DOI:
10.1039/D3RA04111E
(Paper)
RSC Adv., 2023,
13, 23745-23753
Six metal–organic architectures from a 5-methoxyisophthalate linker: assembly, structural variety and catalytic features†
Received
19th June 2023
, Accepted 31st July 2023
First published on 7th August 2023
Abstract
A methoxy-functionalized isophthalic acid, 5-methoxy isophthalic acid (H2mia), was used a versatile linker for assembling six new metal(II) compounds under hydrothermal conditions. The obtained products were [Cu2(μ2-mia)2(phen)2(H2O)2]·2H2O (1), [Mn(μ3-mia)(phen)]n (2), [Co(μ2-mia)(2,2′-bipy)(H2O)]n·nH2O (3), [Co(μ3-mia)(μ2-4,4′-bipy)]n·nH2O (4), [Co(μ3-mia)(py)2]n (5), and [Cd(μ2-mia)(py)(H2O)2]n·nH2O (6), where phen(1,10-phenanthroline), 2,2′-bipy(2,2′-bipyridine), 4,4′-bipy(4,4′-bipyridine) or py(pyridine) were incorporated as auxiliary ligands. The crystal structures of 1–6 range from 0D (1) and 1D (2, 3, 5, 6) CPs to a 2D network (4) with a variety of topological types. The catalytic behavior of 1–6 was studied in the cyanosilylation reaction between trimethylsilyl cyanide and aldehydes, resulting in up to 99% yields of products under optimized conditions. Various reaction parameters as well as catalyst recycling and substrate scope were investigated. This study widens the use of H2mia as a versatile dicarboxylate linker for assembling a diversity of functional metal–organic architectures with remarkable structural features and catalytic properties.
Introduction
Coordination polymers (CPs) are currently the most explored crystalline materials in the fields of inorganic and coordination chemistry. Owing to their almost infinite structural and topological diversity,1–4 and plethora of unique properties, these compounds have attracted tremendous attention. The research on CPs opened up potential applications of CPs in gas adsorption and separation,5–9 sensing,10–13 heterogeneous catalysis,14–20 and many other areas.21–25
The synthesis of CPs can often be guided for producing definite structural and topological types toward various uses.26–30 Despite the significant progress made in this field, structural prediction is not always facile, especially when exploring self-assembly protocols under hydro- or solvothermal conditions. The crystallization of CPs and their final crystal structures might be affected by different factors, including the type of organic linkers and auxiliary ligands,31–33 coordination chemistry of the metal ions involved,34,35 type of solvent,36,37 reaction temperature38,39 and pH.40,41
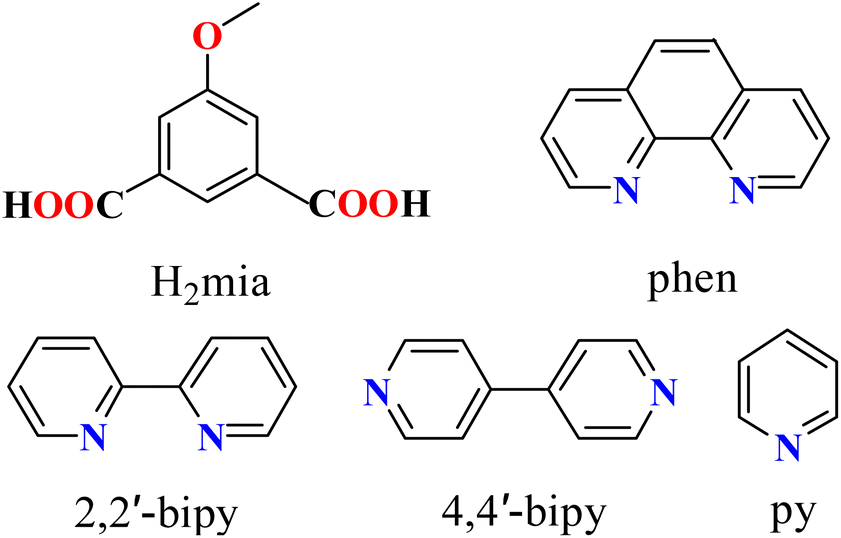
Aromatic polycarboxylic acids are the most commonly used organic linkers for the synthesis of CPs, because of their adaptable coordination behavior, varying charge and degree of deprotonation, excellent thermal stability, and participation in noncovalent interactions.19,20,35,38 Recently, our groups have been focused on designing functional CPs based on multicarboxylate blocks.20,42,43 As an extension of this general research direction, herein we selected an underexplored methoxy-functionalized isophthalate ligand, 5-methoxy isophthalic acid (H2mia, Scheme 1). The organic compound can potentially act as a versatile flexible linker for the synthesis of coordination polymers. The selection of the ligand has relied on the following considerations. (1) The H2mia ligand has a lot of coordination modes and diverse deprotonated types (Scheme 2) according to our survey of the Cambridge Crystallographic Database, which is beneficial for the implementation of unique and interesting networks, such as porous frameworks. (2) The dicarboxylic acid is stable under hydrothermal synthetic conditions. (3) The H2mia ligand results in diverse structures with excellent luminescent, adsorbing and separating, and magnetic properties.44–51 However, only a H2mia-based network with catalytic properties has been reported.52 Thus the present study gave us a good opportunity to develop this field.
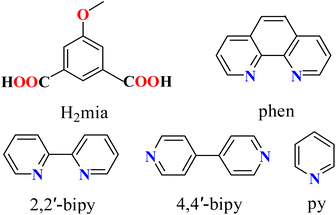 |
| | Scheme 1 Structures of H2mia and auxiliary ligands. | |
 |
| | Scheme 2 Coordination modes of mia2− linkers. | |
This work reports on the hydrothermal synthesis, isolation, characterization, crystal structures, topological features, thermal stability, and catalytic properties of six new coordination compounds prepared from metal(II) chlorides, H2mia, and various supporting ligands (Scheme 1). The obtained compounds differ from 0D (1) and 1D (2, 3, 5, 6) CPs to a 2D network (4), namely [Cu2(μ2-mia)2(phen)2(H2O)2]·2H2O (1), [Mn(μ3-mia)(phen)]n (2), [Co(μ2-mia)(2,2′-bipy)(H2O)]n·nH2O (3), [Co(μ3-mia)(μ2-4,4′-bipy)]n·nH2O (4), [Co(μ3-mia)(py)2]n (5), and [Cd(μ2-mia)(py)(H2O)2]n·nH2O (6).
Experimental section
General methods
5-Methoxy isophthalic acid (H2mia) was acquired from Jinan Henghua Sci. & Tec. Co., Ltd. C/H/N analyses were obtained using an Elementar Vario EL elemental analyzer. FTIR spectra (KBr) were recorded on a Bruker EQUINOX 55 spectrometer. TGA measurements were performed on a LINSEIS STA PT1600 thermal analyzer (10 °C min−1 heating rate, N2 flow). PXRD analyses were run on a Rigaku-Dmax 2400 diffractometer (Cu Kα radiation, λ = 1.54060 Å). Magnetic susceptibility data were collected in the 2–300 K temperature range with a Quantum Design SQUID Magnetometer MPMS XL-7 with a field of 0.1 T. A correction was made for the diamagnetic contribution prior to data analysis. Solution 1H NMR spectra were measured on a JNM ECS 400 M spectrometer.
Synthesis of coordination polymers 1–6
The compounds 1–6 were prepared by hydrothermal synthesis, starting from the reaction mixtures composed of copper(II), manganese(II), cobalt(II) or cadmium(II) chlorides, dicarboxylic acid linker, auxiliary ligand, sodium hydroxide, and water as solvent (Table 1). After hydrothermal treatment at 160 °C for three days, the reaction mixtures were gradually (10 °C h−1) cooled to room temperature for crystallization of CPs. Analytical data and detailed synthesis procedures for 1–6 are provided in ESI.†
Table 1 Summary of synthetic conditions and assembled productsa
| Product formula |
Metal(II) precursor |
ALb |
M2+/linker/AL/NaOH molar ratio |
Dimensi-onality |
| Hydrothermal synthesis: stainless steel reactor (25 mL volume; Teflon-lined), MCl2 (0.2 mmol), H2O (10 mL), 3 days at 160 °C, 10 °C h−1 cooling rate. AL, N-donor auxiliary ligand. The oxidation state of the metals in compounds 1–5 was determined by the values of magnetic moment (Fig. S3 and Table S3). |
| [Cu2(μ2-mia)2(phen)2(H2O)2]·2H2O (1) |
CuCl2·2H2O |
Phen |
1/1/1/2 |
0D |
| [Mn(μ3-mia)(phen)]n (2) |
MnCl2·4H2O |
Phen |
1/1/1/2 |
1D |
| [Co(μ2-mia)(2,2′-bipy)(H2O)]n·nH2O (3) |
CoCl2·6H2O |
2,2′-Bipy |
1/1/1/2 |
1D |
| [Co(μ3-mia)(μ2-4,4′-bipy)]n·nH2O (4) |
CoCl2·6H2O |
4,4′-Bipy |
1/1/1/2 |
2D |
| [Co(μ3-mia)(py)2]n (5) |
CoCl2·6H2O |
py |
1/1/31/— |
1D |
| [Cd(μ2-mia)(py)(H2O)2]n·nH2On (6) |
CdCl2·H2O |
py |
1/1/31/— |
1D |
Crystal structures
Bruker Smart CCD or an Agilent SuperNova diffractometers (Cu Kα radiation, λ = 1.54178 Å) were used for single-crystal X-ray data collection. SADABS was applied for semiempirical absorption correction. Structure solution by direct methods and refinement by full-matrix least-squares on F2 was performed on SHELXS-97 and SHELXL-97.53 All atoms (except for hydrogen atoms) were refined anisotropically by full-matrix least-squares methods on F2. Hydrogen atoms riding at carbon centers were placed in calculated positions with fixed isotropic thermal parameters and included in final structure factor calculations. Other hydrogen atoms were positioned by difference maps and constrained to ride on their parent O atoms. Some highly disordered solvent molecules in 4 were removed using SQUEEZE routine in PLATON.54 The final amount of solvent molecules was estimated from the data of elemental and thermogravimetric analyses. Crystal data are given in Table 2. ESI contains selected bonding and H-bonding parameters (Tables S1 and S2†).
Table 2 Crystal data for 1–6
| R1 = Σ‖Fo| − |Fc‖/|Fo|. wR2 = {Σ[w(Fo2 − Fc2)]2/Σ[w(Fo2)]2}1/2. |
| Compound |
1 |
2 |
3 |
4 |
5 |
6 |
| Chemical formula |
C42H36Cu2N4O14 |
C42H28Mn2N4O10 |
C19H18CoN2O7 |
C19H16CoN2O6 |
C19H16CoN2O5 |
C14H17CdNO8 |
| Formula weight |
947.83 |
858.56 |
445.28 |
427.25 |
411.27 |
439.68 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
Monoclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c |
P21/c |
P |
C2/c |
P |
| a/Å |
7.3455(7) |
38.823(4) |
8.8603(3) |
10.0273(10) |
16.0459(6) |
9.3015(5) |
| b/Å |
10.4021(9) |
10.4967(8) |
17.4505(5) |
10.0728(9) |
12.0320(4) |
10.1706(6) |
| c/Å |
12.8273(10) |
18.7612(12) |
12.4228(4) |
10.7050(14) |
19.4535(9) |
10.2299(5) |
| α/° |
96.431(7) |
90 |
90 |
74.711(9) |
90 |
68.453(5) |
| β/° |
92.558(7) |
105.531(8) |
97.718(3) |
66.547(11) |
105.900(4) |
81.292(4) |
| γ/° |
101.886(8) |
90 |
90 |
76.120(8) |
90 |
67.661(5) |
| V/Å3 |
950.75(15) |
7366.3(11) |
1903.37(11) |
945.6(2) |
3612.1(3) |
832.50(9) |
| T/K |
293(2) |
293(2) |
293(2) |
293(2) |
293(2) |
293(2) |
| Z |
1 |
8 |
4 |
2 |
8 |
2 |
| Dc/g cm−3 |
1.655 |
1.548 |
1.554 |
1.437 |
1.513 |
1.754 |
| μ/mm−1 |
2.071 |
6.161 |
7.480 |
7.399 |
7.748 |
10.901 |
| F(000) |
486 |
3504 |
916 |
418 |
1688 |
440 |
| Refl. measured |
3319 |
6861 |
3367 |
3216 |
3136 |
2869 |
| Unique refl. (Rint) |
2810 (0.0239) |
3204 (0.0933) |
2795 (0.0237) |
2712 (0.0329) |
2533 (0.0260) |
2740 (0.0390) |
| GOF on F2 |
1.065 |
0.988 |
1.079 |
1.017 |
1.048 |
1.030 |
| R1[I > 2σ(I)]a |
0.0434 |
0.0806 |
0.0378 |
0.0536 |
0.0421 |
0.0353 |
| wR2[I > 2σ(I)]b |
0.1153 |
0.1956 |
0.0937 |
0.1351 |
0.1028 |
0.0951 |
PXRD patterns for the microcrystalline samples of compounds 1–6 were obtained at room temperature and are given in Fig. S2 (ESI),† along with the diffractograms simulated from CIF files. Both experimental and simulated patterns are in good agreement with what confirms a phase purity of 1–6.
Crystal structures were also analyzed from topological perspective53 by employing the concept of underlying net.53,55 Such simplified nets were built by omitting terminal ligands and contracting bridging ligands to centroids, while preserving the connectivity. CCDC-2269625–2269630 contain the supplementary crystallographic data for 1–6.
Catalytic activity in cyanosilylation of benzaldehydes
In a typical procedure for the cyanosilylation of benzaldehydes, a suspension comprising 4-nitrobenzaldehyde (0.50 mmol), trimethylsilyl cyanide (TMSCN, 1.0 mmol), and catalyst (typically 3.0 mol%) in dichloromethane (2.5 mL) was stirred at 35 °C for a desired time. Then, the catalyst was isolated by centrifugation. The obtained solution was evaporated under reduced pressure, resulting in crude solid. This was dissolved in CDCl3 and analyzed by 1H NMR spectroscopy (JNM ECS 400 M spectrometer) for product quantification (further details are given in Fig. S4, ESI†). In the catalyst recycling tests, the catalyst was separated by centrifugation, washed with CH2Cl2, dried at 25 °C, and then reused in subsequent runs. Substrate scope using other aldehydes was investigated following the above-described procedure.
Results and discussion
Hydrothermal preparation of 1–6
To explore 5-methoxy isophthalic acid (H2mia) as a linker in the design of coordination polymers incorporating metal(II) centers (Cu, Mn, Co, or Cd), we performed a number of hydrothermal reactions between the corresponding metal(II) chlorides, H2mia, NaOH, and auxiliary ligands selected from phen, 2,2′-bipy, 4,4′-bipy, or py. The reactions were carried out for 3 days at 160 °C, followed by crystallization of CPs through a slow cooling of the reaction mixtures. Several reaction attempts led to positive results, permitting the generation and full characterization of six new coordination compounds (Tables 1 and 2). These were isolated in good yields as microcrystalline solids, and characterized by C/H/N analysis, FTIR, TGA, PXRD, and single-crystal X-ray diffraction. A variety of structure types observed in 1–6 can be ascribed to differences in coordination preferences of metal(II) centers and the presence of different auxiliary ligands that mediate the crystallization process.
Description of structures
[Cu2(μ2-mia)2(phen)2(H2O)2]·2H2O (1). Compound 1 shows a discrete dinuclear structure with its asymmetric unit contains one Cu(II) atom, one μ2-mia2− ligand, one phen moiety, one H2O ligand and one lattice water molecule. The Cu1 atom is five-coordinate and shows a distorted square pyramidal {CuN2O3} environment that is made of two carboxylate O donors from two μ2-mia2−, one O donor from the H2O ligand, and two Nphen atoms (Fig. 1a). In 1, the mia2− blocks act as μ2-linkers (mode I, Scheme 2) with monodentate COO− groups. Which connect copper(II) centers into a Cu2 molecular unit having a Cu⋯Cu distance of 8.240(3) Å. These discrete Cu2 units are interconnected by means of O–H⋯OH–bonds to give a 3D supramolecular network (Fig. 1b and Table S2, ESI†).
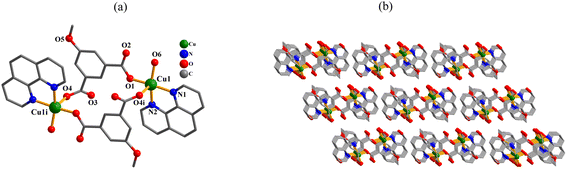 |
| | Fig. 1 Structural fragments of 1. (a) Coordination environment around Cu(II) centers; H atoms are omitted. Symmetry code: (i) = −x + 2, −y + 2, −z + 2. (b) 2D supramolecular network; view along the a axis. | |
[Mn(μ3-mia)(phen)]n (2). This structure possesses a 1D chain structure (Fig. 2). The asymmetric unit bears two Mn(II) centers (Mn1, Mn2), two μ3-mia2− linkers, and a pair of phen moieties. Both Mn atoms are six-coordinate with distorted octahedral {MnN2O4} environments (Fig. 2a), which are occupied by four O atoms from three μ3-mia2− linkers and two Nphen donors. The mia2− ligands behave as μ3-linkers (mode II, Scheme 2), with two COO− functionalities adopting bidentate or bridging bidentate modes. The μ3-mia2− linkers connect the Mn(II) centers to generate a 1D ladder-like metal–organic chain (Fig. 2b). The 1D ladder-like chains in this structure are defined as a mononodal 3-connected net (Fig. 2c) with a (4,4)(0,2) topology and a point symbol of (42·6).
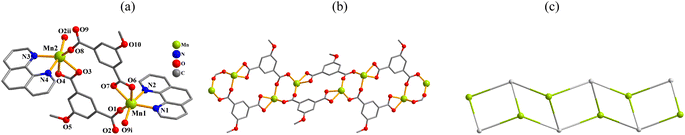 |
| | Fig. 2 Structural fragments of 2. (a) Coordination environment around Mn(II) atoms; H atoms are omitted. Symmetry codes: (i) = x, −y + 2, z − 1/2; (ii) = x, −y + 2, z + 1/2. (b) 1D metal–organic network; view along the b axis. (c) Topological representation of a mononodal 3-connected ladder chain with a (4,4)(0,2) topology; view along the b axis; 3-linked Mn centers (yellow), centroids of 3-linked μ2-mia2− linkers (gray). | |
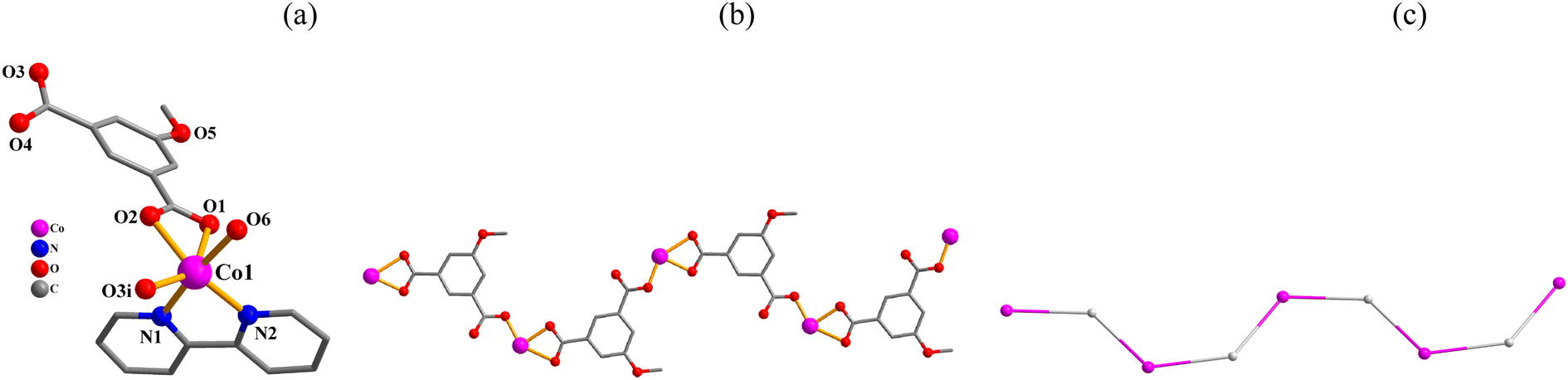
[Co(μ2-mia)(2,2′-bipy)(H2O)]n·nH2O (3). This 1D coordination polymer is composed of one distinct Co(II) center, one μ2-mia2− linker, one 2,2′-bipy moiety, one H2O ligand and one lattice water molecule per asymmetric unit (Fig. 3). The Co1 atom is six-coordinate and shows a distorted octahedral {CoN2O4} environment that is made of three carboxylate O donors from two μ2-mia2−, one O atom from the H2O ligand, and two N2,2′-bipy atoms (Fig. 3a). The mia2− ligand functions as a μ2-linker (mode III, Scheme 2) with its carboxylate groups being monodentate or bidentate. The mia2− blocks link adjacent cobalt(II) atoms into a 1D metal–organic zigzag chain (Fig. 3b). This structure discloses a mononodal 2-connected network with a 2C1 topology (Fig. 3c).
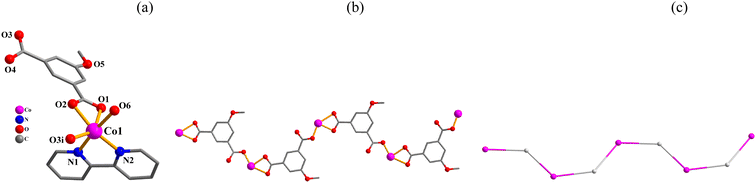 |
| | Fig. 3 Structural fragments of 3. (a) Coordination environment around Co(II) center; H atoms are omitted. Symmetry code: (i) = −x, y − 1/2, −z + 3/2. (b) 1D metal–organic chain; view along the a axis. (c) Topological representation of a mononodal 2-linked chain with a 2C1 topology; 2-connected Co1 nodes (pink), centroids of 2-connected μ2-mia2− nodes (gray); view along the a axis. | |
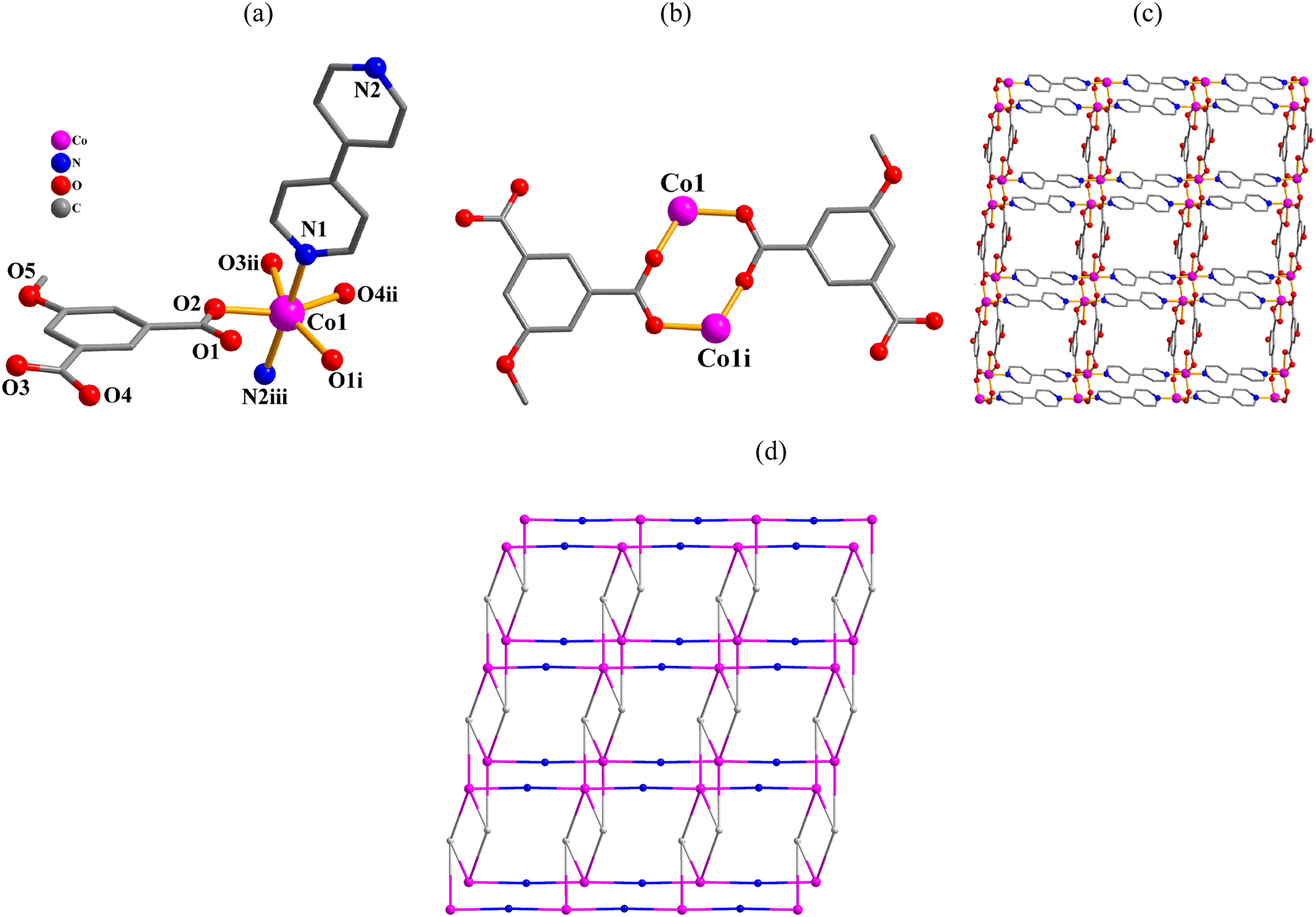
[Co(μ3-mia)(μ2-4,4′-bipy)]n·nH2O (4). This structure features a 2D sheet coordination polymer (Fig. 4). An asymmetric unit possesses one Co atom, one μ3-mia2− linker, one 4,4′-bipy ligand, and one lattice water molecule (Fig. 4a). Co1 center is 6-coordinate and unveils a distorted octahedral {CoN2O4} environment. It is completed by four carboxylate O atoms from three μ3-mia2− linkers and two N4,4′-bipy donors from two different 4,4′-bipy moieties (Fig. 4a). The mia2− block acts as a μ3-linker (mode II, Scheme 2) with two COO− groups being bidentate or bridging bidentate. The 4,4′-bipy moieties adopt a bridging coordination mode. Carboxylate groups of the μ3-mia2− blocks link neighbouring Co(II) centers to form a dimeric Co2 subunit with a Co⋯Co distance of 4.088(3) Å (Fig. 4b). These Co2 subunits are further connected by the μ3-mia2− linkers and μ2-4,4′-bipy ligands into a 2D network (Fig. 4c). The 2D network in this structure is built from the 5-linked Co1, 3-linked μ3-mia2− nodes and 2-linked μ2-4,4′-bipy nodes (Fig. 4d). It can be defined as a 3-nodal net with a new topology and point symbol of (42·6.86·12)(42·6)(8).
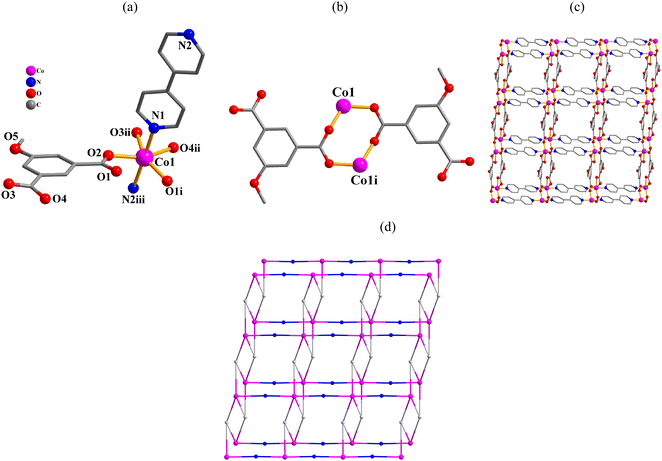 |
| | Fig. 4 Structural fragments of 4. (a) Coordination environment around Co(II) center; H atoms are omitted. Symmetry codes: (i) = −x + 1, −y + 1, −z + 1; (ii) = x, y + 1, z; (iii) = x − 1, y, z + 1. (b) Co2 subunit. Symmetry code: (i) = −x + 1, −y + 1, −z + 1. (c) 2D metal–organic network; view along the c axis. (d). Topological representation of a3-nodal network with a new topology; 5-connected Co1 nodes (pink), centroids of 3-connected μ3-mia2− nodes (gray), centroids of 2-connected μ2-4,4′-bipy linkers (blue), view along the c axis. | |
[Co(μ3-mia)(py)2]n (5). This structure features a 1D ladder chain coordination polymer (Fig. 5). An asymmetric unit possesses one Co atom, one μ3-mia2− linker, and two py ligands. Co1 center is 6-coordinate and shows a distorted octahedral {CoN2O4} environment, which is completed by four carboxylate O atoms from three μ3-mia2− blocks and two Nipy donors (Fig. 5a). The mia2− ligand acts as a μ3-linker (mode II, Scheme 2) with two COO− groups being bidentate or bridging bidentate, giving a 1D ladder chain (Fig. 5b). The 1D ladder-like chain in this structure is defined as a mononodal 3-connected net (Fig. 5c) with a (4,4)(0,2) topology and a point symbol of (42·6).
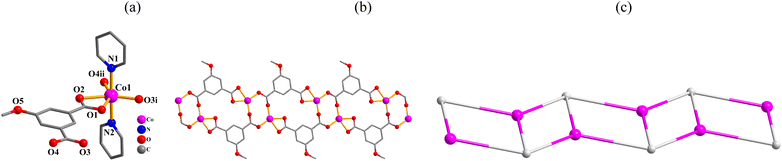 |
| | Fig. 5 Structural fragments of 5. (a) Coordination environment around Co(II) center; H atoms are omitted. Symmetry codes: (i) = −x + 2, −y + 1, −z + 1 (ii) = x + 1/2, y + 1/2, z. (b) 1D metal–organic ladder chain; view along the c axis. (c) Topological representation of a mononodal 3-connected ladder chain with a (4,4)(0,2) topology; view along the c axis; 3-connected Co1 nodes (pink), centroids of 3-connected μ3-mia2− nodes (gray). | |
[Cd(μ2-mia)(py)(H2O)2]n·nH2O (6). This structure also discloses a 1D metal–organic chain (Fig. 6). In the asymmetric unit, there is one Cd(II) atom, a μ2-mia2− linker, a py moiety, two H2O ligands, and one lattice water molecule. The cobalt(II) center is six-coordinate and features a distorted octahedral {CdNO5} environment, which is built by three carboxylate O atoms from two μ2-mia2− blocks, two O donors from two H2O ligands, and one Npy donor (Fig. 6a). The mia2− block functions as a μ2-linker (mode II, Scheme 2), in which one COO− group is monodentate while another group is bidentate. The μ2-mia2− blocks connect Cd(II) centers to form a 1D zigzag chain (Fig. 6b). This structure discloses a mononodal 2-connected network with a 2C1 topology (Fig. 6c).
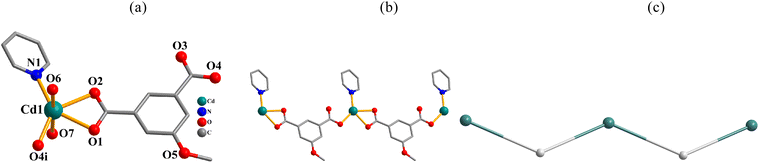 |
| | Fig. 6 Structural fragments of 6. (a) Coordination environment around Cd(II) center; H atoms are omitted. Symmetry code: (i) = x, y, z − 1. (b) 1D metal–organic chain; view along the b axis. (c) Topological representation of a mononodal 2-connected network with a 2C1 topology; view along the b axis; 2-connected Cd1 nodes (turquoise balls), centroids of 2-connected μ2-mia2− nodes (gray). | |
Structural comparison of 1–6
In the obtained compounds 1–6, 5-methoxy isophthalic acid (H2mia) act as mia2− ligand showing three distinct coordination fashions, respectively (Scheme 2). Complexes 1 and 2 were synthesized under the same reaction conditions, except for metal chlorides (CuCl2·2H2O for 1 and MnCl2·4H2O for 2). Their structural differences unambiguously indicate that the assembly process is metal ion-dependent. The structural change between 3 and 4 caused by adding different auxiliary ligands (2,2′-bipy in 3 and 4,4′-bipy in 4) under the same reaction conditions. These results imply that the reaction conditions including metal ions and auxiliary ligands have significant effects on the formation and structures of the resulting complexes. Despite similarity of synthesis conditions (Table 1), the obtained compounds show distinct compositions, dimensionalities, and topologies.
Thermal stability
Thermogravimetric analysis (TGA) for 1–6 was carried out under N2 atmosphere with a heating rate of 10 °C min−1 (Fig. 7). For 1, there is a loss of two crystallization water molecules and two H2O ligands in the 62–194 °C range (exptl, 7.3%; calcd, 7.6%), followed by a decomposition above 240 °C. CP 2 does not contain crystallization solvent or temperature-sensitive ligands, maintaining stability up to 398 °C. For 3, a release of one lattice water molecule and one water ligand is observed in the 108–158 °C range (exptl, 7.8%; calcd, 8.1%); the remaining solid maintains stability until 217 °C. For 4, a loss of crystallization H2O molecule is seen in the 65–157 °C interval (exptl, 4.0%; calcd, 4.2%); the metal–organic network is stable up to 358 °C. The TGA curve of 5 shows a release of two py ligands between 184 and 288 °C (exptl, 38.2%; calcd, 38.5%), followed by a decomposition above 363 °C. For 6, there is a loss of one crystallization water molecule, two H2O ligands and one py ligand in the 32–311 °C range (exptl, 30.0%; calcd, 30.3%), followed by a decomposition above 337 °C.
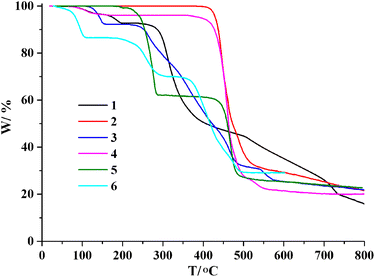 |
| | Fig. 7 TGA plots of 1–6 (10 °C min−1, 25–800 °C, N2 atmosphere). | |
Catalytic studies
Considering an ability of some coordination polymers to catalyze the cyanosilylation of aldehydes with TMSCN,56–59 we tested the obtained products 1–6 as heterogeneous catalysts in this reaction. 4-Nitrobenzaldehyde and TMSCN were used as model substrates for initial screening. The reactions were carried out in CH2Cl2 medium at 35 °C to generate 2-(4-nitrophenyl)-2-[(trimethylsilyl)oxy]acetonitrile (Scheme 3 and Table 3). The effects of various reaction parameters were investigated, including the reaction time, type of solvent, catalyst loading and recycling, and substrate scope.
 |
| | Scheme 3 Cyanosilylation of model substrate (4-nitrobenzaldehyde). | |
Table 3 Cyanosilylation of 4-nitrobenzaldehyde with TMSCNa
| Entry |
Catalyst |
Reaction time, h |
Catalyst loading, mol% |
Solvent |
Product yieldb (%) |
| Conditions: 4-nitrobenzaldehyde (0.5 mmol), TMSCN (1.0 mmol), catalyst (2–3 mol%), solvent (2.5 mL), 35 °C. Yield based on 1H NMR analysis: [moles of product per mol of aldehyde substrate] × 100%. |
| 1 |
1 |
1 |
3.0 |
CH2Cl2 |
37 |
| 2 |
1 |
2 |
3.0 |
CH2Cl2 |
68 |
| 3 |
1 |
4 |
3.0 |
CH2Cl2 |
80 |
| 4 |
1 |
6 |
3.0 |
CH2Cl2 |
89 |
| 5 |
1 |
8 |
3.0 |
CH2Cl2 |
96 |
| 6 |
1 |
10 |
3.0 |
CH2Cl2 |
>99 |
| 7 |
1 |
10 |
2.0 |
CH2Cl2 |
95 |
| 8 |
1 |
10 |
3.0 |
CH3CN |
88 |
| 9 |
1 |
10 |
3.0 |
THF |
85 |
| 10 |
1 |
10 |
3.0 |
CH3OH |
97 |
| 11 |
1 |
10 |
3.0 |
CHCl3 |
95 |
| 12 |
2 |
10 |
3.0 |
CH2Cl2 |
86 |
| 13 |
3 |
10 |
3.0 |
CH2Cl2 |
90 |
| 14 |
4 |
10 |
3.0 |
CH2Cl2 |
81 |
| 15 |
5 |
10 |
3.0 |
CH2Cl2 |
84 |
| 16 |
6 |
10 |
3.0 |
CH2Cl2 |
80 |
| 17 |
Blank |
10 |
— |
CH2Cl2 |
5 |
| 18 |
CuCl2·2H2O |
10 |
3.0 |
CH2Cl2 |
9 |
| 19 |
H2mia |
10 |
3.0 |
CH2Cl2 |
6 |
As compound 1 disclosed the highest activity with a quantitative conversion of 4-nitrobenzaldehyde to 2-(4-nitrophenyl)-2-[(trimethylsilyl)oxy]acetonitrile (Table 3), it was selected as a preferable catalyst in further optimization studies. In particular, the reaction time has a significant effect, as attested by the product yield growth from 37 to >99% on extending the reaction from 1 to 10 h (Table 3, entries 1–6 and Fig. S5, ESI†). Catalyst amount was studied, disclosing a slight improvement in the yield from 95 to >99% when changing the catalyst loading from 2 to 3 mol% (entries 6 and 7). Besides acetonitrile, methanol, tetrahydrofuran, and chloroform were screened as solvents but revealed inferior suitability (85–97% product yields).
It is important to indicate that the cyanosilylation of 4-nitrobenzaldehyde is much less efficient in the absence of catalyst (only 5% product yield) or when using H2mia (6% yield), CuCl2·2H2O (9% yield, entries 17–19, Table 3). Despite there is no clear relation between catalyst structure and activity, a slightly superior activity of 1 can be associated with its coordinative unsaturation and better accessibility of active sites.20,60,61
After the above optimization of reaction conditions (3.0 mol% 1, CH2Cl2, 10 h), different substituted benzaldehydes were screened for investigating the substrate scope in the cyanosilylation reaction with TMSCN. The corresponding products were obtained in the yields ranging from 27 to >99% (Table 4). Benzaldehydes with a strong electron-withdrawing substituent (e.g., –NO2 or –Cl) disclosed the best efficacy (entries 2–5, Table 4), which can be regarded to increased electrophilicity of these substrates. The use of benzaldehydes with an electron-donating group (e.g., –CH3 or –OCH3) resulted in inferior product yields (entries 7 and 8, Table 4).
Table 4 Substrate scope for Cu-catalyzed cyanosilylation of substituted benzaldehydes with TMSCNa
| Entry |
Substituted benzaldehyde (R-C6H4CHO) |
Product yieldb (%) |
| Conditions: aldehyde (0.5 mmol), TMSCN (1.0 mmol), catalyst 1 (3.0 mol%), CH2Cl2 (2.5 mL), 35 °C. Yield on the basis of 1H NMR analysis: [moles of product per mol of aldehyde substrate] × 100%. |
| 1 |
R = H |
96 |
| 2 |
R = 2-NO2 |
98 |
| 3 |
R = 3-NO2 |
>99 |
| 4 |
R = 4-NO2 |
>99 |
| 5 |
R = 4-Cl |
94 |
| 6 |
R = 4-OH |
61 |
| 7 |
R = 4-CH3 |
56 |
| 8 |
R = 4-OCH3 |
27 |
Finally, the recycling of catalyst 1 was attempted. The obtained results (Fig. S6 and S7, ESI†) indicate that the compound 1 preserves its activity for at least five reaction cycles, as evidenced by similar product yields. Furthermore, the PXRD patterns confirm that the structure of 1 is retained, despite the observation of several novel signals or widening of some parent peaks. These alterations are common for a recycled catalyst, and can be associated with a decrease in crystallinity and presence of impurities.
The achieved catalytic performance of compound 1 in the cyanosilylation of 4-nitrobenzaldehyde with TMSCN is comparable62,63 or superior64–69 to those exhibited by the other heterogeneous catalysts based on other metal–carboxylate coordination compounds (Table S4†).
Conclusions
In summary, six new Cu(II), Mn(II), Co(II), and Cd(II) coordination compounds were generated by facile hydrothermal synthesis and completely characterized. Catalytic properties of the generated products were screened in the cyanosilylation of 4-nitrobenzaldehyde with TMSCN, resulting in close to quantitative yields of product yields, good substrate scope, and possibility of catalyst recycling.
Author contributions
X. X. Fan, J. Z. Gu and J. J. Xue designed the project. H. Y. Wang, B. Zhang and X. Q. Kang performed the synthesis and related measurements. J. Z. Gu carried out the structural characterization. X. X. Fan, J. Z. Gu and J. J. Xue prepared the manuscript. All authors participated in the discussion of experimental results.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the 111 Project of MOE (111-2-17).
References
- K. Wang, Y. Li, L.-H. Xie, X. Li and J.-R. Li, Chem. Soc. Rev., 2022, 51, 6417–6441 RSC.
- J. Chen, R. Abazari, K. A. Adegoke, N. W. Maxakato, O. S. Bello, M. Tahir, S. Tasleem, S. Sanati, A. M. Kirillov and Y. Zhou, Coord. Chem. Rev., 2022, 469, 214664 CrossRef CAS.
- G. Maurin, C. Serre, A. Cooper and G. Fereyd, Chem. Soc. Rev., 2017, 46, 3104–3107 RSC.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- W. Fan, S. Yuan, W. Wang, L. Feng, X. Liu, X. Zhang, X. Wang, Z. Kang, F. Dai, D. Yuan, D. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2020, 142, 8728–8737 CrossRef PubMed.
- R. Vismara, G. Tuci, N. Mosca, K. V. Domasevitch, C. Di Nicola, C. Pettinari, G. Giambastiani, S. Galli and A. Rossin, Inorg. Chem. Front., 2019, 6, 533–545 RSC.
- W. Fan, X. Wang, X. Liu, B. Xu, X. Zhang, W. Wang, X. Wang, Y. Wang, F. Dai, D. Yuan and D. Sun, ACS Sustainable Chem. Eng., 2019, 7, 2134–2140 CrossRef CAS.
- X. Zhao, Y. Wang, D. S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef.
- B. Zheng, X. Luo, Z. Wang, S. Zhang, R. Yun, L. Huang, W. Zeng and W. Liu, Inorg. Chem. Front., 2018, 5, 2355–2363 RSC.
- N. Patel, P. Shukla, P. Lama, S. Das and T. K. Pal, Cryst. Growth Des., 2022, 22, 3518–3564 CrossRef CAS.
- J. Li, J. F. Tian, H. H. Yu, M. Y. Fan, X. Li, F. B. Liu, J. Sun and Z. M. Su, Cryst. Growth Des., 2022, 22, 2954–2963 CrossRef CAS.
- A. M. Alsharabasy, A. Pandit and P. Farras, Adv. Mater., 2021, 33, 2003883 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS.
- T. Zhang, Z. Zhang, H. Chen, X. Zhang and Q. Li, Cryst. Growth Des., 2022, 22, 304–312 CrossRef CAS.
- L. Zhao, Z. Du, G. Ji, Y. Wang, W. Cai, C. He and C. Duan, Inorg. Chem., 2022, 61, 7256–7265 CrossRef CAS.
- K. Li, Y.-F. Liu, X.-L. Lin and G.-P. Yang, Inorg. Chem., 2022, 61, 6934–6942 CrossRef CAS.
- X. Ma, F. Liu, Y. Helian, C. Li, Z. Wu, H. Li, H. Chu, Y. Wang, Y. Wang, W. Lu, M. Guo, M. Yu and S. Zhou, Energy Convers. Manage., 2021, 229, 113760 CrossRef CAS.
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS.
- J. Z. Gu, M. Wen, Y. Cai, Z. F. Shi, A. S. Arol, M. V. Kirillova and A. M. Kirillov, Inorg. Chem., 2019, 58, 2403–2412 CrossRef CAS PubMed.
- S. Q. Zhao and J. Z. Gu, Chin. J. Inorg. Chem., 2022, 38, 161–170 CAS.
- Y. Zhu, N. Xin, Z. Qiao, S. Chen, L. Zeng, Y. Zhang, D. Wei, J. Sun and H. Fan, Adv. Healthcare Mater., 2020, 9, 2000205 CrossRef CAS PubMed.
- T. Simon-Yarza, A. Mielcarek, P. Couvreur and C. Serre, Adv. Mater., 2018, 30, 1705189 CrossRef PubMed.
- J. Feng, W.-X. Ren, F. Kong and Y.-B. Dong, Inorg. Chem. Front., 2021, 8, 848–879 RSC.
- Y.-B. Lu, J. Huang, X.-R. Yuan, S.-J. Liu, R. Li, H. Liu, M.-P. Liu, H.-R. Wen, S.-D. Zhu and Y.-R. Xie, Cryst. Growth Des., 2022, 22, 1045–1053 CrossRef CAS.
- T. Neumann, M. Ceglarska, L. S. Germann, M. Rams, R. E. Dinnebier, S. Suckert, I. Jess and C. Naether, Inorg. Chem., 2018, 57, 3305–3314 CrossRef CAS PubMed.
- W.-H. Huang, X.-X. Zhang and Y.-N. Zhao, Dalton Trans., 2021, 50, 15–28 RSC.
- C. P. Raptopoulou, Materials, 2021, 14, 310 CrossRef CAS PubMed.
- X.-D. Zheng and T.-B. Lu, CrystEngComm, 2010, 12, 324–336 RSC.
- Q. Guan, L.-L. Zhou and Y.-B. Dong, Chem. Soc. Rev., 2022, 51, 6307–6416 RSC.
- M. P. Merkel, C. E. Anson, G. E. Kostakis and A. K. Powell, Cryst. Growth Des., 2021, 21, 3179–3190 CrossRef CAS.
- G. Zhan, W. Zhong, Z. Wei, Z. Liu and X. Liu, Dalton Trans., 2017, 46, 8286–8297 RSC.
- G. Fan, L. Hong, X. Zheng, J. Zhou, J. Zhan, Z. Chen and S. Liu, RSC Adv., 2018, 8, 35314–35326 RSC.
- Z. Lei, L. Hu, Z.-H. Yu, Q.-Y. Yao, X. Chen, H. Li, R.-M. Liu, C.-P. Li and X.-D. Zhu, Inorg. Chem. Front., 2021, 8, 1290–1296 RSC.
- Y. Yang, C. Lu, H. Wang and X. Liu, Dalton Trans., 2016, 45, 10289–10296 RSC.
- J. Z. Gu, Y. H. Cui, X. X. Liang, J. C. Wu, D. Y. Lv and A. M. Kirillov, Cryst. Growth Des., 2016, 16, 4658–4670 CrossRef CAS.
- B. Ramezanpour, M. Mirzaei, V. Jodaian, M. N. Shahrak, A. Frontera and E. Molins, Inorg. Chim. Acta, 2019, 484, 264–275 CrossRef CAS.
- N. Wei, M.-Y. Zhang, X.-N. Zhang, G.-M. Li, X.-D. Zhang and Z.-B. Han, Cryst. Growth Des., 2014, 14, 3002–3009 CrossRef CAS.
- Y. Li, J. Wu, J.-Z. Gu, A. Qiu Wen and A.-S. Feng, Chin. J. Struct. Chem., 2020, 39, 727–736 CAS.
- X. X. Zou, J. Wu, J. Z. Gu, N. Zhao, A. S. Feng and Y. Li, Chin. J. Inorg. Chem., 2019, 35, 1705–1711 CAS.
- J. Z. Gu, Z. Q. Gao and Y. Tang, Cryst. Growth Des., 2012, 12, 3312–3323 CrossRef CAS.
- D.-C. Zhong, W.-G. Lu and J.-H. Deng, CrystEngComm, 2014, 16, 4633–4640 RSC.
- J. Z. Gu, S. M. Wan, W. Dou, M. V. Kirillova and A. M. Kirillov, Inorg. Chem. Front., 2021, 8, 1229–1242 RSC.
- X. Y. Cheng, L. R. Guo, H. Y. Wang, J. Z. Gu, Y. Yang, M. V. Kirillova and A. M. Kirillov, Inorg. Chem., 2022, 61, 12577–12590 CrossRef CAS.
- K. Wang, Y. Li, L.-H. Xie, X. Li and J.-R. Li, Chem. Soc. Rev., 2022, 51, 6417–6441 RSC.
- L. McCormick, S. Morris, S. Teat, M. McPherson, A. Slawina and R. Morris, Dalton Trans., 2015, 44, 17686–17695 RSC.
- L.-F. Ma, B. Liu, L.-Y. Wang, C.-P. Li and M. Du, Dalton Trans., 2010, 39, 2301–2308 RSC.
- X.-H. Chang, J.-H. Qin, L.-F. Ma and L.-Y. Wang, J. Solid State Chem., 2014, 212, 121–127 CrossRef CAS.
- O. Qazvini, V.-J. Scott, L. Bondorf, M. Ducamp, M. Hirscher, F.-X. Coudert and S. Telfer, Chem. Mater., 2021, 33, 8886–8894 CrossRef CAS.
- Z. Zhang, J. Zhou, H.-X. Sun, M. He, W. Li, L. Du, M. Xie and Q.-H. Zhao, Cryst. Growth Des., 2021, 21, 5086–5099 CrossRef CAS.
- V.-M. Au, K. Nakayashiki, H. Huang, S. Suginome, H. Sato and T. Aida, J. Am. Chem. Soc., 2019, 141, 53–57 CrossRef CAS PubMed.
- L.-F. Ma, J.-W. Zhao, M.-L. Han, L.-Y. Wang and M. Du, Dalton Trans., 2012, 41, 2078–2083 RSC.
- J. Lai, Y. Han, H.-M. Li, J. Wang, C.-L. Wang, L. Suo, Y. Sun and K.-L. Wang, J. Cluster Sci., 2020, 31, 1389–1398 CrossRef CAS.
-
(a) V. A. Blatov. IUCr CompComm Newsletter, 2006, vol. 7, pp. 4–38 Search PubMed;
(b) V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- J.-J. Du, X. Zhang, X.-P. Zhou and D. Li, Inorg. Chem. Front., 2018, 5, 2772–2776 RSC.
- Y.-M. Xi, Z.-Z. Ma, L.-N. Wang, M. Li and Z.-J. Li, J. Cluster Sci., 2019, 30, 1455–1464 CrossRef CAS.
- L. M. Aguirre-Díaz, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and M. Á. Monge, CrystEngComm, 2013, 15, 9562–9571 RSC.
- J. Amaro-Gahete, D. Esquivel, J. R. Ruiz, C. Jiménez-Sanchidrián and F. J. Romero-Salguero, Appl. Catal., A, 2019, 585, 117190 CrossRef CAS.
- E. Loukopoulos and G. E. Kostakis, J. Coord. Chem., 2018, 71, 371–410 CrossRef CAS.
- L.-P. Xue, Z.-H. Li, T. Zhang, J.-J. Cui, Y. Gao and J.-X. Yao, New J. Chem., 2018, 42, 14203–14209 RSC.
- A. Yadav, S. Kumari, P. Yadav, A. Hazra, A. Chakraborty and P. Kanoo, Dalton Trans., 2022, 51, 15496–15506 RSC.
- W. Jiang, J. Yang, Y.-Y. Liu, S.-Y. Song and J.-F. Ma, Inorg. Chem., 2017, 56, 3036–3043 CrossRef CAS PubMed.
- F. Li, R. Ma, Z. Xia, Q. Wei, S. Chen and S. Gao, J. Solid State Chem., 2021, 301, 122337 CrossRef CAS.
- A. Karmakar, A. Paul, G. Rúbio, M. Silva and A. Pombeiro, Eur. J. Inorg. Chem., 2016, 5557–5567 CrossRef CAS.
- J.-M. Gu, W.-S. Kim and S. Huh, Dalton Trans., 2011, 40, 10826–10829 RSC.
- F.-Z. Jin, C.-C. Zhao, H.-C. Ma, G.-J. Chen and Y.-B. Dong, Inorg. Chem., 2019, 58, 9253–9259 CrossRef CAS PubMed.
- P. Wu, J. Wang, Y. Li, C. He, Z. Xie and C. Duan, Adv. Funct. Mater., 2011, 21, 2788–2794 CrossRef CAS.
- Y. Cao, Z. Zhu, J. Xu, L. Wang, J. Sun, X. Chen and Y. Fan, Dalton Trans., 2015, 44, 1942–1947 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical data and synthesis for 1–6, FTIR spectra (Fig. S1), PXRD patterns (Fig. S2), temperature dependence of χMT vs. T for compounds 1–5 (Fig. S3), additional catalysis data (Fig. S4–S7) and structural parameters (Tables S1 and S2). The magnetic moments of compounds 1–5 (Table S3), and comparison of catalytic activity (Table S4†). CCDC 2269625–2269630. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra04111e |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Ji-jun Xue*
* and
Ji-jun Xue*