DOI:
10.1039/D3RA04656G
(Paper)
RSC Adv., 2023,
13, 27359-27362
Development of subtype-selective estrogen receptor modulators using the bis(4-hydroxyphenyl)silanol core as a stable isostere of bis(4-hydroxyphenyl)methanol†
Received
12th July 2023
, Accepted 30th August 2023
First published on 12th September 2023
Abstract
In this study, we synthesized and evaluated silanol-based bisphenol derivatives as stable isosteres of bis(4-hydroxyphenyl)methanol. The developed silanols exhibited estrogen receptor (ER)-modulating activity. Among them, bis(4-hydroxyphenyl)(methyl)silanol (5a) showed a characteristic ER subtype selectivity, namely, antagonistic activity toward ERα and agonistic activity toward ERβ. Docking simulation indicated that the silanol moiety plays a key role in this selectivity. Our results suggest that silanol-based bisphenols offer a unique scaffold for biologically active compounds.
1 Introduction
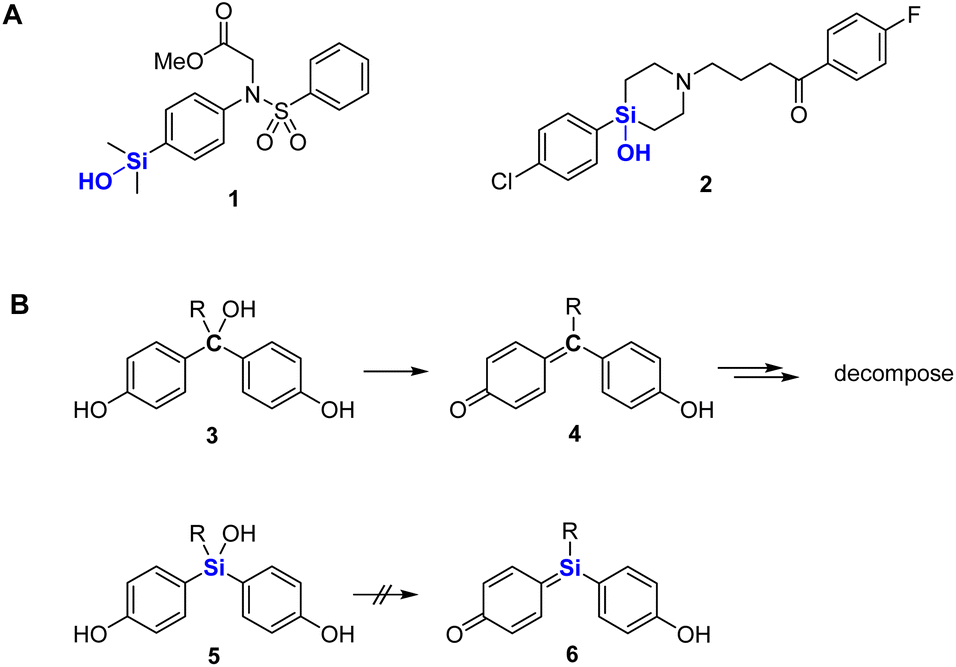
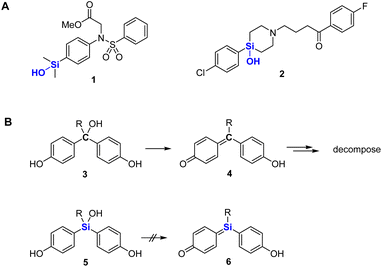
Silanols are widely used in various fields, including materials chemistry and synthetic chemistry.1 For example, they undergo cross-coupling reactions and are key components of various silicone materials and ceramics.2 In the field of biologically active compounds, silanols have been utilized in place of alcohols,3 since they are more acidic and hydrophobic than the corresponding alcohols, and therefore have distinct profiles of biological activity. We have developed silanol derivatives such as 1 with unique target selectivity.4 In addition, Tacke and coworkers have developed sila-haloperidol (2) and related silicon analogs of dopamine receptor antagonists in order to avoid the formation of neurotoxic metabolites, which are generated by elimination of water during the metabolism of the parent carbon analogs (Fig. 1A).5 We considered that silanols might be more widely applicable, and here we report the development and biological evaluation of silanol-based bisphenol derivatives as candidate estrogen receptor (ER) ligands.
 |
| | Fig. 1 (A) Structures of some silanol-based biologically active compounds. (B) Structures and properties of bis(4-hydroxyphenyl)methanols (3) and bis(4-hydroxyphenyl)silanols (5). Compounds 3 are easily converted to quinone methides 4, whereas silanols 5 cannot generate the corresponding quinone congeners. | |
2 Results and discussion
2.1 Molecular design
Bis(4-hydroxyphenyl)methanol (3) is a versatile scaffold of functional molecules such as fluorescein.6 However, bis(4-hydroxyphenyl)methanols, especially in case of aliphatic substituents (R = alkyl), are readily converted to electrophilic quinone methides (4) by elimination of water, and therefore are not available as biologically active compounds (Fig. 1B). On the other hand, bisphenol is a promising core structure of biologically active compounds,7 particularly modulators of nuclear estrogen receptors (ERs).8 The ERs are members of the nuclear receptor superfamily of ligand-dependent transcription factors.9
There are two subtypes, namely ERα and ERβ,10 which have distinct target tissue distributions and functional activities, and may have divergent roles. For example, activation of ERα results in the progression of estrogen-dependent breast cancer, whereas ERβ inhibits proliferation and invasion of breast cancer cells. Thus, the development of subtype-selective ER modulators is an attractive approach for anticancer drug discovery.11 Bisphenol derivatives function as modulators of ERs, and the substituents on the central carbon atom of bisphenols greatly influence their ER subtype selectivity.8 Therefore, introduction of a hydroxyl group on the bisphenol core structure is a reasonable approach to develop novel subtype-selective ER modulators. However, since bis(4-hydroxyphenyl)methanols (3) are insufficiently stable, we aimed to synthesize bis(4-hydroxyphenyl)silanols (5) as more stable isosteres of bis(4-hydroxyphenyl)methanols (3), and to investigate their potency as ER modulators.
2.2 Synthesis
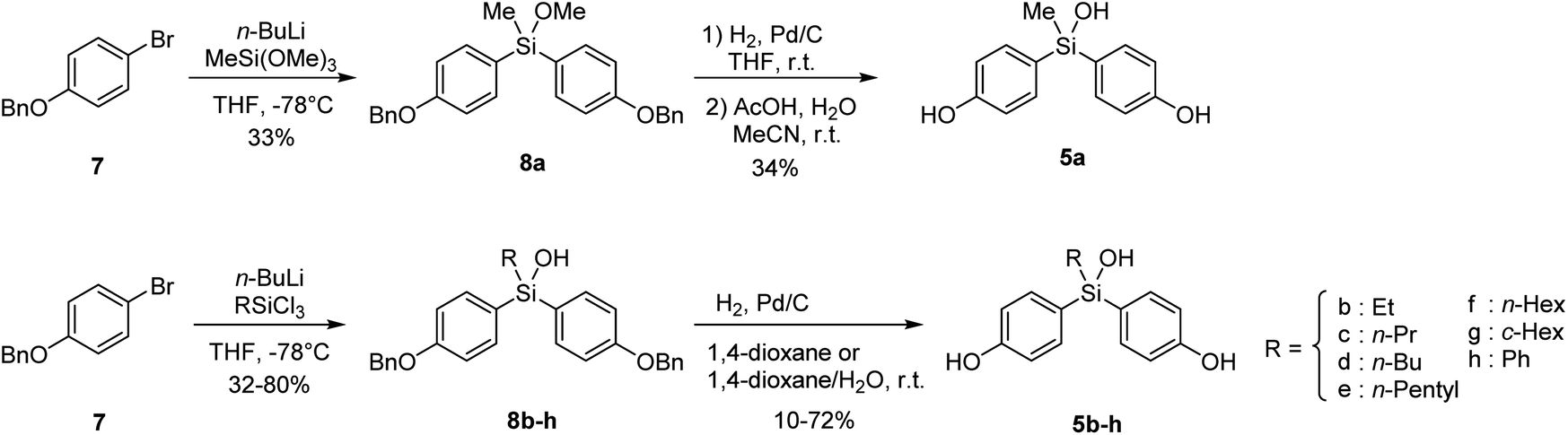
Synthesis of the designed bis(4-hydroxyphenyl)silanols 5a–5h is summarized in Scheme 1. From 4-benzyloxybromobenzene (7) as the starting material, lithiation and arylation with trimethoxymethylsilane gave methoxysilane 8a, and removal of the benzyl groups and methyl group afforded the desired silanol 5a. Silanol derivatives 5b–5h were synthesized similarly, using alkyltrichlorosilane instead of trimethoxysilane. Namely, lithiation of 7 and arylation with alkyltrichlorosilane gave the corresponding silanols 8b–8h, and removal of the benzyl groups afforded the desired silanols 5b–5h, respectively (Scheme 1).
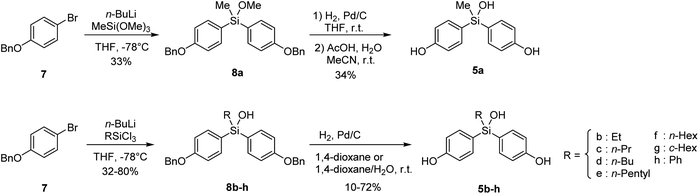 |
| | Scheme 1 Synthesis of the designed bis(4-hydroxyphenyl)silanols 5a–5h. | |
2.3 ER modulating activity
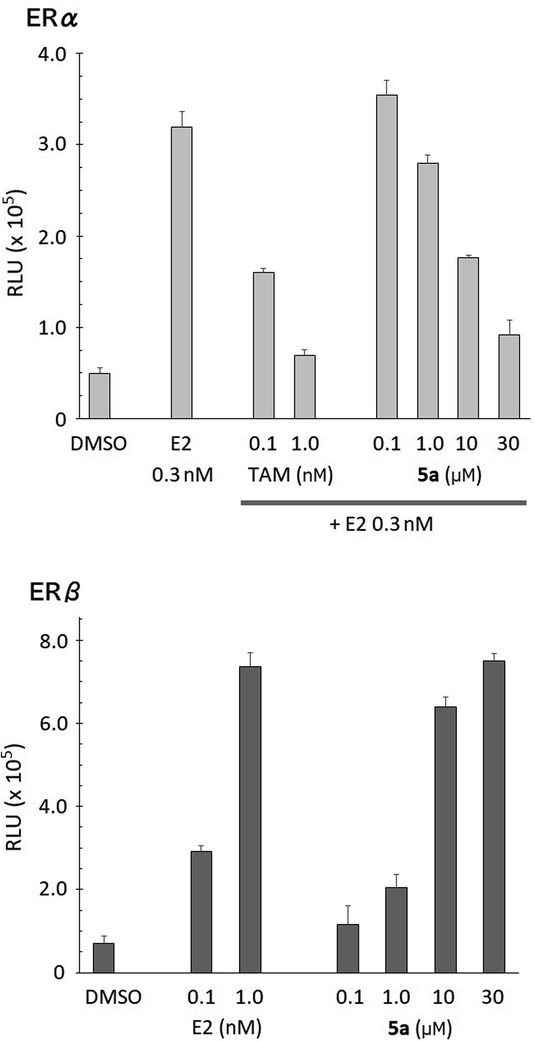
The ER-modulating activities of the synthesized bis(4-hydroxyphenyl)silanol derivatives were evaluated by means of luciferase reporter gene assay using HEK293 cells, and the results are summarized in Table 1. Agonistic activities toward each ER subtype were assessed in terms of the increase in luciferase activity and are shown as 50% effective concentration (EC50). Antagonistic activities were assessed in terms of the decrease in the luciferase activity induced by 0.3 nM estradiol (E2) and are shown as 50% inhibitory concentration (IC50). All the synthesized compounds exhibited ER-modulating potency, and compounds 5d–5g bearing a large aliphatic substituent at the silicon atom exhibited significant antagonistic activity toward both ERα and ERβ. Compound 5h bearing a phenyl group showed moderate antagonistic activity. Decrease in the bulkiness of the substituent, interestingly, had different effects on the two subtypes. The methyl (5a) and propyl (5c) derivatives exhibited antagonistic activity towards ERα with potency similar to those of 5d–5g. On the other hand, propyl derivative 5c exhibited weak antagonistic activity and partial agonistic activity towards ERβ. Methyl (5a) and ethyl (5b) derivatives did not exhibit ERβ-antagonistic activity, but showed ERβ-agonistic activity. Thus, the methyl derivative 5a serves as a unique ER subtype-selective modulator, i.e., an antagonist toward ERα and a full agonist toward ERβ (Fig. 2). Although the activity of 5a was weaker than the developed ER modulators, the unique subtype selectivity profile of 5a is interesting and suggestive for development of novel ER modulators.
Table 1 ER agonistic and antagonistic activity of silanol derivatives determined by means of luciferase reporter gene assay using HEK293 cells transiently transfected with pCX-GAL4-hER-LBD and MH100x4-tk-LUC vectors. Data are presented as mean ± SD
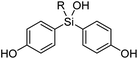
|
| Compd |
R |
ERα |
ERβ |
| EC50a (μM) |
IC50b (μM) |
EC50a (μM) |
IC50b (μM) |
| Concentration showing 50% of the maximal activity of E2. Concentration inhibiting the activity of 0.3 nM E2 by 50%. Percentage of the maximal activation of E2 at 30 μM. Percent inhibition at 30 μM. NA: no activity. |
| 5a |
Me |
NA |
9.85 ± 1.71 |
3.35 ± 0.74 |
NA |
| 5b |
Et |
(28 ± 7%) c |
(40 ± 10%)d |
7.87 ± 2.36 |
NA |
| 5c |
n-Pr |
(19 ± 5%) c |
11.2 ± 2.57 |
(30 ± 4%)c |
(17% ± 15)d |
| 5d |
n-Bu |
NA |
9.63 ± 2.78 |
NA |
21.5 ± 33 |
| 5e |
n-Pen |
NA |
10.0 ± 4.93 |
NA |
3.62 ± 3.5 |
| 5f |
n-Hex |
NA |
6.09 ± 1.86 |
NA |
4.80 ± 3.85 |
| 5g |
c-Hex |
NA |
10.5 ± 5.56 |
NA |
0.59 ± 0.31 |
| 5h |
Ph |
NA |
(27 ± 6%)d |
NA |
22.2 ± 16.1 |
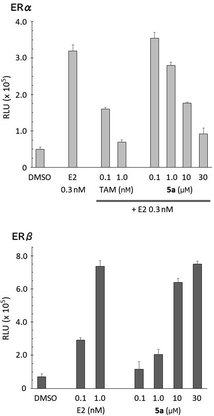 |
| | Fig. 2 Dose–response relationship of 5a in reporter gene assay using HEK293 cells. Compound 5a induced transcription of ERβ, but suppressed the transcription of ERα induced by E2. Data are presented as mean ± SD. TAM: tamoxifen. | |
2.4 Docking simulation
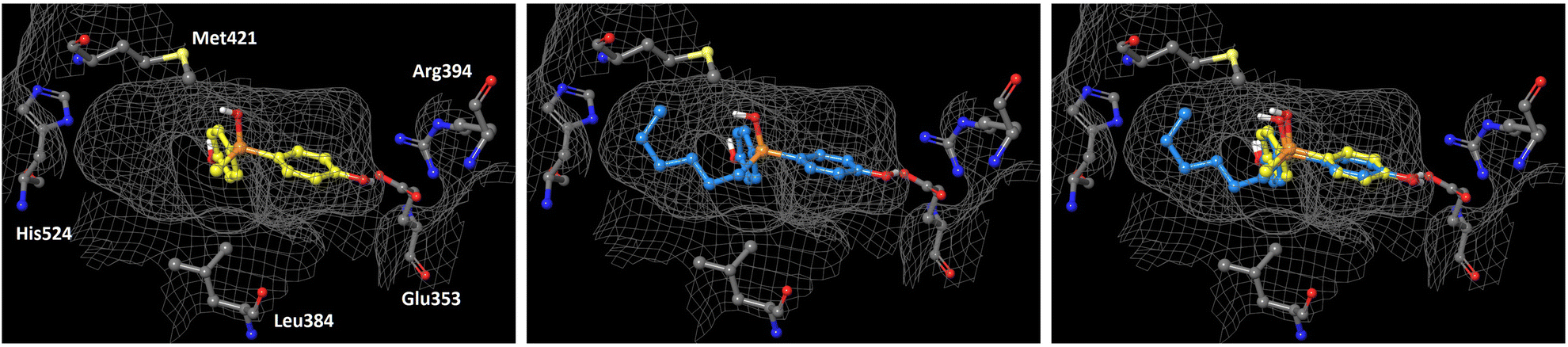
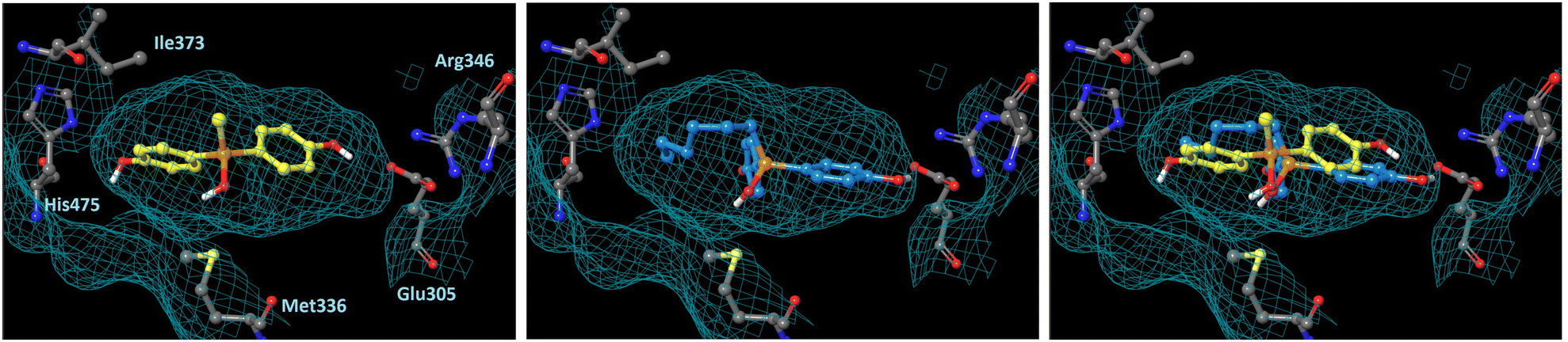
In order to understand the subtype-selective activity of silanol 5a, docking simulation using the crystal structures of human ERα (PDB ID: 3ERT)12 and ERβ (PDB-ID: 3OLS)13 was performed (Fig. 3 and 4). The homology of the ligand-binding domains of the two ER subtypes is very high, and only two residues in the ligand-binding pocket are different, namely, Leu (Leu384) and Met (Met421) in ERα are replaced by Met (Met336) and Ile (Ile373) in ERβ, respectively. Therefore, we postulated that the interaction with these residues would be the key to the subtype-selective action of 5a. In the case of ERα, one of the phenolic hydroxyl groups of 5a interacts with Glu353 and Arg394, which form hydrogen-bonds with the 3-OH group of estradiol. The OH group of silanol is located near Met421. Although the hydrogen bonding between His524 and a ligand is important for ERα agonistic activity, 5a did not interact with His524 in the calculated structure (Fig. 3 left). On the other hand, in the docked structure with ERβ, compound 5a forms hydrogen bonds with Glu305 and Arg346, which correspond to Glu353 and Arg394 in ERα, respectively, as well as with His475, which corresponds to His524 in ERα. The OH group of silanol is also located near Met336 (Fig. 4 left). Thus, compound 5a can form suitable hydrogen bonds for agonistic activity with ERβ. These results suggests that the silanol moiety of 5a plays a key role in recognizing the structural difference between ERα and ERβ, and the loss of hydrogen bonding with His524 results in the ERα-antagonistic activity of 5a.
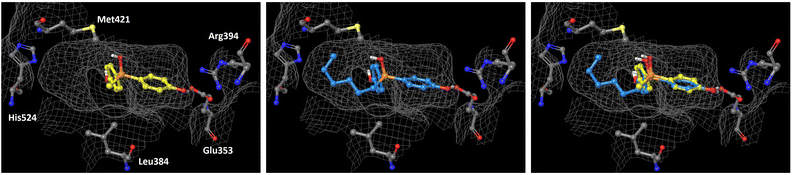 |
| | Fig. 3 Docking models of silanol 5a and 5f with hERα LBD (PDB ID: 3ERT) obtained with AutoDock 4.2.14 The protein surface is indicated as a grey mesh. (Left) Docking model of 5a (yellow) in the hERα LBD. (Center) Docking model of 5f (cyan) in the hERα LBD. (Right) Superimposition of the docking models of 5a and 5f. | |
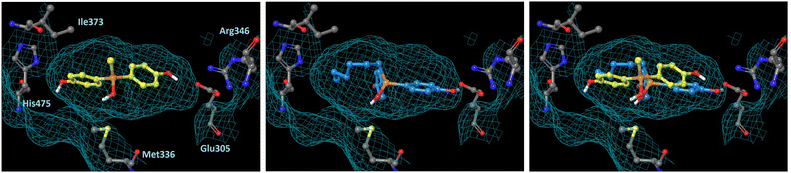 |
| | Fig. 4 Docking models of silanol 5a and 5f with hERβ LBD (PDB ID: 3OLS) obtained with AutoDock 4.2. The protein surface is indicated as a blue mesh. (Left) Docking model of 5a (yellow) in the hERβ LBD. (Center) Docking model of 5f (cyan) in the hERβ LBD. (Right) Superimposition of the docking models of 5a and 5f. | |
We also performed docking simulation of n-hexyl derivative 5f which acted as an antagonist towards both ERα and ERβ. In the docked structure with ERα, the bisphenol core of compound 5f takes a similar conformation to that of 5a, in which the OH group of silanol is located near Met421, and the n-hexyl group occupies the hydrophobic cavity (Fig. 3 center). In the docked structure with ERβ, on the other hand, the conformation of compound 5f is quite different from that of 5a. One phenolic OH group of 5f interacts with Glu305 and Arg346, but there is no interaction with His475, and the n-hexyl group occupies the cavity that is occupied by the phenol group of 5a (Fig. 4 center). This calculated structure of 5f in ERβ is similar to that in ERα, which may explain why the silanol derivatives 5d–5h bearing a bulky substituent function as antagonists toward both ERα and ERβ. These results suggests that the loss of hydrogen bonding with His524 is responsible for the ERα-antagonistic activity of 5a.
We performed docking simulation of imaginary methyl and hexyl carbinol analogs with hERα LBD. These carbon analogs adopt conformation similar to the corresponding silanols 5a and 5f, respectively (Fig. S1†). However, these carbon analogs are inherently unstable. Thus, the silanol moiety plays a key role in realization of the characteristic activity.
3 Conclusions
In order to expand the utility of silicon-based molecular construction in medicinal chemistry, we designed and synthesized a series of silanol-based bisphenol derivatives as stable isosteres of bis(4-hydroxyphenyl)methanol derivatives, which readily undergo dehydration to form electrophilic quinone methide species. The synthesized silanol derivatives served as ligands of ERα and ERβ, and methyl silanol 5a exhibited a unique subtype selectivity profile, namely, antagonist activity toward ERα and agonist activity toward ERβ. Docking simulation suggested that the interaction of the silanol moiety with methionine (Met421 in ERα or Met336 in ERβ) and hydrophobic interaction of the alkyl substituent are key factors. Since both antagonistic activity toward ERα and agonistic activity toward ERβ are desirable for anticancer activity,11 the compounds developed in this study may be promising lead compounds for cancer drug discovery.
4 Experimental
The detailed procedures for synthesis of compounds, biological evaluation and docking simulation is given in the ESI file.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by Grants in-Aid for Scientific Research from JSPS (KAKENHI Grant No. 17H03997, 19K22486 and 23K06046 (SF)), Takeda Science Foundation (SF), and Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP23ama121043.
Notes and references
-
(a) V. Chandrasekhar, R. Boomishankar and S. Nagendran, Chem. Rev., 2004, 104, 5847–5910 CrossRef CAS PubMed;
(b) M. Jeon, J. Han and J. Park, ACS Catal., 2012, 2, 1539–1549 CrossRef CAS.
-
(a) S. E. Denmark and A. Ambrosi, Org. Process Res. Dev., 2015, 19, 982–994 CrossRef CAS PubMed;
(b) S. E. Denmark, J. Org. Chem., 2009, 74, 2915–2927 CrossRef CAS PubMed.
-
(a) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS PubMed;
(b) S. Fujii and Y. Hashimoto, Future Med. Chem., 2017, 9, 485–505 CrossRef CAS PubMed;
(c) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
-
(a) H. Toyama, S. Sato, H. Shirakawa, M. Komai, Y. Hashimoto and S. Fujii, Bioorg. Med. Chem. Lett., 2016, 26, 1817–1820 CrossRef CAS PubMed;
(b) H. Toyama, H. Shirakawa, M. Komai, Y. Hashimoto and S. Fujii, Bioorg. Med. Chem., 2018, 26, 4493–4501 CrossRef CAS PubMed.
-
(a) R. Tacke, T. Heinrich, R. Bertermann, C. Burschka, A. Hamacher and M. U. Kassack, Organometallics, 2004, 23, 4468–4477 CrossRef CAS;
(b) R. Tacke, F. Popp, B. Müller B, B. Theis, C. Burschka, A. Hamacher, M. U. Kassack, D. Schepmann, B. Wünsch, U. Jurva and E. Wellner, ChemMedChem, 2008, 3, 152–164 CrossRef CAS PubMed.
- X. Chen, T. Pradhan, F. Wang, J.-S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS PubMed.
- S. Hosoda, D. Matsuda, H. Tomoda and Y. Hashimoto, Mini-Rev. Med. Chem., 2009, 9, 572–580 CrossRef CAS PubMed.
- K. Maruyama, M. Nakamura, S. Tomoshige, K. Sugita, M. Makishima, Y. Hashimoto and M. Ishikawa, Bioorg. Med. Chem. Lett., 2013, 23, 4031–4036 CrossRef CAS PubMed.
-
(a) H. Gronemeyer, J.-A. Gustafsson and V. Laudet, Nat. Rev. Drug Discovery, 2004, 3, 950–964 CrossRef CAS PubMed;
(b) D. J. Mangelsdorf, C. Thummel, M. Beato, P. Herrlich, G. Schütz, K. Umesono, B. Blumberg, P. Kastner, M. Mark, P. Chambon and R. M. Evans, Cell, 1995, 83, 835–839 CrossRef CAS PubMed;
(c) R. M. Evans, Science, 1988, 240, 889–894 CrossRef CAS PubMed.
-
(a) N. Heldring, A. Pike, S. Andersson, J. Matthews, G. Cheng, J. Hartman, M. Tujague, A. Ström, E. Treuter, M. Warner and J.-A. Gustafsson, Physiol. Rev., 2007, 87, 905–931 CrossRef CAS PubMed;
(b) B. J. Deroo and K. S. Korach, J. Clin. Invest., 2006, 116, 561–570 CrossRef CAS PubMed;
(c) C. Thomas and J.-Å. Gustafsson, Nat. Rev. Cancer, 2011, 11, 597–608 CrossRef CAS PubMed;
(d) B. S. Katzenellenbogen, M. M. Montano, T. R. Ediger, J. Sun, K. Ekena, G. Lazennec, P. G. Martini, E. M. McInerney, R. Delage-Mourroux, K. Weis and J. A. Katzenellenbogen, Recent Prog. Horm. Res., 2000, 55, 163–193 CAS.
-
(a) I. Paterni, C. Granchi, J. A. Katzenellenbogen and F. Minutolo, Steroids, 2014, 90, 13–29A CrossRef CAS PubMed;
(b) K. Shiau, D. Barstad, J. T Radek, M. J. Meyers, K. W. Nettles, B. S. Katzenellenbogen, J. A. Katzenellenbogen, D. A. Agard and G. L. Greene, Nat. Struct. Biol., 2002, 9, 359–364 Search PubMed.
- A. K. Shiau, D. Barstad, P. M. Loria, L. Cheng, P. J. Kushner, D. A. Agard and G. L. Greene, Cell, 1998, 95, 927–937 CrossRef CAS PubMed.
- S. Mocklinghoff, R. Rose, M. Carraz, A. Visser, C. Ottmann and L. Brunsveld, ChemBioChem, 2010, 11, 2251–2254 CrossRef PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 16, 2785–2791 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b