
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLocalized surface plasmon resonance shift of biosynthesized and functionalized quasi-spherical gold nanoparticle systems†
Ricky Kristan M. Raguindin * and
Candy C. Mercado
* and
Candy C. Mercado
Department of Mining, Metallurgical and Materials Engineering, College of Engineering, University of the Philippines Diliman, Quezon City 1101, Philippines. E-mail: rmraguindin1@up.edu.ph
First published on 14th August 2023
Abstract
Rapid and more environment-friendly means of gold nanoparticle synthesis is necessary in many applications, as in ion detection. Leaf extracts have become effective and economical reducing agents for gold nanoparticle formation, however, effects of extract combinations have not been thoroughly investigated. With the exploitation of combined extract effects, gold nanoparticles were synthesized then functionalized and investigated to produce selected nanoparticle systems which are capable of detecting aqueous lead(II) ions with minimum detection limits of 10–11 ppm. The measured localized surface plasmon resonance absorption peaks of the gold nanoparticles were 541–800 nm for the synthesis and 549 nm for the functionalization. The diameters of different gold nanoparticle systems were 17–37 nm. These were mostly quasi-spherical in morphology with some rod-, triangular-, and hexagonal plate-like particles. The biosynthesis used polyphenols and acids present in the extracts in the reduction of gold ions into gold nanoparticles, and in the nanoparticle capping and stabilization. Functionalization replaced the capping compounds with alliin, S-allylcysteine, allicin, and ajoene. Gold nanoparticle stability in aqueous systems was verified for two weeks up to five months. The investigations concluded the practicability of the gold nanoparticles in lead(II) ion detection with selectivity initially verified for other divalent cations.
Introduction
Gold (Au) nanoparticles—having promising viability for technological advancements in a multitude of fields such as in optoelectronics, sensing, biomedicine, and nanoelectronics due to their tuneable and morphology-dependent properties—are of great significance to scientific research.1 With the wide range of possibilities that this material can offer, it is imperative to provide and explore more ways of synthesizing or processing Au nanoparticles especially using natural compounds which pose negligible harm to our health and environment.In the past years, researchers made use of environment-friendly biosynthesis routes of creating Au nanoparticles. Biosynthesis is the use of biological media, such as plants or bacteria etc., in order to create chemicals or materials. Plants, termed as the ‘chemical factories’ of nature, and their extracts seem to be a promising option for nanoparticle synthesis because of their cost efficiency and low maintenance aside from the fact that they generally produce nanoparticles with good stability and appropriate dimensions due to a single-step fabrication process.2 Generally, leaf extracts are also capable of producing Au nanoparticles at room temperature, hence energy-saving benefits add up to their significance.
However, there are still research gaps regarding the effects of extract combination in Au nanoparticle synthesis. In this study, the combinatory extract type effects on the Au nanoparticles were investigated. For the functionalization, a common practice for previous studies is to use specific compounds;3–5 meanwhile, this study utilized Allium sativum (AS) extract with various sulfur (S) containing compounds to highlight the strong Au–S bond. The target application for the synthesis is making the spectroscopically silent lead(II) ion (Pb2+) be detectable in the visible light region for monitoring purposes in aqueous systems.
Lead is toxic which can damage respiratory, cardiovascular, urinary, neurological, and cognitive functions of the human body by means of oxidative, immune-modulation, and inflammatory mechanisms.6 One of the ways that Pb2+ enters the body is through the digestive system which occurs when we eat or drink lead-contaminated food and liquids. The absorption rate of Pb2+ from drinking water is 35–50% for adults, while it goes beyond 50% for children.7 The action level, the point wherein further actions must be implemented, of the United States Environmental Protection Agency (US EPA) for Pb2+ in water directed to public drinking water systems is 15 parts per billion (ppb) or 15 μg L−1, but their maximum contaminant level goal is zero since even trace amounts can be detrimental.8,9 Meanwhile, the recommended maximum concentration of Pb2+ in irrigation water by Ayers and Westcot (1985) is 5 ppm.10 This value was used as basis of Farid et al. (2015)11 in assessing environmental waters such as canal, ground, and waste water samples. For the National Oceanic and Atmospheric Administration (NOAA), the indicated concentration of Pb2+ as the severe effect level is 250 ppm.12
Rigorous assessments of water bodies and sources must be carried out for the presence of Pb2+. Inductively coupled plasma mass spectrometry (ICP-MS), atomic absorption spectroscopy (AAS) and inductively coupled plasma optical emission spectrometry (ICP-OES), are some of the techniques used, with high accuracy and advanced instrumentation requiring operation expertise.13–16 The use of the localized surface plasmon resonance (LSPR) property of Au nanoparticles for intermediate concentration levels is a useful, easy and economical alternative. This can be done by functionalizing or capping the surface of Au nanoparticles with compounds that will interact with varying Pb2+ concentrations. According to studies, there is aggregation of gallic acid stabilized Au nanoparticles upon exposure to Pb2+.17,18 The resulting Au nanoparticles capped with the carboxylic acid functional group (–COOH), more especially with gallic acid, is seen to detect Pb2+. Lagerstroemia speciosa (LS) and Moringa oleifera (MO) leaf extracts both contain gallic acid,19,20 and some major compounds of aqueous Muntingia calabura (MC) leaf extract are citric and gallic acids.21 Moreover, a major AS clove compound that contain the S element is alliin or S-allyl-l-cysteine sulfoxide22 which can also be exploited for its affinity to Au (due to the Au–S bond) and Pb2+ (due to the –COOH). The Pb2+-induced aggregation of functionalized Au nanoparticles causes a redshift in their LSPR which can provide a correlation with the Pb2+ concentration.
Experimental
Preparation of aqueous extracts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/v ratio) then crushed and allowed to stand for an additional 30 minutes at room temperature. Finally, the Allium sativum cloves aqueous extract (designated sample code: AS) was filtered and stored at 4 °C for further use.
:1 w/v ratio) then crushed and allowed to stand for an additional 30 minutes at room temperature. Finally, the Allium sativum cloves aqueous extract (designated sample code: AS) was filtered and stored at 4 °C for further use.Au nanoparticle biosynthesis using various leaf extract combinations
The combined leaf extracts, as reducing agents, were added to 5 mM HAuCl4 at room temperature with rapid swirling. The initial time of conversion, based on brown to purple color change, was recorded for each of the systems. The colloids were incubated in dark conditions overnight at about 4 °C to allow complete conversion. The formation of Au nanoparticles was confirmed by the appearance of absorbance peaks due to plasmon resonance. The colloids were centrifuged at 15200 rpm for 30 minutes then rinsed and dispersed in ultrapure water with short ultrasonication. The Au nanoparticles in ultrapure water were stored at 4 °C. The four resulting colloidal Au nanoparticle systems are indicated in Table 1.
| Combined extracts | Extract-to-HAuCl4 ratio | Designated sample code |
|---|---|---|
| MCLS | 1:1 |
Au-MCLS-1 |
| LSMO | 1:1 |
Au-LSMO-1 |
| MCMO | 1:1 |
Au-MCMO-1 |
| MLM | 1:1 |
Au-MLM-1 |
Functionalization of Au-MCLS-1 nanoparticles using AS extract
The Au-MCLS-1 nanoparticle system, synthesized with equal parts of MCLS extract and HAuCl4, was chosen as the initial combination for the functionalization with AS extract. First, Au-MCLS-1 was centrifuged at 15200 rpm for 30 minutes followed by rinsing and redispersion with ultrapure water. The redispersed nanoparticles were ultrasonicated for 30–60 s. The entire process was repeated three times before AS extract (1 g AS/mL H2O) was used as the dispersion medium for the nanoparticles. The colloidal system was stored for 68 hours at 4 °C in order to allow the substitution of capping agents by organosulfur compounds. Finally, another cycle of centrifugation-rinsing-redispersion was done to remove the excess AS extract, and ultrapure water was reintroduced as the colloidal dispersion medium. The AS-functionalized Au nanoparticle sample was designated as Au-AS-1 and stored at 4 °C.
Stability assessments
All of the biosynthesized and functionalized Au nanoparticle systems (Au-MCLS-1, Au-LSMO-1, Au-MCMO-1, Au-MLM-1, and Au-AS-1) were immersed in ultrapure water over a period of two weeks to test the stability. From each of the diluted stock colloidal dispersions of Au nanoparticle systems, absorbance spectra of samples were collected. The remaining stock colloidal dispersions were stored at about 4 °C in the dark. After two weeks, another set of the diluted Au nanoparticle systems were analyzed in which the generated spectra were compared to their initial spectra. The zeta potential values of the Au nanoparticles were measured via dynamic light scattering (Malvern Zetasizer Nano ZSP) at 25.0 °C after five months in the storage conditions mentioned.Lead detection experiments and determination of detection limits
The detection experiments were done for both Au-MCLS-1 and Au-AS-1 nanoparticle systems by comparing the absorbance spectra after exposure to an analyte. The analytes were blank (ultrapure water) and solutions with different Pb2+ concentrations (95, 189, 284, 378, 473, 568, 662, 757, and 851 μM). In each test vial, equal amounts of the Au nanoparticle system and analyte were combined, then the absorbance spectrum was taken; the peak wavelength versus Pb2+ concentration was plotted and the upper and lower limits of Pb2+ detection were determined.Characterization of extracts and Au nanoparticles
000–150000× magnifications with an accelerating voltage of 5.0 kV. ImageJ software was used to obtain the particle sizes. Au-AS-1 nanoparticles were imaged and analyzed using TEM (Transmission Electron Microscopy) with SAED (Selected Area Electron Diffraction) (JEOL JEM-2100F). The Au nanoparticles were deposited in such a way that a drop of the colloidal Au sample was placed on a TEM grid (Ted Pella Carbon Type-B, 200 mesh, Copper with an approximate grid hole size of 97 μm) and then allowed to air-dry prior to the TEM analysis.Results and discussion
Effects of leaf extract combinations on Au nanoparticles
Absorbance spectra of the Au nanoparticle systems are shown in Fig. S3.† The spectra of the extracts do not overlap with Au nanoparticle peaks. The LSPR peak of Au-MCLS-1 is at 541 nm. There are two peaks for Au-LSMO-1 which are at 543.75 and 730 nm. Meanwhile, Au-MCMO-1 exhibited a peak at 548.75 nm and Au-MLM-1 at 541.75 nm. It is important taking into consideration that Au-MLM-1 also showed a second less intense peak near the 800 nm wavelength mark. The major peaks correspond to quasi-spherical nanoparticles, while the secondary peaks for Au-LSMO-1 and Au-MLM-1 were attributed to the nanoparticles of different shapes. The order of Au nanoparticles based on LSPR peak wavelength is Au-MCMO-1 > Au-LSMO-1 > Au-MLM-1 > Au-MCLS-1. The LSPR bands as determined by absorbance spectroscopy can also provide information regarding the size of nanoparticles. Increasing sizes of nanoparticles lead to redshifting of the LSPR.25
Comparisons among the 1H NMR peaks of Au-MCLS-1, AU-LSMO-1, Au-MCMO-1, and Au-MLM-1 are summarized in Table 2. The peaks present in all of the Au nanoparticle systems that were biosynthesized using varying combination leaf extracts are those corresponding to hydrogens attached to a carbon bonded with OH of a cyclic alcohol structure, and hydrogen atoms belonging to methoxy and methyl groups. The peak for hydrogen attached to an aromatic group was observed in all of the Au nanoparticle systems except for Au-LSMO-1. Only Au-MCLS-1 and Au-MCMO-1 have peaks which could possibly correspond to hydrogen atoms attached to a junction of a cyclic structure. Moreover, Au-MCLS-1 was the sole Au nanoparticle system to exhibit a peak that is attributed to hydrogen bonded to an alkene group. For the peak which could be probably designated to the hydrogen attached to a carbon atom bonded to another R group of a cyclic structure, only Au-MCLS-1 and Au-MLM-1 were able to display such.
| a Black regions indicate absence of peaks per Au nanoparticle system. |
|---|
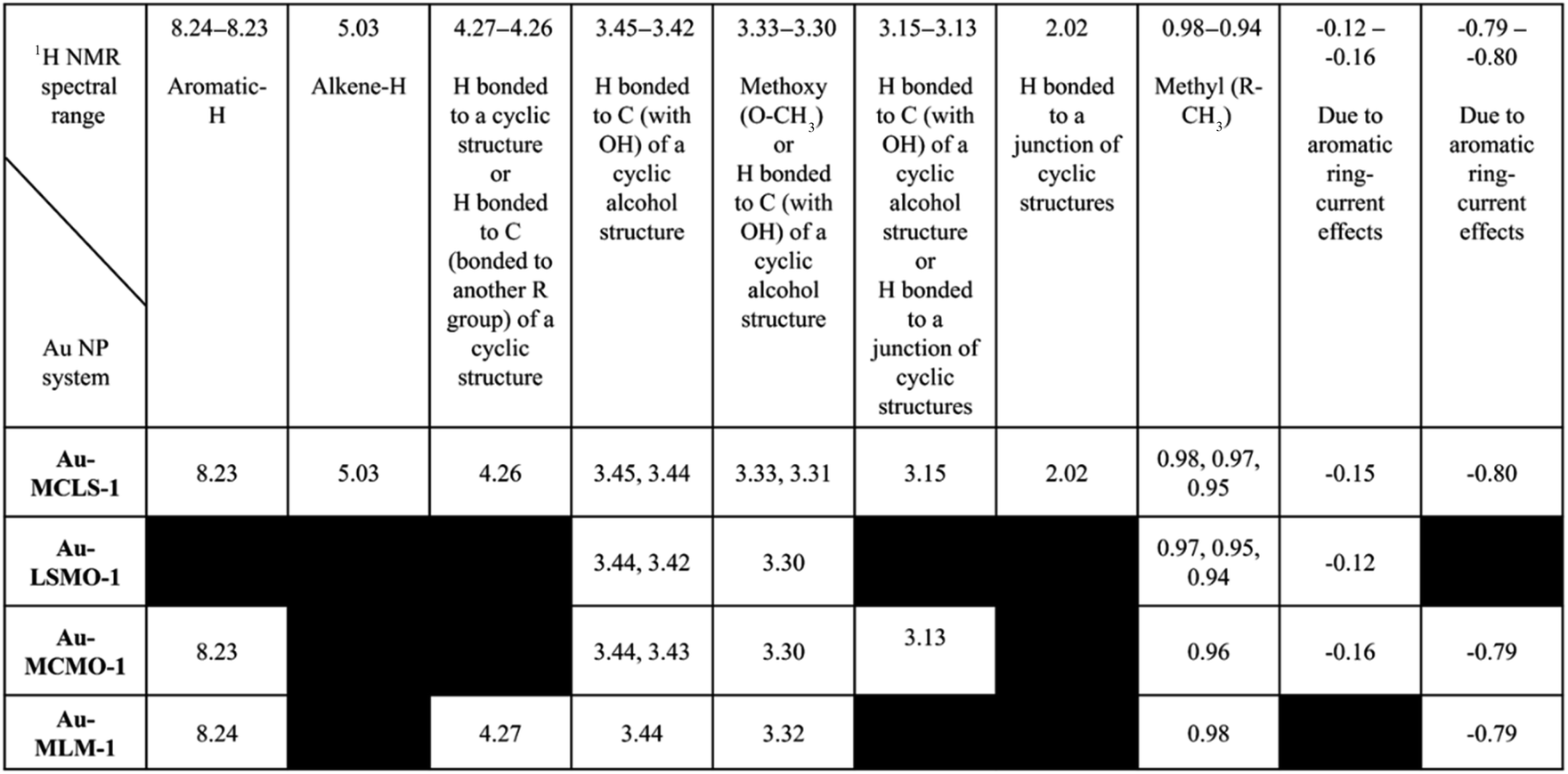 |
To complement the 1H NMR analysis, FTIR spectroscopy was also done to the Au nanoparticle systems that were biosynthesized using different combinations of leaf extracts. FTIR spectra of Au-MCLS-1, Au-LSMO-1, Au-MCMO-1, and Au-MLM-1 are shown in Fig. 1. Important FTIR peaks were attributed to certain functional groups which provide information regarding the capping compounds of the Au nanoparticles.
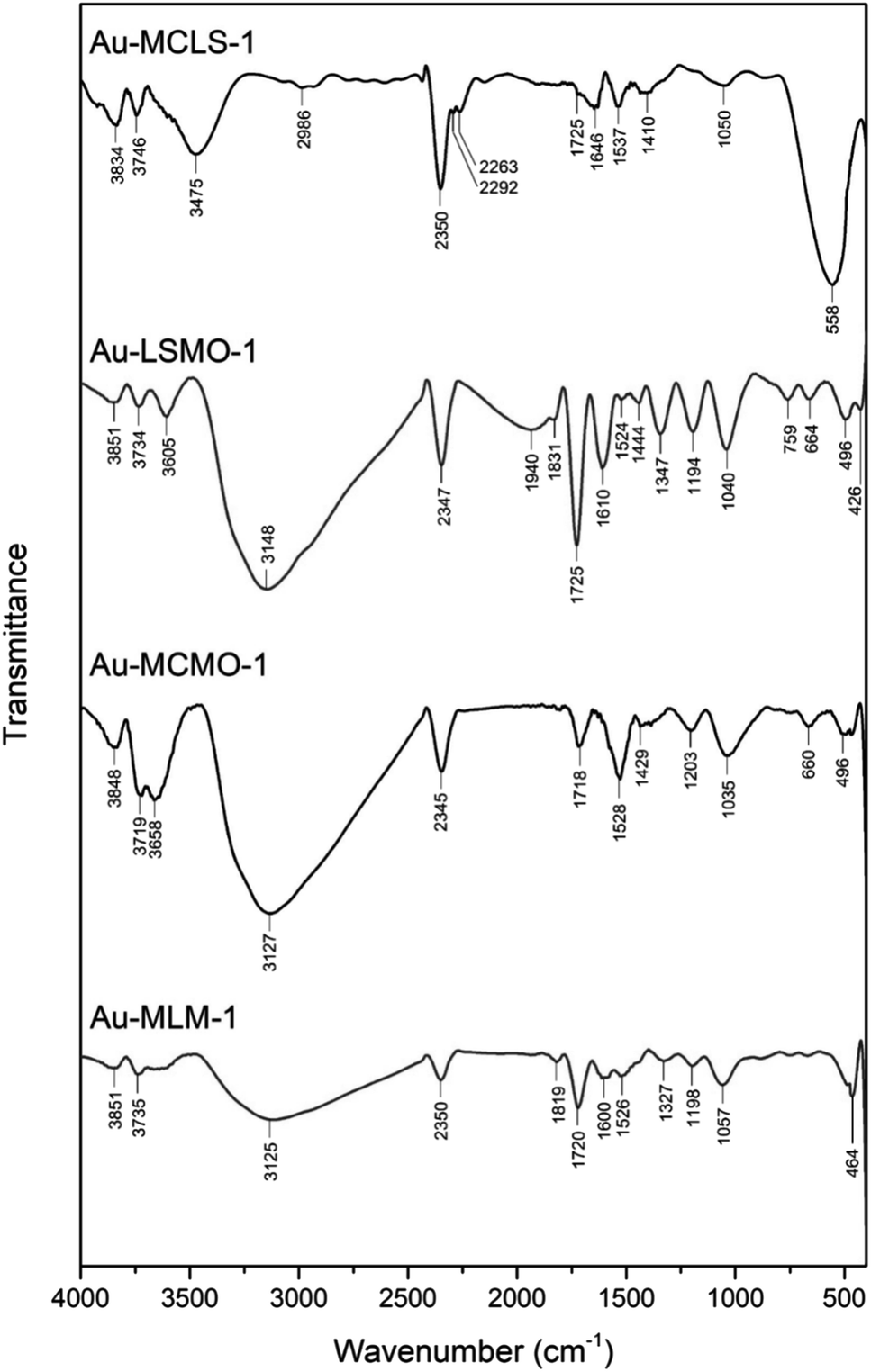 | ||
| Fig. 1 Functional groups as represented by FTIR spectral peaks of biosynthesized Au nanoparticle systems using various combined leaf extracts (Au-MCLS-1, Au-LSMO-1, Au-MCMO-1, and Au-MLM-1). The corresponding functional groups are detailed in the ESI:† participating biochemical compounds and Table S2.† | ||
The comparison of the summaries of functional groups for the Au nanoparticle systems are shown in Table S2.† The recorded 1H NMR and FTIR spectral peaks could be tentatively attributed to various polyphenols and organic acids such as: gallic acid, citric acid, and flavonoid glycosides from MC;21 caffeic acid, p-coumaric acid, corosolic acid, gallic acid, 4-hydroxybenzoic acid, kaempferol, ellagic acid, quercetin, 3-O-methylprotocatechuic acid, and isoquercitrin from LS,28 and chlorogenic acid, gallic acid, and caffeic acid from MO.29
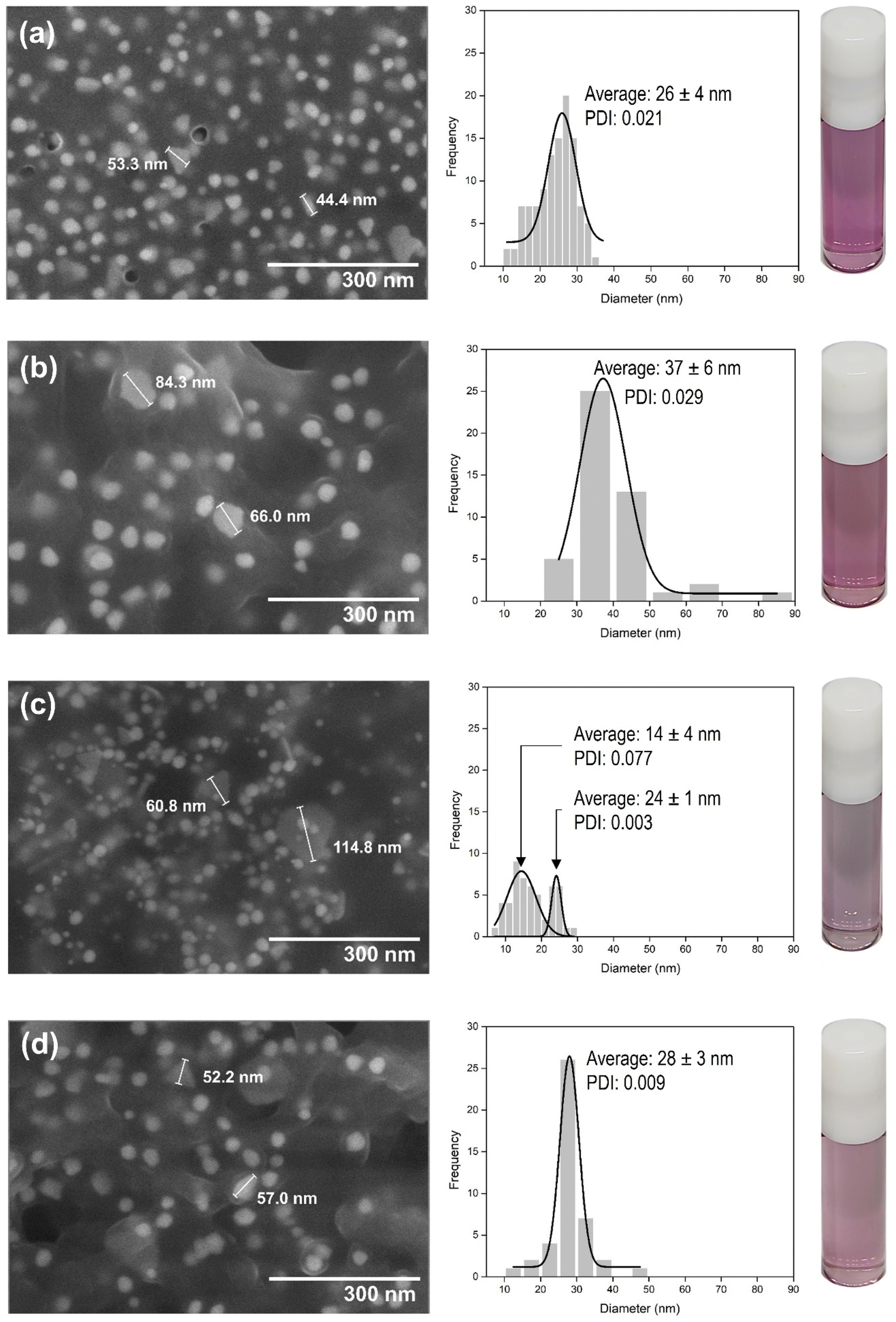 | ||
| Fig. 2 SEM image analysis of biosynthesized Au nanoparticle systems using various combined leaf extracts: (a) Au-MCLS-1; (b) Au-LSMO-1; (c) Au-MCMO-1, and (d) Au-MLM-1. Except for Au-MCMO-1, all other systems can be considered as monodipersed. The vials contain the Au nanoparticles dispersed in ultrapure water. | ||
The stability of Au-MCLS-1, Au-LSMO-1, Au-MCMO-1, and Au-MLM-1 nanoparticle systems was determined from LSPR spectrum evolution with time (Fig. 3). The Au nanoparticle systems did not exhibit significant changes on their spectra after two weeks. Based on this, the stability of all the Au nanoparticle systems were considered to withstand two weeks without apparent colloidal aggregation. Additionally, the measured zeta potential values of Au-MCLS-1, Au-LSMO-1, Au-MCMO-1, and Au-MLM-1 after five months were −21.3, −22.3, −20.1, and −22.0 mV, respectively and are comparable.
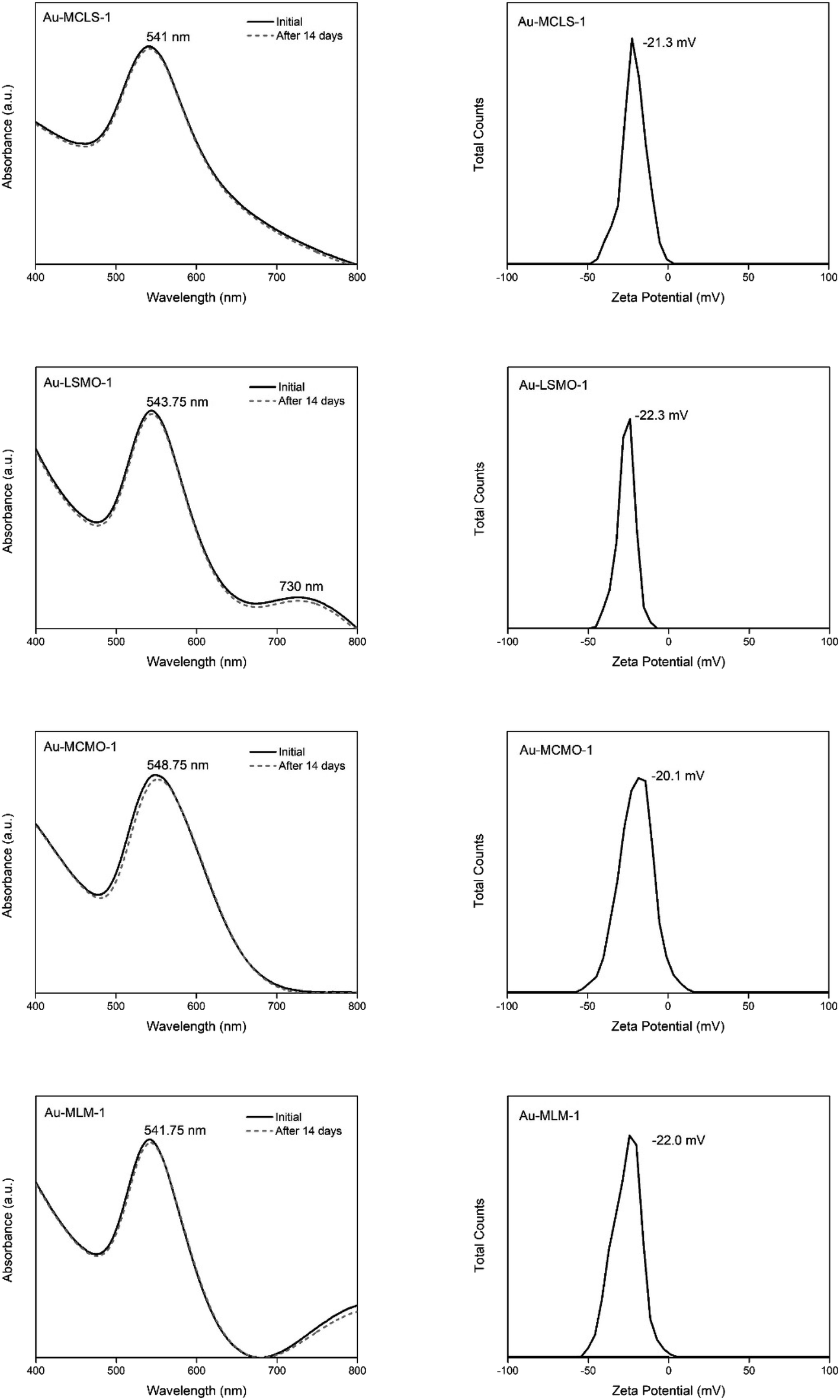 | ||
| Fig. 3 Analysis of the stability by absorbance spectral comparison and zeta potential of the biosynthesized Au nanoparticle systems using various combination of leaf extracts. All the synthesized nanoparticle systems can be considered stable. | ||
The Au nanoparticles can still be considered as stable after five months based on their zeta potential values. The zeta potential provides information regarding the electrochemical equilibrium at the interface of the liquid medium and Au nanoparticles. Higher magnitudes indicate higher resistance of the particles to aggregation, hence better colloidal stability. For the intended application of Au nanoparticles in this study, good stability of particles prior to ion exposure is beneficial since the mechanism of detection is via ion-induced particle attractions. Moreover, the Au nanoparticles exhibited a negative charge while dispersed in an aqueous medium. The negative charge makes the interparticle attractions after cation exposure possible.
The stability is attributed to the capping compounds, such as oxidized polyphenols and organic acids, of the Au nanoparticle systems which also acted as stabilizing agents. Polyphenolic compounds can act as both reducing and stabilizing agents, thus there is no need to introduce other capping agents.30
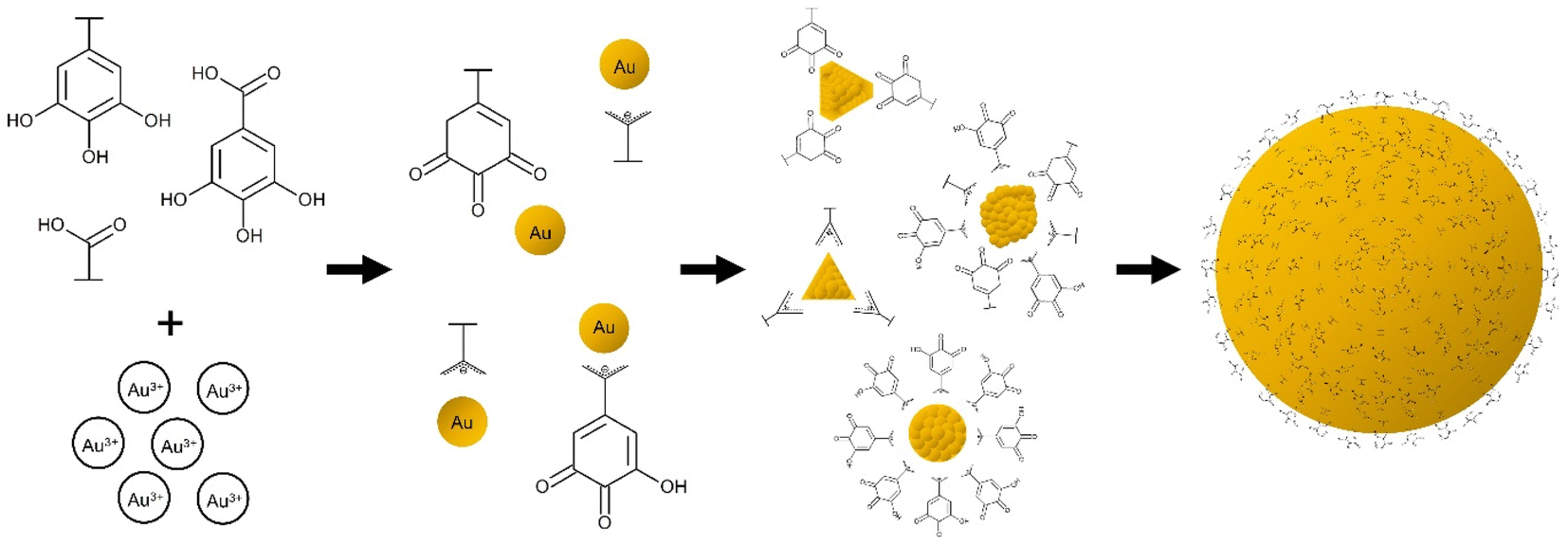 | ||
| Fig. 4 Graphical representation of proposed biosynthesis and capping mechanism of Au nanoparticles by polyphenols and organic acids. Polyphenols and organic acids can act in the reduction and growth of the biosynthesis of Au nanoparticles in which their interactions can dictate the morphology of the resulting nanoparticles. | ||
Functionalization viability from Au-MCLS-1 to Au-AS-1
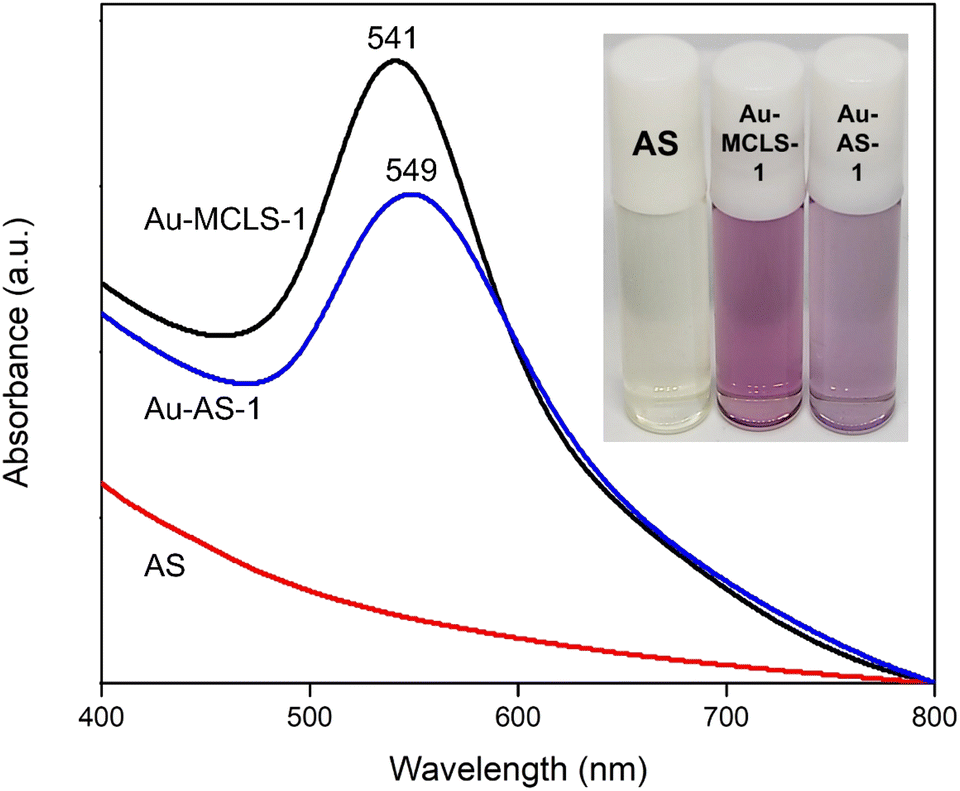 | ||
| Fig. 5 Comparisons of color and absorbance spectra of AS extract, and the Au nanoparticle systems before (Au-MCLS-1) and after (Au-AS-1) functionalization. A red shift after functionalization signals the substitution of previous capping compounds by AS organosulfur compounds. | ||
 | ||
| Fig. 6 Analysis and comparison of Au-MCLS-1 and Au-AS-1 compounds: (a) corresponding organosulfur compounds of AS extract obtained from 1H NMR spectral features of Au-AS-1 nanoparticle system in comparison to the 1H NMR spectra of Au-MCLS-1 as inset, and (b) FTIR spectral peaks corresponding to functional groups of Au-MCLS-1 before and after functionalization with AS extract to produce Au-AS-1. Differences in peak positions and features imply functionalization via substitution/displacement from Au-MCLS-1 to Au-AS-1. | ||
For comparisons, the 1H NMR spectra for Au-MCLS-1 is presented as inset in Fig. 6. The most intense Au-MCLS-1 peak at 8.23 ppm that was attributed to hydrogen atoms bonded to an aromatic group was also apparently missing in the spectra of Au-AS-1. Table S4† summarizes the 1H NMR peak differences between Au-MCLS-1 and Au-AS-1. Based on the comparison, almost complete conversion from Au-MCLS-1 to Au-AS-1 was achieved. This is due to their differences in peak positions in which only a minor peak with very faint signal was detected in Au-AS-1 which was considered to be a residue from Au-MCLS-1. This residual peak was attributed to hydrogen atoms attached to either a cyclic structure or to a carbon atom bonded to another R group of a cyclic structure as stated previously on the discussion based on Ahmed et al. (2015)26 and Al-Majed et al. (2018).27 This could be due to polycyclic organic acid compounds found in MC or LS. Aside from this peak, all other 1H NMR peaks of Au-AS-1 were different from those of Au-MCLS-1.
IR transmittance identify important functional groups of the organosulfur capping compounds. Additional information regarding the assessment of the viability of transforming Au-MCLS-1 to Au-AS-1 was also shown. The FTIR spectra comparison of Au-MCLS-1 an Au-AS-1 are shown in Fig. 6b. Both Au-MCLS-1 and Au-AS-1 have peaks at 3834 and 1537 cm−1 which were designated to the stretching of free hydroxyl which could be from phenolic structures and to the deformation of –NH, respectively. These peaks were considered as residual peaks from the capping compounds of Au-MCLS-1. Previously, a residual peak in 1H NMR was due to hydrogen atoms attached to either a cyclic structure or to a carbon atom bonded to another R group of a cyclic structure. In the employed 1H NMR spectroscopy, hydroxyl and –NH groups become undetected since deuterium from the deuterated water solvent exchanges with the hydrogen atoms of the hydroxyl and –NH groups. The hydrogens in NH, OH, COOH, and NH2 groups, and those bonded to S atoms are exchangeable by deuterium.39 By connecting the information from both analyses, it could be determined that the residual capping compounds from Au-MCLS-1 could be phenolic molecules having cyclic and amine groups in their structure or other similar derivatives. The FTIR peak of both Au-MCLS-1 and Au-AS-1 at 3834 cm−1 were of similar weak intensity, while in the 1H NMR spectra, the peak at 4.26–4.27 ppm was extremely weak for Au-AS-1 as compared to Au-MCLS-1. However, the 1537 cm−1 peak from Au-MCLS-1 was of a significantly less degree as compared to that of Au-AS-1. Since two identified organosulfur compounds, alliin and S-allylcysteine, also have –NH groups in their structures, the increase in peak intensity could be attributed to these compounds. In general, it could still be considered that almost complete substitution or displacement by organosulfur compounds occurred in the processing of Au-MCLS-1 to form Au-AS-1. This could be further validated since aside from the FTIR peaks at 3834 and 1537 cm−1, no other peaks were the same for the two Au nanoparticle systems.
The peaks at 3739, 3604, 3580, and 2934 cm−1 were due to the stretching of the free hydroxyl which can be assigned to the carboxylic acid functional groups. In addition to these, the 1436 cm−1 was attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in the carboxylic structure. Meanwhile, at the 3268 and 1243 cm−1 peaks, the stretching of N–H and the amine C–N bond can be assigned, respectively. These peaks representing the carboxylic acid and amine functional groups can be associated with the presence of alliin and S-allylcysteine as substituting capping compounds. The presence of other organosulfur compounds such as ajoene, which can have (E)- and (Z)-isomers, and allicin can be related to the peaks at 500 and 463 cm−1 which are designated to the S–S bond. The SO stretching at 1040 cm−1 can be attributed to alliin, allicin, and the two isomers of ajoene. The stretching of alkenyl CC was assigned to the peak at 1663 cm−1 and the out-of-plane bending of alkene C–H was ascribed to the 940 cm−1 peak. Both the peaks at 818 and 673 cm−1 were due to the C–S bond stretching. The alkenyl CC, alkene C–H, and C–S bonds are all characteristic features that are found in the structures of alliin, S-allylcysteine, (E)-ajoene, (Z)-ajoene, and allicin. Similar with all of the other Au nanoparticle systems investigated via FTIR analysis, Au-AS-1 also exhibited carbon dioxide adsorption capability as indicated by the peak at 2346 cm−1 which represents its asymmetric stretching. The comparison of the functional group summaries of Au-MCLS-1 and Au-AS-1 as determined by FTIR analysis are presented in Table S5.†
O group in the carboxylic structure. Meanwhile, at the 3268 and 1243 cm−1 peaks, the stretching of N–H and the amine C–N bond can be assigned, respectively. These peaks representing the carboxylic acid and amine functional groups can be associated with the presence of alliin and S-allylcysteine as substituting capping compounds. The presence of other organosulfur compounds such as ajoene, which can have (E)- and (Z)-isomers, and allicin can be related to the peaks at 500 and 463 cm−1 which are designated to the S–S bond. The SO stretching at 1040 cm−1 can be attributed to alliin, allicin, and the two isomers of ajoene. The stretching of alkenyl CC was assigned to the peak at 1663 cm−1 and the out-of-plane bending of alkene C–H was ascribed to the 940 cm−1 peak. Both the peaks at 818 and 673 cm−1 were due to the C–S bond stretching. The alkenyl CC, alkene C–H, and C–S bonds are all characteristic features that are found in the structures of alliin, S-allylcysteine, (E)-ajoene, (Z)-ajoene, and allicin. Similar with all of the other Au nanoparticle systems investigated via FTIR analysis, Au-AS-1 also exhibited carbon dioxide adsorption capability as indicated by the peak at 2346 cm−1 which represents its asymmetric stretching. The comparison of the functional group summaries of Au-MCLS-1 and Au-AS-1 as determined by FTIR analysis are presented in Table S5.†
 | ||
| Fig. 7 Quasi-spherical, triangular, and rod-shaped nanoparticles are seen on the (a) TEM image of Au-AS-1, and (b) the (002), (022), (113), and (004) reflections of Au are shown via electron diffraction. | ||
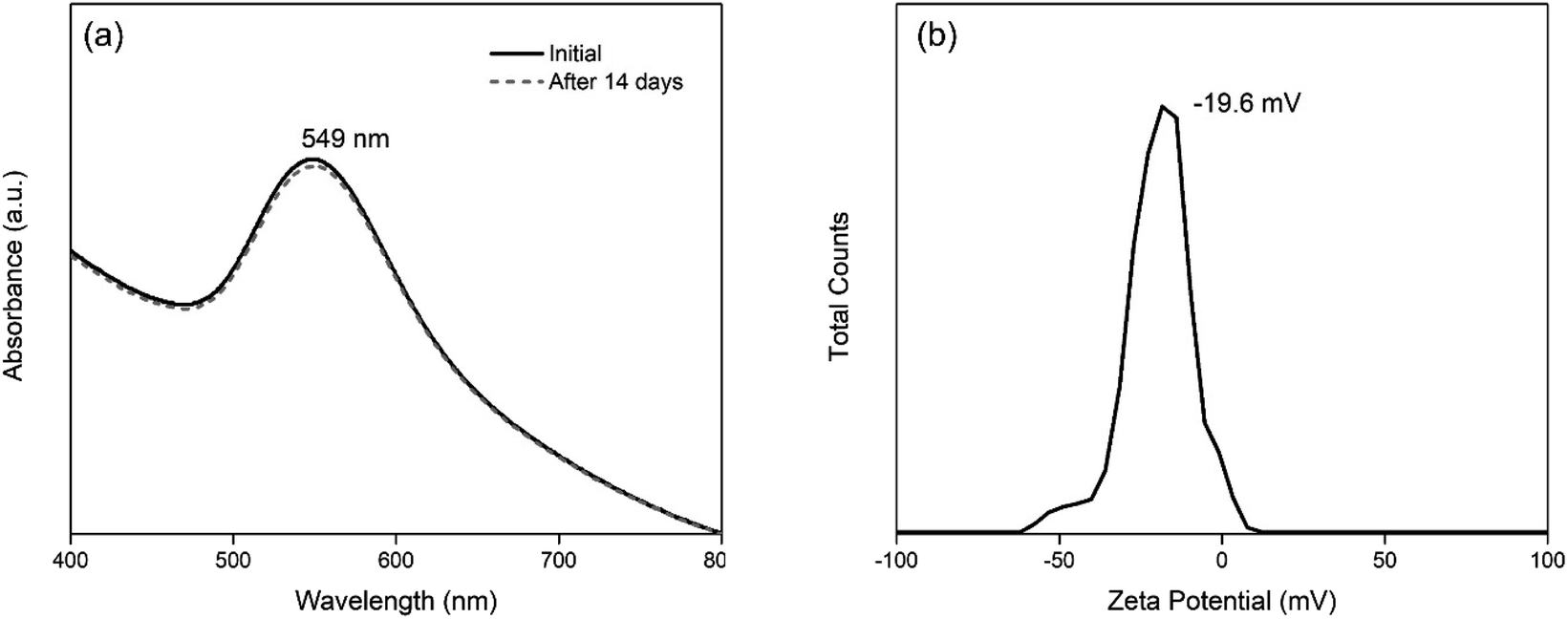 | ||
| Fig. 8 Stability of Au-AS-1 nanoparticle system as shown by its (a) absorbance spectra over a two-week period, and (b) zeta potential value after five months. | ||
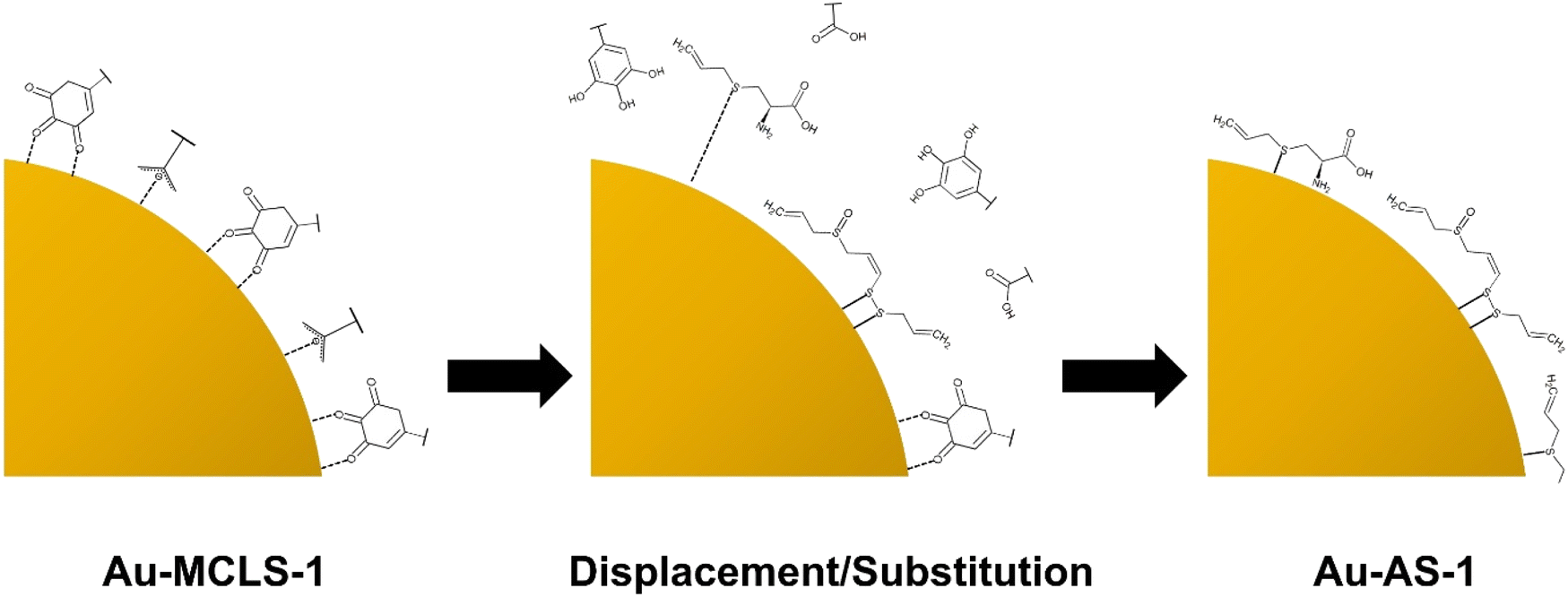 | ||
| Fig. 9 Graphical representation of the displacement/substitution of Au-MCLS-1 capping compounds by alliin, SAC, (E)-ajoene, (Z)-ajoene, and allicin (not drawn to scale) showing the preference of Au to bind with S. | ||
Au nanoparticle and ion interaction experiments
The interaction between Au-MCLS-1 nanoparticles and Pb2+ can be described by two distinct regions of their LSPR peak shift (nm) versus concentration (μM) plot as shown in Fig. 10a. The first region (0–283.78 μM) exhibits a linearly increasing relationship of Pb2+ concentration with LSPR peak shift. Meanwhile, the second region (283–851 μM) shows a quasi-plateau trend with fluctuations between Pb2+ concentrations with LSPR peak wavelength. The quasi-plateau trend could signify that the excess of Pb2+ does not significantly contribute to the LSPR peak shift of Au-MCLS-1, which could be attributed to the saturation of available hydroxyl functional groups that supposedly interact with the ions within the 283–851 μM range. In addition, the observed decrease of the absorbance with accompanying redshifting of the plasmon band could be attributed to the nanoparticle aggregation as caused by increasing Pb2+ concentrations.44,45
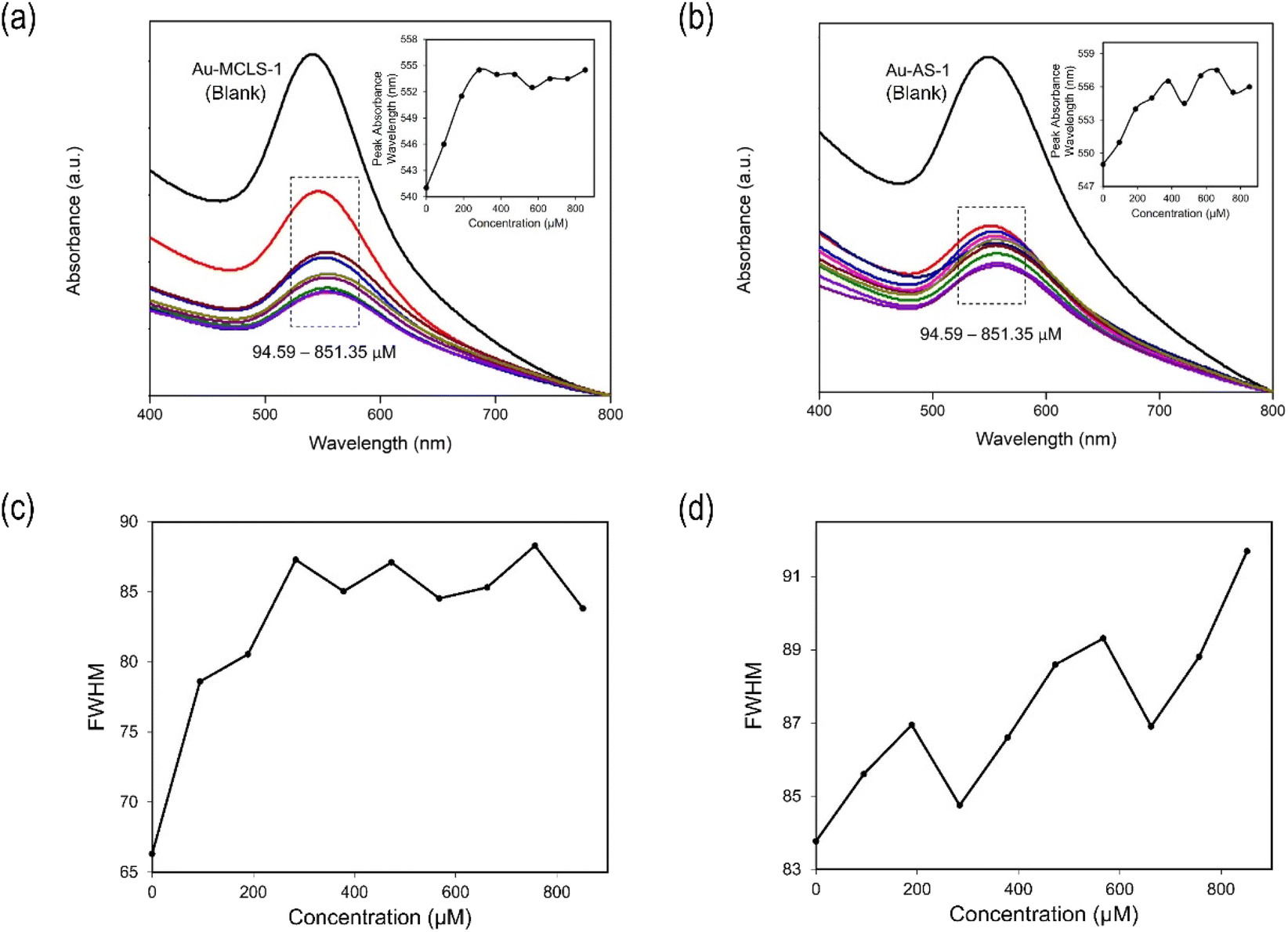 | ||
| Fig. 10 Absorbance of (a) Au-MCLS-1 and (b) Au-AS-1 with varying Pb2+ concentrations and their corresponding plot of absorbance peak wavelength vs. concentration as inset. Full width at half maximum (FWHM) values of the absorbance spectra of (c) Au-MCLS-1 show a linearly increasing then a fluctuating trend with Pb2+, while those of (d) Au-AS-1 indicate a fluctuating and generally increasing trend with Pb2+. The FWHM values were obtained using the Origin software via Gaussian fitting. | ||
The linear region (Fig. 10) is used to identify the Pb2+-containing AuNP solution. However, the Pb2+ quantification would require at least a UV-vis spectrophotometer to measure peak shifts since color tint variations are not easily distinguishable visually.
Similar to Au-MCLS-1, the detection of Pb2+ by Au-AS-1 nanoparticle system can be observed from the reduction of violet colloidal coloration intensity after the addition of ions as shown in Fig. S6b.† However, similarly, associating a specific color tint to the Pb2+ concentration with which the Au-AS-1 nanoparticle system is exposed with is difficult. Due to this, only the presence, but not the quantity, of Pb2+ can be determined via naked-eye observation.
There are also two distinct regions for the LSPR peak shift (nm) versus concentration (μM) plot (Fig. 10b) of the interaction between Au-AS-1 nanoparticles and Pb2+: linearly increasing behavior (0–378 μM) and fluctuating trend (378–851 μM). The peak shifts caused by Pb2+ are also accompanied by absorbance suppression which is in accordance to the observed decrease in colloidal color intensity. The region where linearity between LSPR peak shift and Pb2+ concentration exists can be used to determine the quantity of exposed ions with the Au-AS-1 nanoparticle system. This is because within the linear region, specific LSPR peak shifts can correspond to certain Pb2+ concentrations. However, with this method of ion quantification, a UV-vis spectrophotometer would be a requisite in order to determine peak shifts since colloidal color tint differences are difficult to observe via naked-eye means.
In general, minimal shifting of LSPR peaks were observed in the experiments. These minimal changes can be due to the ion concentrations which were not high enough to cause drastic shifts or even immediate aggregations. Further increasing the concentrations, may give rise to larger shifts and/or separate peaks due to aggregations.
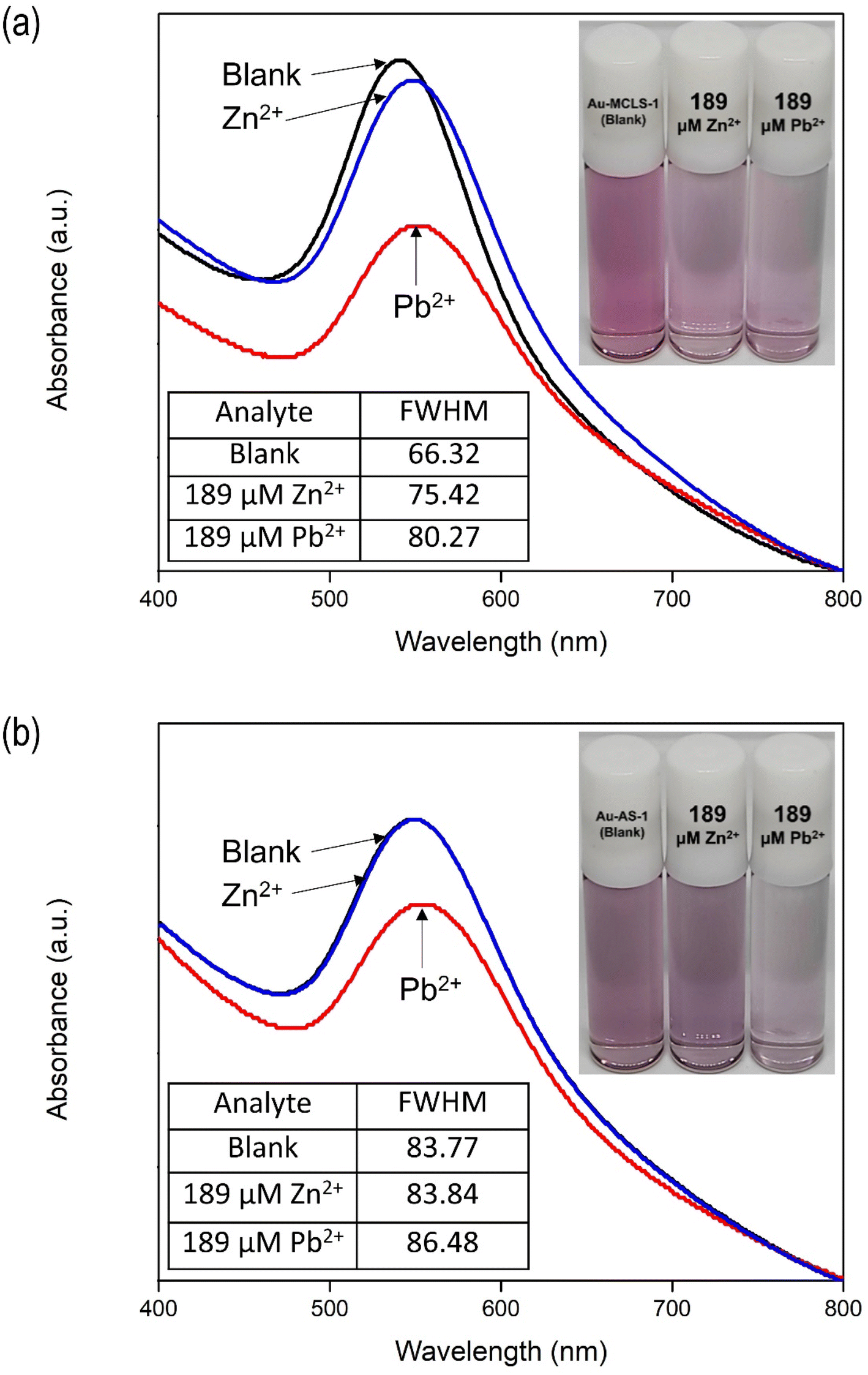 | ||
| Fig. 11 Absorbance spectra and color comparisons of (a) Au-MCLS-1 and (b) Au-AS-1 nanoparticle systems upon exposure to ultrapure water and 189 μM Pb2+ or Zn2+. FWHM values of the absorbance of Au-MCLS-1 increased due to Pb2+ and Zn2+. FWHM of the absorbance of Au-AS-1 increased due to Pb2+ while the change due to Zn2+ can be considered insignificant. Au-AS-1 is more selective to Pb2+ than Zn2+ as compared to Au-MCLS-1. The FWHM values were obtained using the Origin software via Gaussian fitting. | ||
The addition of Zn2+ to Au-AS-1 did not change the colloidal color, while Pb2+ exposure reduced the intensity. The LSPR of Au-AS-1 nanoparticles shifted significantly upon the exposure to Pb2+, while very minimal or negligible change was observed in its absorbance spectrum after Zn2+ were introduced into the system as shown in Fig. 11b. This means that the Au-AS-1 nanoparticle can differentiate between Pb2+ and Zn2+.
The decrease in observed intensity can also be explained in terms of the Pb2+- and Zn2+-induced destabilization of Au-MCLS-1 nanoparticles, and Pb2+-induced destabilization of Au-AS-1 nanoparticles, wherein the net negative charge on the surface of nanoparticles are destabilized.46,47 The ions cause the destabilization or particle aggregation that manifests as absorption peak broadening accompanied by redshifting. Initial response of the Au nanoparticle systems is decrease in bulk color intensity attributed to agglomeration due to the attractive forces caused by either or both ions to the capping compounds of Au nanoparticles. When the nanoparticles form networks, the absorption peak of the colloidal system becomes less pronounced. Meanwhile, after prolonged periods of time, the Pb2+-spiked colloidal systems and Zn2+-spiked Au-MCLS-1 system become clear with suspended dark violet to blackish aggregated particles.
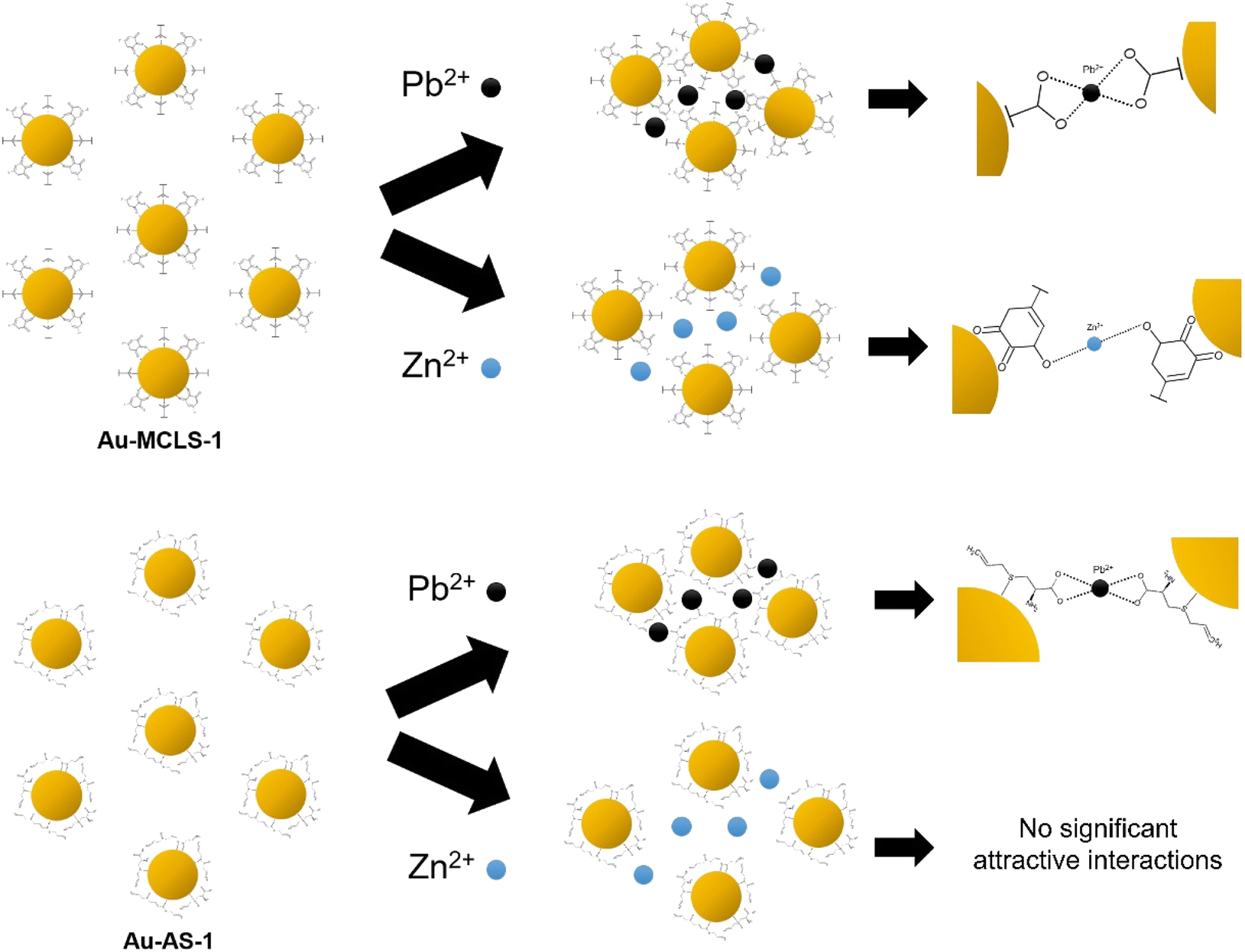 | ||
| Fig. 12 Graphical representation of the effects of Pb2+ or Zn2+ on the interparticle distances of Au-MCLS-1 and Au-AS-1 nanoparticles (not drawn to scale). Au-AS-1 has more selectivity to Pb2+ as compared to Au-MCLS-1 due to the selective affinity of capping compounds.44,48–51 | ||
The degree of aggregation can be dependent on the concentration of ions. As previously discussed, there are certain concentration ranges wherein the LSPR peak shift has linear relationship with Pb2+ concentration. These ranges could be exploited for the quantification of ions. Above these concentration ranges, erratic fluctuations in detection could occur. But even if ion quantification is not possible for the fluctuating regions, qualitative ion detection could still be done: Au-MCLS-1 could be used for confirming the presence of Pb2+ and/or Zn2+, then Au-AS-1 can be used to selectively confirm Pb2+ presence.
In order to determine the lower limits of detection of Au-MCLS-1 and Au-AS-1, linear regression analysis was performed on the LSPR peak shift versus log of Pb2+ concentration plots. The lower limits of detection are the extrapolated concentration values after obtaining the linear equations. These values correspond to the maximum Pb2+ concentration with which no LSPR peak shift is exhibited. The lower limits of detection for Au-MCLS-1 and Au-AS-1 are 49 μM (10 ppm) and 55 μM (11 ppm), respectively. Fig. 13a and b show the plot of peak shift vs. concentration and linear regression analysis of shift in peak vs. logarithm of concentration for Au-MCLS-1 and Au-AS-1, respectively. In comparison to literature, the lower limits of Pb2+ detection of Au-MCLS-1 (49.44 μM) and Au-AS-1 (55.18 μM) are significantly greater than that of the Au nanoparticles prepared in the study of Chai et al.53 and Mao et al.,54 having 0.1 μM of minimum detection limit. Despite this, the prepared Au nanoparticles could still be used in ion detection applications of relatively higher permissible limits of Pb2+.
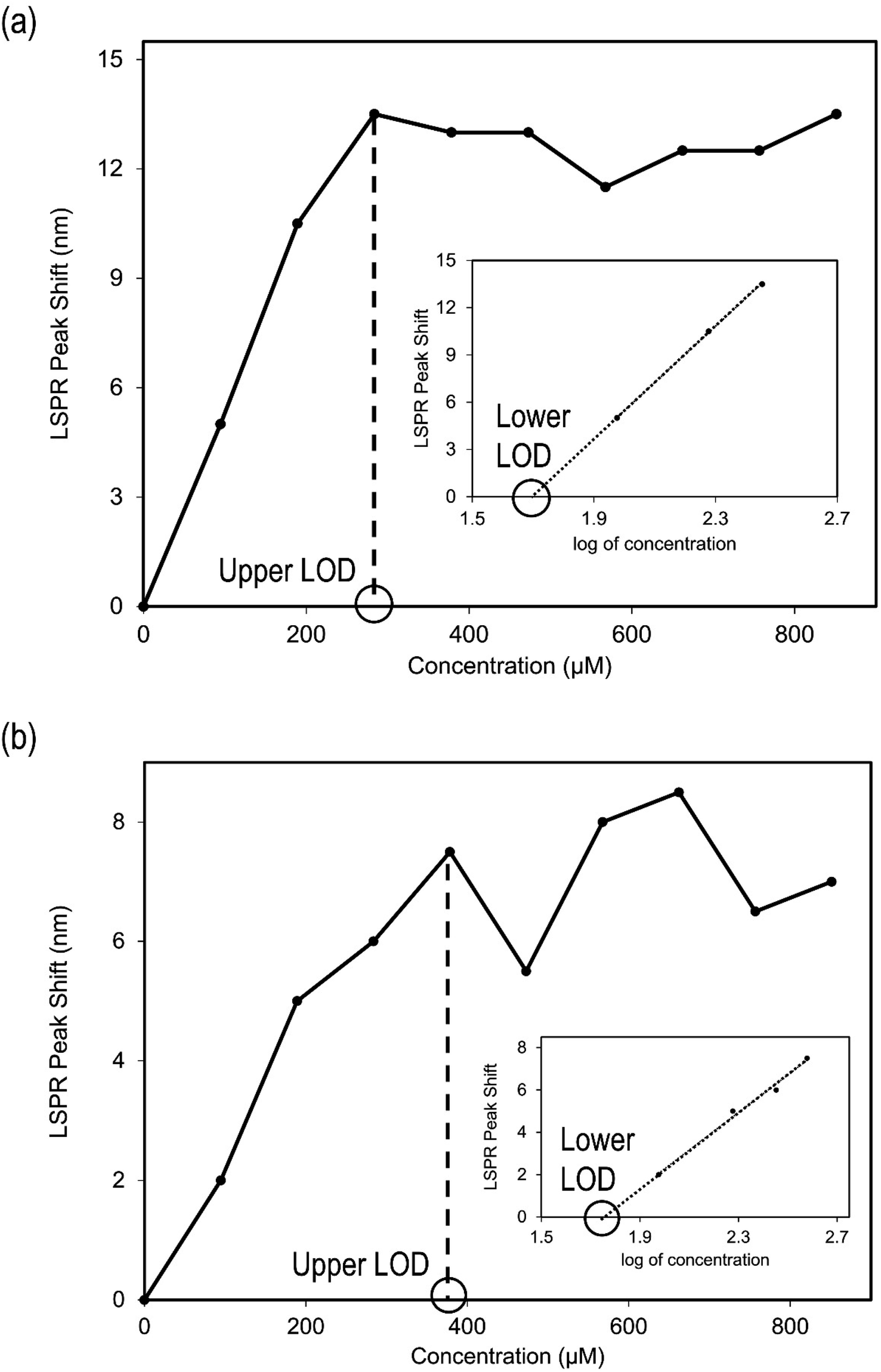 | ||
| Fig. 13 Plots of peak shift vs. concentration and linear regression analysis of shift in peak vs. log concentration plots for (a) Au-MCLS-1: upper LOD is 284 μM (59 ppm) and the lower LOD is 49 μM (10 ppm), and (b) Au-AS-1: upper LOD is 378 μM (78 ppm) and the lower LOD is 55 μM (11 ppm). | ||
Ayers and Westcot (1985) recommended 5 ppm as the permissible amount of lead in irrigation water,10 and this value was used by Farid et al. (2015)11 as basis for assessing environmental waters. Although the lower limits of detection for Au-MCLS-1 (10.24 ppm) and Au-AS-1 (11.43 ppm) are above this value, the differences can still be considered as not too pronounced. Moreover, Au-MCLS-1 and Au-AS-1 nanoparticle systems could still be used for initial screening of water samples.
In addition, the Au-AS-1 nanoparticle system have the potential to be used for the assessment of environmental water remediation effectiveness especially in achieving Pb2+ concentrations below the severe effect level of 250 ppm as stated by NOAA.12
Conclusions
Stable and nearly-monodisperse Au nanoparticle systems were biosynthesized via a relatively rapid one-pot approach at room temperature with manual agitation using different combinations of leaf extracts (MCLS, LSMO, MCMO, and MLM), and functionalized using heat-treated AS cloves extract. The sizes of the quasi-spherical Au nanoparticles have significant differences among different combinations of leaf extracts. Organic acids and polyphenols from leaf extracts were found to be of major contribution to the Au nanoparticles' makeup. In the functionalization, almost complete displacement of Au-MCLS-1 capping and stabilizing compounds occurred due to substitution by the biochemical compounds from AS extract such as alliin, S-allylcysteine, allicin, and ajoene. The interaction of Au-MCLS-1 to Pb2+ is almost similar to its interaction with Zn2+, while Au-AS-1 tends to be more selective to Pb2+ as compared to Zn2+. Based on the investigations, the highlighted straightforward biosynthesis and functionalization methods are viable for Au nanoparticle system processing, and the Au-MCLS-1 and Au-AS-1 nanoparticle systems have the potential to be used in the detection of divalent ions (i.e. Pb2+ or Zn2+) and Pb2+, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Department of Science and Technology – Science Education Institute and Engineering Research and Development for Technology for the funding.References
- V. Dhumale, R. Gangwar and N. Pande, Mater. Res. Innovations, 2020, 25, 354–362 CrossRef.
- K. Parveen, V. Banse and L. Ledwani, AIP Conf. Proc., 2016, 1724, 020048 CrossRef.
- L. Bertel Garay, F. Martínez Ortega and S. Méndez-Sanchez, J. Mex. Chem. Soc., 2018, 62, 1–9 CrossRef.
- J. Wai and S. New, RSC Adv., 2020, 10, 1088–1094 RSC.
- N. Komova, K. Serebrennikova, A. Berlina, S. Pridvorova, A. Zherdev and B. Dzantiev, The 1st International Electronic Conference on Chemical Sensors and Analytical Chemistry, 2021, 5, p. 6 Search PubMed.
- M. Balali-Mood, K. Naseri, Z. Tahergorabi, M. Khazdair and M. Sadeghi, Front. Pharmacol., 2021, 12, 1–19 CrossRef PubMed.
- P. Tchounwou, C. Yedjou, A. Patlolla and D. Sutton, Exper. Suppl., 2012, 133–164 CrossRef PubMed.
- U.S. Environmental Protection Agency, Lead and Copper Rule, https://www.epa.gov/dwreginfo/lead-and-copper-rule#:%7E:text=The1991Rule,-In1991%2CEPA%26text=Theruleestablishedamaximum,copperwithinthedistributionsystem, (accessed May 2022) Search PubMed.
- U.S. Environmental Protection Agency, Lead and Copper Rule: Summary of Revisions, http://nepis.epa.gov/Exe/ZyPDF.cgi?Dockey=P1005884.txt, (accessed May 2022) Search PubMed.
- R. Ayers and D. Westcot, Water quality for agriculture, Food and Agriculture Organization, Rome, 1985 Search PubMed.
- G. Farid, N. Sarwar, Saifullah, A. Ahmad, A. Ghafoor and M. Rehman, Adv. Crop Sci. Technol., 2015, 4, 1–7 CrossRef.
- National Oceanic and Atmospheric Administration, Screening Quick Reference Tables, https://response.restoration.noaa.gov/sites/default/files/SQuiRTs.pdf, (accessed May 2022) Search PubMed.
- J. Anderson, E. Bowden and P. Pickup, Anal. Chem., 1996, 68, 379R–444R CrossRef.
- A. Burlingame, R. Boyd and S. Gaskell, Anal. Chem., 1996, 599R–651R CrossRef CAS PubMed.
- K. Jackson and G. Chen, Anal. Chem., 1996, 68, 231R–256R CrossRef.
- T. Lin and M. Chung, Sensors, 2008, 8, 582–593 CrossRef CAS PubMed.
- K. Yoosaf, B. Ipe, C. Suresh and K. Thomas, J. Phys. Chem. C, 2007, 111, 12839–12847 CrossRef CAS.
- M. McDonald, I. Mila and A. Scalbert, J. Agric. Food Chem., 1996, 44, 599–606 CrossRef CAS.
- N. Modi, T. Goel, T. Das, S. Malik, S. Suri, A. Rawat, S. Srivastava, R. Tuli, S. Malhotra and S. Gupta, Indian J. Med. Res., 2013, 137, 540–548 Search PubMed.
- G. Oboh, A. Ademiluyi, A. Ademosun, T. Olasehinde, S. Oyeleye, A. Boligon and M. Athayde, Biochem. Res. Int., 2015, 1–8 Search PubMed.
- Z. Zakaria, N. Mahmood, S. Mamat, N. Nasir and M. Omar, Front. Pharmacol., 2018, 8, 982 CrossRef PubMed.
- S. Santhosha, P. Jamuna and S. Prabhavathi, Food Biosci., 2013, 3, 59–74 CrossRef CAS.
- J. Imai, N. Ide, S. Nagae, T. Moriguchi, H. Matsuura and Y. Itakura, Planta Med., 1994, 60, 417–420 CrossRef CAS PubMed.
- F. Rajathi, R. Arumugam, S. Saravanan and P. Anantharaman, J. Photochem. Photobiol., Bc, 2014, 135, 75–80 CrossRef PubMed.
- G. Wang and W. Sun, J. Phys. Chem. B, 2006, 110, 20901–20905 CrossRef CAS PubMed.
- Y. Ahmed, Z. Yaakob, P. Akhtar, M. Rahman and F. Islam, Int. J. Pharmacol., 2015, 11, 821–827 CAS.
- A. Al-Majed, A. Bakheit, R. Alharbi and H. Abdel Aziz, Profiles of Drug Substances, Excipients and Related Methodology, 2018, pp. 209–254 Search PubMed.
- N. Bai, K. He, M. Roller, B. Zheng, X. Chen, Z. Shao, T. Peng and Q. Zheng, J. Agric. Food Chem., 2008, 56, 11668–11674 CrossRef CAS PubMed.
- M. Vergara-Jimenez, M. Almatrafi and M. Fernandez, Antioxidants, 2017, 6, 91 CrossRef PubMed.
- S. Dash, R. Majumdar, A. Sikder, B. Bag and B. Patra, Appl. Nanosci., 2014, 4, 485–490 CrossRef CAS.
- K. Kumar, B. Mandal, H. Kiran Kumar and S. Maddinedi, Spectrochim. Acta, Part A, 2013, 116, 539–545 CrossRef PubMed.
- H. Liu, X. Zhang, Z. Xu, Y. Wang, Y. Ke, Z. Jiang, Z. Yuan and H. Li, Nanotechnology, 2020, 31, 415601 CrossRef CAS PubMed.
- S. Moreno-Álvarez, G. Martínez-Castañón, N. Niño-Martínez, J. Reyes-Macias, N. Patiño-Marin, J. Loyola-Rodriguez and F. Ruiz, J. Nanopart. Res., 2010, 12, 2741–2746 CrossRef.
- M. Ouellette, F. Masse, M. Lefebvre-Demers, Q. Maestracci, P. Grenier, R. Millar, N. Bertrand, M. Prieto and E. Boisselier, Sci. Rep., 2018, 8, 14357 CrossRef PubMed.
- L. Jayathilaka, S. Gupta, J. Huang, J. Lee and B. Lee, J. Biomol. Tech., 2014 Search PubMed.
- J. Debouzy, J. Neumann, M. Hervé, D. Daveloose, J. Viret and R. Apitz-Castro, Eur. Biophys. J., 1989, 17, 211–216 CrossRef CAS PubMed.
- L. Small, J. Bailey and C. Cavallito, J. Am. Chem. Soc., 1947, 69, 1710–1713 CrossRef CAS PubMed.
- A. Roseblade, A. Ung and M. Bebawy, Acta Pharmacol. Sin., 2017, 38, 1353–1368 CrossRef CAS PubMed.
- G. Chen, I. Daaro, B. Pramanik and J. Piwinski, J. Mass Spectrom., 2009, 44, 203–213 CrossRef CAS PubMed.
- Y. Yulizar, H. Ariyanta and L. Abduracman, Bull. Chem. React. Eng. Catal., 2017, 12, 212–218 CrossRef CAS.
- L. Yang, X. Zhang, J. Wang, H. Sun and L. Jiang, Microchim. Acta, 2018, 185, 472 CrossRef PubMed.
- R. Nuzzo, B. Zegarski and L. Dubois, J. Am. Chem. Soc., 1987, 109, 733–740 CrossRef CAS.
- G. Perera, S. Athukorale, F. Perez, C. Pittman, Jr and D. Zhang, J. Colloid Interface Sci., 2017, 511, 335–343 CrossRef PubMed.
- V. Le, T. Duong, V. Le, T. Phan, T. Nguyen, T. Chau and V. Doan, RSC Adv., 2021, 11, 15438–15448 RSC.
- T. Mostowtt, J. Munoz and B. McCord, The Analyst, 2019, 144, 6404–6414 RSC.
- M. Annadhasan, T. Muthukumarasamyvel, V. R. Sankar Babu and N. Rajendiran, ACS Sustainable Chem. Eng., 2014, 2, 887–896 CrossRef CAS.
- E. Priyadarshini and N. Pradhan, Sci. Rep., 2017, 7, 1–8 CrossRef CAS PubMed.
- N. Ratnarathorn, O. Chailapakul and W. Dungchai, Talanta, 2015, 132, 613–618 CrossRef CAS PubMed.
- K. Filipiak, M. Hidalgo, J. Silvan, B. Fabre, R. Carbajo, A. Pineda-Lucena, A. Ramos, B. de Pascual-Teresa and S. de Pascual-Teresa, Food Funct., 2014, 5, 381–389 RSC.
- J. Pang, J. Yen, H. Wu and S. Huang, Int. J. Mol. Sci., 2017, 18, 1354 CrossRef PubMed.
- Y. Kim, R. C. Johnson and J. T. Hupp, Nano Lett., 2001, 1, 165–167 CrossRef CAS.
- L. Chen, J. Li and L. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 15897–15904 CrossRef CAS PubMed.
- F. Chai, C. Wang, T. Wang, L. Li and Z. Su, ACS Appl. Mater. Interfaces, 2010, 2, 1466–1470 CrossRef CAS PubMed.
- X. Mao, Z.-P. Li and Z.-Y. Tang, Front. Mater. Sci., 2010, 5, 322–328 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04092e |
| This journal is © The Royal Society of Chemistry 2023 |