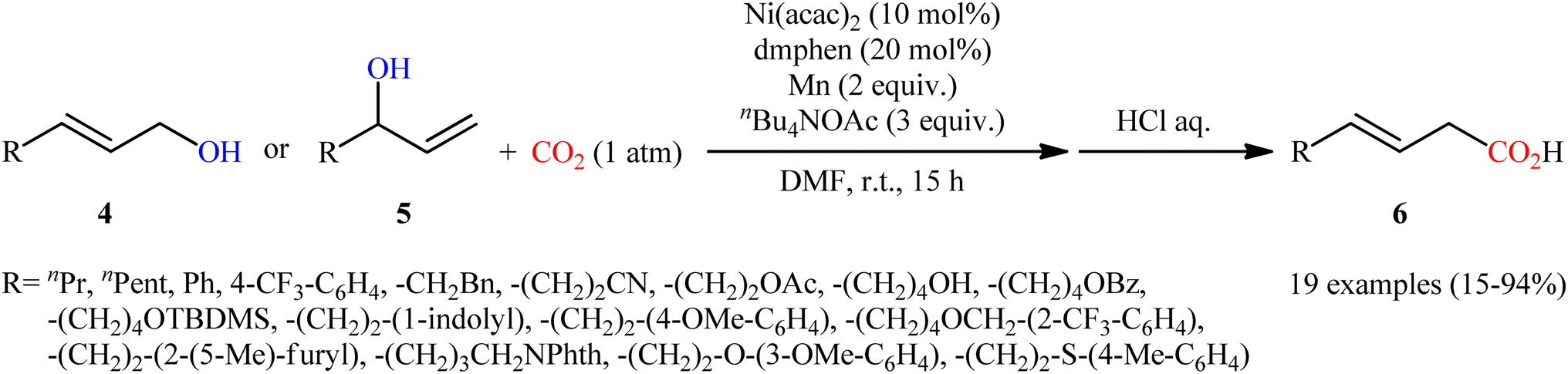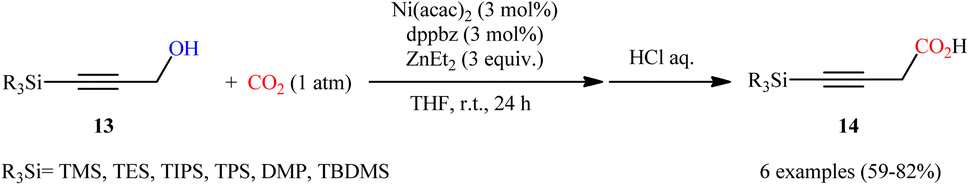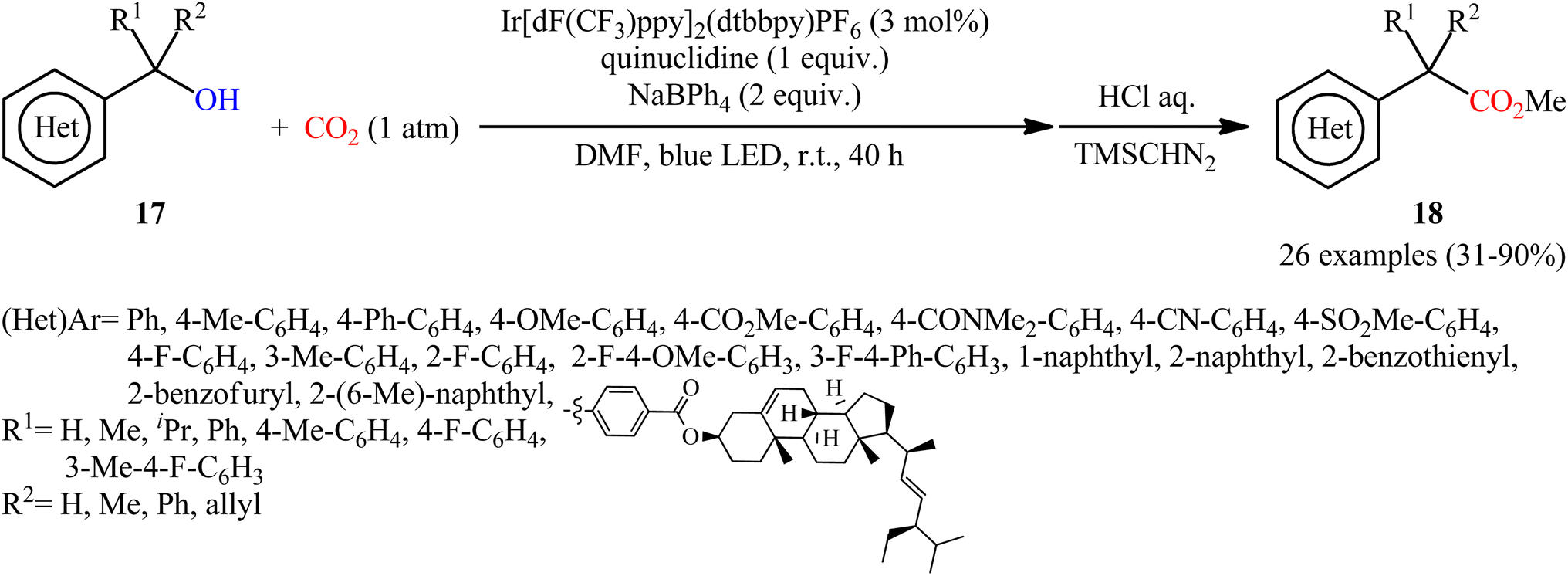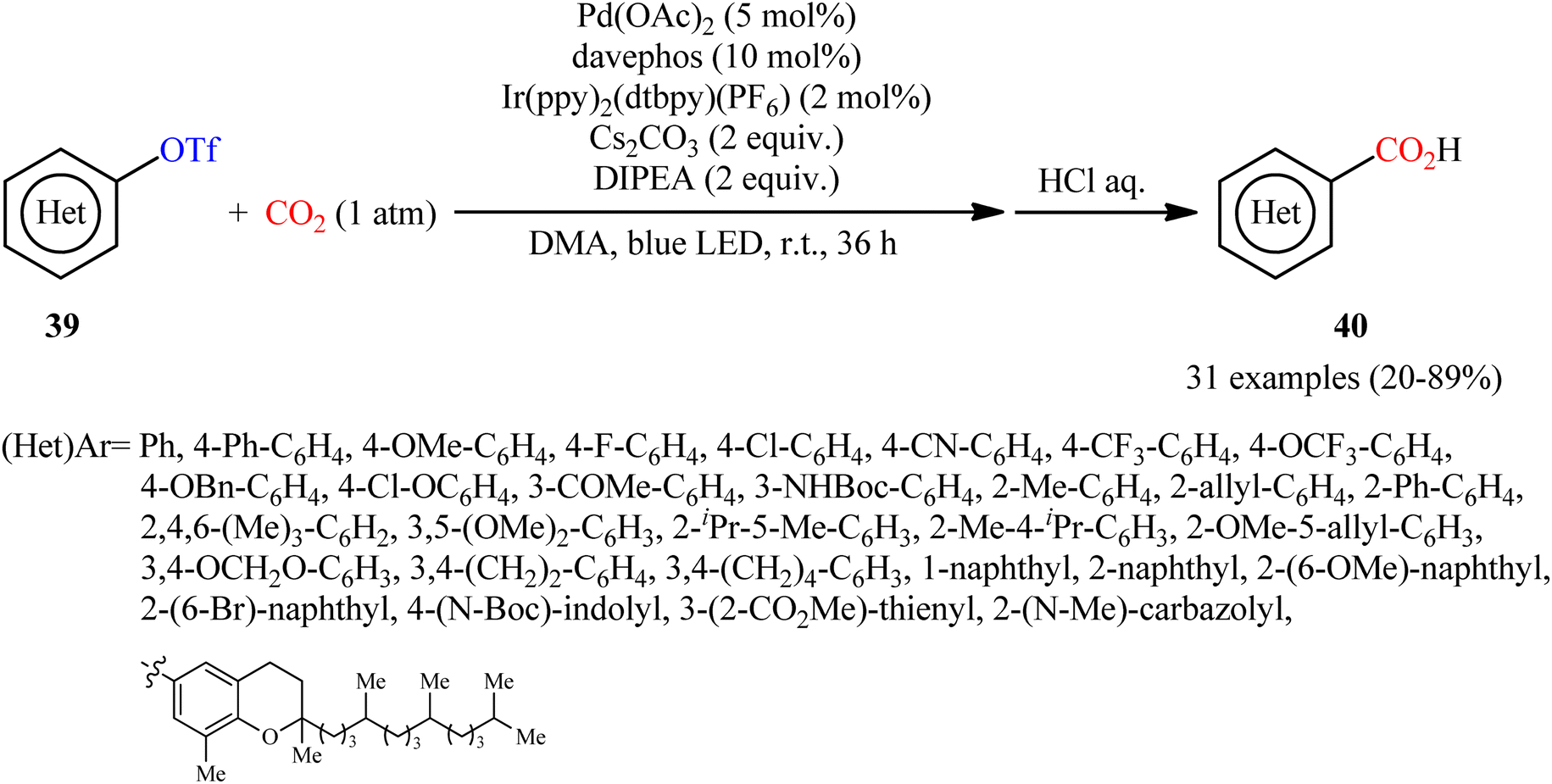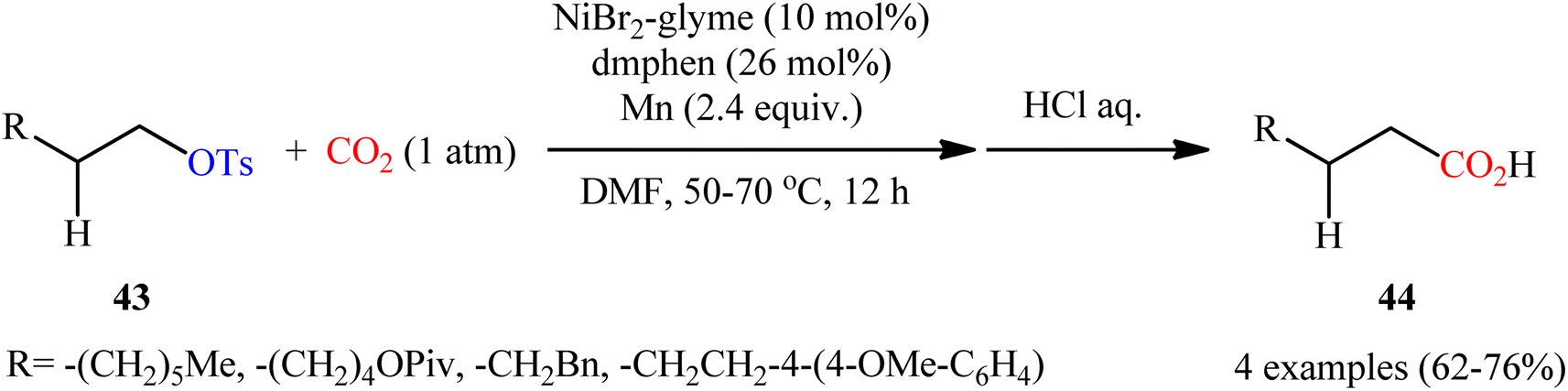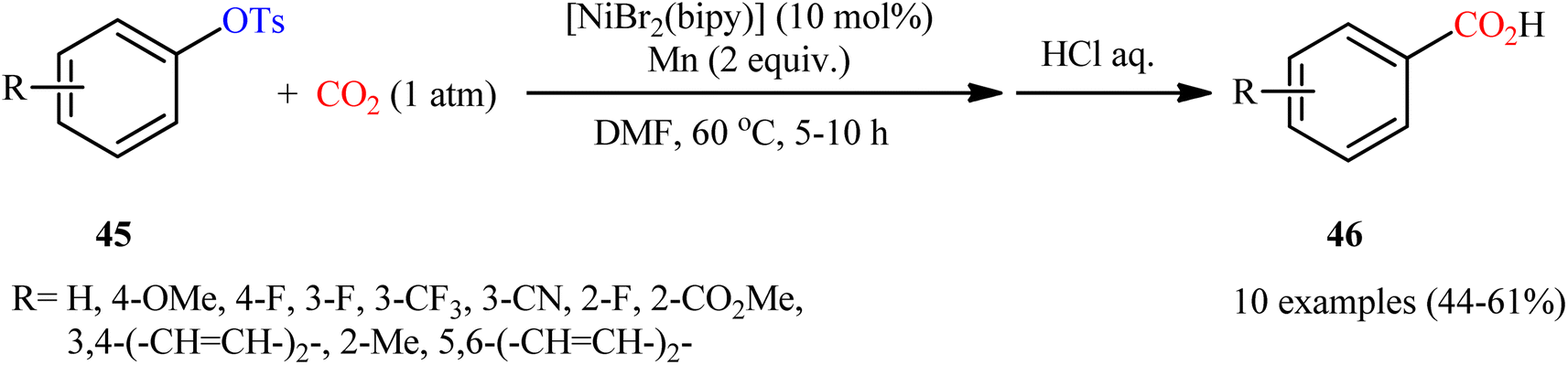DOI:
10.1039/D3RA04073A
(Review Article)
RSC Adv., 2023,
13, 32502-32517
Recent progress in reductive carboxylation of C–O bonds with CO2
Received
19th June 2023
, Accepted 22nd September 2023
First published on 3rd November 2023
Abstract
Transformation of carbon dioxide (CO2) into value-added organic compounds has attracted increasing interest of scientific community in the last few decades, not only because CO2 is the primary greenhouse gas that drives global climate change and ocean acidification, but also because it has been regarded as a plentiful, nontoxic, nonflammable and renewable one-carbon (C1) feedstock. Among the various CO2-conversion processes, carboxylation reactions represent one of the most beautiful and attractive research topics in the field, since it offers the possibility for the construction of synthetically and biologically important carboxylic acids from various easily accessible (pseudo)halides, organosilicon, and organoboron compounds. The purpose of this review is to summarize the available literature on deoxygenative carboxylation of alcohols and their derivatives utilizing CO2 as a carboxylative reagent. Depending on the C–O compounds employed, the paper is divided into five major sections. The direct dehydroxylative carboxylation of free alcohols is discussed first. This is followed by reductive carboxylation of carboxylates, triflates, and tosylates. In the final section, the only reported example on catalytic carboxylation of fluorosulfates will be covered. Notably, special attention has been paid on the mechanistic aspects of the reactions that may provide new insights into catalyst improvement and development, which currently mainly relies on the use of transition metal catalysts.
1. Introduction
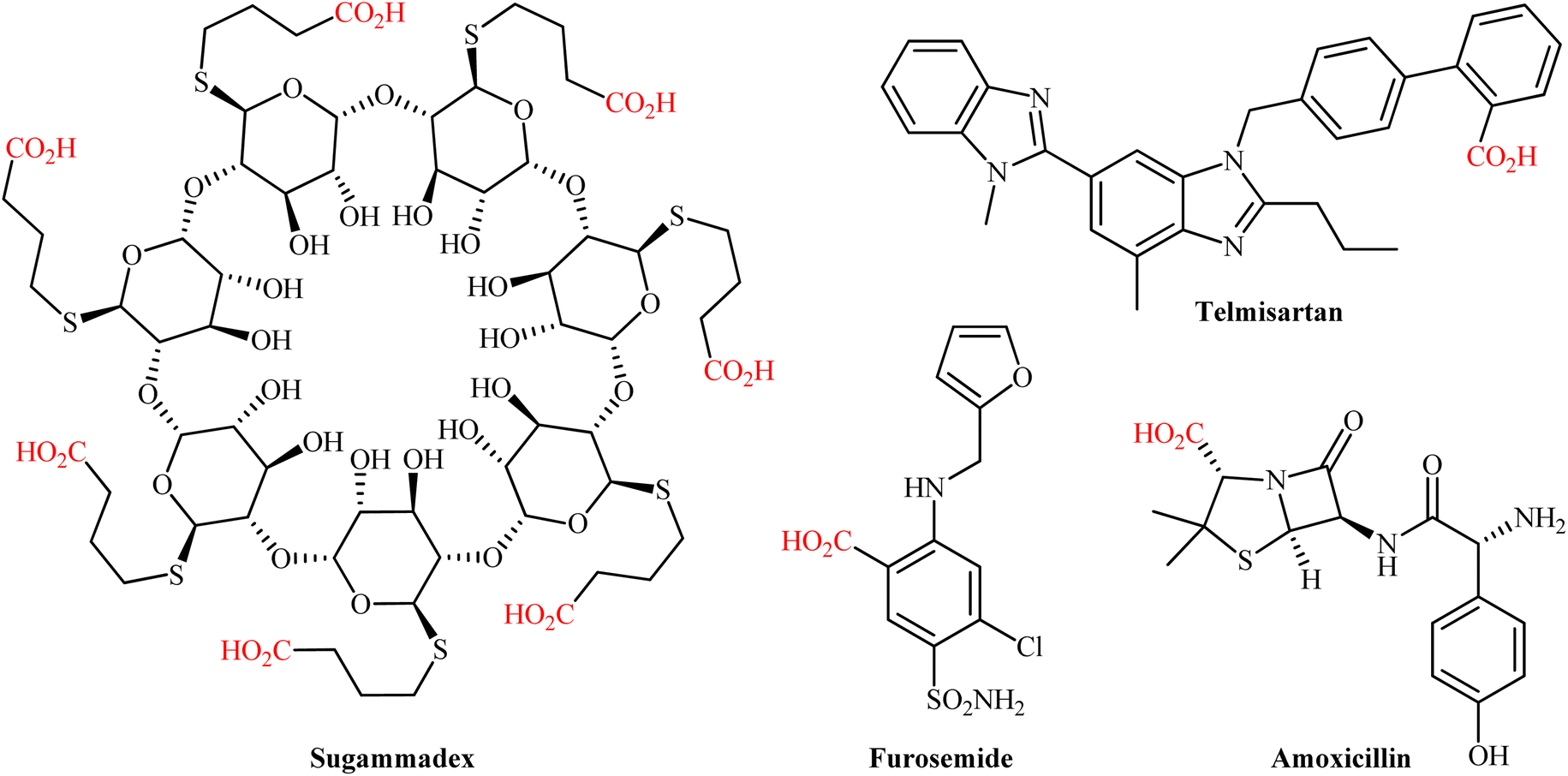
The catalytic fixation of carbon dioxide (CO2) into value-added organic compounds is an extremely important and hot research topic in synthetic organic chemistry and green chemistry from the viewpoint of developing this plentiful greenhouse gas as an inexpensive, nontoxic, and renewable one-carbon (C1) feedstock.1 Among the various CO2-fixation reactions,2 carboxylation and decarboxylation reactions represent one of the most attractive and promising transformations, since it offers the possibility for the construction of valuable carboxylic acids, from various organic substrates in one step and eco-friendly manner.3 Needless to say that compounds containing the carboxylic acid functional group play a cardinal role in drug discovery programs due to their broad spectrum of biological activities including diuretic, antibacterial, antiretroviral, antihypertensive, anti-inflammatory, antiarrhythmic, antipsychotic, and antihistamine activities (Scheme 1). The classical synthesis of carboxylic acids from CO2 involves the stoichiometric conversion of an electrophilic organic halide to a nucleophilic Grignard reagent followed by carboxylation and subsequent acidic workup of the resulted halomagnesium carboxylate.4 However, poor compatibility of Grignard reagents with a verity of functional groups, limited the utility of this synthetic procedure. In order to bypass the need for highly reactive and difficult to handle Grignard reagents, the direct catalytic carboxylation of organohalides with CO2 has thus been developed as one of the most sustainable and efficient synthesis strategies to carboxylic acid derivatives.5 Although, considerable successes have been achieved using this synthetic approach, some inherent limitations such as the high cost of organic bromides and iodides, low reactivity of organic chlorides, and environmental toxicity of some organic halides, might limit the application profile of this page of carboxylic acid synthesis.6
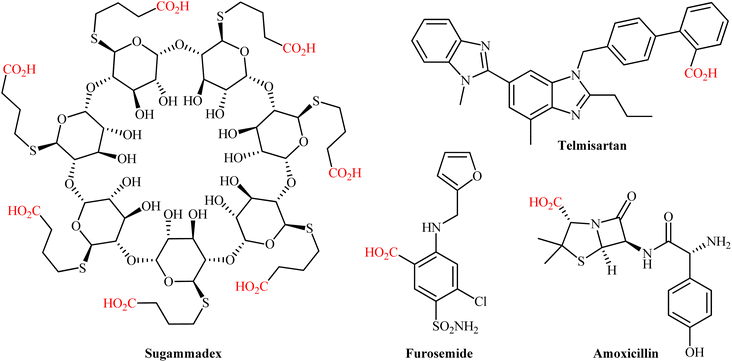 |
| | Scheme 1 Selected examples of FDA-approved drugs containing carboxylic acid moiety. | |
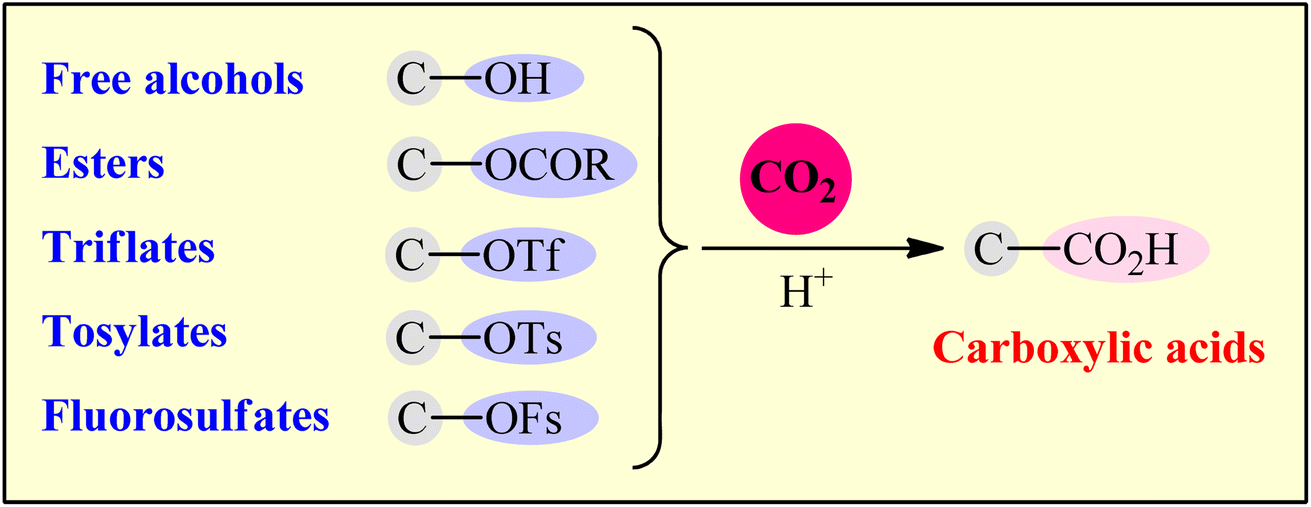
Considering the above considerations, from a sustainable chemistry point of view, there is a need for new protocols for the preparation of carboxylic acids from CO2 that are truly efficient, high yielding, eco-friendly, byproduct-free, and responsive to mild reaction conditions. In this regard, alcohols and their activated derivatives (e.g., carboxylates, tosylates, and fluorosulfates), which, as compared to organohalides, offer more sustainable starting materials because most of them are easily accessible from biomass,7 have recently applied as viable alternative to halides in CO2-fixation reactions. Needless to say that not all activated alcohols meet the criteria of green chemistry and may also be potentially toxic (e.g., triflates, due to the formation of genotoxic triflic acid).
Although several nice reviews articles were briefly covered some aspects of this topic,8 a comprehensive review on this highly active research arena has not appeared in the literature to date. As a part of our ongoing review papers of the CO2-fixation reactions,2b,c,3c–e,9 herein, we will highlight the most important developments on the preparation of carboxylic acid derivatives through the reductive carboxylation of respective (activated) alcohols with CO2 (Fig. 1).
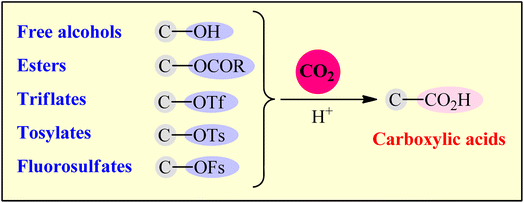 |
| | Fig. 1 Reductive carboxylation of C–O bonds with CO2. | |
2. Carboxylation of free alcohols
The direct dehydroxylative functionalization of free alcohols has recently become one of the most impactful areas of research in synthetic chemistry because it is an economic, powerful and eco-friendly strategy for the fabrication of various valuable functionalized organic compounds from inexpensive, environmentally benign, and naturally abundant feedstocks without pre-functionalization of starting materials and isolation of intermediates.10
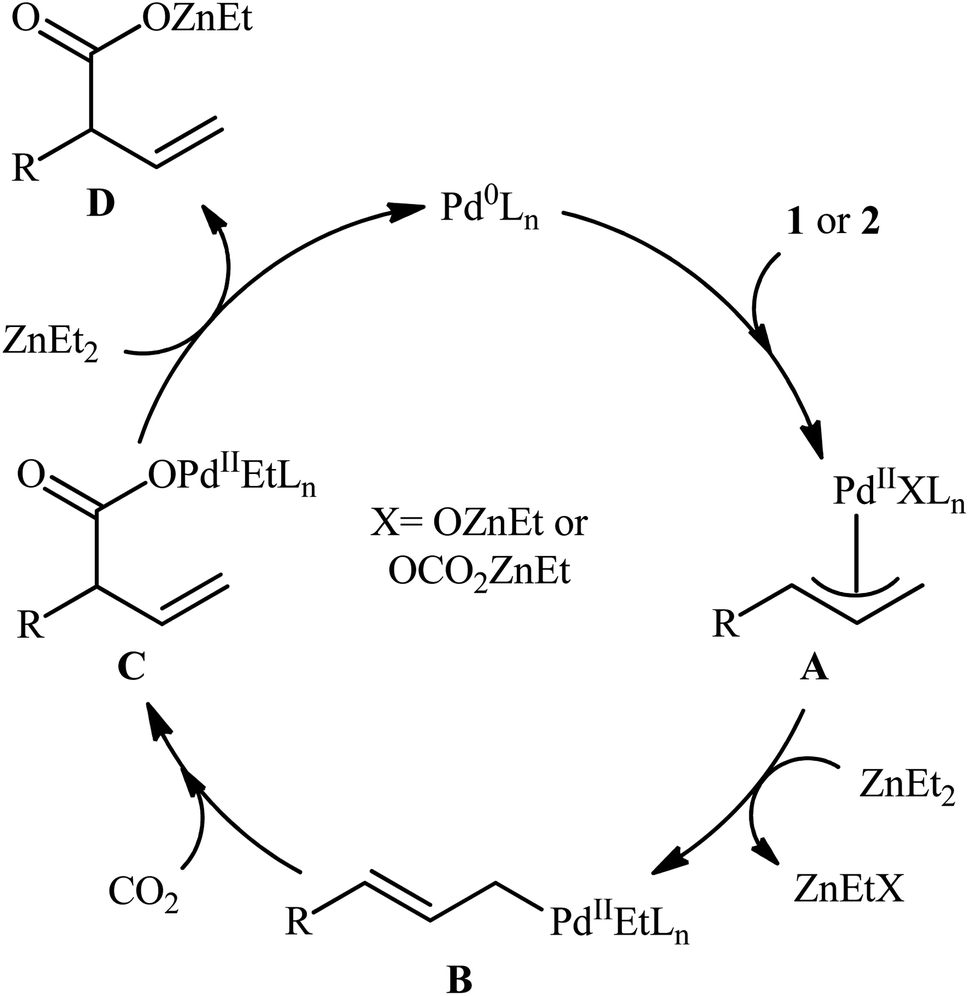
One of the earliest reports of the direct dehydroxylative carboxylation of allylic alcohols using CO2 as the carboxyl source was published by Mita, Higuchi, and Sato in 2015,11 who unfolded that the treatment of allylic alcohols with a catalytic amount of PdCl2/PPh3 under a CO2 atmosphere in the presence of ZnEt2 as a stoichiometric transmetalation reagent/reductant, resulted in the regioselective formation of branched α,β-unsaturated carboxylic acids in good to excellent yields after hydrolysis. To facilitate the isolation process, the obtained carboxylic acids were derivatized into their methyl esters by methyl esterification with TMSCHN2. Thus, a library of branched α,β-unsaturated methyl esters 3 were efficiently synthesized through the sequential carboxylation of various linear and branched allylic alcohols 1 and 2 with CO2 and subsequent methyl esterification of the generated carboxylate with TMSCHN2 (Scheme 2). In order to demonstrate the synthetic applicability of their mythology, the authors performed the synthesis of baclofen, a pain-relieving medication, starting from 2-(4-chlorophenyl)prop-2-en-1-ol using their carboxylation strategy as a key step. A possible mechanism for this Pd-catalyzed dehydroxylative carboxylation is illustrated in Scheme 3. The reaction starts with the generation of η3-allylpalladium A through the activation of allylic alcohol 1/2 by Lewis acidic ZnEt2 or by carbonate formation with CO2. Subsequently, this intermediate undergoes transmetalation with ZnEt2 to give the nucleophilic η1-allylpalladium B, which after nucleophilic attack to CO2 at the γ-position affords palladium carboxylate C. Finally, reduction of this complex by ZnEt2 leads to the formation of zinc carboxylate D and regenerates Pd0.
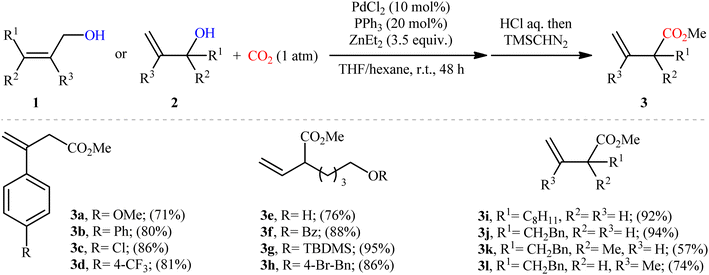 |
| | Scheme 2 Pd-catalyzed carboxylation of allylic alcohols 1 and 2 with CO2 developed by Mita et al. | |
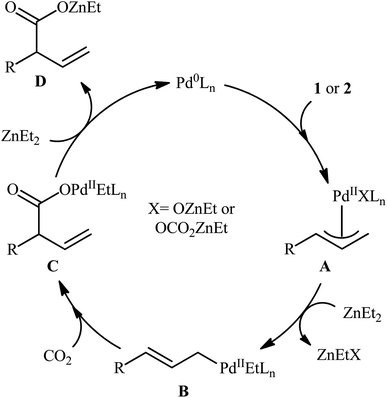 |
| | Scheme 3 Plausible mechanism for the reaction in Scheme 2. | |
Two years later, Mei's research group described an alternative carboxylation of allylic alcohols with atmospheric CO2 in the absence of pyrophoric Et2Zn using the ability of nBu4NOAc to facilitate C–O bond-cleavage.12 Thus, by using Ni(acac)2/2,9-dimethyl-1,10-phenanthroline (dmphen)/Mn powder/nBu4NOAc combination as a catalytic system in DMF, various linear and branched allylic alcohols 4 and 5, respectively, carboxylated regioselectively to afford branched α,β-unsaturated carboxylic acids 6 exclusively (Scheme 4). Notably, the reaction displayed high degree of stereoselectivity in which both cis-allylic and trans-allylic alcohols predominantly provided (E)-configured carboxylic acids, which suggests that the reaction occurred via π-allylnickel intermediates. Interestingly, the electronic effects of the substituents had no significant influence on the outcome of the reaction. On the other hand, the steric effect was very strong, as the allylic alcohols wherein the alkene is conjugated with an arene or is trisubstituted afforded low yields of the desired products. In this study, the authors also provided further examples of α,β-unsaturated carboxylic acids preparation via hydrogenation of propargylic alcohols to allylic alcohols using water as an H atom source, followed by carboxylation under the identical conditions.
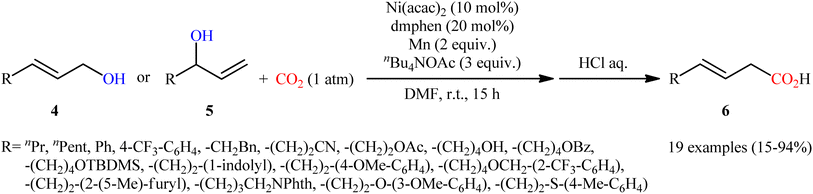 |
| | Scheme 4 Ni-catalyzed carboxylation of allylic alcohols 4 and 5 with CO2. | |
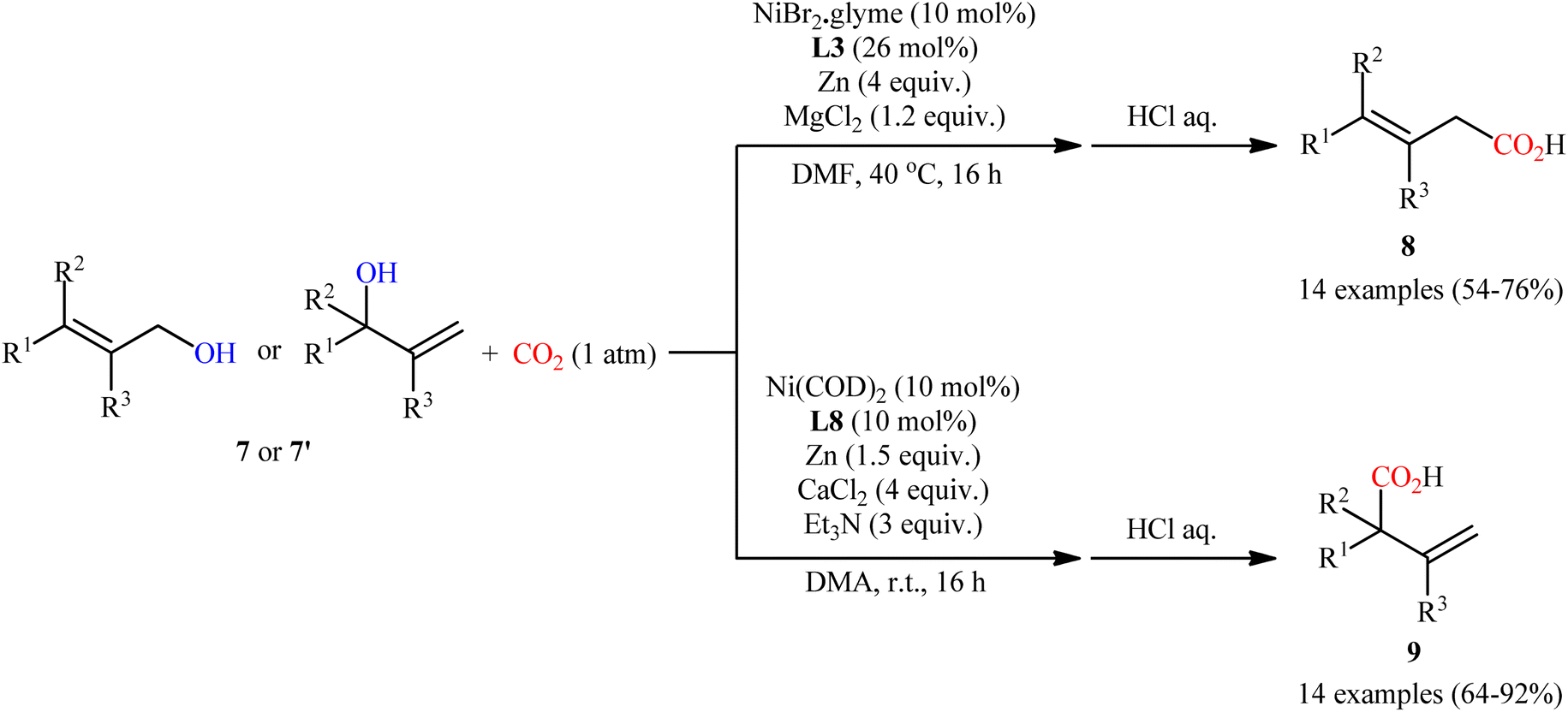
Concurrently, Martin and co-workers screened a series of bi- and tridentate ligands for the ligand-controlled, site-selective Ni-catalyzed carboxylation of allylic alcohols 7 (or 7′) with CO2.13 Their results revealed that different ligands can control site-selectivity pattern. Therefore, the use of bidentate ligands was selective toward linear products 8. Among the bidentate ligands examined, commercially available bathocuproine (2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline) L3 afforded the highest regioselectivity towards linear derivatives. On the other hand, tridentate 4,4′′,6,6′′-tetramethyl-2,2′:6′,2′′-terpyridine L8 provided exclusively branched products 9 (Scheme 5).
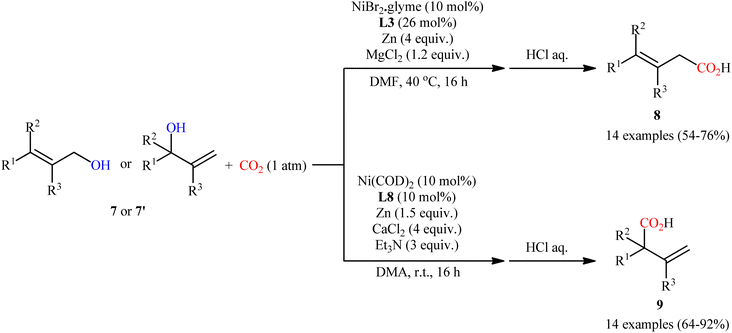 |
| | Scheme 5 Ni-catalyzed, switchable site-selective carboxylation of allylic alcohols 7 with CO2 by using either a Ni/L3 or Ni/L8 couple. | |
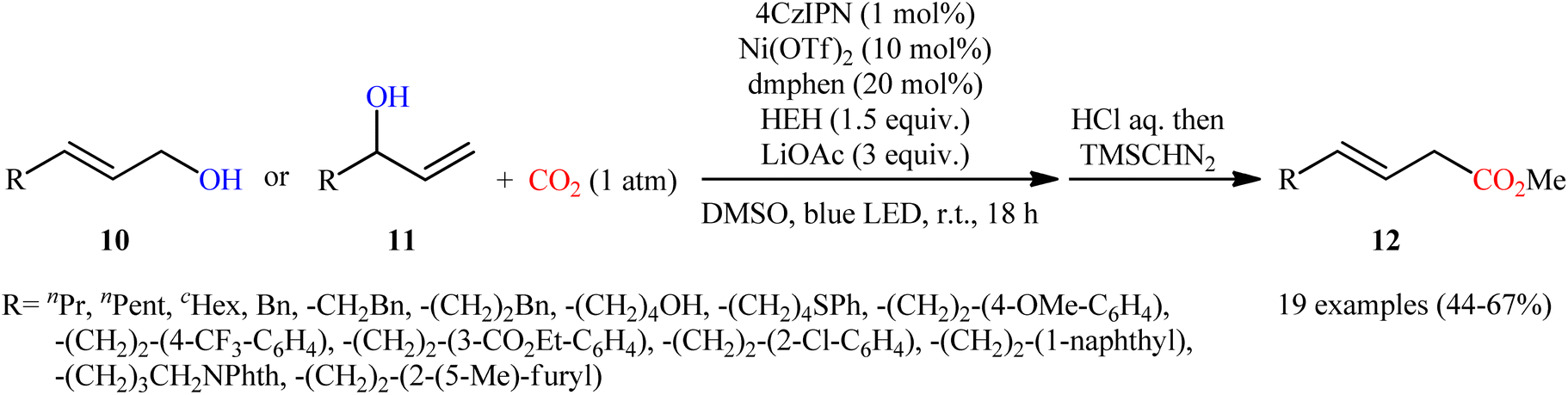
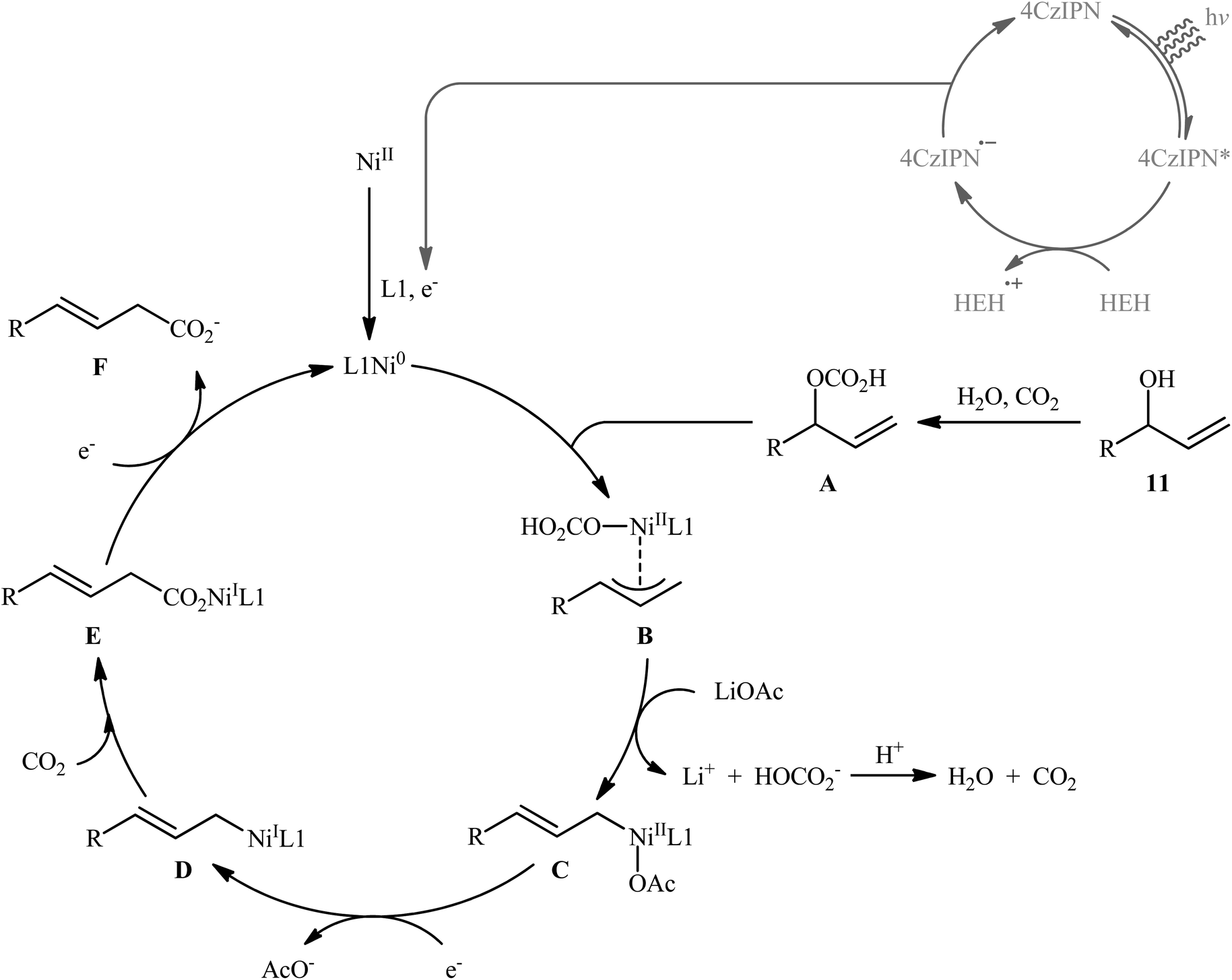
Very recently, Xi and co-workers disclosed that the merge of a Ni catalyst with a photoredox catalysis can be used as effective metallaphotoredox catalytic system for regioselective carboxylation of allylic alcohols with CO2 without consuming any stoichiometric metallic reductant.14 The optimal reaction employed 4CzIPN/Ni(OTf)2 as the metallaphotoredox catalyst, dmphen as a ligand, Hantzsch ester (HEH) as the reductant and LiOAc as an additive. Under optimized conditions, a variety of linear α,β-unsaturated carboxylic acids 12 were prepared in moderate to good yields from the corresponding mono-substituted allylic alcohols 10 and 11 (Scheme 6). However, π-conjugated substrates were sluggish to participate in this protocol. Based on several control experiments, the authors suggested that this C–C bond forming reaction proceeds through the following key steps (Scheme 7): (i) excitation of 4CzIPN under visible light irradiation to form excited state of 4CzIPN* (ii) reductive quenching of 4CzIPN* by HEH to generate 4CzIPN˙−; (iii) reduction of NiII in the presence of L1 through a single electron transfer (SET) from 4CzIPN˙− to form to L1Ni0 and regenerate ground state 4CzIPN; (iv) preactivation of allylic alcohol to the corresponding allylic hydrogen carbonate A in the presence of water and CO2; (v) coordination and oxidative addition of active catalytic species to the allylic hydrogen carbonate A to give π-allylnickel intermediate B; (vi) ligand exchange of B with LiOAc to generate intermediate C; (vii) reduction of complex C by 4CzIPN˙− to produce NiI intermediate D; (viii) insertion of CO2 into the Ni–C bond of D to afford nickel carboxylate E; and (ix) single electron reduction of E to provide the corresponding carboxylate F and regenerate the catalytic Ni0.
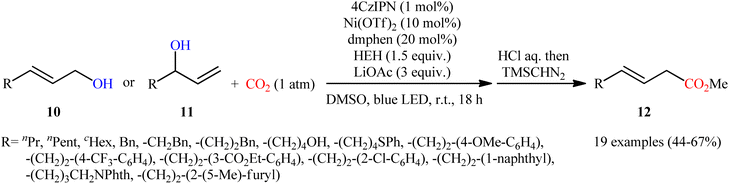 |
| | Scheme 6 Visible-light-driven carboxylation of allylic alcohols 10 and 11 by the combined use of nickel and photoredox catalysts. | |
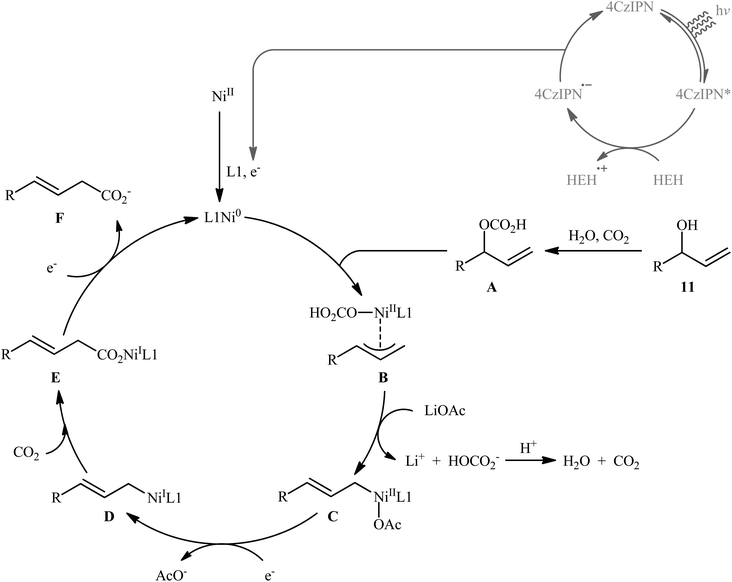 |
| | Scheme 7 Possible mechanism of dual photoredox/nickel-catalyzed carboxylation of allylic alcohols 11 with CO2. | |
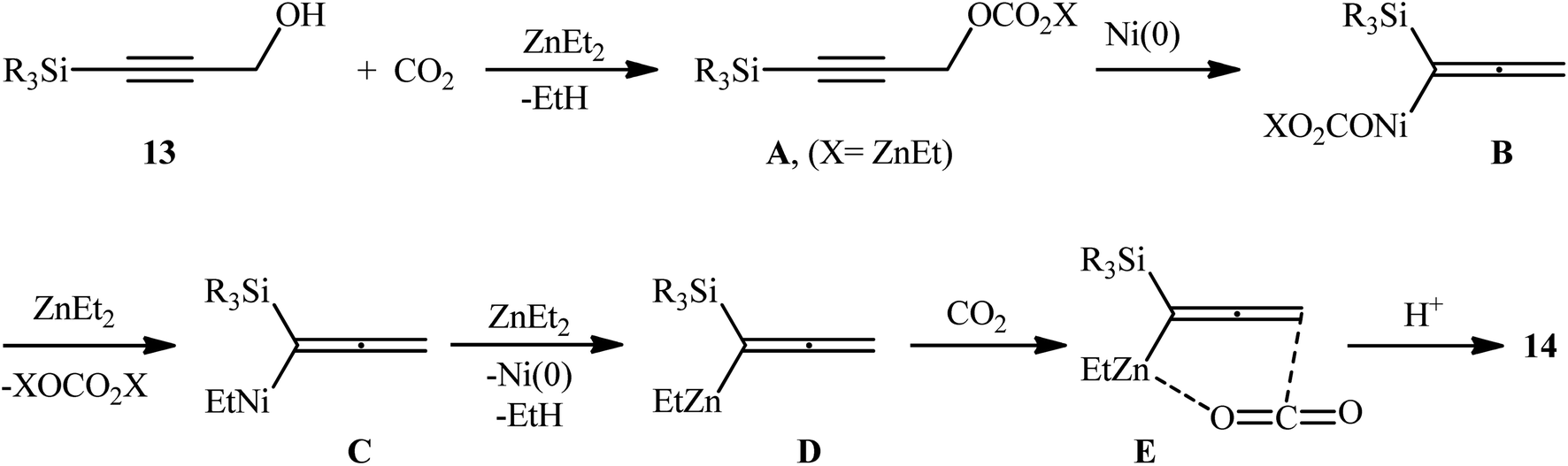
In a significant contribution in this field, Kimura and co-workers found that treatment of propargylic alcohols 13 containing a silyl group at the terminal position with CO2 (1 atm) in the presence of Ni(acac)2/dppbz/Et2Zn combination as a catalytic system, resulted in the formation of the corresponding β,γ-alkynyl carboxylic acids 14 in high yields within 24 h (Scheme 8).15 The results indicated that the presence of a silyl group at the terminal acetylenic carbon atom was crucial for this coupling reaction. No products were obtained by 3-phenyl and 3-ethyl substituted propargyl alcohols under the same reaction conditions. Furthermore, the presence of functionalities at the C1-position of 3-silylated propargyl alcohols prevented carboxylation, and no reaction proceeded at all. According to the author proposed mechanism (Scheme 9), this reaction proceeds through the formation of an allenyl metal intermediate and the presence of silyl group is important for stabilization of this key intermediate. Moreover, it was suggested that CO2 playing a dual role in this transformation; a C1 source and an accelerator for the C–O bond cleavage.
 |
| | Scheme 8 Ni-catalyzed carboxylation of propargylic alcohols 13 with CO2. | |
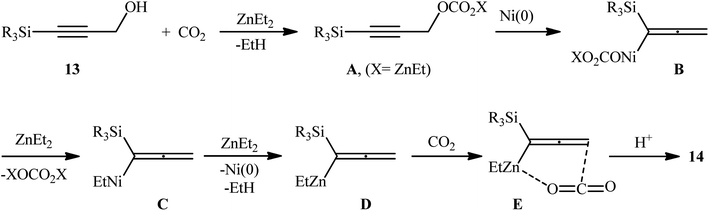 |
| | Scheme 9 Proposed mechanistic pathway for the formation of β,γ-alkynyl carboxylic acids 14. | |
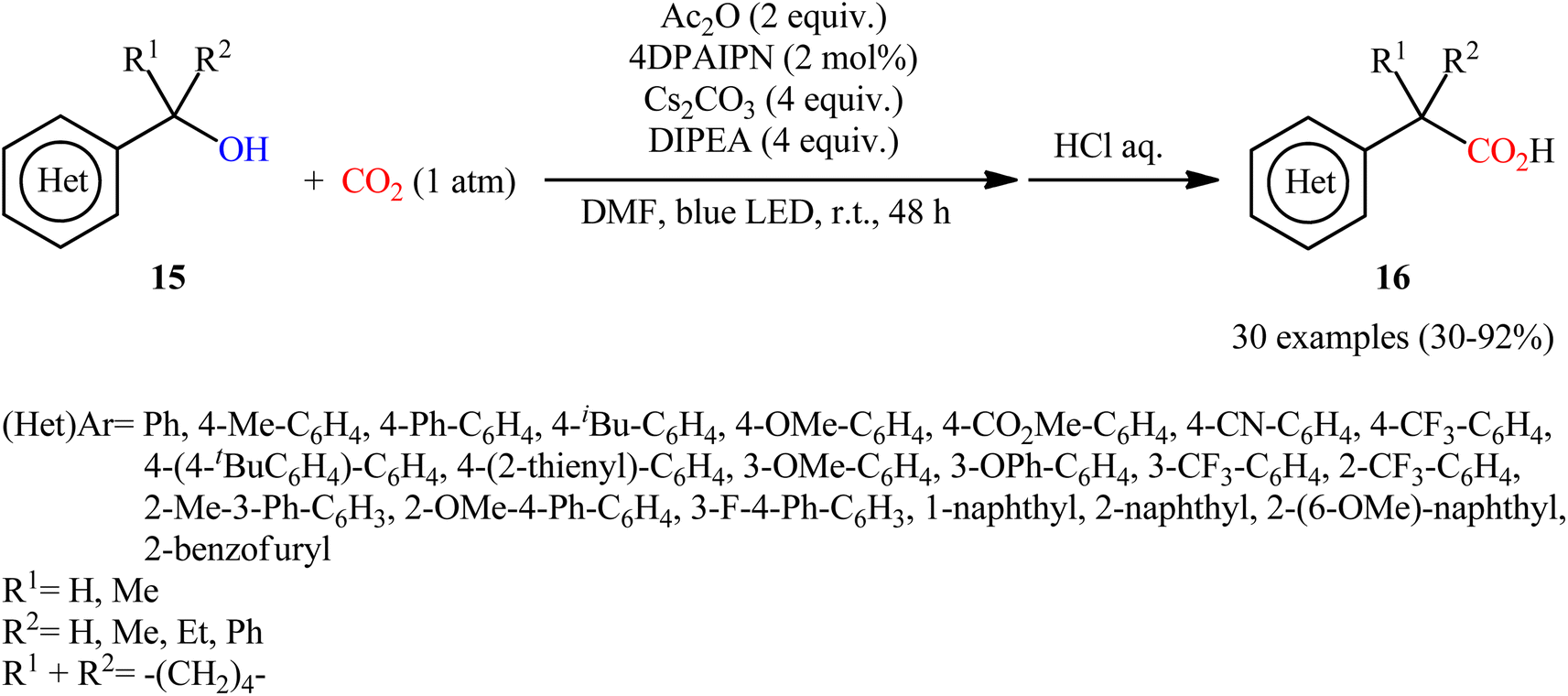
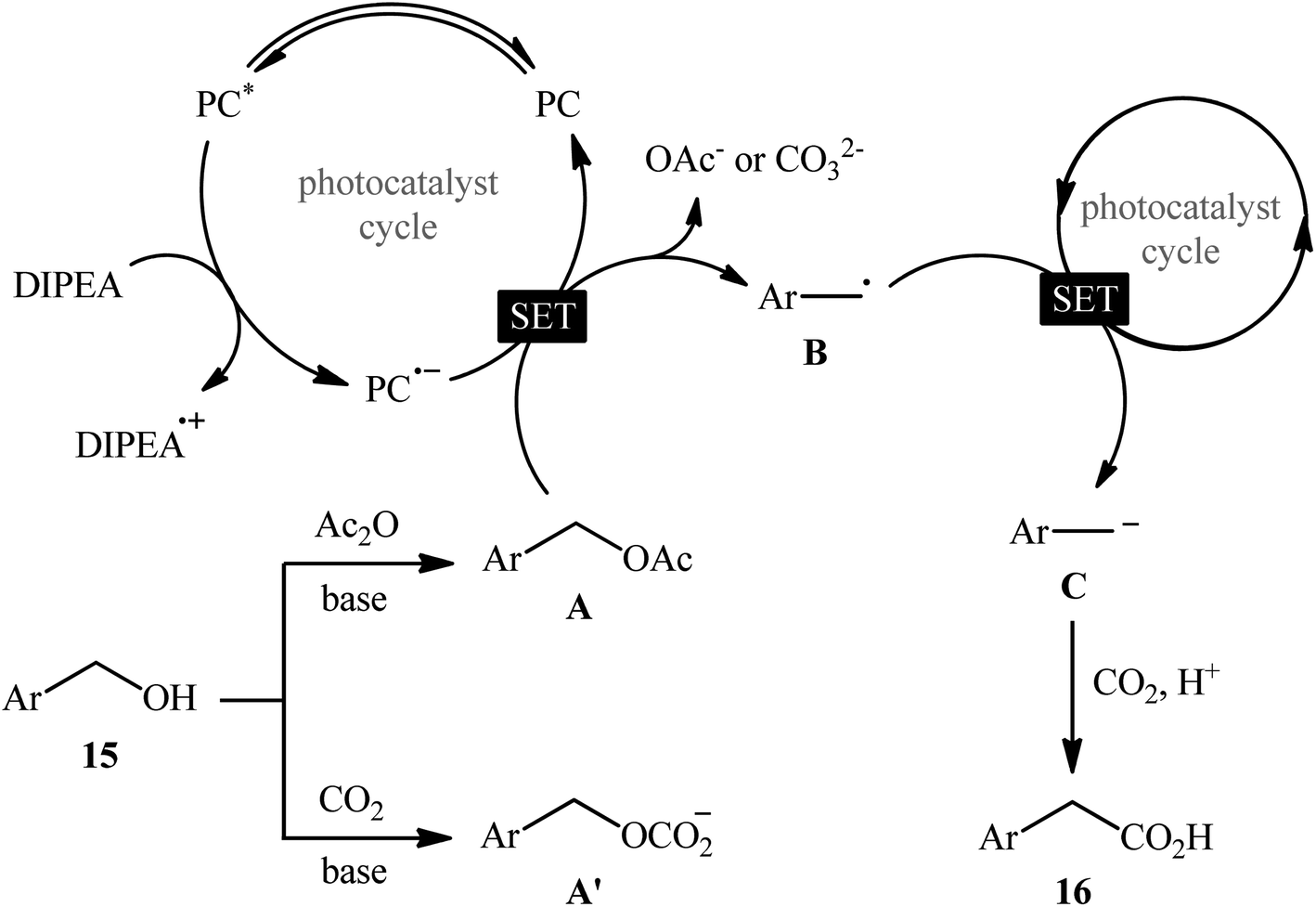
In 2022, Liao, Yu and co-workers described an elegant visible-light photoredox-catalyzed dehydroxylative carboxylation of benzyl alcohols 15 with atmospheric CO2 under ambient temperature.16 No transition-metal was used and only 2 mol% of organic dye 2,4,5,6-tetrakis(diphenylamino)isophthalonitrile (4DPAIPN) was enough for preparation of a panel of 30 functionalized aryl acetic acids 16 in 30–92% yields (Scheme 10). Various primary-, secondary-, as well as tertiary benzylic alcohols were well tolerated under the reaction conditions, thus indicating the broad applicability of this synthetic strategy. It is worthwhile to note that beside benzylic alcohols, benzylic carbonates were also successfully applied as C–O compounds under the identical conditions. Mechanistically, after a series of control experiments, it was confirmed that this CO2-fixation reaction most likely proceeds via a radical pathway through the key benzylic radical intermediate B (Scheme 11).
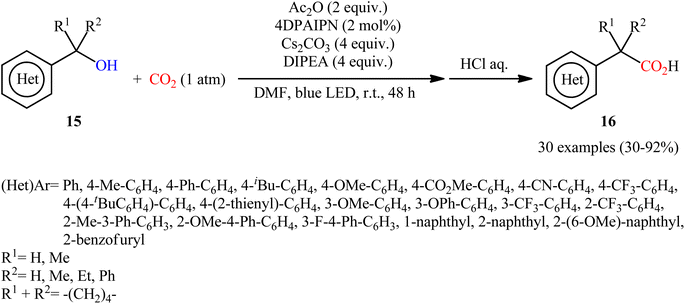 |
| | Scheme 10 Visible-light photoredox-catalyzed carboxylation of benzylic alcohols 15 with CO2. | |
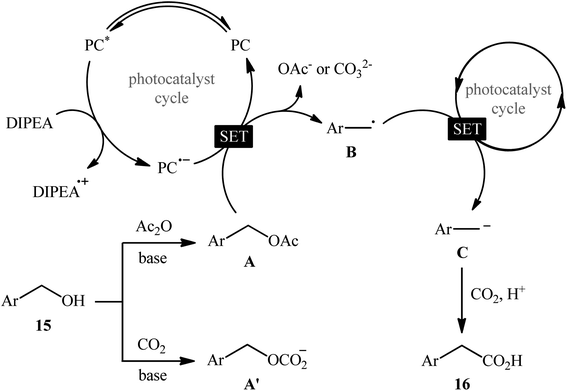 |
| | Scheme 11 Mechanistic explanation for the formation of aryl acetic acids 16. | |
Another independent benzylic alcohol carboxylation method was published by Lan, Xia and co-workers via activation of free alcohols with neutral diphenyl boryl radical (Ph2B˙) generated from sodium tetraphenylborate (NaBPh4) under mild visible light photoredox conditions.17 They showed that the treatment of various benzyl alcohols 17 with NaBPh4 under CO2 atmosphere and visible light irradiation in the presence of a catalytic amount of Ir[dF(CF3)ppy]2(dtbbpy)PF6 afforded the corresponding carboxylic acid derives 18 in modest to high yields within 40 h (Scheme 12). Interestingly, all the three kinds of benzyl alcohols (primary, secondary, and tertiary benzyl alcohols) were applicable to this reaction. Intriguingly, this synthetic strategy has also been successfully applied for the preparation of various commercialized drugs (i.e., flurbiprofen, naproxen, and felbinac) from the corresponding free alcohols. However, nonbenzylic alcohols, 1-phenylpropan-2-ol, and 2-methyl-1-phenylpropan-2-ol, did not furnish the desired products.
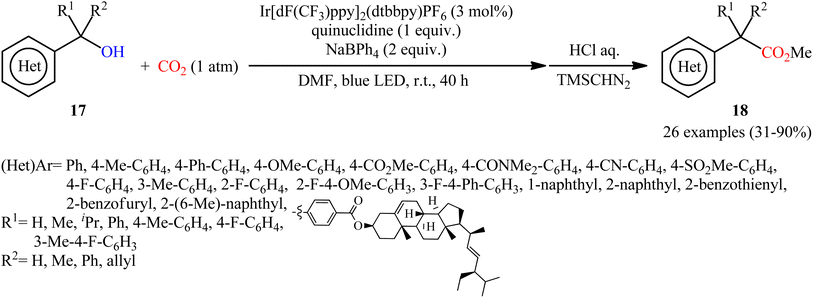 |
| | Scheme 12 Lan–Xia's synthesis of carboxylic acid derivatives 18. | |
In summary, despite significant progress in recent years, numerous challenges remain in this field of expertise. For example, the majority of methodologies are relayed on the use of Ni and Pd catalysts. Therefore, these approaches cannot be attractive from an industrial perspective because Pd and Ni are expensive and toxic.18 Although in order to overpass these limitations, recently, Liao, Yu and co-workers16 developed an innovative transition metal-free method for the photoredox-catalyzed dehydroxylative carboxylation of free alcohols. However, their synthetic strategy appears to be limited to only benzylic alcohol derivatives. Thus, further research should be focused on development of inexpensive, non-toxic, and highly active catalytic systems with much broader substrate scope. Along this line, first-row transition metals are low-cost and naturally abundant and could be promising candidates for catalyst development in dehydroxylative carboxylation of free alcohols.
3. Carboxylation of esters
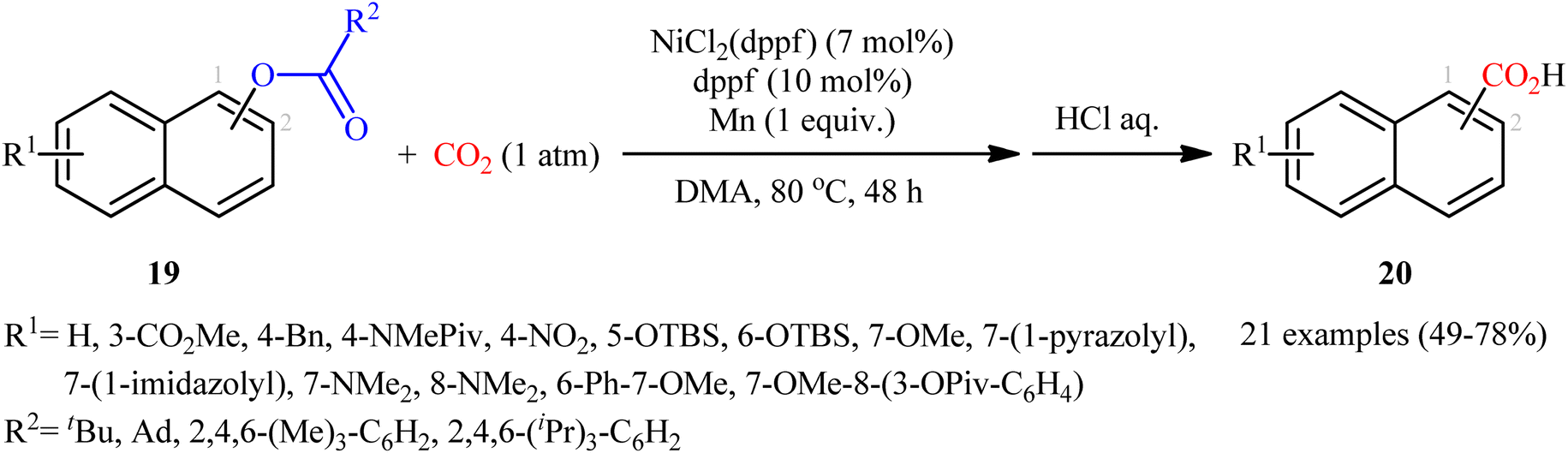
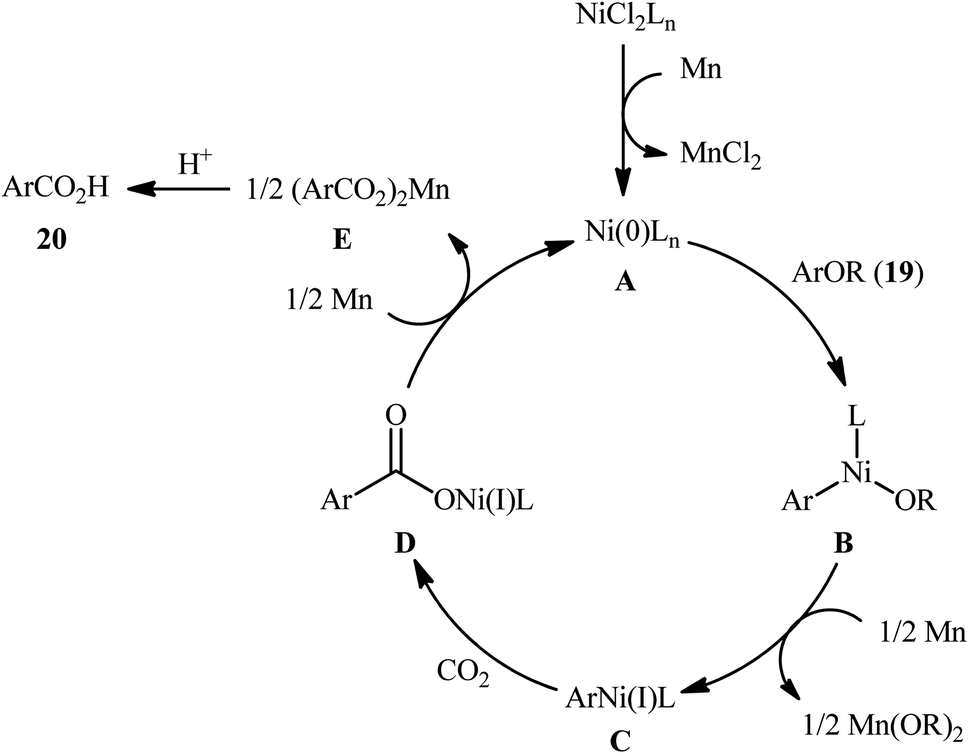
Reductive functionalization of alcohols through the preactivation of C–OH bonds by converting to the reactive esters is a well-documented and useful synthetic protocol.19 In 2014, the group of Martin documented one of the earliest methods for the reductive carboxylation of esteric C–O bonds with CO2 to generate carboxylic acids.20 Here, NiCl2(dppf) [dppf = 1,1′-bis(diphenylphosphino)ferrocene] was used as the catalyst and Mn powder as a reducing reagent. The reaction scope appears to be broad as various aromatic esters 19 with either electron-donating or electron-withdrawing substituents were tolerated and provided the corresponding carboxylic acids 20 in moderate to good yields (Scheme 13). However, the results indicated that the steric effects of the substituents on the acyl terminus played a crucial role on the success of this transformation. Generally, the presence of a bulky substituent (e.g., tbutyl, adamantyl) on the acyl terminus is necessary for the reaction to proceed efficiently; esters bearing less hindered acyl substrates such as acetate and carbamate failed to enter into this C–C bond forming reaction. The authors explained this observation by possible stabilization of the transient Ni species within the catalytic cycle by the bulkier substituent on the acyl terminus which might prevent decomposition pathways. Notably, carboxylation of more challenging C(sp3)–O bonds were also found to be feasible by this synthetic strategy, as exemplified by the preparation of 28 benzylic carboxylic acids from the respective benzyl esters. It should be mentioned that unlike the carboxylation of C(sp2)–O bonds that was found to be specific for sterically demanding ester derivatives, the nature of the leaving group on the carboxylation of C(sp3)–O bonds did not have such a profound effect on reactivity. Scheme 14 depicted the possible mechanistic cycle for this reaction. First, reduction of the Ni(II) precatalyst with Mn generates active catalytic species A, which, undergo oxidative addition into the C–O bond of ester 19 to afford the Ni(II) intermediate B. Subsequently, reduction of the newly formed complex B in the presence of Mn leads to the formation of more nucleophilic Ni(I) species C. Next, the insertion of CO2 into the nickel–carbon bond of complex C yields the nickel carboxylate D. Finally, transmetalation of intermediate D with Mn regenerates the active Ni(0)L species A and the manganese carboxylate E that upon hydrolytic workup delivers the expected carboxylic acid 20.
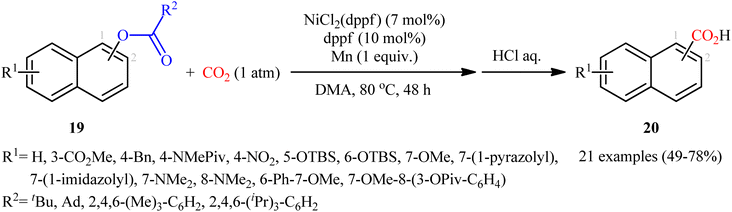 |
| | Scheme 13 Ni-catalyzed carboxylation of esteric C–O bonds with CO2. | |
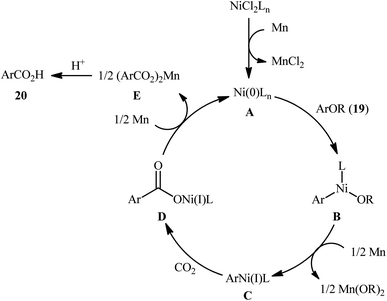 |
| | Scheme 14 Proposed mechanism for the formation of aromatic carboxylic acids 20. | |
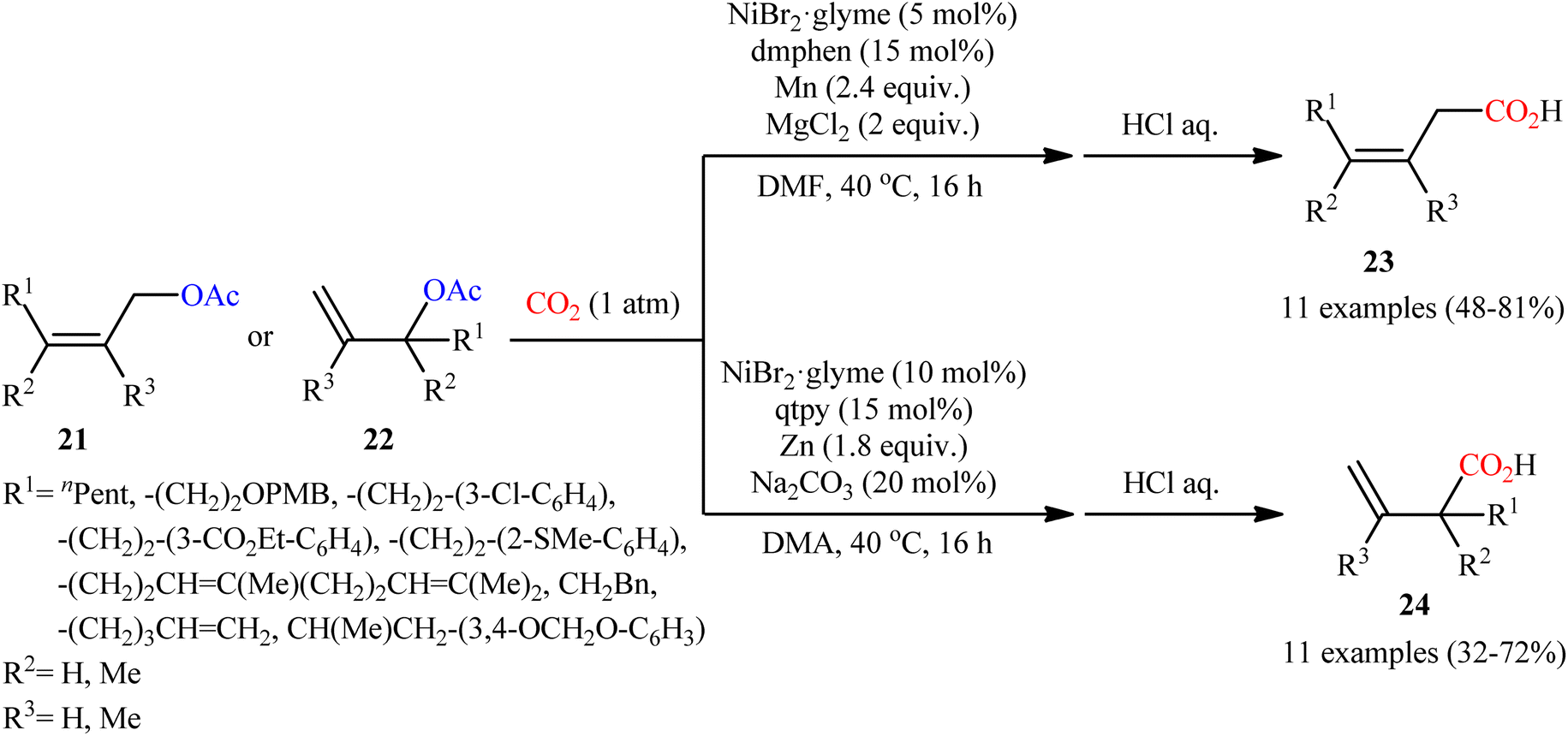
Shortly afterwards, the same research team reported an interesting Ni-catalyzed ligand-controlled regiodivergent reductive carboxylation of allyl acetates 21 and 22 with atmospheric CO2 which allow the introduction of the carboxylic motif at any site of the allyl terminus depending on the coordination geometry of the ligand employed.21 Utilizing oct-2-en-1-yl acetate as the reactant and NiBr2·glyme as a catalyst, optimization revealed two sets of suitable reaction conditions: (i) the bidentate ligand dmphen in the presence of Mn (a reductant) and MgCl2 (an additive); and (ii) the tetradentate ligand 2,2′:6′,2′′:6′′,2′′′-quaterpyridine (qtpy) in the presence of Zn (a reductant) and Na2CO3 (an additive). The first system was found to be highly selective toward linear products 23. While performing the process under the second catalytic system resulted in the predominant formation of α-branched carboxylic acids 24 regardless of whether linear or α-branched allyl acetates were utilized (Scheme 15). Unfortunately, a plausible mechanistic scheme to explain this regiodivergent process has not been proposed.
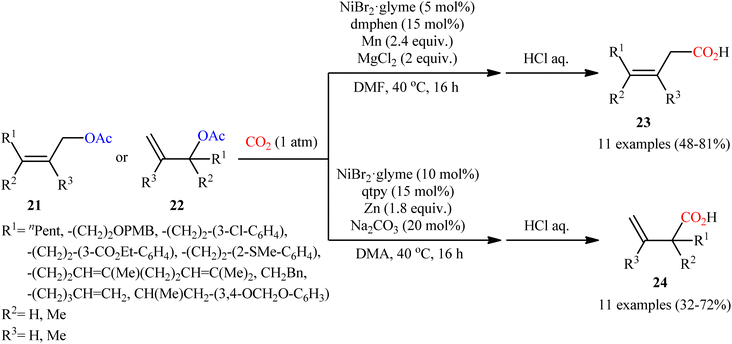 |
| | Scheme 15 Ni-catalyzed ligand-controlled regiodivergent reductive carboxylation of allyl acetates 21 and 22 with CO2. | |
Concurrently, the combination of CoI2(phen) and Mn was found by Fujihara, Tsuji, and co-workers as efficient catalytic system for carboxylation of propargyl acetates with atmospheric CO2 at room temperature.22 In this reaction, various α-unsubstituted, α-mono-substituted, and α,α-disubstituted propargyl acetates 25 proceeded well to afford the desired 3-butynoic acid derivatives 26 with fair to excellent yields after hydrolysis (Scheme 16). The best product yields were obtained for the substrates possessing a silyl group on the alkyne terminus. The authors demonstrated that the silyl group (TMS) can be easily removed in a single step procedure by protodesilylation in the presence of a suitable base and the obtained terminal alkynes then could subjected to Sonogashira coupling reactions. In this study, an optically pure propargyl acetate was also subjected to the carboxylation. However, chirality-transfer products were not observed, and the carboxylated product was obtained as a racemic form.
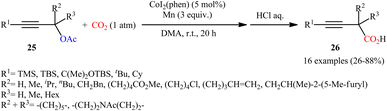 |
| | Scheme 16 Co-catalyzed carboxylation of propargyl acetates 25 with atmospheric CO2. | |
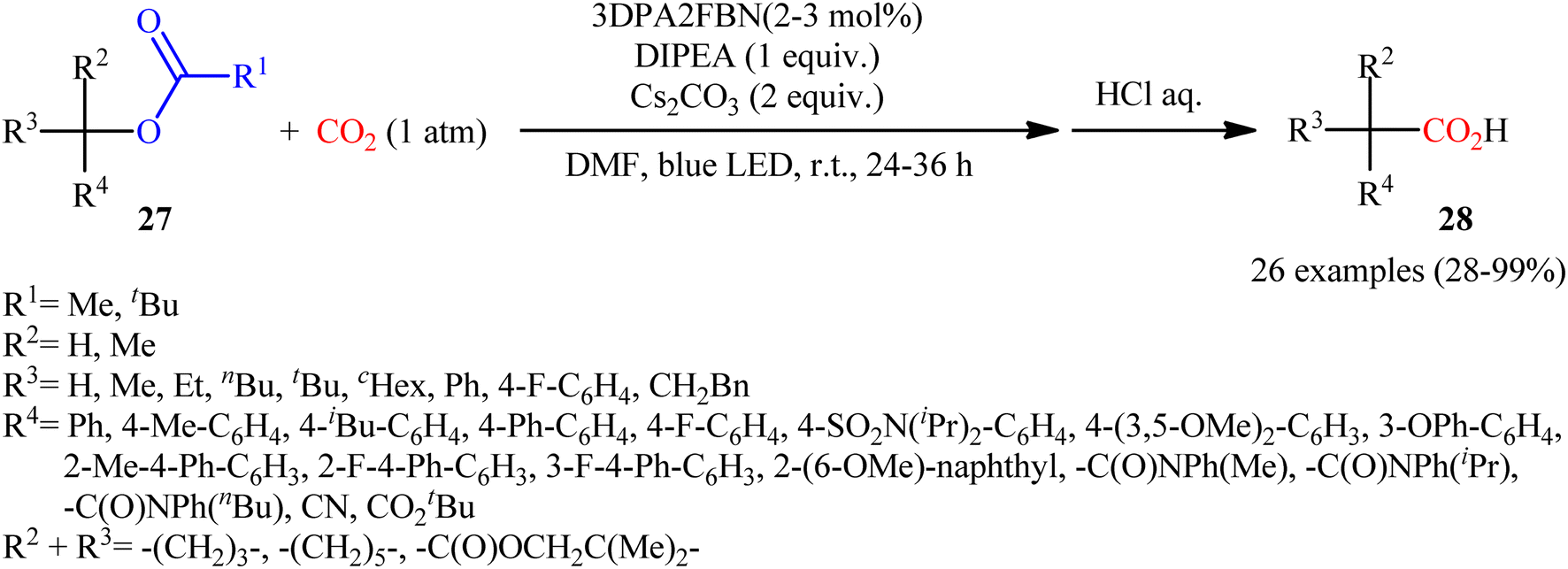
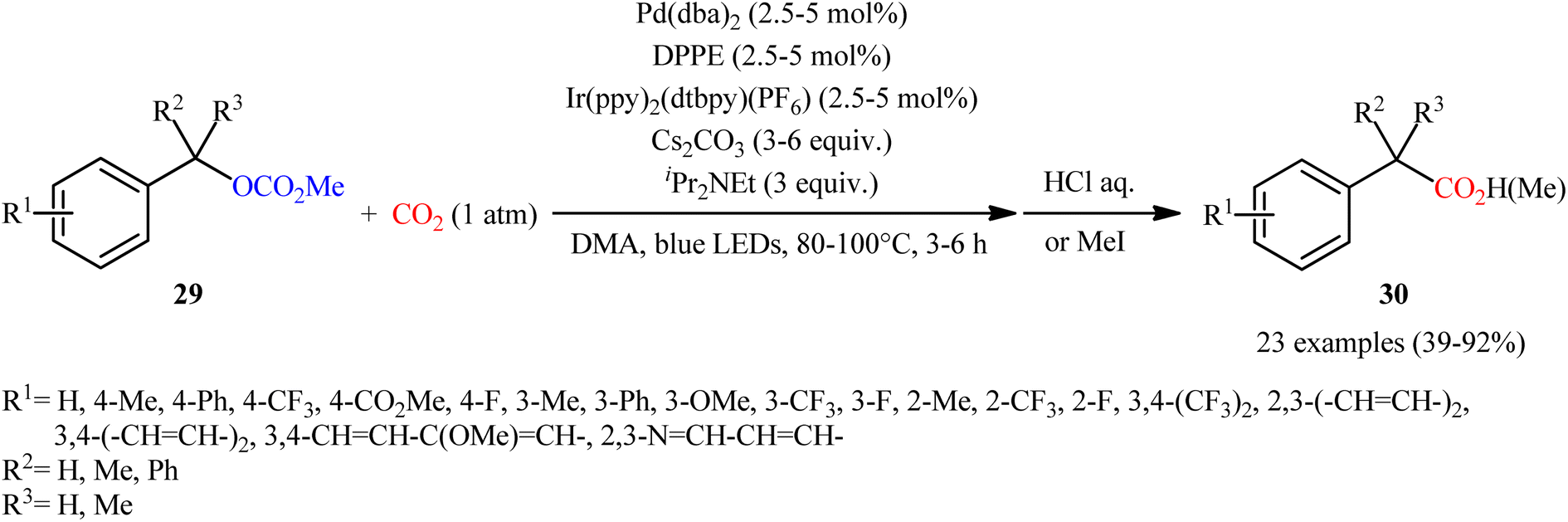
Quite recently, in the same paper describing the direct dehydroxylative carboxylation of benzylic alcohols to aryl acetic acids in the presence of catalytic amounts of an organo dye, Liao, Yu and co-workers reported the successful visible-light photoredox-catalyzed preparation of aliphatic carboxylic acids from the corresponding carboxylates.16 Thus, in the presence of catalytic amount of 2,4,6-tris-(diphenylamino)-3,5-difluorobenzonitrile (3DPA2FBN) under irradiation of visible light, deoxygenative carboxylation of various alkyl/benzyl carboxylates 27 with atmospheric CO2 furnished the corresponding aliphatic acids 28 in moderate to quantitative yields (Scheme 17). A variety of primary, secondary, and tertiary benzylic carboxylates were compatible under the reaction conditions, indicating the general applicability of this synthetic procedure. Furthermore, some drug molecules such as ibuprofen, (±)naproxen, fenoprofen, and flubiprofen were also effectively constructed under the optimal reaction. Beside good yields and broad substrate scope, scale ability can be considered as the advantages of this photoredox-catalyzed carboxylation process. Another independent visible-light-enabled benzyl carboxylates reductive carboxylation method was published by Iwasawa and co-workers using a palladium/iridium dual catalyst under visible light irradiation.23 However, in this method, the reaction was heated at 80–100 °C using DMA as solvent (Scheme 18).
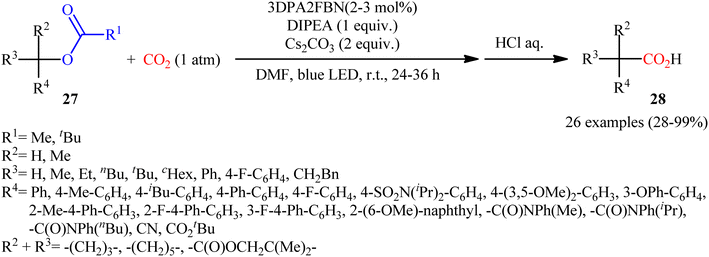 |
| | Scheme 17 Visible-light photoredox-catalyzed carboxylation of alkyl/benzyl carboxylates 27 with CO2. | |
 |
| | Scheme 18 Visible-light-enabled Pd/Ir-catalyzed carboxylation of benzyl alcohol derivatives 29 with CO2. | |
4. Carboxylation of triflates
Due to the excellent leaving ability of trifluoromethanesulfonate anion (triflate, TfO−), triflates are known as one of the superior C–O electrophiles and have widely been employed as alternatives and/or replacements for organic halides.24 Despite their several inherent drawbacks such as instability and toxicity,25 they are still among the most widely used electrophiles in the pharmaceutical industry.26
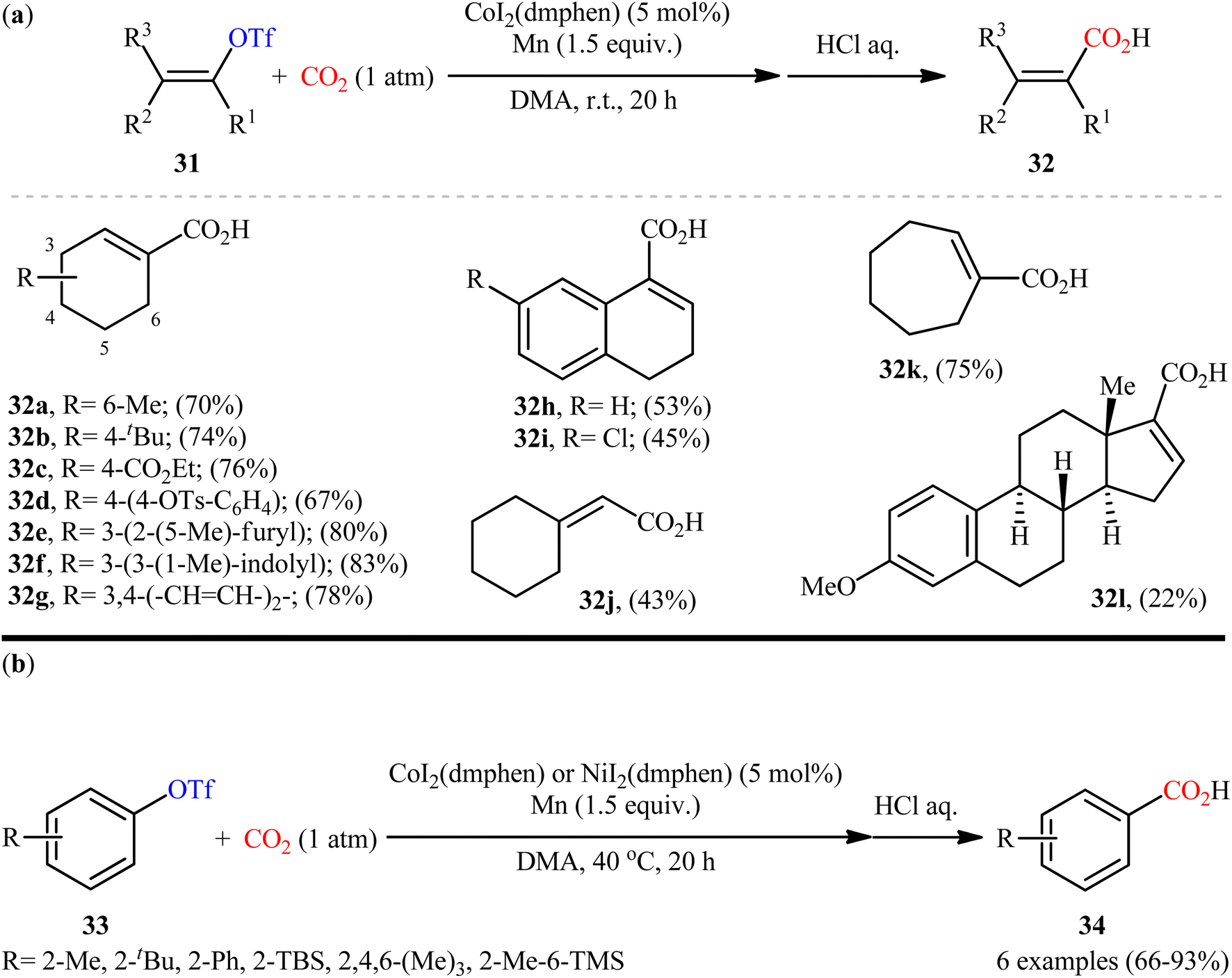
After pioneering work by Tsuji and co-workers on the synthesis of a small library (two examples) of benzoic acid derivatives through the Ni-catalyzed carboxylation of aryl triflates employing CO2,27 the first general and practical example of the carboxylation of triflates with CO2 was published by the same research team in 2015.28 In this study, twelve α,β-unsaturated carboxylic acids 32 were selectively synthesized through the treatment of various cyclic alkenyl triflates 31 with atmospheric CO2 in the presence of CoI2(dmphen)/Mn combination as the catalytic system in DMA at room temperature (Scheme 19a). Intriguingly, the triflate moiety was selectively carboxylated even in the presence of tosylate and chloro functionalities which could be potentially reactive under the reductive carboxylation conditions. However, owing to extensive side reactions, oct-1-en-2-yl trifluoromethanesulfonate did not take part in the reaction and therefore no other exo-methylene containing substrates were examined in the protocol. It is worthwhile to note that carboxylation of a series of electron-rich aryl triflate derivatives 33 was also achieved with use of similar reaction conditions (Scheme 19b). Unfortunately, the authors did not unravel the plausible mechanistic cycle for this CO2-fixation reaction.
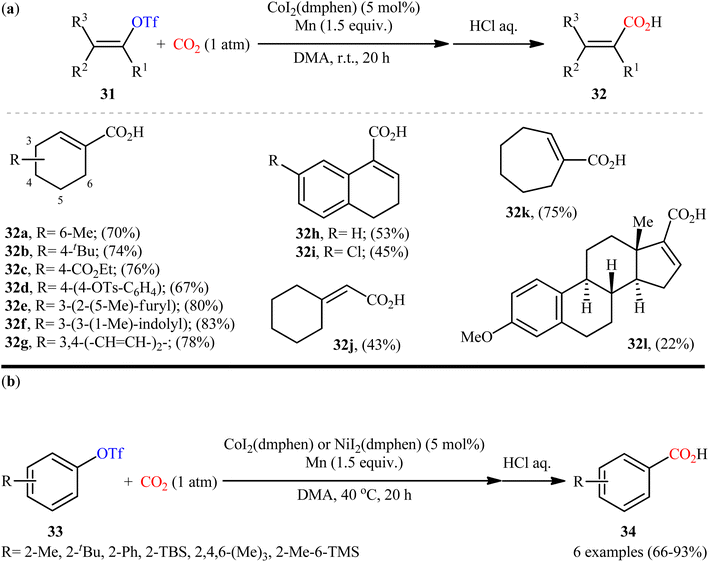 |
| | Scheme 19 Co-catalyzed carboxylation of (a) alkenyl triflates 31 and (b) aryl triflates 33 with CO2. | |
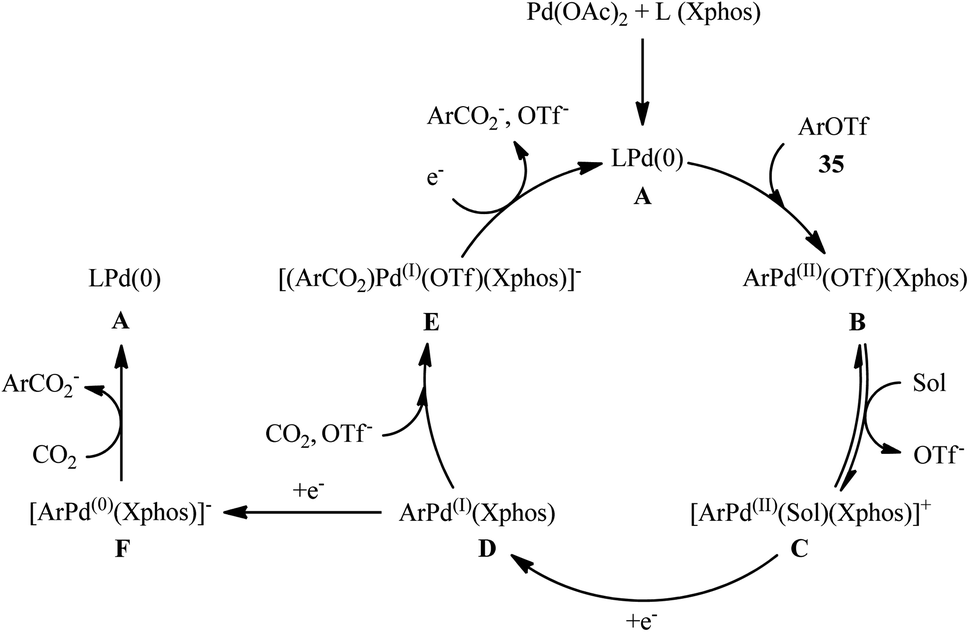
Four years later, the innovative research group of Iwasawa realized an efficient metallaphotoredox-catalyzed carboxylation of aryl triflates 35 employing atmospheric CO2 under ambient conditions.29 In this protocol, the combination of Ir(ppy)2(dtbpy)(PF6) as a photocatalyst and Pd(OAc)2 as a transition-metal-catalyst upon irradiation of blue LEDs was found to be significant for this carboxylation reaction. The presented methodology exhibited good functional group compatibility and the target carboxylic acid derivatives 36 were obtained in moderate to high yields within 4 h (Scheme 20a). Inspired by this success, the authors further extended the scope of their methodology to the more challenging alkenyl triflates. It was shown that under the identical conditions, a panel of 15 cyclic and acyclic alkenyl triflates 37 underwent selective carboxylation and subsequent methylation to afford the desired α,β-unsaturated methyl esters 38 in satisfactory yields (Scheme 20b). Importantly, exo-methylene containing alkenyl triflates which were not compatible substrates under the condition developed by Tsuji, were successfully carboxylated in this protocol. Recently, the same authors conducted a detailed DFT study to understand the reaction mechanism of this palladium-catalyzed visible light-driven carboxylation.30 The computations revealed that the active species for photoredox-catalyzed reduction was cationic ArPd(II)+ species to generate nucleophilic ArPd(I) or its further reduced ArPd(0)− species, which reacted with CO2 to give carboxylic acids. Based on these findings and experimental results, they suggested that the reaction may proceeds through the catalytic cycle as depicted in Scheme 21, which was started with the oxidative addition of ArOTf to Pd(0)(Xphos) A to form neutral ArPd(II)(OTf)(Xphos) B, followed by ligand exchange to produce cationic [ArPd(II)(Sol)(Xphos)]+ C. Then, one-electron reduction of this complex by Ir(II) species generated nucleophilic ArPd(I)(Xphos) D or its X−-coordinated anionic ArPd(I) species. Subsequently, this Pd(I) species reacted with CO2 to yield the corresponding (ArCO2)Pd(I)(Xphos) species E, which is finally reduced to Pd(0)(Xphos) A with release of ArCO2−. In another possibility, intermediated D would be further reduced to anionic [ArPd(0)(Xphos)]− F and the carboxylation would be proceeded with F.
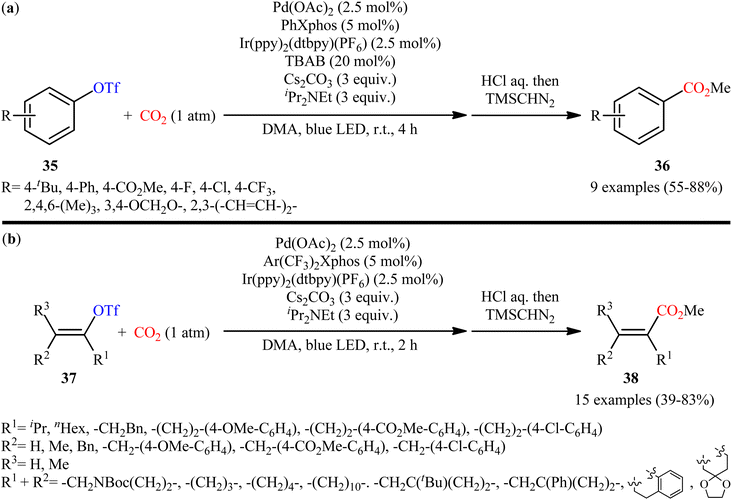 |
| | Scheme 20 (a) Metallaphotoredox-catalyzed carboxylation of aryl triflates 31 and (b) alkenyl triflates 33 with atmospheric CO2. | |
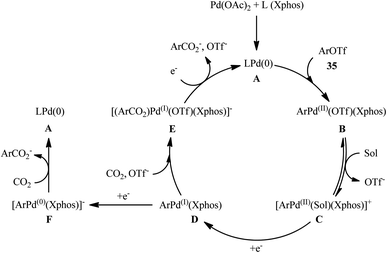 |
| | Scheme 21 Proposed mechanism for the reaction in Scheme 20. | |
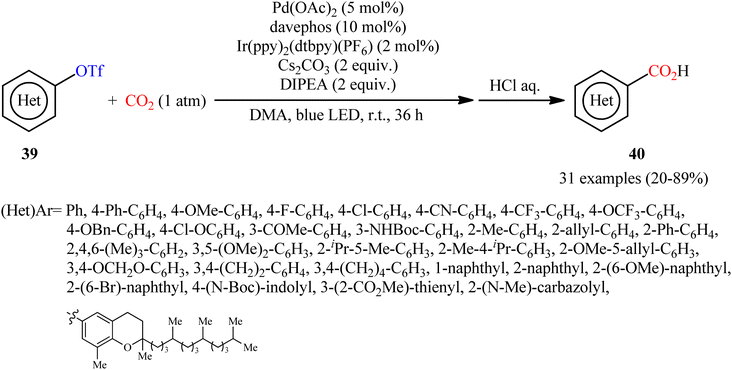
In a closely related investigation, Jana and co-workers also described that a diverse range of (hetero)aromatic triflates 39 were converted to the corresponding (hetero)aryl carboxylic acids 40, via carboxylation with CO2 using the same palladium and visible-light-photoredox catalysts and replacing PhXphos with davephos ligand (Scheme 22).31 In this transformation, a wide panel of important functional groups, such as halide, cyano, amine, ketone, ether, and ester functionalities were well tolerated, thus provided the opportunity to further functionalize the carboxylic acid products using other convenient reactions. In addition, a tolerance for an alkenyl triflate was also demonstrated. To demonstrate the practical utility of this methodology, one-pot version of this reaction using an in situ generated aryl triflate from the corresponding phenol was performed and the desired product was obtained in good yield. Unfortunately, neither benzylic nor allylic triflates were effectively reacted under the conditions employed. Furthermore, when tosylate, mesylate, and nonaflates were employed under the optimal reaction conditions, only trace amounts of carboxylated products were observed.
 |
| | Scheme 22 Jana's synthesis of (hetero)aryl carboxylic acids 40. | |
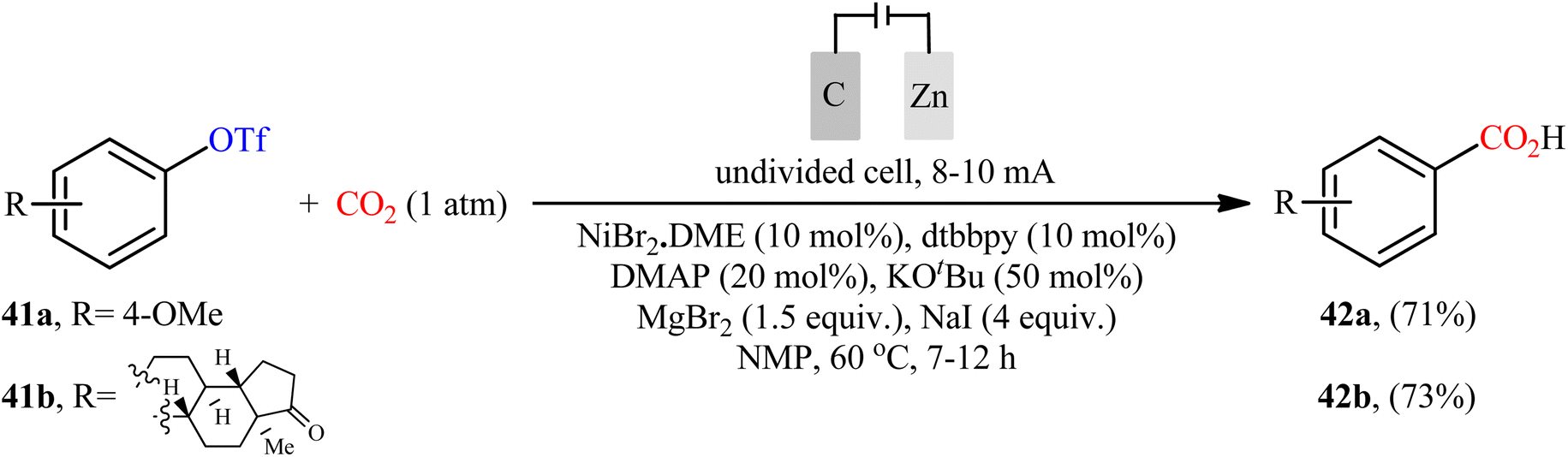
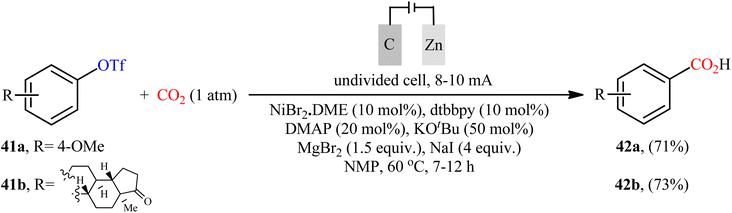
Recently, Sun et al. developed an interesting electrochemically enabled nickel-catalyzed methodology for carboxylation of aryl triflates 41 with CO2 using electron as the clean reductant.32 The efficient combination of NiBr2·DME, dtbbpy, DMAP, KOtBu, MgBr2, and NaI with a carbon felt anode and a Zn plate cathode in an undivided cell, under constant current of 8–10 mA, provided the benzoic acid derivatives 42 in good yields (Scheme 23). Apart from triflates, other activated alcohols such as tosylates and mesylates were also compatible with this scenario. Although only two examples were disclosed, this paper represents the first example of the electrocatalytic carboxylation of triflates with CO2.
 |
| | Scheme 23 Electrochemical carboxylation of aryl triflates 41 with CO2. | |
5. Carboxylation of tosylates
Tosylates (OTs) are one of the viable C–O electrophiles and widely used in certain organic reactions such as nucleophilic substitution and cross-coupling reactions. Although they are more attractive than triflates in terms of cost, stability, and availability of reagents, but they tend to be less reactive than triflates as electrophiles.33
Compared with triflates, carboxylation of tosylates with CO2 is considerably less explored. In fact, to the best of our knowledge, only two practical examples of such a CO2-fixation reaction were reported in literature thus far. In 2014, Martin and colleagues realized the first metal-catalyzed carboxylation of tosylates with CO2 by the combination of NiBr2·glyme, dmphen, and Mn powder as catalyst, ligand, and reducing agent, respectively.34 The transformation proceeded under atmospheric pressure of CO2 in DMF, tolerated a series of alkyl tosylates 43 containing other potentially reactive functionality such as OPiv and OMe groups, and provided the corresponding aliphatic carboxylic acids 44 in good yields, ranging from 62% to 76% (Scheme 24). Notably, the carboxylation of alkyl mesylates and trifluoroacetates was also feasible, albeit in lower yields.
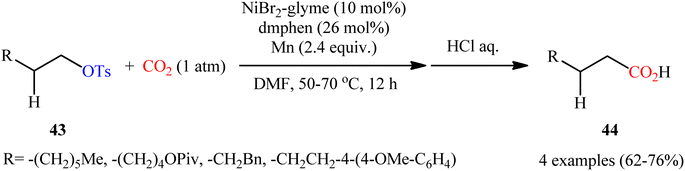 |
| | Scheme 24 Ni-catalyzed carboxylation of alkyl tosylates 43 with CO2. | |
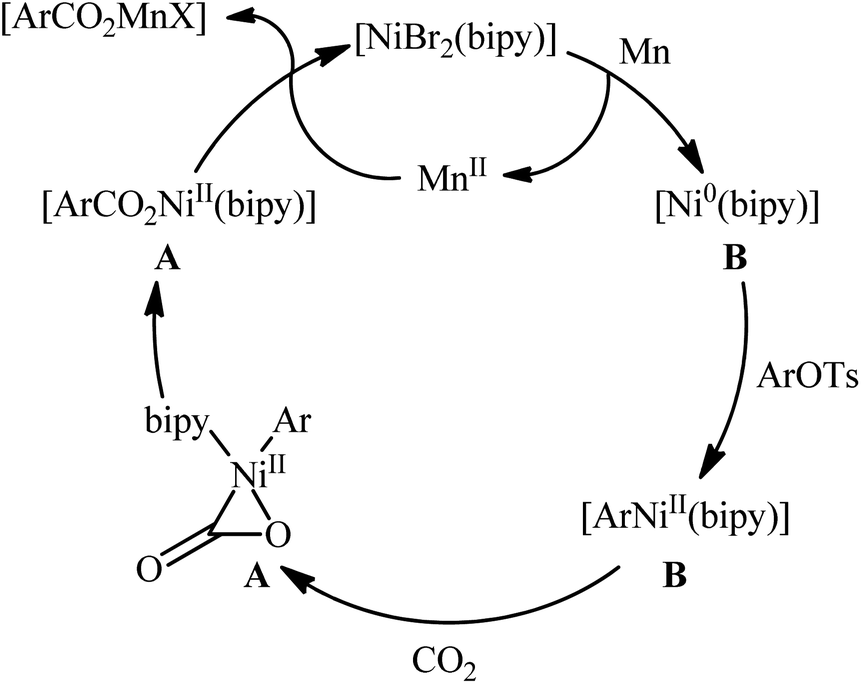
Two years later, the group of Durandetti extended the scope of this chemistry to carboxylation of aryl tosylates employing [NiBr2(bipy)]/Mn combination as a catalytic system and DMF as the solvent.35 A variety of electron deficient- and donating-substituted aryl tosylates 45 easily took part in this transformation and produced the desired carboxylic acid derivatives 46 in moderate to good yields (Scheme 25); in addition, a tolerance for aryl iodide as well as aryl bromide was also demonstrated. In this report, several control experiments, such as electrochemical investigations, DFT calculation and others, were conducted for the insight of the reaction mechanism, which indicated the formation of key Aresta-type Ni-η2-CO2 complex in the catalytic cycle (Scheme 26).
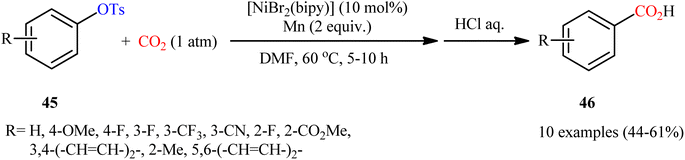 |
| | Scheme 25 Ni-catalyzed carboxylation of aryl tosylates 45 with CO2. | |
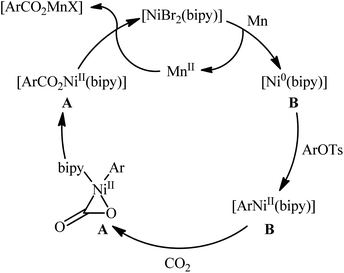 |
| | Scheme 26 Mechanistic proposal for the formation of carboxylic acid derivatives 46. | |
6. Carboxylation of fluorosulfates
Fluorosulfates (OFs) as a more stable and eco-friendly alternative of triflates, have recently gained increasing attention as versatile electrophilic partners in cross-coupling reactions.36 They are easily and conveniently prepared from alcohols using sulfuryl fluoride (SO2F2) as an inexpensive sulfonyl fluoride provider.
In 2019, Mei's research group demonstrated for the first time the usefulness of aryl fluorosulfates as coupling partners in carboxylation reactions using CO2, providing a novel opportunity to construction of carboxylic acids.37 To evaluate various parameters, such as catalyst and ligand, 4-(tert-butyl)phenylfluorosulfate was chosen as the model reactant. The combination of Ni(PPh3)2Cl2 with dmphen and Mn powder was found to be more effective catalytic system, which afforded a better yield of the desired 4-(tert-butyl)benzoic acid. Under the optimized conditions, 24 benzoic acid derivatives 48 were obtained in high to almost quantitative yields from the respective aryl fluorosulfonates 47 (Scheme 27). Apart from aryl fluorosulfonates, heteroaryl derivatives (e.g., pyridine, quinoline) were also compatible with this scenario. Notably, the authors also disclosed one-pot version of the same reaction where the requisite aryl fluorosulfonates were prepared in situ from the corresponding phenols and SO2F2.
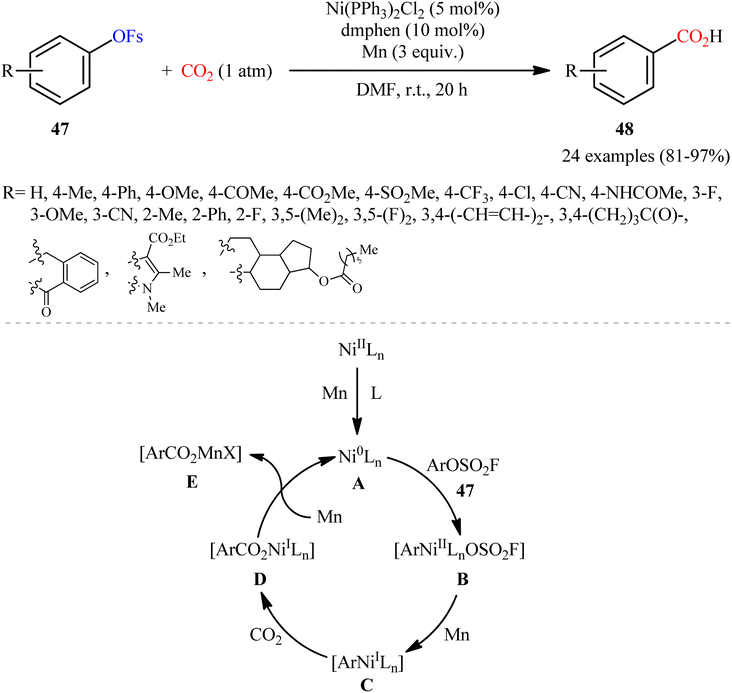 |
| | Scheme 27 Ni-catalyzed carboxylation of aryl fluorosulfates 47 with CO2 developed by Mei. | |
7. Conclusion
The ubiquity of carboxylic acids in nature and a myriad of applications in the fields of agriculture, pharmaceuticals, medicine, food, and other industries inspire constant development of complementary and alternative routes for their synthesis, which are green and sustainable. In this regard, catalytic carboxylation of alcohols and some of their derivatives with CO2 via C–O bond cleavage has received remarkable attention. This page of carboxylic acid synthesis not only benefits from the use of CO2 as an inexpensive, nontoxic, and renewable one-carbon (C1) feedstock, but also allows the use of alcohol derivatives as far more sustainable and safer alternatives of the conventionally employed organohalides. Intriguingly, without exception, all deoxygenative carboxylations covered in this review were performed under atmospheric pressure of CO2 and majority of them at room temperature. Interestingly, some of these reactions were performed under metal-free condition which is highly desirable from the standpoint of green chemistry. These results clearly indicate the potential application of this page of carboxylic acid synthesis in industry. Despite notable recent advances, this interesting research arena is still in infancy and there are still many challenges to be addressed in this synthetic strategy. Some of these are listed below: (i) the application of this methodology in the enantioselective synthesis of chiral carboxylic acids remains elusive, and therefore development of asymmetric catalytic systems will be still highly desirable task in this field; (ii) the direct dehydroxylative carboxylation of free alcohols are mainly limited to the use of only aliphatic alcohols. Therefore, the development of new catalytic systems that being able to catalyze carboxylation of (hetero)aromatic alcohols is highly desirable; and (iii) the utilized activated alcohols are mainly limited to esters, triflates, tosylates, and fluorosulfates. Therefore, of course, expanding of the scope of C–O reagents are necessary.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347–3357 RSC;
(b) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed;
(c) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30, DOI:10.22034/crl.2018.85117.
-
(a) X. Wang, W. Ping, A. G. Ebadi, S. Majedi, Z. Hossaini and M. Toughani, J. CO2 Util., 2021, 50, 101592 CrossRef CAS;
(b) L. Wang, S. Que, Z. Ding and E. Vessally, RSC Adv., 2020, 10, 10806 RSC;
(c) M. Li, S. Abdolmohammadi, M. S. Hoseininezhad-Namin, F. Behmagham and E. Vessally, J. CO2 Util., 2020, 38, 220–231 CrossRef CAS;
(d) X. Zhao, S. Yang, S. Ebrahimiasl, S. Arshadi and A. Hosseinian, J. CO2 Util., 2019, 33, 37–45 CrossRef CAS;
(e) A. Monfared, R. Mohammadi, A. Hosseinian, S. Sarhandi and P. D. K. Nezhad, RSC Adv., 2019, 9, 3884–3899 RSC;
(f) A. Hosseinian, S. Ahmadi, R. Mohammadi, A. Monfared and Z. Rahmani, J. CO2 Util., 2018, 27, 381–389 CrossRef CAS;
(g) N. Salehi and B. Azizi, J. Chem. Lett., 2021, 2, 2–8, DOI:10.22034/jchemlett.2021.275293.1023;
(h) N. A. A. Salman, A. H. Adhab, H. Bahir, M. H. Sami and R. Sadeghzadeh, Chem. Rev. Lett., 2023, 6, 139–149, DOI:10.22034/crl.2023.390701.1214.
-
(a) Y. Cao, H. A. Dhahad, H. M. Hussen, A. E. Anqi, N. Farouk, A. Issakhov and M. R. P. Heravi, J. CO2 Util., 2021, 52, 101664 CrossRef CAS;
(b) S. Nan, W. Hai-Bin, G. Li, Z. Jing-Yao, G. Jian-Feng, W. Fang and A. Ebadi, J. CO2 Util., 2021, 48, 101522 CrossRef CAS;
(c) W. Xu, D. Guo, A. G. Ebadi, M. Toughani and E. Vessally, J. CO2 Util., 2021, 45, 101403 CrossRef CAS;
(d) W. Xu, A. G. Ebadi, M. Toughani and E. Vessally, J. CO2 Util., 2021, 43, 101358 CrossRef CAS;
(e) L. Feng, X. Li, B. Liu and E. Vessally, J. CO2 Util., 2020, 40, 101220 CrossRef CAS;
(f) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed. Engl., 2018, 57, 15948–15982 CrossRef CAS PubMed;
(g) S. Pradhan, S. Roy, B. Sahoo and I. Chatterjee, Chem.–Eur. J., 2021, 27, 2254–2269 CrossRef CAS PubMed;
(h) T. Fujihara and Y. Tsuji, Front. Chem., 2019, 7, 430 CrossRef CAS PubMed;
(i) S. Wang, G. Du and C. Xi, Org. Biomol. Chem., 2016, 14, 3666–3676 RSC.
- A. Correa and R. Martin, Angew. Chem., Int. Ed., 2009, 48, 6201–6204 CrossRef CAS PubMed.
- M. Börjesson, T. Moragas, D. Gallego and R. Martin, ACS Catal., 2016, 6, 6739–6749 CrossRef PubMed.
- M. Abdoli and H. Saeidian, J. Sulfur Chem., 2015, 36, 556–582 CrossRef CAS.
-
(a) M. Granda, C. Blanco, P. Alvarez, J. W. Patrick and R. Menendez, Chem. Rev., 2014, 114, 1608–1636 CrossRef CAS PubMed;
(b) H. Xu, F. Ma, N. Wang, W. Hou, H. Xiong, F. Lu, J. Li, S. Wang, P. Ma, G. Yang and R. A. Lerner, Adv. Sci., 2019, 6, 1901551 CrossRef CAS PubMed.
-
(a) Y. You and T. Mita, Asian J. Org. Chem., 2022, 11, 202200082 CrossRef;
(b) Y. G. Chen, X. T. Xu, K. Zhang, Y. Q. Li, L. P. Zhang, P. Fang and T. S. Mei, Synthesis, 2018, 50, 35–48 CrossRef CAS;
(c) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS PubMed;
(d) D. Yu, S. P. Teong and Y. Zhang, Coord. Chem. Rev., 2015, 293, 279–291 CrossRef.
- Selected examples:
(a) S. Farshbaf, L. Z. Fekri, M. Nikpassand, R. Mohammadi and E. Vessally, J. CO2 Util., 2018, 25, 194–204 CrossRef CAS;
(b) A. Hosseinian, S. Farshbaf, R. Mohammadi, A. Monfared and E. Vessally, RSC Adv., 2018, 8, 17976–17988 RSC;
(c) E. Vessally, R. Mohammadi, A. Hosseinian, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 24, 361–368 CrossRef CAS;
(d) K. Didehban, E. Vessally, M. Salary, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 23, 42–50 CrossRef CAS;
(e) E. Vessally, A. Hosseinian, L. Edjlali, M. Babazadeh and K. Didehban, Mini-Rev. Org. Chem., 2018, 15, 315–323 CrossRef CAS;
(f) E. Vessally, K. Didehban, M. Babazadeh, A. Hosseinian and L. Edjlali, J. CO2 Util., 2017, 21, 480–490 CrossRef CAS;
(g) E. Vessally, M. Babazadeh, A. Hosseinian, S. Arshadi and L. Edjlali, J. CO2 Util., 2017, 21, 491–502 CrossRef CAS;
(h) E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and M. Babazadeh, J. CO2 Util., 2017, 21, 342–352 CrossRef CAS;
(i) S. Arshadi, E. Vessally, A. Hosseinian, S. Soleimani-amiri and L. Edjlali, J. CO2 Util., 2017, 21, 108–118 CrossRef CAS;
(j) S. Arshadi, E. Vessally, M. Sobati, A. Hosseinian and A. Bekhradnia, J. CO2 Util., 2017, 19, 120–129 CrossRef CAS.
-
(a) W. L. Hu, X. G. Hu and L. Hunter, Synthesis, 2017, 49, 4917–4930 CrossRef CAS;
(b) Y. Cao, R. Ahmadi, M. R. P. Heravi, A. Issakhov, A. G. Ebadi and E. Vessally, RSC Adv., 2021, 11, 39593–39606 RSC.
- T. Mita, Y. Higuchi and Y. Sato, Chem.–Eur. J., 2015, 21, 16391–16394 CrossRef CAS PubMed.
- Y. G. Chen, B. Shuai, C. Ma, X. J. Zhang, P. Fang and T. S. Mei, Org. Lett., 2017, 19, 2969–2972 CrossRef CAS PubMed.
- M. van Gemmeren, M. Börjesson, A. Tortajada, S. Z. Sun, K. Okura and R. Martin, Angew. Chem., Int. Ed., 2017, 129, 6658–6662 CrossRef.
- Z. Fan, S. Chen, S. Zou and C. Xi, ACS Catal., 2022, 12, 2781–2787 CrossRef CAS.
- T. Yamahira, G. Onodera, T. Fukuda and M. Kimura, Chem. Lett., 2021, 50, 853–855 CrossRef CAS.
- C. K. Ran, Y. N. Niu, L. Song, M. K. Wei, Y. F. Cao, S. P. Luo, Y. M. Yu, L. L. Liao and D. G. Yu, ACS Catal., 2021, 12, 18–24 CrossRef.
- W. D. Li, Y. Wu, S. J. Li, Y. Q. Jiang, Y. L. Li, Y. Lan and J. B. Xia, J. Am. Chem. Soc., 2022, 144, 8551–8559 CrossRef CAS PubMed.
-
(a) K. Kunz, U. Scholz and D. Ganzer, Synlett, 2003, 2428–2439 CrossRef CAS;
(b) E. Sperotto, G. P. van Klink, J. G. de Vries and G. van Koten, J. Org. Chem., 2008, 73, 5625–5628 CrossRef CAS PubMed.
- P. Villo, A. Shatskiy, M. D. Kärkäs and H. Lundberg, Angew. Chem., Int. Ed., 2023, 135, 202211952 CrossRef.
- A. Correa, T. Leon and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062–1069 CrossRef CAS PubMed.
- T. Moragas, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 17702–17705 CrossRef CAS PubMed.
- K. Nogi, T. Fujihara, J. Terao and Y. Tsuji, Chem. Commun., 2014, 50, 13052–13055 RSC.
- Y. Jin, N. Toriumi and N. Iwasawa, ChemSusChem, 2022, 15, 202102095 CrossRef PubMed.
- A. Suzuki, J. Org. Chem., 1999, 576, 147–168 CrossRef CAS.
- A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfared and Z. Rahmani, RSC Adv., 2018, 8, 33828–33844 RSC.
- C. M. So, O. Y. Yuen, S. S. Ng and Z. Chen, ACS Catal., 2021, 11, 7820–7827 CrossRef CAS.
- T. Fujihara, K. Nogi, T. Xu, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2012, 134, 9106–9109 CrossRef CAS PubMed.
- K. Nogi, T. Fujihara, J. Terao and Y. Tsuji, J. Org. Chem., 2015, 80, 11618–11623 CrossRef CAS PubMed.
- K. Shimomaki, T. Nakajima, J. Caner, N. Toriumi and N. Iwasawa, Org. Lett., 2019, 21, 4486–4489 CrossRef CAS PubMed.
- N. Toriumi, K. Shimomaki, J. Caner, K. Murata, R. Martin and N. Iwasawa, Bull. Chem. Soc. Jpn., 2021, 94, 1846–1853 CrossRef CAS.
- S. K. Bhunia, P. Das, S. Nandi and R. Jana, Org. Lett., 2019, 21, 4632–4637 CrossRef CAS PubMed.
- G. Q. Sun, W. Zhang, L. L. Liao, L. Li, Z. H. Nie, J. G. Wu, Z. Zhang and D. G. Yu, Nat. Commun., 2021, 12, 7086 CrossRef CAS PubMed.
-
(a) C. M. So and F. Y. Kwong, Chem. Soc. Rev., 2011, 40, 4963–4972 RSC;
(b) J. Albaneze-Walker, R. Raju, J. A. Vance, A. J. Goodman, M. R. Reeder, J. Liao, M. T. Maust, P. A. Irish, P. Espino and D. R. Andrews, Org. Lett., 2009, 11, 1463–1466 CrossRef CAS PubMed.
- Y. Liu, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 11212–11215 CrossRef CAS PubMed.
- F. Rebih, M. Andreini, A. Moncomble, A. Harrison-Marchand, J. Maddaluno and M. Durandetti, Chem.–Eur. J., 2016, 22, 3758–3763 CrossRef CAS PubMed.
- S. K. Saraswat, R. Seemaladinne, H. Zaini, N. Ahmad, N. Ahmad and E. Vessally, RSC Adv., 2023, 13, 13642–13654 RSC.
- C. Ma, C. Q. Zhao, X. T. Xu, Z. M. Li, X. Y. Wang, K. Zhang and T. S. Mei, Org. Lett., 2019, 21, 2464–2467 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Shakir Mahmood Saiedc,
Maha Dhurgham Azeezd,
Russul R. Abbasse,
Ayat Hussein Adhabf and
Esmail Vessally
b,
Shakir Mahmood Saiedc,
Maha Dhurgham Azeezd,
Russul R. Abbasse,
Ayat Hussein Adhabf and
Esmail Vessally