
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the mechanism of acceptorless dehydrogenation of N-heterocycles catalyzed by tBuOK: a computational study†
Lishuang Ma a,
Wenxu Fenga,
Shidong Zhaoa,
Chuangye Wanga,
Yanyan Xib and
Xufeng Lin*ab
a,
Wenxu Fenga,
Shidong Zhaoa,
Chuangye Wanga,
Yanyan Xib and
Xufeng Lin*ab
aCollege of Chemistry and Chemical Engineering, China University of Petroleum (East China), Qingdao 266580, P. R. China. E-mail: hatrick2009@upc.edu.cn
bState Key Laboratory of Heavy Oil Processing, China University of Petroleum (East China), Qingdao 266580, P. R. China
First published on 11th July 2023
Abstract
The catalytic acceptorless dehydrogenation (ADH) of saturated N-heterocycles has recently gained considerable attention as a promising strategy for hydrogen release from liquid organic hydrogen carriers (LOHCs). Recently, a simple tBuOK base-promoted ADH of N-heterocycles was developed by Yu et al. (Adv. Synth. Catal. 2019, 361, 3958). However, it is still open as to how the tBuOK plays a catalytic role in the ADH process. Herein, our density functional study reveals that the tBuOK catalyzes the ADH of 1,2,3,4-tetrahydroquinoline (THQ) through a quasi-metal–ligand bifunctional catalytic channel or a base-catalyzed pathway with close energy barriers. The hydride transfer in the first dehydrogenation process is determined to be the rate determining step, and the second dehydrogenation can proceed directly from 34DHQ regulated by the tBuOK. In addition, the computational results show that the cooperation of a suitable alkali metal ion with the tBuO− group is so critical that the tBuOLi and the isolated tBuO− are both inferior to tBuOK as a dehydrogenation catalyst.
1. Introduction
Liquid organic hydrogen carriers (LOHCs) have been recognized as promising hydrogen storage media, due to their high hydrogen capacity (5–7.4 wt%), reversibility, moderate dehydrogenation kinetics, relative safety and compatibility with existing infrastructure for fuels.1 The utilization of N-containing LOHCs has particularly attracted growing attention in recent years as the introduction of N atoms can benefit the dehydrogenation process, both thermodynamically and kinetically, compared to the carbocycle compounds.2,3 However, the H2 release process itself is entropically favoured but thermodynamically uphill.2 In addition, some key factors such as catalytic activity, selectivity, durability as well as heating demand strongly depend on the performance of catalysts. Thus, there is a highly desire to develop efficient catalysts and catalytic strategies for the N-containing LOHC systems.3The catalytic acceptorless dehydrogenation (ADH) is a cornerstone achievement in organometallic catalysis as reviewed by Milstein,4 which is not only atom-economical with liberation of H2 and no waste generation, but also environmentally benign without the need of any stoichiometric oxidants or sacrificial acceptors. Given these advantages, the catalytic ADH has been increasingly applied in the LOHC-based hydrogen storage, and significant efforts have been dedicated to developing diverse homogenous and heterogeneous catalysts for ADH of N-heterocycles.2c,d,5 To date, a series of transition metal catalysts,5 including the homogenous ones such as Cp*Ir(III),6,7 Fe(II)-PNP,8a Co(II)-PNP,8b CpNi(NHC),9 Ru(II)-NNC,10 Ru(II)-CNN(H)11 and Os(IV or II)12 metal-complex catalysts, and the heterogeneous ones such as Fe,13 Co14 and Mn15 based single-atom and nanoparticle catalysts, have been developed for ADH reactions of N-heterocycles. With the aim of providing low-cost and environmentally benign protocols, the transition-metal-free catalysts represent appealing alternatives, although such catalysts are less exploited, especially under acceptor-free conditions. The heterogenous metal-free carbocatalysts such as nitrogen-assembly carbons (NCs)16a and reduced graphene oxides (rGOs)16b were demonstrated efficient with high reusability and stability even under ambient temperature. The homogenous B(C6F5)3 catalyst was used by Kanai et al. in 2016, to catalyze the ADH of N-heterocycles with high functional group tolerance.17a The B(C6F5)3 was also applied in the frustrated Lewis pair catalyzed ADH of the N-protected indolines, in which a weak Lewis acid acts as the hydride shuttle.17b In 2019, the ADHs of 1,2,3,4-tetrahydroquinoline (THQ), indoline and their derivatives promoted by only a simple tBuOK base were efficiently realized by Yu and co-workers, without using any other catalysts or additives in o-xylene at 140 °C (see Scheme 1),18 which offers a transition-metal-free and operationally simple alternative.
 | ||
| Scheme 1 Acceptorless dehydrogenation of THQ promoted by tBuOK.18 Numbering scheme is shown in red for THQ. | ||
Despite the remarkable progress in the experimental investigations on the ADH of N-heterocycles, of equivalent importance is the fundamental understanding to serve for rational catalyst design and optimization.19,20 Previously, several computational studies demonstrated that the ADH of N-heterocycles catalyzed by Cp*Ir20a and Fe-PNP20b,c transition metal complexes can operate effectively by the cooperation of metal and ligand, via stepwise or concerted proton/hydride transfer pathways. However, the simple tBuOK catalyzed dehydrogenation additionally complicates the mechanism,18 since the intrinsic catalytic activities of bases such as tBuOK were usually neglected in the presence of a transition metal catalyst.21 In addition, it was previously reported by Berkessel and Muller that the isolated alkali base can catalyze the hydrogenation of ketones, which was proposed to occur via a six-membered transition state in which H2 is activated by bridging ketone and alkoxide (see TSBC in Scheme 2), based on kinetic studies22 and inspiration of the metal–ligand bifunctional catalytic mechanism developed by Noyori (see TSMLC in Scheme 2).23 This base catalytic mechanism was further revised by Dub et al., who suggested a stepwise pathway in solution where the H–H bond is first activated by tBuOK to afford a KH intermediate.24 These raise the questions of how the tBuOK plays a catalytic role in the dehydrogenation of N-heterocycles, and whether it behaves like a bifunctional transition metal catalyst or that in the base catalyzed hydrogenation reaction.22–24 Herein, we therefore employed the density functional theory (DFT) method to provide a comprehensive understanding of the ADH pathways of 1,2,3,4-tetrahydroquinoline (THQ) catalyzed by a tBuOK base catalyst.
 | ||
| Scheme 2 Key transition states in mechanisms of the metal–ligand bifunctional catalysis (TSMLC) and the base-catalyzed hydrogenation (TSBC).22,23 | ||
2. Computational methodology
In the present study, DFT calculations were performed to investigate the ADH mechanism of THQ catalyzed by the tBuOK base catalyst. All minima of the reactants, products as well as intermediates were first obtained by full system optimization, using the B3LYP hybrid density functional25 including empirical dispersion correction computed with Grimme's D3 formula (B3LYP-D3).26 The def2-TZVP27 basis set was applied for all the atoms. The transition states on the energy profiles were obtained by employing the conventional approach of transition state optimization at the same level. Vibrational frequency analysis calculations were also performed for all the optimized structures, to ensure that each stationary point truly represented a local minimum or a saddle point, and to derive the thermochemical corrections for the enthalpies and free energies. To inspect the influence of the calculation level on the relative energies, the energies of some rate-determining intermediates and transition states were refined by single-point energy calculations at the B3LYP-D3/def2-QZVP level of theory. To consider solvent effects, the solvation model based on density (SMD) for o-xylene (ε = 2.5454) was employed for all the calculated points at a temperature of 298.15 K. All calculations were carried out by using the Gaussian 09 program package.28 The non-covalent interaction (NCI) and extended transition state-natural orbitals for chemical valence (ETS-NOCV) analyses were performed by using the Multiwfn programme.293. Results and discussion
3.1. Dehydrogenation from THQ to DHQ
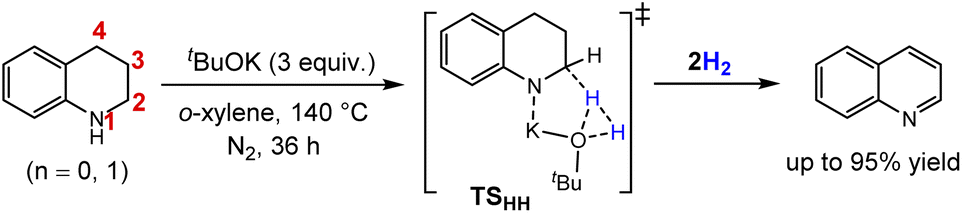
The tetrameric cubane-type cluster (tBuOK)4 was proposed as the reactive catalytic species in several reports,30 however, the cubic cluster can dissolve into different small aggregates in protic tert-butanol according to the AIMD simulations reported by Dub et al.,24a and it can be speculated that the result would turn out to be very complicated under high temperature up to 140 °C experimentally.18 Indeed, it has proven to be practical to study the base catalytic mechanism using a single tBuOK as the model catalyst.31 Thus, we commenced this work with an isolated tBuOK as the catalyst to understand the base catalyzed ADH process of THQ. As shown in Fig. 1a, an encounter complex M1 can be initially formed via an N–H⋯O hydrogen bond with a stabilization energy of 4.1 kcal mol−1. Subsequently, proton migration from N1 to the oxygen atom of tBuOK occurs by overcoming a tiny free energy barrier (1.8 kcal mol−1) via TS1. An ion-pair complex M2 is thus generated after proton transfer, and is 7.6 kcal mol−1 in free energy more stable than the separated species. Note that the optimized M2 is more stable than TS1 in terms of electronic energy, but higher after thermal corrections. As a result, this process leads to the shortened distance between K+ and H2a (M1: 3.82 Å → M2: 3.60 Å) as well as the concomitant slightly elongation of C2–H2a bond (1.11 Å in M1 versus 1.09 Å in THQ), which is helpful in preparing for the subsequent hydride transfer.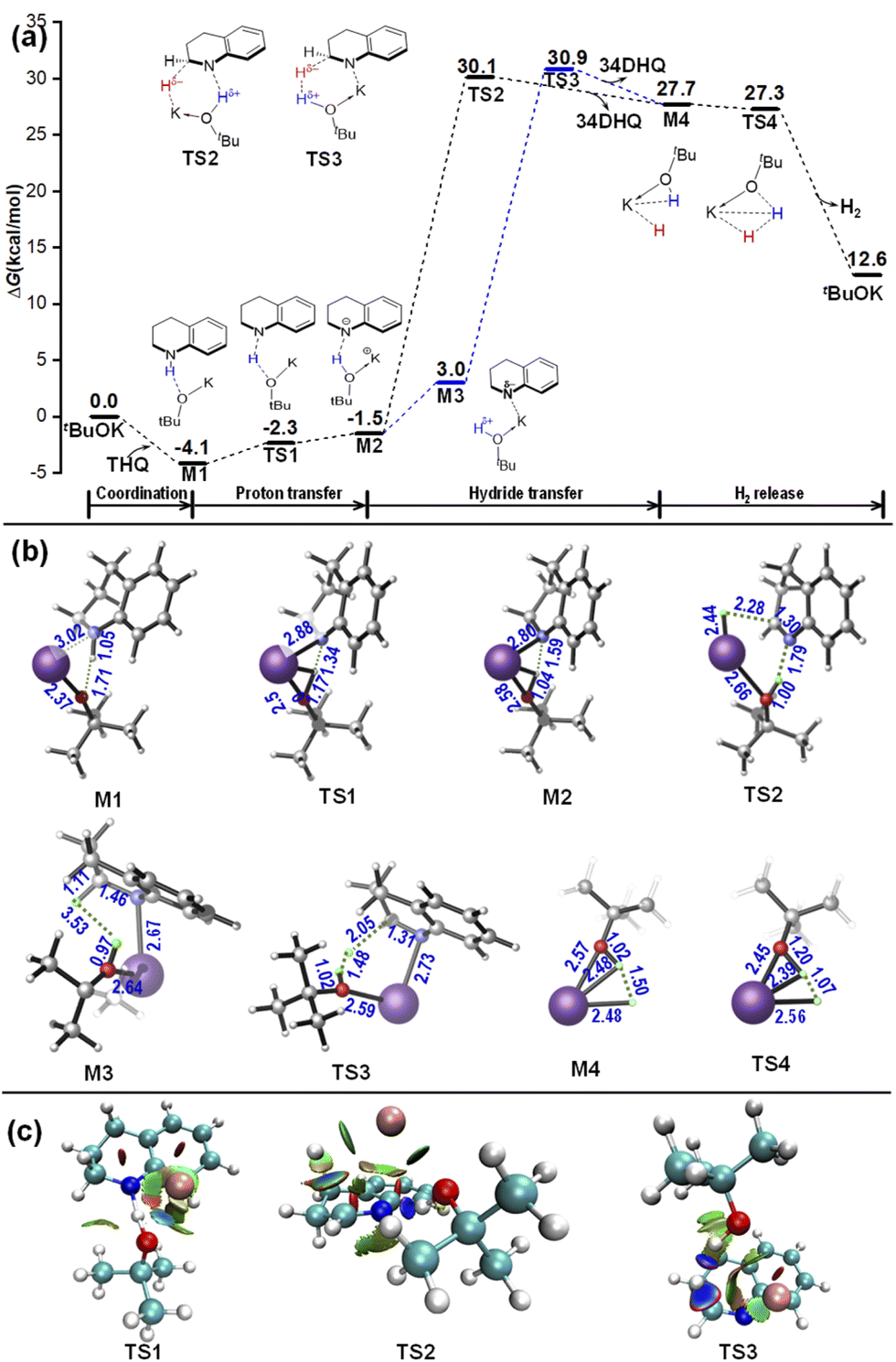 | ||
| Fig. 1 (a) Relative free energy profiles for the dehydrogenation reaction of THQ catalyzed by tBuOK in o-xylene. (b) Geometrical information. (c) NCI analyses of TS1-3. Energies are in kcal mol−1, bond distances are in angstroms. | ||
Starting from M2, hydride transfer occurs from C2(sp3)–H2a to K+, via the six-membered transition state TS2 with a sizeable energetic barrier of 34.2–34.5 kcal mol−1 (obtained by using B3LYP-D3 functional with def2-TZVP and def2-QZVP basis sets), relative to M1 according to the energetic span model.32 In contrast, hydride transfer pathway from the equatorial C2(sp3)–H2e bond to K+ was calculated to afford an energy barrier 2.9 kcal mol−1 higher than the former one (see Fig. S1†), indicating that the hydride abstraction is prone to take place from the axial C2–H2a bond.
As shown in Fig. 1c, NCI analysis shows that TS2 is mainly stabilized by N⋯H–O hydrogen-bonding and C⋯H⋯K+ weak interactions. Taking the above proton and hydride transfer steps together, and regarding the K+ as a metal center and the tBuO− as a ligand like in the well-established bifunctional transition metal complex catalysts, the six-membered-ring TS2 is structurally similar with the Noyori-type metal–ligand bifunctional transition state (TSMLC in Scheme 2) along an outer-sphere pathway.23,33,34 The first dehydrogenation process through sequential TS1 and TS2 can be attributed to be a stepwise (proton then hydride) pathway, where a proton migrates to tBuO− and a hydride transfers to K+. Note that both the concerted and stepwise proton/hydride transfer pathways were taken into consideration at first. However, all attempts to locate a transition state for the concerted mechanism failed. In addition, the transition state for sequential hydride and proton transfer, i.e. the H2a on C2 first shifts to the tBuO− before proton transfer, could also not be located. Therefore, the only feasible mechanism is the stepwise outer-sphere proton and hydride transfer pathway.
Besides the above-mentioned quasi-metal–ligand functional pathway via TS2, another competing base-catalyzed type pathway was also identified. In this case, a precursor intermediate M3 stabilized by the Nδ−⋯Kδ+ coordination should be first formed, and is 4.5 kcal mol−1 higher than M2. Then, hydride migration can take place through TS3. This calculated pathway was initially inspired by the mechanism proposed by Liu et al.18 However, different from the transition state TSHH (i.e. C in ref. 19, see Scheme 1), there is no O⋯H⋯C hydrogen-bonding interaction found in TS3, but instead the Nδ−⋯Kδ+ weak interaction and the Oδ−–Hδ+⋯Hδ−⋯Cδ+ hydrogen-bonding interaction play vital roles in stabilizing the TS3 as indicated by NCI analyses (see Fig. 1c). Interestingly, this H–H bridging transition state is structurally similar with the H–H bond cleavage transition state in based-catalyzed hydrogenation (TSBC in Scheme 2) proposed by Berkessel et al.,22,35 and more specifically it is analogous with the reverse pathway of the stepwise tBuOK catalyzed hydrogenation calculated by Dub et al.24 In this case, the hydride is abstracted by the proton on tBuOK(Hδ+) unit, while the K+ is coordinated with N atom stabilizing the TS3 according to the NCI diagram. The corresponding energy barrier is calculated to be 0.8 kcal mol−1 (0.7 kcal mol−1 refined at B3LYP-D3/def2-QZVP level of theory) higher than that via TS2, indicating that the two Noyori- and base-catalyzed type hydride transfer pathways may coexist and the former one is slightly more superior. No matter in which case the reaction takes place, with the departure of H2a, the configuration of C2 atom changes from sp3 into sp2, resulting in the 3,4-dihydroquinoline (34DHQ) and the dihydride species KOtBu(H)(H), i.e. the M4 in Fig. 1, composed of a contact ion pair of KH and a tert-butanol. This dihydride species is also the precursor for the hydrogen release in the next step.
The H2 elimination from O and K+ sites within the dihydride species occurs along a downhill pathway, regenerating the tBuOK base catalyst. The sum of free energies of the final products (34DHQ + tBuOK + H2) are calculated to be endothermic by 12.6 kcal mol−1 relative to the separated species (THQ + tBuOK). Although the overall dehydrogenation reaction is thermodynamically unfavorable, it is generally entropically favored due to the H2 liberation.
3.2. The second dehydrogenation from dihydroquinoline (DHQ) to quinoline (Q)
Once the 34DHQ is produced, the second dehydrogenation may subsequently occur on the C3–C4 bond site. Alternatively, it may also take place on the N1 and C4 sites when 34DHQ is converted to 14DHQ by isomerization, or on the N1–C2 bond again if the N1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2 double bond in 34DHQ can be shifted to the C3–C4 position to give the 12DHQ, the details are shown as follows.
C2 double bond in 34DHQ can be shifted to the C3–C4 position to give the 12DHQ, the details are shown as follows.
 | ||
| Fig. 2 Relative free energy profiles for the dehydrogenation reaction of 34DHQ (a) and 12DHQ (b) catalyzed by tBuOK in o-xylene. (c) Geometrical information. Energies are in kcal mol−1, bond distances are in angstroms. | ||
Then starting from M6, hydride migrates from C4 site via the base-catalyzed type H–H bridging TS6, which is structurally similar with TS3 in the first dehydrogenation reaction, but is evidently more stable with an energetic span of 27.2 kcal mol−1. Moreover, H2 elimination takes place synchronously with the hydride transfer, without the formation of a dihydride intermediate. Alternatively, the ion-pair complex M6 can rearrange to be the N⋯H–O hydrogen bonded complex M7 by releasing energy of 3.5 kcal mol−1. The hydride transfer subsequently occurs along the quasi-metal–ligand bifunctional pathway via TS7, the energetic span of which is slightly smaller (2.1 kcal mol−1) than TS6, indicating that the two pathways my also coexist and the Noyori-type one is slightly more facile. The quinoline (Q) is obtained after passing through TS7, delivering the dihydride intermediate M4. Finally, H2 liberation and catalyst regeneration can be achieved via a barrierless pathway as that in the first dehydrogenation process.
The 14DHQ can be obtained through proton transfer from tBuOH to the N1 site within the intermediate M7, where the tBuOH acts as a proton shuttle to facilitate the tautomerization between 34DHQ and 14DHQ (see Fig. S2 of ESI†). Then, to carry out the dehydrogenation from 14DHQ, a reverse proton transfer from N1 site of 14DHQ to tBuOK should occur to form the ion-pair complex M7 again, and the subsequent hydride transfer is same as discussed above for 34DHQ. In other words, an additional reversible proton transfer subprocess between 14DHQ and M7 is embedded in this case compared with the dehydrogenation pathway of 34DHQ.
According to the above calculated results in Sections 3.1 and 3.2, the rate determining step of the dehydrogenation of THQ is the hydride transfer from C2 of THQ to tBuOK during the first dehydrogenation process, which may proceed along a quasi-metal–ligand bifunctional pathway via a six-membered-ring rate-determining TS2, or along a base-catalyzed pathway via a H–H bridging rate-determining TS3, which are analogous with the reverse processes of the hydrogenation mechanisms proposed by Noyori23 and Berkessel,22 respectively. The second dehydrogenation from DHQ is found to be achieved directly from 34DHQ without the need of isomerization, and is both kinetically and thermodynamically more favourable due to the enhanced aromaticity. As a significant result, once dehydrogenation of THQ is triggered, only the fully dehydrogenated product Q will be delivered, as observed experimentally.18
Compared with the dehydrogenation mechanism of THQ mediated by transition metal catalysts such as Fe-PNP and Cp*Ir complexes as reported by Wang20a and Surawatanawong20b et al., on one hand, hydride transfer in the first dehydrogenation is the rate-determining step in all the three systems, though the hydride shift occurs before the proton transfer when using the Cp*Ir catalyst. On the other hand, proton transfer in this tBuOK catalyzed system is much more facile than the other two cases, which can be attributed to the strong protonophilic ability of tBuO− group. Subsequently, as a result that for the second dehydrogenation from DHQ, isomerization between 34DHQ and 12DHQ via disproportionation is required to shift the double bond from N–C2 to the C3–C4, using the Cp*Ir catalyst, because proton abstraction from C3 of 34DHQ is unlikely with high energy barrier; while it can take place directly from 34DHQ with a proton-transfer rate-determining step, using the Fe-PNP catalyst; and in this work with a tBuOK as the catalyst, we found it can occur also directly from 34DHQ, but hydride migration is the rate-determining step.
3.3. Effect of nitrogen substituent for THQ dehydrogenation
To explore the regulation effect of the nitrogen substituent, similar process of 1,2,3,4-tetrahydronaphthalene (THNap) catalyzed by tBuOK was also investigated, as shown in Fig. 3. Different from the case for THQ, the first dehydrogenation of THNap is so slow that an energetic span as high as 43.1 to 43.6 kcal mol−1 along the two type pathways should be overcome. On one side, a moderate energy barrier of 15.1 kcal mol−1 was determined for the proton transfer from C1 on THNap to oxygen atom on tBuOK, which becomes more difficult compared to that for THQ due to the N–H bond in THQ is weaker than the C–H bond in THNap.2c On the other hand, the hydride transfer from C2 of THNap becomes more thermodynamically unfavorable than that for THQ, because the C2–H bond adjacent to the N1 site in THQ also gets weaker. In short, the incorporation of N-heteroatom can facilitate both the proton transfer and especially the hydride transfer processes. In addition, compared with the dehydrogenation of THNap catalyzed by the Fe(II) complex in which the proton transfer step was reported to be the rate-determining step with an energy barrier of 42.2 kcal mol−1,20b the rate-determining step in this case is the hydride transfer step whereas the proton transfer is relatively feasible. The total results show that the nitrogen substituent is also necessary for dehydrogenation reaction promoted by the tBuOK base catalyst. | ||
| Fig. 3 (a) Relative free energy profiles for the dehydrogenation reaction of 1,2,3,4-tetrahydronaphthalene (THNap) catalyzed by tBuOK in o-xylene. (b) Geometrical information. Energies are in kcal mol−1, bond distances are in angstroms. | ||
3.4. Dehydrogenation of THQ catalyzed by tBuO− and tBuOLi
To further explore the effect of alkali ion, the reaction pathways catalyzed an isolated tBuO− anion (see Fig. S3†) and by a tBuOLi base were also calculated (see Fig. 4). Given that the first dehydrogenation of THQ is rate-determining, only the pathways of first dehydrogenation from THQ in these cases were studied.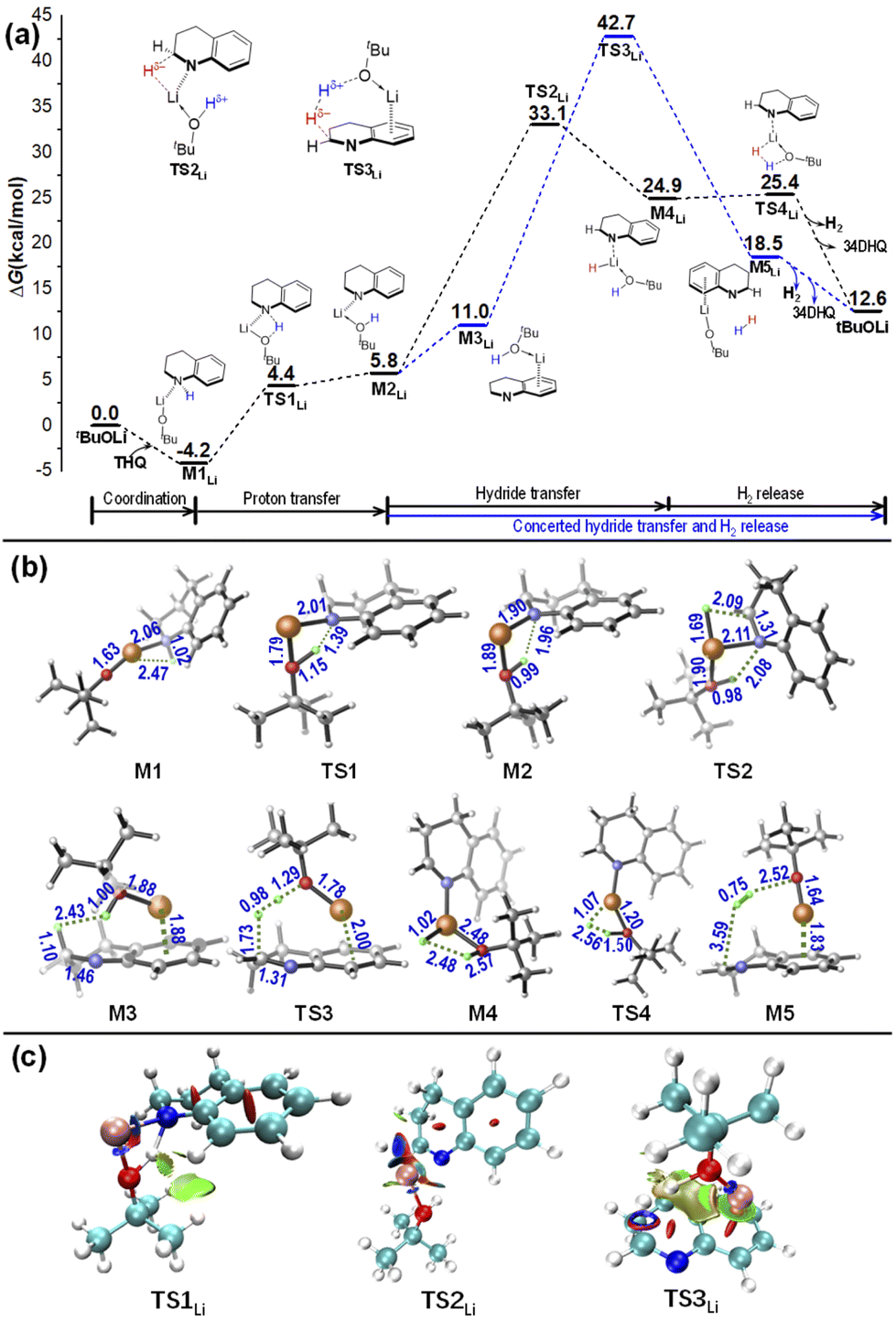 | ||
| Fig. 4 (a) Relative free energy profiles for the dehydrogenation reaction of THQ catalyzed by tBuOLi in o-xylene. (b) Geometrical information. (c) NCI analyses of TS1-3. Energies are in kcal mol−1, bond distances are in angstroms. | ||
The relative free energy profiles for the dehydrogantion of THQ catalyzed by an isolated tBuO− is plotted in Fig. S3.† Note that without the involvement of any alkali ion, only the base-catalyzed mechanism was considered in this case. The calculated energetic span is as high as 40 kcal mol−1 via TS2t, suggesting that the K+ ion plays vital role in stabling the transition state and facilitating the dehydrogenation reaction.
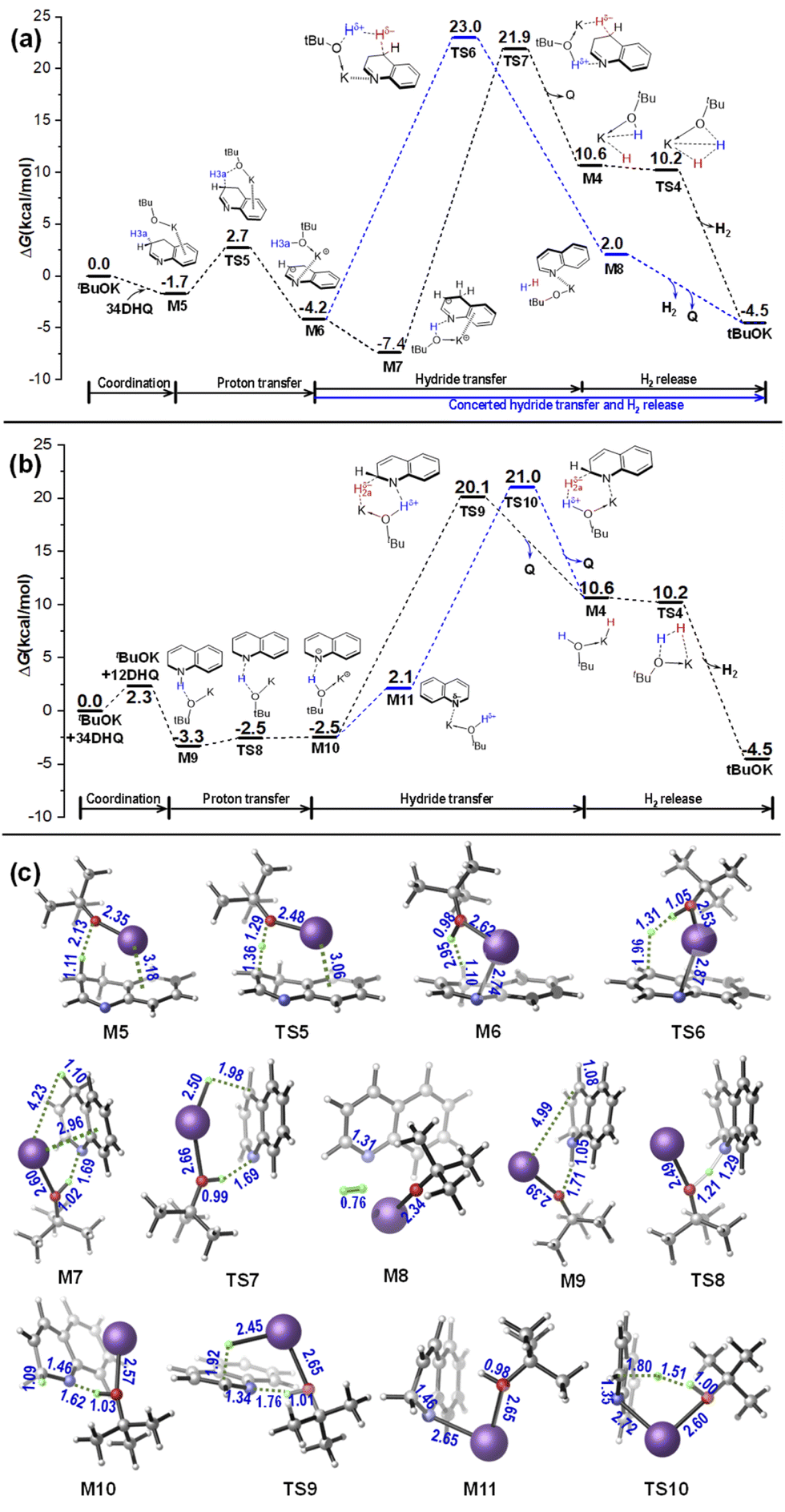
As shown in Fig. 4, in the case of using tBuOLi as the catalyst, the associated complexes M1Li is stabled by Li+⋯N covalent interaction, and no presence of N–H⋯O hydrogen bond like the cases of using tBuOK as the catalyst. As a result, the barrier for proton transfer becomes obviously higher. In addition, different from the six-membered-ring quasi-Noyori-type TS2 along the metal–ligand bifunctional pathway mediated by tBuOK, the TS2Li exhibits a four-membered-ring interaction according to the structural information and NCI analyses in Fig. 4 versus that in Fig. 2. Further bonding analyses were also done using the ETS-NOCV method. The NOCV analyses illustrated in Fig. S5† reveal that there is evident binding interaction between N atom and Li+ (in M2Li and TS2Li) but not with the case of tBuOK+, indicating that the small size as well as strong Lewis acidity of Li+ allows for effective binding to N atom and prevention of non-covalent interaction for hydride transfer.37 Correspondingly, the energy barrier for hydride transfer via the four-membered TS2Li is determined to be 37.3–38.8 kcal mol−1 (obtained by using B3LYP-D3 functional with def2-TZVP and def2-QZVP basis sets), and is 3.1–4.3 kcal mol−1 higher than that via the six-membered TS2 catalyzed by tBuOK.
Another possible process along the base-catalyzed-type pathway was also examined. The results show that a high energy barrier of 46.9–48.9 kcal mol−1 (obtained by using B3LYP-D3 functional with def2-TZVP and def2-QZVP basis sets) is required by using a tBuOLi catalyst. Structurally different from the TS3(K), the Li+ binds to phenyl ring of THQ via cation⋯π interaction with TS3Li (see Fig. 3). In addition, it takes places along a concerted hydride transfer and H2 release channel via the H–H bridging transition state TS3Li, and C–H and O–H bonds are simultaneously broken, leading to a remarkably high energy barrier. Interestingly, it was reported by Dub et al. that in the ketone hydrogenation reaction promoted by tBuOK, the H–H bond cleavage and the hydride transfer show a concert behaviour in gas phase, while it is stepwise with a lower energy barrier in solution phase.24a This work also suggests that the stepwise behaviour in the presence of tBuOK can facilitate the reaction in the framework of the base-catalyzed type mechanism.
Based on the above data and discussions, a mechanism diagram can be drawn for comparison of the dehydrogenation of THQ catalyzed by tBuOK vs. tBuOLi, as shown in Scheme 3. It is clearly seen that both the quasi-metal–ligand pathway and the base-catalyzed type channel catalyzed by tBuOLi are energetically higher than that mediated by tBuOK. In particular, the base-catalyzed type one via TS3Li is infeasible with a remarkably large energetic span. These calculated results provide a clearly physical picture to explain why the experimentally observed yield was sharply dropped when using tBuOLi (19%) instead of tBuOK (99%) as the catalyst.18
 | ||
| Scheme 3 Summary of catalytic cycles for the dehydrogenation from THQ mediated by the tBuOK (A) and tBuOLi (B) base catalysts. | ||
4. Conclusions
This DFT computational study provides mainly the following insights into the ADH of THQ catalyzed by an alkali catalyst of tBuOK versus tBuOLi as a case study:(1) This work reveals that the dehydrogenation mediated by tBuOK can proceed efficiently along a quasi-metal–ligand bifunctional catalytic channel or a base-catalyzed pathway with close energy barriers.
(2) No matter which pathway the reaction follows, the hydride transfer in the first dehydrogenation process is the rate determining step. The second dehydrogenation is found to be achieved directly from 34DHQ regulated by a tBuOK catalyst, without the need of further isomerisation.
(3) In contrast, for the case in presence of the tBuOLi catalyst, the concerted hydride transfer and H2 elimination along a base-catalyzed pathway is infeasible with a significantly high energy barrier. Thus, the dehydrogenation can only proceed along the quasi-metal–ligand bifunctional pathway with a relatively higher barrier, due to binding of N atom weakens the affinity of Li+ for hydride abstraction. This is consistent with the experimental observation that tBuOLi is inferior to tBuOK as a dehydrogenation catalyst.
In short, this work contributes to a comprehensive understanding of the ADH catalyzed by a simple base catalyst, which may provide help in the rational design of new catalysts for the ADH reactions as well as the LOHC systems.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful to the supports from the National Natural Science Foundation of China (NSFC22003076, NSFC21576291), the Natural Science Foundation of Shandong Province (ZR2020MB023), the Fundamental Research Funds for the Central Universities (22CX06012A, 20CX06031A) and the Qingdao Postdoctoral Applied Research Project (QDYY20190035).References
- (a) M. Niermann, S. Timmerberg, S. Drünert and M. Kaltschmitt, Renewable Sustainable Energy Rev., 2021, 135, 110171 CrossRef CAS; (b) P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 CrossRef CAS PubMed; (c) Z. X. Giustra, J. S. A. Ishibashi and S.-Y. Liu, Coord. Chem. Rev., 2016, 314, 134–181 CrossRef CAS; (d) M. Niermann, A. Beckendorff, M. Kaltschmitt and K. Bonhoff, Int. J. Hydrogen Energy, 2019, 44, 6631–6654 CrossRef CAS.
- (a) E. Clot, O. Eisenstein and R. H. Crabtree, Chem. Commun., 2007, 2231–2233 RSC; (b) A. Moores, M. Poyatos, Y. Luo and R. H. Crabtree, New J. Chem., 2006, 30, 1675–1678 RSC; (c) R. H. Crabtree, Energy Environ. Sci., 2008, 1, 134–138 RSC; (d) R. H. Crabtree, ACS Sustainable Chem. Eng., 2017, 5, 4491–4498 CrossRef CAS.
- (a) Y. Zhang, J. Wang, F. Zhou and J. Liu, Catal. Sci. Technol., 2021, 11, 3990–4007 RSC; (b) K. C. Tan, T. He, Y. S. Chua and P. Chen, J. Phys. Chem. C, 2021, 125, 18553–18566 CrossRef CAS.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed.
- T. Shimbayashi and K.-I. Fujita, Tetrahedron, 2020, 76, 130946 CrossRef CAS.
- (a) R. Yamaguchi, C. Ikeda, Y. Takahashi and K. Fujita, J. Am. Chem. Soc., 2009, 131, 8410–8412 CrossRef CAS PubMed; (b) K. Fujita, Y. Tanaka, M. Kobayashi and R. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 4829–4832 CrossRef CAS PubMed; (c) K. Fujita, T. Wada and T. Shiraishi, Angew. Chem., Int. Ed., 2017, 56, 10886–10889 CrossRef CAS PubMed.
- (a) D. Talwar, A. Gonzalez-de-Castro, H. Y. Li and J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 5223–5227 CrossRef CAS PubMed; (b) J. Wu, D. Talwar, S. Johnston, M. Yan and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 6983–6987 CrossRef CAS PubMed; (c) B. Maji, A. Bhandari, D. Bhattacharya and J. Choudhury, Organometallics, 2022, 41(13), 1609–1620 CrossRef CAS.
- (a) R. Xu, S. Chakraborty, H. Yuan and W. D. Jones, ACS Catal., 2015, 5, 6350–6354 CrossRef CAS; (b) S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136, 8564–8567 CrossRef CAS PubMed.
- O. R. Luca, D. L. Huang, M. K. Takase and R. H. Crabtree, New J. Chem., 2013, 37, 3402 RSC.
- Q. F. Wang, H. N. Chai and Z. K. Yu, Organometallics, 2018, 37, 584–591 CrossRef CAS.
- P. Sánchez, M. Hernández-Juárez, N. Rendón, J. López-Serrano, L. L. Santos, E. Álvarez, M. Paneque and A. Suárez, Dalton Trans., 2020, 49, 9583–9587 RSC.
- (a) M. A. Esteruelas, V. Lezáun, A. Martínez, M. Oliván and E. Oñate, Organometallics, 2017, 36, 2996–3004 CrossRef CAS; (b) M. L. Buil, M. A. Esteruelas, M. P. Gay, M. Gómez-Gallego, A. I. Nicasio, E. Oñate, A. Santiago and M. A. Sierra, Organometallics, 2018, 37, 603–617 CrossRef CAS.
- G. Jaiswal, V. G. Landge, D. Jagadeesan and E. Balaraman, Nat. Commun., 2017, 8, 1–13 CrossRef CAS PubMed.
- (a) Y. Han, Z. Wang, R. Xu, W. Zhang, W. Chen, L. Zheng, J. Zhang, J. Luo, K. Wu, Y. Zhu, C. Chen, Q. Peng, Q. Liu, P. Hu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 11262–11266 CrossRef CAS PubMed; (b) G. Jaiswal, M. Subaramanian, M. K. Sahoo and E. Balaraman, ChemCatChem, 2019, 11, 2449–2457 CrossRef CAS.
- Z. Zhang, W. Liu, Y. Zhang, J. Bai and J. Liu, ACS Catal., 2021, 11, 313–322 CrossRef CAS.
- (a) H. T. Hu, Y. Q. Nie, Y. W. Tao, W. Y. Huang, L. Qi and R. F. Nie, Sci. Adv., 2022, 8, 9478 CrossRef PubMed; (b) A. Mollar-Cuni, D. Ventura-Espinosa, S. Martín, H. García and J. A. Mata, ACS Catal., 2021, 11, 14688–14693 CrossRef CAS PubMed.
- (a) M. Kojima and M. Kanai, Angew. Chem., Int. Ed., 2016, 128, 12412–12415 CrossRef; (b) S. Tamke, Z.-W. Qu, N. A. Sitte, U. Flörke, S. Grimme and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 12219–12223 CrossRef PubMed.
- T. Liu, K. Wu, L. Wang and Z. Yu, Adv. Synth. Catal., 2019, 361, 3958–3964 CrossRef CAS.
- H. Li and Z. Wang, Sci. China: Chem., 2012, 55, 1991–2008 CrossRef CAS.
- (a) H. Li, J. Jiang, G. Lu, F. Huang and Z.-X. Wang, Organometallics, 2011, 30, 3131–3141 CrossRef CAS; (b) B. Sawatlona and P. Surawatanawong, Dalton Trans., 2016, 45, 14965 RSC; (c) S. M. Bellows, S. Chakraborty, J. B. Gary, W. D. Jones and T. R. Cundari, Inorg. Chem., 2017, 56, 5519–5524 CrossRef CAS PubMed.
- A. Bera, S. Bera and D. Banerjee, Chem. Commun., 2021, 57, 13042–13058 RSC.
- A. Berkessel, T. J. S. Schubert and T. N. Muller, J. Am. Chem. Soc., 2002, 124, 8693–8698 CrossRef CAS PubMed.
- (a) M. Yamakawa, H. Ito and R. Noyori, J. Am. Chem. Soc., 2000, 122, 1466–1478 CrossRef CAS; (b) R. Noyori, Adv. Synth. Catal., 2003, 345, 15–32 CrossRef CAS.
- (a) P. A. Dub and N. V. Tkachenko, J. Phys. Chem. A, 2021, 125, 5726–5737 CrossRef CAS PubMed; (b) P. A. Dub, Eur. J. Inorg. Chem., 2021, 47, 4884–4889 CrossRef.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- (a) A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef; (b) K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian Inc., Wallingford, CT, 2013 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- (a) I. D. Jenkins and E. H. Krenske, ACS Omega, 2020, 5, 7053–7058 CrossRef CAS PubMed; (b) M. G. Stanton, C. B. Allen, R. M. Kissling, A. L. Lincoln and M. R. Gagné, J. Am. Chem. Soc., 1998, 120, 5981–5989 CrossRef CAS; (c) P. Hima, M. Vageesh, M. Tomasini, A. Poater and R. Dey, Mol. Catal., 2023, 542, 113110 CrossRef.
- (a) L. Zhang, H. Yang and L. Jiao, J. Am. Chem. Soc., 2016, 138, 7151–7160 CrossRef CAS PubMed; (b) G. Nocera, A. Young, F. Palumbo, K. J. Emery, G. Coulthard, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2018, 140, 9751–9757 CrossRef CAS PubMed.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2010, 44, 101–110 CrossRef PubMed.
- (a) C. Hou, Z. Zhang, C. Zhao and Z. Ke, Inorg. Chem., 2016, 55, 6539–6551 CrossRef CAS PubMed; (b) K. T. Tseng, J. W. Kampf and N. K. Szymczak, ACS Catal., 2015, 5, 5468–5485 CrossRef CAS; (c) G. Zeng, S. Sakaki, K. Fujita, H. Sano and R. Yamaguchi, ACS Catal., 2014, 4, 1010–1020 CrossRef CAS.
- P. A. Dub and J. C. Gordon, Nat. Chem. Rev., 2018, 2, 396–408 CrossRef CAS.
- B. Chan and L. Radom, J. Am. Chem. Soc., 2005, 127, 2443–2454 CrossRef CAS PubMed.
- X.-B. Zhang and X. Zhao, Phys. Chem. Chem. Phys., 2011, 13, 3997–4004 RSC.
- Y.-W. Zhao, H.-R. Zhu, S.-Y. Sung, D. J. Wink, J. M. Zadrozny and T. G. Driver, Angew. Chem., Int. Ed., 2021, 60, 19207–19213 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04305c |
| This journal is © The Royal Society of Chemistry 2023 |