 | 0.96 | 2.95 | 3.886(10) | 165.2 | x, 1 + y, z | |
| C21–H21B⋯O6 | 0.96 | 2.791 | 3.44(2) | 125.6 | x, y, 1 + z | |
| C20–H20B⋯O6 | 0.96 | 2.811 | 3.46(2) | 125.2 | x, y, 1 + z | |
| 4 | O3–H3⋯O1 | 0.84(2) | 2.05(4) | 2.855(3) | 159(10) | 1 − x, 1 − y, 1 − z |
| O3–H3⋯O2 | 0.84(2) | 2.44(7) | 3.049(3) | 130(7) | 1 − x, 1 − y, 1 − z | |
| O5A–H5A⋯O4 | 0.850(5) | 2.000(5) | 2.835(6) | 167(4) | ||
| O5B–H5B⋯O3 | 0.861(5) | 2.014(3) | 2.874(5) | 176.7(7) | ||
| C1–H1B⋯O5B | 0.96 | 2.42 | 3.371(9) | 169.9 | 1 − x, 1 − y, 1 − z | |
| C9–H9⋯O12 | 0.93 | 2.53 | 3.441(7) | 166.6 | 1/2 − x, 3/2 − y, −z | |
| C11B–H11B⋯O5B | 0.98 | 1.78 | 2.568(16) | 135.3 | 1 − x, 1 − y, −z | |
| C12–H12B⋯O2 | 0.96 | 2.49 | 3.345(6) | 147.9 | x, 1 − y, z |
First, we have analyzed the MEP surfaces of complexes 1, 2 and 4 in order to investigate the relative basicity of the oxoanionic cavity. It should be emphasized that the manganese(III) complexes are positive, and consequently in these calculations we have included the counterion to keep the global system neutral. The MEP surfaces are plotted in Fig. 4, evidencing that in all cases the global MEP minimum is located at the perchlorate anion (ranging −60 to −69 kcal mol−1) and the maximum at the coordinated water protons (ranging +65 to +75 kcal mol−1). In all cases, the MEP is negative at the region under the influence of both phenolate oxygen atoms, in spite of the global positive charge of the manganese(III) complex. The presence of the extra oxygen atoms from the methoxy (2) and ethoxy (4) groups significantly increases the ability of the O4-cavity to interact with electron poor atoms. Finally, the MEP surface analysis of complex 1 also revels the presence of a σ-hole at the bromine atoms (+19 kcal mol−1), as highlighted in Fig. 4a (bottom-right) using a reduced MEP scale (+19 to 0 kcal mol−1).
 | ||
| Fig. 4 MEP surfaces of complexes 1 (a), 2 (b) and 4 (c) at the RI-BP86-D3/def2-TZVP level of theory. The values at selected points of the surfaces are given in kcal mol−1. Isovalue 0.001 a.u. | ||
Fig. 5a shows the QTAIM/NCIplot analysis of the interaction of the perchlorate with the manganese(III) complex, disclosing the formation of a bifurcated HaB, that is characterized by two bond critical points (CPs, red spheres) and bond paths (orange lines) connecting two oxygen atoms of the anion to the bromine atom. The interaction is further characterized by a green RDG (reduced density gradient) isosurface (green color is used herein to indicate weak and attractive NCI) that is located between the bromine-atom of both oxygen atoms of the anion. In order to estimate the strength of the bifurcated HaB free from the pure coulombic attraction between the counterions, we have used the method proposed by Bartashevich and Tirelson73 that is based on the value of the potential energy density Vr at the bond CP. As a result, the HaB estimation is −1.20 kcal mol−1, in line with the green color of the RDG isosurface and revealing its modest strength. This HaB has been also studied from an orbital point of view using the natural bond orbital (NBO) analysis and focusing on the second order perturbation analysis, since it is very convenient to analyze orbital donor–acceptor interactions. Interestingly, the NBO method discloses an electron transfer from the lone pairs at the oxygen-atoms of the perchlorate to the antibonding σ*(C–Br) orbital with a concomitant stabilization energy of 0.57 kcal mol−1, and confirming the σ-hole nature of the interaction. The NBOs involved in one of both LP(O) → σ*(C–Br) interactions is shown in Fig. 5B.
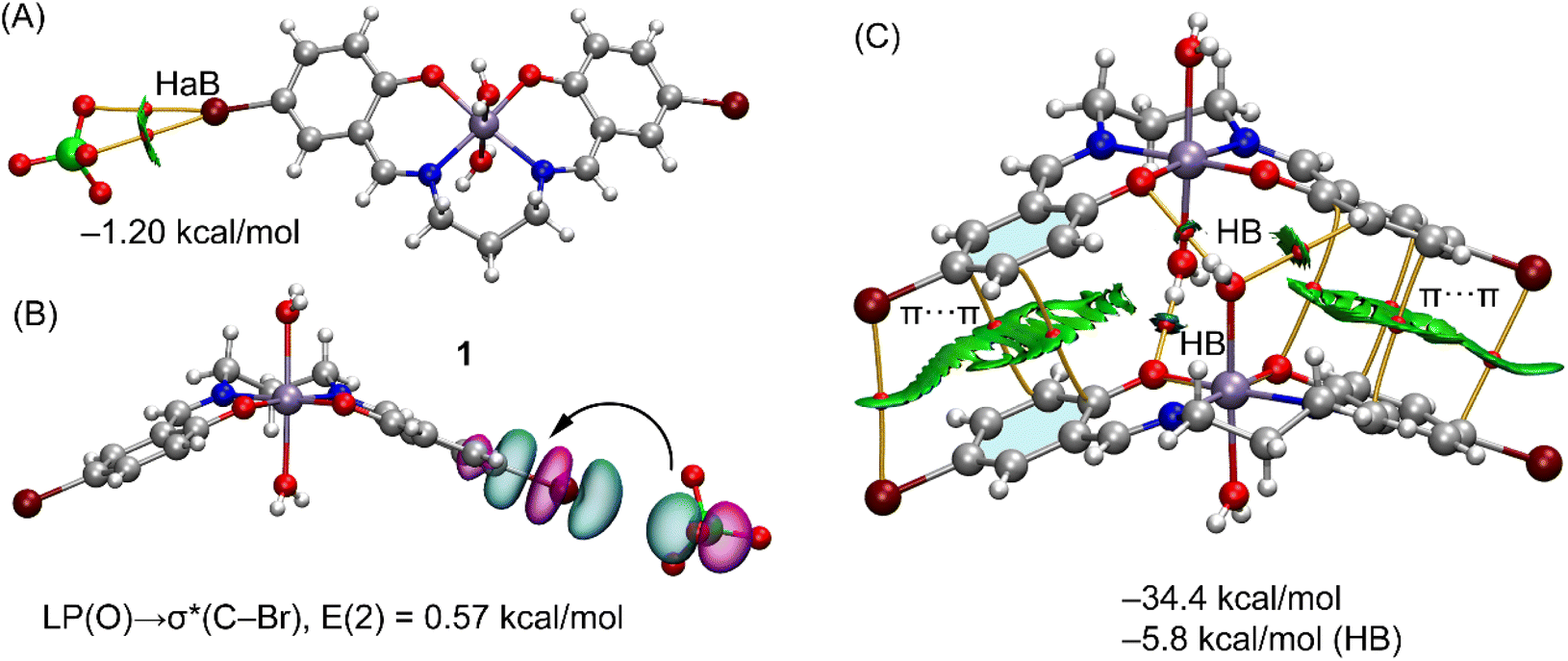 | ||
| Fig. 5 (A) QTAIM (bond CPS in red and paths as orange lines) and NICPlot (see computational method for details) characterization of the HaB interaction. (B) NBOs involved in the LP(O) → σ*(C–Br) interaction. (C) QTAIM/NCIplot analysis of the self-assembled dimer in 1. Only intermolecular interactions are represented. The counterions are omitted in the representation of (c), though they were taken into consideration for the calculation of the interaction energies. The HB contribution has been estimated using the Vr values at the bond CPs that characterize the HBs. | ||
In the crystal the packing of complex 1 shows a polymer formed by successive hydrogen bonds involving the coordinated water molecules and the phenolate oxygen atoms formed along the crystallographic a axis (Table 4). A model has been abstracted, shown in Fig. 5C of two adjacent molecules in these polymers in which the coordinated water molecule, O(3), of one manganese(III) complex forms donor hydrogen bonds to the acceptor phenolate oxygen atoms of the adjacent molecule {symmetry −1/2 + x, 3/2 − y, 1 − z}. The second axial water molecule, O(4), forms similar donor hydrogen bonds in this dimer. Also involved is the water molecule, O(10), which acts as an acceptor to the coordinated water molecules and as a donor to two perchlorate oxygen atoms. Using the dimeric model, three H-bonds are established, each one characterized by the corresponding bond CP, bond path and green-blue isosurface. The total contribution of the HBs using the Vr energy predictor is −5.8 kcal mol−1. The formation of the dimer is further supported by π-stacking interactions, characterized by several bond CPs and bond paths interconnecting the aromatic rings with a distance of 3.92 Å between centroids. Moreover, the interactions are further characterized by extended green isosurfaces that embrace the whole aromatic surfaces and the C–Br bonds. The total interaction energy is very large (−34.4 kcal mol−1) due to the electrostatic contribution (ion pair attraction), between the perchlorate anions (not shown in Fig. 5C for clarity) and the cationic manganese(III) complexes.
Both complexes 2 and 4 also form polymers but for simplicity only dimers are considered in the calculations and these are shown in Fig. 6, including their QTAIM/NCI plot analysis. Both dimers show extended RDG isosurfaces between the aromatic ring, disclosing the existence of π-stacking interactions. These are also evidenced by several bond CPs and bond path interconnecting the rings. Both dimers exhibit very large dimerization energies, due to the electrostatic attraction between the counterions. In complex 2, one of the water molecules, O(4), establishes four H-bonds with the four oxygen-atoms of the electron rich O4-cavity from the adjacent molecule {symmetry −x + 3/2, y − 1/2, −z + 3/2}. In this dimer, the second water molecule, O(3), only forms two hydrogen bonds to the two uncoordinated methoxy atoms, O(5) and O(6). Presumably as a consequence of the formation of so many hydrogen bonds involving the coordinated water molecules, listed in Table 4, there are no such bonds involving the perchlorate oxygen atoms. The other water molecule involved in the formation of this dimer establishes only two H-bonds with the methoxy groups. The overall contribution of the six H-bonds in the dimer of 2 is −21.1 kcal mol−1, confirming the importance of the OH⋯O interactions and supporting the relevance of this motif in the solid state.
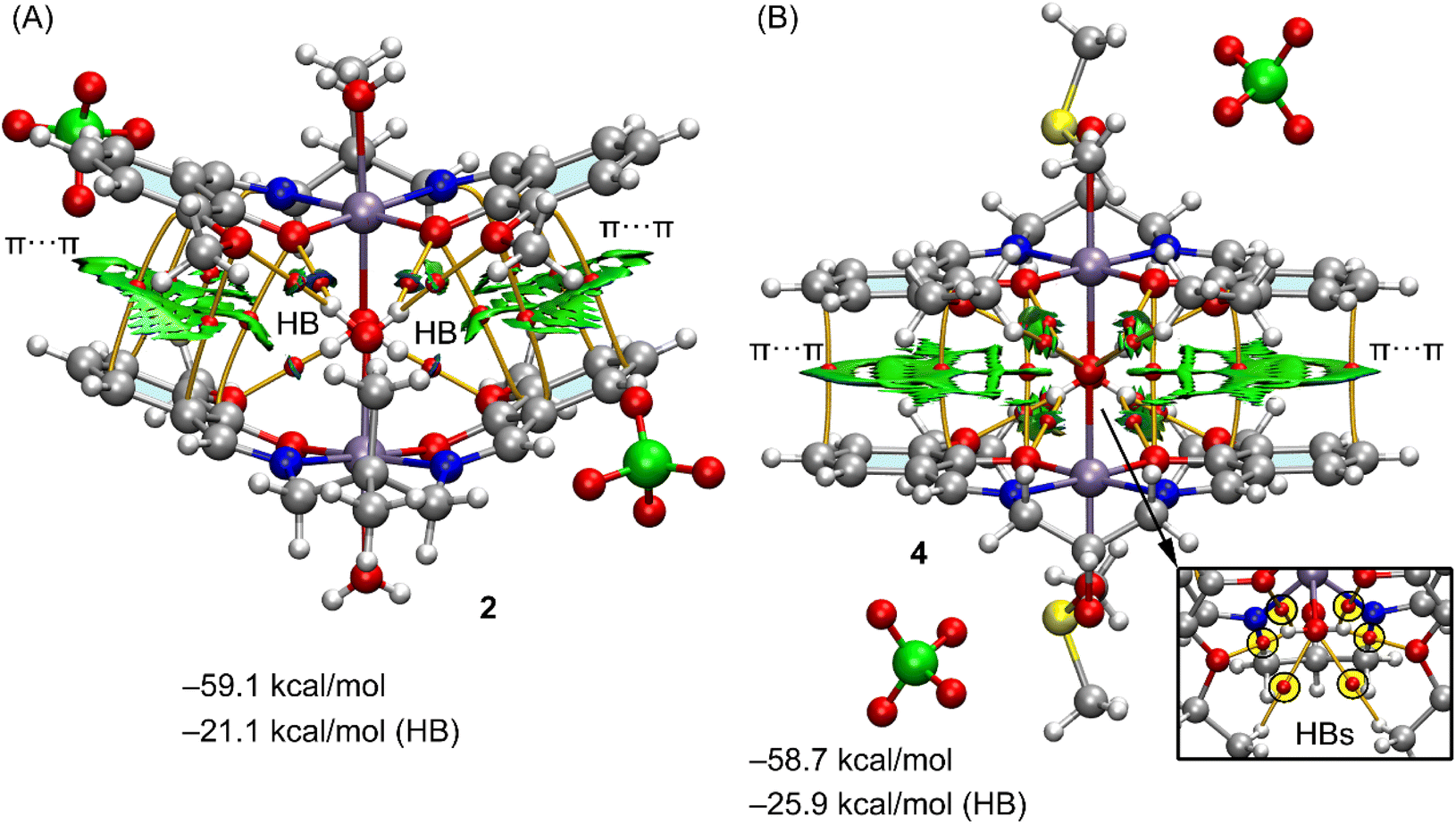 | ||
| Fig. 6 (A) QTAIM (bond CPS in red and paths as orange lines) and NICPlot (see computational method for details) analysis of the self-assembled dimers in 2 (A) and 4 (B). Only intermolecular interactions are represented. The HB contributions have been estimated using the Vr values at the bond CPs that characterize the HBs. | ||
In complex 4, the metal has mirror symmetry, and the coordinated water molecule, O(3), establishes four donor H-bonds (see Fig. 6B, bottom-right), to O(1)*2 and O(2)*2 in the electron rich O4-cavity of a neighboring (1 − x, y, 1 − z) symmetry related molecule to form the dimer. An additional H-bond is found from the disordered CH–OH group in the ligand. There are two alternative positions for the oxygen, named O(5A) and O(5B) with respective populations of 0.35(2), 0.65(2) and the coordinated water molecule, O(3) forms an acceptor H-bond to O(5B) while the O(4), from DMSO, forms an acceptor H-bond to O(4B). All H-bond dimensions are given in Table 4.
Consequently the contribution of the H-bonds (−25.9 kcal mol−1) is larger in 4 than in the dimer of 2 (−21.1 kcal mol−1). The large contribution of the H-bonds and the formation of the four H-bonds between the water molecule and the electron rich cavity and hydrogen-atoms of the ethoxy groups reveals a strong complementarity between the water molecules and the cavity and explains the formation of such binding motif in the solid state. It is also worth mentioning such motifs are governed by the cooperation of the H-bonds and π-stacking interactions, even in the presence of stronger but non-directional electrostatic forces. It is interesting to note that as in 2, which also contains alkoxy groups, there are no hydrogen bonds involving the perchlorate anion.
Finally, in complex 3, the coordinated water molecule forms two donor hydrogen bonds, one to perchlorate oxygen atom, O(5) and the other to a ligand oxygen, O(2) of a neighboring symmetry related (2 − x, 1 − y, 1 − z) complex molecule. However the most notable interactions are observed where the perchlorate anion bridges two manganese(III) complexes (symmetry transformations, x, y, z and x, 1 + y, z) by means of the formation of two concurrent σ-holes (HaB and ChB). In particular, one oxygen-atom, O(6), in the perchlorate is located opposite to the SO bond of the coordinated DMSO molecule and also opposite to the C(16)–Cl(2) bond, thus establishing both ChB and HaB interactions. Both contacts have been confirmed by QTAIM analysis, showing bond CPs and bond paths connecting one oxygen-atom of the anion to the chlorine and sulfur atoms of the Schiff-base and DMSO ligands, respectively, as detailed in Fig. 7A. Two CH bonds (C20–H20B and C21–H21B) of the DMSO are also connected with the oxygen atom, O(6), thus forming two additional CH⋯O interactions. All interactions are also revealed by the NCIplot analysis and characterized by green colored RDG isosurfaces. Fig. 7B shows the NBOs involved in the interactions, showing two contributions: LP(O) → σ*(C–Cl) and LP(O) → σ*(S–O), thus confirming the σ-hole nature of both interactions. Moreover, the second order stabilization energies disclose that the ChB is slightly stronger (E(2) = 1.75 kcal mol−1) than the HaB (E(2) = 1.59 kcal mol−1).
 | ||
| Fig. 7 (A) QTAIM (bond CPs in red and paths as orange lines) and NICPlot (see computational method for details) characterization of the HaB and ChB interactions in complex 3. (B) NBOs involved in the LP(O) → σ*(C–Cl) and LP(O) → σ*(S–O) interactions. | ||
Hirshfeld surface analysis
Hirshfeld surface analysis of complexes 1–4 was performed to identify and emphasize the significant intermolecular interactions that exist in their solid-state structures. The surfaces were kept transparent to visualize the molecular moiety clearly. Fig. S1 (ESI†) illustrates the mapping of the Hirshfeld surface for each complex, utilizing dnorm in the range of −0.1 Å to 1.5 Å, as well as shape index and curvedness. The Hirshfeld surfaces mapped by dnorm, display individual spikes representing the corresponding interactions, where unique spikes indicate intermolecular interactions. The noticeable marks present on the Hirshfeld surfaces of the complexes are indicative of the contacts Br⋯H/H⋯Br (18.4%), O⋯H/H⋯O (24.2%), and C⋯H/H⋯C (6.6%) for complex 1, O⋯H/H⋯O (32.1%) and C⋯H/H⋯C (8.6%) for complex 2, Cl⋯H/H⋯Cl (17.9%), O⋯H/H⋯O (22.9%), C⋯H/H⋯C (9.4%) and S⋯H/H⋯S (3.1%) for complex 3 respectively. The Hirshfeld surface of complex 4 is composed of different proportions of O⋯H/H⋯O, C⋯H/H⋯C, and S⋯H/H⋯S interactions, which account for 23.0%, 7.6%, and 3.9% respectively. The 2D fingerprint plots clearly depict intermolecular interactions in the form of distinct spikes and these are shown in the Fig. 8–11 for complexes 1–4 respectively. | ||
| Fig. 8 Fingerprint plot of complex 1: full and resolved into Br⋯H/H⋯Br, O⋯H/H⋯O, and C⋯H/H⋯C contacts contributed to the total Hirshfeld surface area. | ||
 | ||
| Fig. 9 Fingerprint plot of complex 2: full and resolved into O⋯H/H⋯O, and C⋯H/H⋯C contacts contributed to the total Hirshfeld surface area. | ||
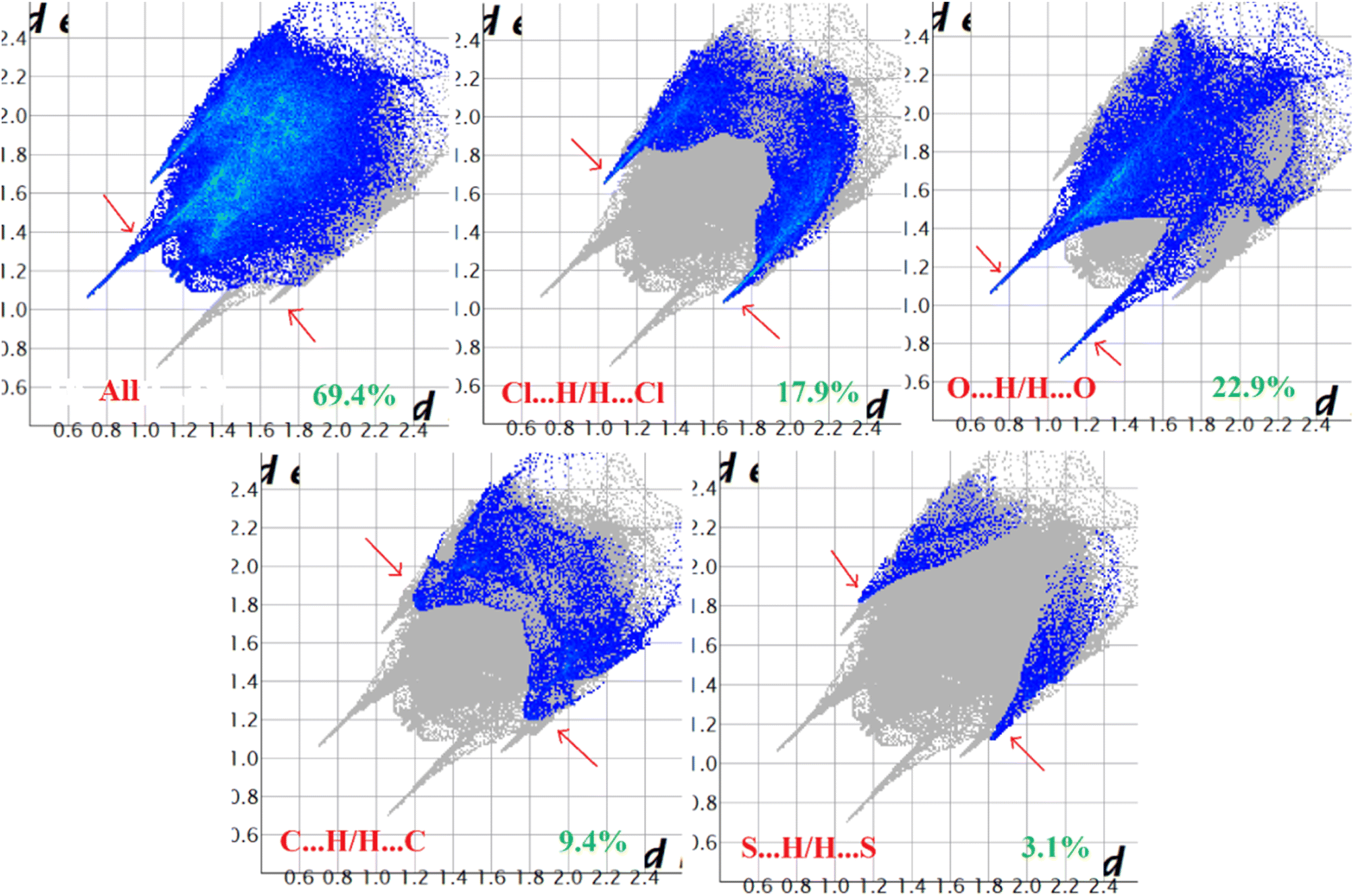 | ||
| Fig. 10 Fingerprint plot of complex 3: full and resolved into Cl⋯H/H⋯Cl, O⋯H/H⋯O, C⋯H/H⋯C, and S⋯H/H⋯S contacts contributed to the total Hirshfeld surface area. | ||
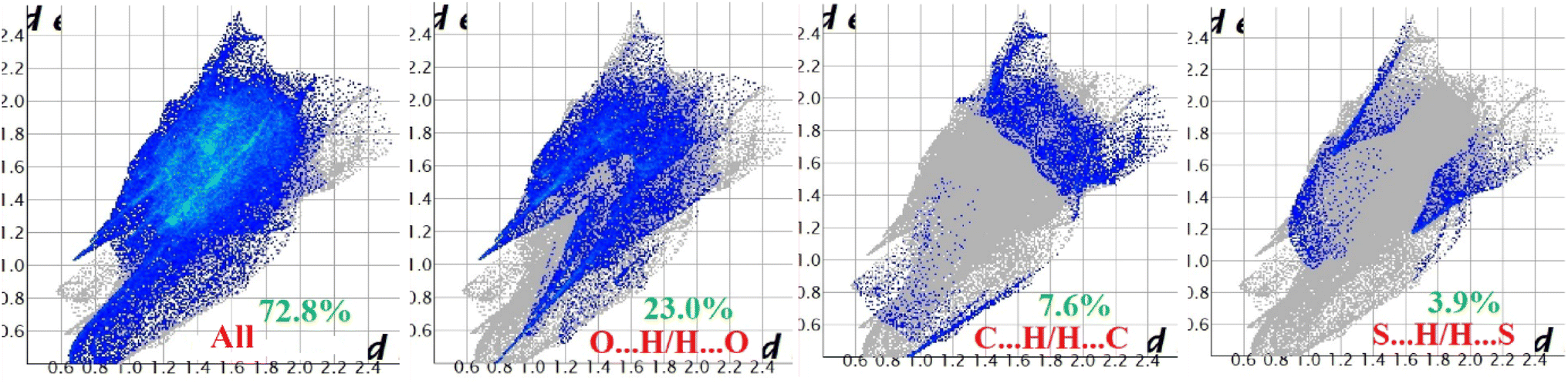 | ||
| Fig. 11 Fingerprint plot of complex 4: full and resolved into O⋯H/H⋯O, C⋯H/H⋯C, and S⋯H/H⋯S contacts contributed to the total Hirshfeld surface area. | ||
IR and electronic spectra and magnetic moment
The IR spectra of complexes 1–4 display consistent and routine observation of broad bands in the 3390–3360 cm−1 range due to the existence of coordinated water. All complexes exhibit the presence of the azomethine (CN) group as indicated by the strong bands around 1608 cm−1. Moreover, the presence of alkyl C–H stretching vibrations is evidenced by the bands appearing within the range 2980–2940 cm−1. Additionally, complexes 1–4 are determined to contain perchlorate ions due to the observation of sharp peaks in the 1080–1040 cm−1 range. The IR spectra of all four complexes are shown in Fig. S2–S5 (ESI†) respectively.
Only one symmetric band (corresponding to 5Tg ← 5Eg electronic transition) is expected for the d4 manganese(III) system under an ideal symmetric octahedral ligand field. However, both static and dynamic Jahn–Teller effects perturb the octahedral symmetry and electronic spectra become complex in nature. In the present case, bands with shoulder have been found around 610–450 nm. The electronic absorption spectra of all four complexes are collected in acetonitrile medium at room temperature. The intense absorption bands in the high energy range of 223–232 nm are ascribed to intra-ligand π → π* transitions, which include the aromatic rings. The absorption bands around 274–290 nm may be assigned to n → π* transitions within the ligands whereas broad absorption bands have been observed in the lower energy region of 382–393 nm due to ligand-to-metal charge transfer transitions.
The magnetic moment of each complex is ∼5.1 B. M. at room temperature. This value is closer to the theoretical value (∼4.90 B. M.) for the magnetically isolated high-spin manganese(III) with four unpaired electrons (S = 2) with t32ge1g electronic configuration.
ESI-MS study
The molecular ion peaks in acetonitrile solution appeared at m/z = 492.7210 (theoretical m/z = 493.06) for complex 1, 423.0227 (theoretical m/z = 423.37) for complex 2, 430.9119 (theoretical m/z = 432.21) for complex 3 and 439.0023 (theoretical m/z = 437.35) for complex 4. The peaks could correspond to [Mn(L)]+ {L = L1, L2, L3 and L4 respectively}. The mass spectra of the complexes have been shown in the Fig. S6–S9 (ESI†).Conclusions
In this work four new manganese(III) complexes have been synthesized and X-ray characterized four manganese(III) complexes with salen type tetradentate N2O2 donor Schiff bases. In each of the complexes, the N2O2 donor atoms of the salen ligands occupy four equatorial positions of octahedral manganese(III) whereas monodentate ligands (either two water molecules, or one water and one DMSO) occupy two axial sites. In the solid state a recurrent self-assembled motif is observed where the hydrogen-atoms of manganese-coordinated water molecules occupies an electron rich cavity and establish several H-bonds with the oxygen-atoms of the Schiff-base ligands. In addition, concurrent π-stacking interactions are also established, reinforcing the supramolecular complexes. Such assemblies have been analyzed theoretically, including their energetic features and the contribution of the H-bonds. Moreover, as the interactions are established between charged systems, the HaB and ChB interactions observed in the solid state of complexes 1 and 3 have been analyzed using QTAIM and the potential energy density (Vr) predictor, NCIplot and NBO analyses. This method is very useful here, as the interactions are established between charged systems. Conventional procedures based on the supramolecular approach would lead to repulsive cation⋯cation interactions in 1 and 2 or to very large and attractive interactions for the anion⋯cation interactions in 3. The evaluation of the CH⋯X (X = C, N, S, Cl) contacts using the QTAIM Vr predictor is free from the effect of pure coulombic forces.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
W. S. thanks the CSIR, India, for awarding a Junior Research Fellowship.References
- N. Sarkar, M. G. B. Drew, K. Harms, A. Bauzá, A. Frontera and S. Chattopadhyay, CrystEngComm, 2018, 20, 1077–1086 RSC.
- P. Bhowmik, H. P. Nayek, M. Corbella, N. Aliaga-Alcalde and S. Chattopadhyay, Dalton Trans., 2011, 40, 7916–7926 RSC.
- P. Seth, M. G. B. Drew and A. Ghosh, J. Mol. Catal. A: Chem., 2012, 365, 154–161 CrossRef CAS.
- P. Kar and A. Ghosh, Inorg. Chim. Acta, 2013, 395, 67–71 CrossRef CAS.
- A. Panja, N. Shaikh, M. Ali and P. Banerjee, Polyhedron, 2003, 22, 1191–1198 CrossRef CAS.
- S. Saha, D. Mal, S. Koner, A. Bhattacherjee, P. Gütlich, S. Mondal, M. Mukherjee and K. Okamoto, Polyhedron, 2004, 23, 1811–1817 CrossRef CAS.
- M. Karmakar and S. Chattopadhyay, J. Mol. Struct., 2019, 1186, 155–186 CrossRef CAS.
- E. Y. Tshuva, I. Goldberg and M. Kol, J. Am. Chem. Soc., 2000, 122, 10706–10707 CrossRef CAS.
- H. Kara, Anal. Sci., 2008, 24, 263–264 Search PubMed.
- M. R. Bermejo, M. I. Fernandez, E. Gomez-Forneas, A. Gonzalez-Noya, M. Maneiro, R. Pedrido and M. J. Rodriguez, Eur. J. Inorg. Chem., 2007, 24, 3789–3797 CrossRef.
- I. C. Hwang and K. Ha, Z. Kristallogr. N. Cryst. Struct., 2006, 221, 363–364 CAS.
- H. Zhou, Y. Wang, X. Shen, Y. Liu and A. Yuan, Inorg. Chim. Acta, 2014, 423, 115–122 CrossRef CAS.
- X. L. Ma and Z. L. You, Transition Met. Chem., 2008, 33, 961–965 CrossRef CAS.
- G. G. Riopedre, M. I. F. Garcia, A. M. G. Noya, M. A. V. Fernandez, M. R. Bermejo and M. Maneiro, Phys. Chem. Chem. Phys., 2011, 13, 18069–18077 RSC.
- C. G. Zhang, G. H. Tian, Z. F. Ma and D. Y. Yan, Transition Met. Chem., 2000, 25, 270–273 CrossRef CAS.
- M. A. V. Fernandez, M. I. F. Garcia, A. M. G. Noya, M. Maneiro, M. R. Bermejo and M. J. R. Douton, Polyhedron, 2012, 31, 379–385 CrossRef.
- C. H. Li, K. L. Huang, J. M. Dou, Y. N. Chi, Y. Q. Xu, L. Shen, D. Q. Wang and C. W. Hu, Cryst. Growth Des., 2008, 8, 3141–3143 CrossRef CAS.
- T. M. Rajendiran, M. T. Caudle, M. L. Kirk, I. Setyawati, J. W. Kampf and V. L. Pecoraro, J. Biol. Inorg. Chem., 2003, 8, 283–293 CrossRef CAS PubMed.
- T. M. Rajendiran, M. L. Kirk, I. A. Setyawati, M. T. Caudle, J. W. Kampf and V. L. Pecoraro, Chem. Commun., 2003, 82, 824–825 RSC.
- M. Watkinson, M. Fondo, M. R. Bermejo, A. Sousa, C. A. McAuliffe, R. G. Pritchard, N. Jaiboon, N. Aurangzebc and M. Naeem, J. Chem. Soc., Dalton Trans., 1999, 1, 31–42 RSC.
- D. Martinez, M. Motevalli and M. Watkinson, Dalton Trans., 2010, 39, 446–455 RSC.
- K. Ha, Z. Kristallogr. N. Cryst. Struct., 2010, 225, 257–258 CAS.
- H. Kargar, Transition Met. Chem., 2014, 39, 811–817 CrossRef CAS.
- (a) A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117–2121 CrossRef CAS; (b) E. C. Sanudo, W. Wernsdorfer, K. A. Abboud and G. Christou, Inorg. Chem., 2004, 43, 4137–4144 CrossRef CAS PubMed; (c) M. Murugesu, J. Raftery, W. Wernsdorfer, G. Christou and E. K. Brechin, Inorg. Chem., 2004, 43, 4203–4209 CrossRef CAS PubMed.
- (a) R. Karmakar, C. R. Choudhury, G. Bravic, J. P. Sutter and S. Mitra, Polyhedron, 2004, 23, 949–954 CrossRef CAS; (b) L. Lecren, W. Wernsdorfer, Y. G. Li, A. Vindigni, H. Miyasaka and R. Clerac, J. Am. Chem. Soc., 2007, 129, 5045–5051 CrossRef CAS PubMed.
- (a) A. Daolio, A. Pizzi, M. Calabrese, G. Terraneo, S. Bordignon, A. Frontera and G. Resnati, Angew. Chem., Int. Ed., 2021, 60, 20723–20727 CrossRef CAS; (b) A. Bauza and A. Frontera, Chem. - Eur. J., 2022, 28, e202201660 CrossRef CAS; (c) A. Daolio, A. Pizzi, G. Terraneo, A. Frontera and G. Resnati, ChemPhysChem, 2021, 22, 2281–2285 CrossRef CAS.
- (a) T. Basak, R. M. Gomila, A. Frontera and S. Chattopadhyay, CrystEngComm, 2021, 23, 2703–2710 RSC; (b) M. Karmakar, A. Frontera, S. Chattopadhyay, T. J. Mooibroek and A. Bauza, Int. J. Mol. Sci., 2020, 21, 7091 CrossRef CAS PubMed; (c) S. Roy, M. G. B. Drew, A. Bauza, A. Frontera and S. Chattopadhyay, New J. Chem., 2018, 42, 6062–6076 RSC.
- S. Thakur, R. M. Gomila, A. Frontera and S. Chattopadhyay, CrystEngComm, 2021, 23, 5087–5096 RSC.
- K. Ghosh, K. Harms, A. Bauza, A. Frontera and S. Chattopadhyay, CrystEngComm, 2018, 20, 7281–7292 RSC.
- S. Thakur, M. G. B. Drew, A. Franconetti, A. Frontera and S. Chattopadhyay, RSC Adv., 2019, 9, 4789–4796 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, SADABS, V2014/5, Software for Empirical Absorption Correction, University of Göttingen, Institute fur Anorganische Chemieder Universitat, Gottingen, Germany, 1999 Search PubMed.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- A. D. Becker, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef PubMed.
- J. P. Perdew, in Electronic Structure of Solids '91, ed. P. Ziesche and H. Eschrig, Akademie Verlag, Berlin, 1991, p. 11 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- J. C. Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef.
- W. Humphrey, A. Dalke and K. Schultan, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0. Theoretical Chemistry Institute, University of Wisconsin, Madison, WI. 2018 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- H. F. Clausen, M. S. Chevallier, M. A. Spackman and B. B. Iversen, New J. Chem., 2010, 34, 193–199 RSC.
- A. L. Rohl, M. Moret, W. Kaminsky, K. Claborn, J. J. McKinnon and B. Kahr, Cryst. Growth Des., 2008, 8, 4517–4525 CrossRef CAS.
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648–652 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Crystal explorer 2.0, University of Western Australia, Perth, 2007, http://hirshfeldsurfacenet.blogspot.com Search PubMed.
- P. Bhowmik, H. P. Nayek, M. Corbella, N. A. Alcalde and S. Chattopadhyay, Dalton Trans., 2011, 40, 7916–7926 RSC.
- K. Ghosh, S. Banerjee and S. Chattopadhyay, CrystEngComm, 2019, 21, 6859–6868 RSC.
- S. Roy, A. Dey, M. G. B. Drew, P. P. Ray and S. Chattopadhyay, New J. Chem., 2019, 43, 5020–5031 RSC.
- S. Roy, M. G. B. Drew, A. Bauza, A. Frontera and S. Chattopadhyay, New J. Chem., 2018, 42, 6062–6076 RSC.
- K. Ghosh, K. Harms, A. Bauzá, A. Frontera and S. Chattopadhyay, Dalton Trans., 2018, 47, 331–347 RSC.
- X. Yang, R. A. Jones, V. Lynch, M. M. Oye and A. L. Holmes, Dalton Trans., 2005, 849–851 RSC.
- F. Pan, Z. M. Wang and S. Gao, Inorg. Chem., 2007, 46, 10221–10228 CrossRef CAS.
- H. Asada, M. Fujiwara and T. Matsushita, Polyhedron, 2000, 19, 2039–2048 CrossRef CAS.
- F. Z. C. Fellah, J. P. Costes, F. Dahan, C. Duhayon and J. P. Tuchagues, Polyhedron, 2007, 26, 4209–4215 CrossRef.
- T. Kajiwara, M. Nakano, K. Takahashi, S. Takaishi and M. Yamashita, Chem. - Eur. J., 2011, 17, 196–205 CrossRef CAS PubMed.
- Y. Yahsi, H. Kara, L. Sorace and O. Buyukgungor, Inorg. Chim. Acta, 2011, 366, 191–197 CrossRef CAS.
- S. Thakurta, R. J. Butcher, C. J. Gomez-Garcia, E. Garribba and S. Mitra, Inorg. Chim. Acta, 2010, 363, 3981–3986 CrossRef CAS.
- A. Cucos, A. Ursu, A. M. Madalan, C. Duhayon, J. P. Sutter and M. Andruh, CrystEngComm, 2011, 13, 375–390 RSC.
- S. Rayati, S. Zakavi, M. Koliaei, A. Wojtczak and A. Kozakiewicz, Inorg. Chem. Commun., 2010, 13, 203–207 CrossRef CAS.
- N. Sarkar, M. G. B. Drew, K. Harms, A. Bauza, A. Frontera and S. Chattopadhyay, CrystEngComm, 2018, 20, 1077–1086 RSC.
- G. Bhargavi, M. V. Rajasekharan, J. P. Costes and J. P. Tuchagues, Polyhedron, 2009, 28, 1253–1260 CrossRef CAS.
- K. R. Reddy, M. V. Rajasekharan and J. P. Tuchagues, Inorg. Chem., 1998, 37, 5978–5982 CrossRef CAS PubMed.
- S. Sen, S. Mitra, D. Luneau, M. S. El Fallah and J. Ribas, Polyhedron, 2006, 25, 2737–2744 CrossRef CAS.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- D. Cremer, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 498–500 CrossRef.
- J. C. A. Boeyens, J. Cryst. Mol. Struct., 1978, 8, 317–320 CrossRef.
- E. V. Bartashevich and V. G. Tsirelson, Russ. Chem. Rev., 2014, 83, 1181–1203 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. Figures from S1–S9, Cif of complexes 1–4. CCDC 2260214–2260217 for complexes 1–4, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra04044e |
| This journal is © The Royal Society of Chemistry 2023 |