
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA modified SELEX approach to identify DNA aptamers with binding specificity to the major histocompatibility complex presenting ovalbumin model antigen†
Yang Lin‡
 a,
Cho-Yi Chen‡b,
Yu-Chia Kub,
Li-Chin Wangb,
Chia-Chien Hungc,
Zhi-Qian Lina,
Bing-Hong Chena,
Jui-Tse Hungd,
Yi-Chen Sunef and
Kai-Feng Hung*ag
a,
Cho-Yi Chen‡b,
Yu-Chia Kub,
Li-Chin Wangb,
Chia-Chien Hungc,
Zhi-Qian Lina,
Bing-Hong Chena,
Jui-Tse Hungd,
Yi-Chen Sunef and
Kai-Feng Hung*ag
aDepartment of Medical Research, Taipei Veterans General Hospital, 201, Section 2, Shi-Pai Road, Taipei 112, Taiwan. E-mail: kfhung@vghtpe.gov.tw; Tel: +886-2-28712121-7382
bInstitute of Biomedical Informatics, National Yang Ming Chiao Tung University, Taipei, Taiwan
cSchool of Computer Science, Georgia Institute of Technology Atlanta, GA, USA
dKang Chiao International School, Taipei, Taiwan
eSchool of Medicine, Tzu-Chi University, Hualien, Taiwan
fDepartment of Ophthalmology, Taipei Tzu Chi Hospital, The Buddhist Tzu Chi Medical Foundation, New Taipei City, Taiwan
gDepartment of Dentistry, School of Dentistry, National Yang Ming Chiao Tung University, Taipei, Taiwan
First published on 6th November 2023
Abstract
Aptamers have sparked significant interest in cell recognition because of their superior binding specificity and biocompatibility. Cell recognition can be mediated by targeting the major histocompatibility complex (MHC) that presents short peptides derived from intracellular antigens. Although numerous antibodies have demonstrated a specific affinity for the peptide–MHC complex, the number of aptamers that exhibit comparable characteristics is limited. Aptamers are usually selected from large libraries via the Systemic Evolution of Ligands by Exponential Enrichment (SELEX), an iterative process of selection and PCR amplification to enrich a pool of aptamers with high affinity. However, the success rate of aptamer identification is low, possibly due to the presence of complementary sequences or sequences rich in guanine and cytosine that are less accessible for primers. Here, we modified SELEX by employing systemic consecutive selections with minimal PCR amplification. We also modified the analysis by selecting aptamers that were identified in multiple selection rounds rather than those that are highly enriched. Using this approach, we were able to identify two aptamers with binding specificity to cells expressing the ovalbumin alloantigen as a proof of concept. These two aptamers were also discovered among the top 150 abundant candidates, despite not being highly enriched, by performing conventional SELEX. Additionally, we found that highly enriched aptamers tend to contain fractions of the primer sequence and have minimal target affinity. Candidate aptamers are easily missed in the conventional SELEX process. Therefore, our modification for SELEX may facilitate the identification of aptamers for more application in diverse biomedical fields. Significance: we modify the conventional method to improve the efficiency in the identification of the aptamer, a single strand of nucleic acid with binding specificity to the target molecule, showing as a proof of concept that this approach is particularly useful to select aptamers that can selectively bind to cells presenting a particular peptide by the major histocompatibility complex (MHC) on the cell surface. Given that cancer cells may express mutant peptide–MHC complexes that are distinct from those expressed by normal cells, this study sheds light on the potential application of aptamers to cancer cell targeting.
Introduction
The nucleic acid aptamer is a class of short single-stranded DNA or RNA molecules capable of binding to specific targets.1,2 Binding affinity and specificity are driven by a three-dimensional structural interaction between cognate targets and aptamers containing a partially complementary sequence with a propensity to form G-quadruplex or hairpin tertiary structures.3,4 Aptamers possess several advantages over antibodies: Aptamers are small in size compared to antibodies (6–30 kDa, 2 nm in diameter vs. 150–180 kDa, >15 nm in diameter), allowing their binding to domains that are inaccessible for antibodies.5–7 Besides, aptamers are synthesized in a chemical setting, which, unlike the production of antibodies that may involve animal and cell colonies, minimizes immunogenicity and contamination risk.7 Moreover, aptamers are easily conjugated with versatile fluorochromes or drugs, further extending their use as molecular imaging systems or therapeutics.8Aptamers are generally selected from large libraries via an iterative in vitro selection process named Systemic Evolution of Ligands by Exponential Enrichment (SELEX).9 An aptamer library usually contains a random region of 20–40 nucleotides, typically comprising up to 1015 to 1016 unique oligonucleotides, franked by two constant primer-binding sites at 5′ and 3′ ends to enable PCR amplification. Traditionally, SELEX involves iterative PCR amplification of aptamers that had undergone positive and negative selection processes until a pool of aptamers with high affinity for target molecules is enriched. Although the concept and procedures of SELEX are straightforward, its success rate remains relatively low,10–12 possibly due to the absence of stable binding motifs in the aptamer library and the presence of complementary sequences within the random regions that act as undesired primers to form PCR by-product.13 In addition, because the sequences rich in guanine and cytosine are less accessible for primers, the aptamers featured with GC-rich sequences may be outcompeted by other aptamers during PCR. Meanwhile, conventional SELEX is time-consuming and often takes months to complete. Therefore, it is crucial to modify conventional SELEX to expedite the identification of aptamers.
Given that the low success rate of SELEX is largely attributed to PCR bias, various alternative approaches to reduce or eliminate the amplification steps have been developed. One such modified approach is known as the non-SELEX method, which involves aptamer selection without any PCR steps.14 Another modified approach is the RNA Aptamer Isolation via Dual cycles SELEX, abbreviated as RAPID-SELEX, which simplifies conventional SELEX by alternately skipping PCR steps.15 In addition to these approaches that modify conventional PCR, a novel variation, termed emulsion PCR (ePCR), has also been incorporated into SELEX. The ePCR encapsulates a small fraction of aptamer with primers and PCR master mix into millions of oil droplets. Because each droplet serves as an isolated compartment for PCR reaction, product–product hybridization is tremendously avoided. Nonetheless, regardless of whether PCR is skipped or reformed, the identification of candidate aptamers still relies on sequence or motif enrichment. While successful identification of aptamers using these modified approaches has been previously reported, the number of aptamers that have been selected by conventional SELEX still exceeds those selected by these alternative approaches. This suggests that there is still room for modifying the SELEX process to improve the efficiency of aptamer identification.
Due to the superior binding specificity and biocompatibility, aptamers have gained significant interest in their use for in vivo cell recognition. The major histocompatibility complex (MHC) is a cell surface molecule that presents short peptides, typically 8 to 20 amino acids in length, derived from intracellular antigens.16 Each cell expresses individual antigens and unique sets of MHC molecules; therefore, peptide–MHC complexes act as a cellular identity card, allowing the immune system to recognize and differentiate between cells.17 Cancer cells present unique peptides derived from mutant proteins, known as neoantigens, using MHC complexes that are shared with their parental normal cells. Cancer cells also frequently express various cancer germline or viral antigens.18 Consequently, a molecule with the ability to differentiate between cancer-associated antigen and self-antigen in an MHC-restricted manner can selectively target tumor cells of a cancer patient. The peptide–MHC complex acts as a ligand for T cell receptor (TCR), and the detection of peptide–MHC complexes can be mediated by TCR-mimic antibody.19,20 To date, many studies have demonstrated the efficacy of targeting peptide–MHC complexes using TCR-mimic antibodies for the detection and treatment of various cancers,20–24 and currently, several peptide–MHC bi-specific molecules, such as IMA401 and IMA202 that, respectively, target MAGE-A4/8 and MAGE-A1 cancer germline antigens in the context of HLA-A2 are under clinical trials.25–27 However, the production of TCR-mimic antibodies is challenging and time-consuming.28 In contrast, aptamers are produced by chemical synthesis at low cost. Accordingly, aptamers that can specifically target the MHC complex presenting a unique peptide amidst the repertoire of normally presented peptide–MHC complexes are appealing tools for cancer imaging and therapy.
Therefore, in this study, we used ovalbumin model antigen to explore whether aptamers with binding specificity to a defined peptide–MHC complex can be identified. To this end, we modified the process and incorporated a new rationale for the selection of candidate aptamers. We also compared the efficiency of our method with that of conventional SELEX, showing that this modified approach is advantageous for the identification of specific aptamers for the peptide–MHC complex. These findings pave the way for further advancements in the development of aptamers for molecular diagnostics and therapies.
Result
Develop a systemic consecutive selection with minimal PCR amplification to facilitate aptamer selection
SELEX consists of iterative cycles of selection and amplification to enrich the aptamers with a high affinity to the target. To improve the efficiency of the aptamer selection process, we modified conventional SELEX by employing continuous selection with minimal PCR amplification. Meanwhile, to examine the potential of aptamers to discriminate between MHC complexes that differ solely by their presenting peptides, we used the mouse H-2Kb MHC class I pentamer bound to the ovalbumin (OVA) 257–264 SIINFEKL peptide as the target molecule. The scheme for the selection of aptamers against the OVA257–264-H-2Kb complex is shown in Fig. 1A. Specifically, the modified approach begins with incubation of the aptamer library with OVA257–264-H-2Kb pentamers, followed by the removal of unbound aptamers and the retrieval of pentamer-bound aptamers. This initial positive selection substantially reduced the size of the library. The aptamers after initial positive selection were amplified by running only 10 PCR cycles, which, based on our preliminary experiments, not only minimize the PCR bias but also sufficiently replenish the aptamer pool after consecutive selections. An aliquot of the PCR product was reserved for sequencing (designated as the sequencing sample #1), while the remaining was refolded for subsequent positive selection that each cycle included incubation with OVA257–264-H-2Kb pentamers, retrieval of bound aptamers and dissociation of the pentamer for aptamer refolding.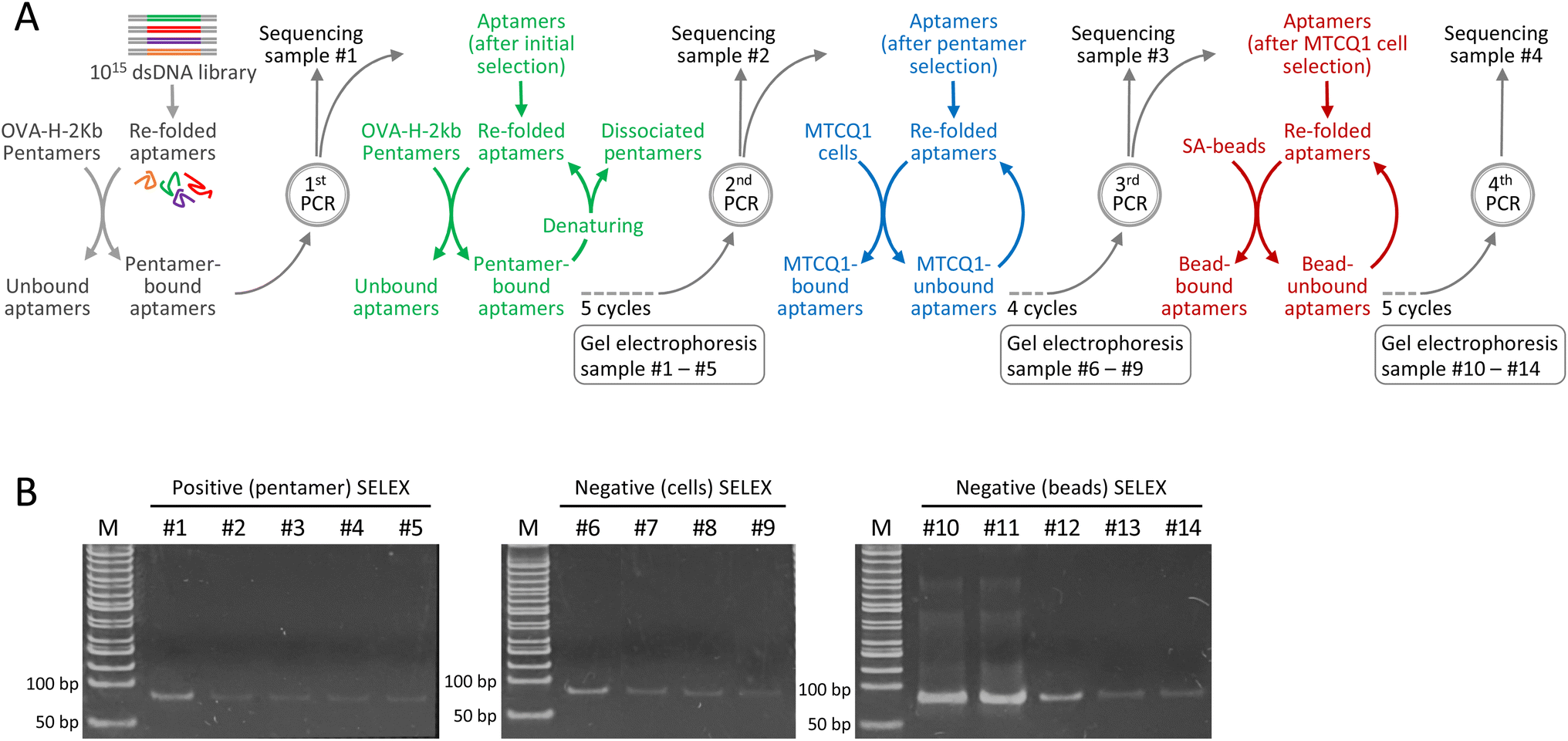 | ||
| Fig. 1 Develop a systemic consecutive selection with minimal PCR amplification for aptamer isolation. (A) Scheme of the modified SELEX consisting of consecutive positive selections (using OVA257–264-H-2Kb pentamers) and negative selections (using MTCQ1 cells and SA-beads) with minimal PCR amplification to identify aptamers against H-2Kb MHC presenting ovalbumin model antigen. (B) Polyacrylamide gel analysis of PCR products derived from modified SELEX. | ||
To determine the optimal number of consecutive selection cycles, we measured the DNA concentration at the end of every selection cycle. We found that each successive cycle of selection led to a substantial decrease in DNA concentration, which remained measurable after five cycles of pentamer-based selections but not after conducting an additional cycle of selection. Consequently, after five consecutive cycles of pentamer-based positive selections, the aptamer pool was amplified. Notably, PCR was run for only 10 cycles because this amplification condition resulted in a sufficient amount of DNA for the subsequent selection process without the accumulation of PCR by-products. An aliquot of the PCR product was reserved for sequencing (designated as sequencing sample #2), while the remaining was subsequently refolded for two different types of negative selection. In the first negative selection, we used MTCQ1 cells (a mouse tongue cancer cell line derived from C57/BL6 mice carrying the H-2Kb MHC I allele) that were not transduced with ovalbumin alloantigen. Approximately 200 ng of the refolded aptamer pool was incubated with MTCQ1 cells, and the unbound aptamers were retrieved and then refolded for the next cycle of selection. Meanwhile, we found that the remaining DNA after five consecutive cycles of cell-based selections was barely measurable. Therefore, after four consecutive cycles of cell-based negative selection, the aptamer pool was amplified by running 10 cycles of PCR. Similarly, an aliquot of the PCR product was reserved for sequencing (designated as sequencing sample #3), and the remaining PCR product was then subjected to the second negative selection with streptavidin-beads (SA-beads) to filter out the aptamers that were unwantedly attached to these magnetic conjugates when the pentamer-bound aptamers were pulled down. Likewise, after five consecutive negative selections, the DNA concentration of aptamer pool was appropriately amplified by running 10 cycles of PCR, and the PCR product was designated as sequencing sample #4.
To monitor the selection process, the samples taken from each selection with pentamers (5 cycles), untransduced MTCQ1 cells (4 cycles), and SA-beads (5 cycles) were subjected to polyacrylamide gel electrophoresis. As shown in Fig. 1B, the intensity of the band corresponding to the size of the aptamers gradually decreased as the consecutive selection cycles progressed, implying that consecutive selections without PCR amplification remove the aptamers that bound nonspecifically or with low affinity to the target molecules. Importantly, 10 cycles of PCR amplification in this consecutive selection protocol did not result in the formation of smear-type or ladder-type artifacts, which are indicative of by-product formation.
Aptamers identified across multiple selection cycles exhibit superior binding affinity to OVA257–264-H-2Kb pentamers
Next, we performed high-throughput sequencing to identify the aptamers in each sequencing sample. Because the consecutive selection process with minimal PCR amplification was used, aptamers with binding specificity were not necessarily highly enriched to dominate the aptamer pool. Instead, we selected the aptamers that are identified in multiple selection cycles based on the rationale that candidate aptamers may survive a multitude of selection processes. We found a total of 124![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 097, 62494, 43661, and 28987 aptamer sequences in samples #1, #2, #3, and #4, respectively. Some aptamer sequences were shared in more than one sequencing sample. For example, while 109988 aptamers only appeared in sample #1, 7520 aptamers simultaneously appeared in both samples #1 and #2; 41 aptamers simultaneously appeared in samples #1, #2, and #3; and two aptamers appeared in all four sequencing samples (Fig. 2A and ESI Table 1†). We focused on the aptamers that simultaneously appeared in more than three samples, including sample #4 and two or more other samples, assuming that candidate aptamers can sustain prior selections until the last cycle of amplification. Consequently, a total of 211 aptamers (including 125 aptamers in samples #1, #2, #4; 28 aptamers in samples #1, #3, #4; 56 aptamers in samples #2, #3, #4; and 2 aptamers in sample #1, #2, #3, #4) were selected for further analysis. We then used the quadruplex forming G-rich sequences (QGRS) mapper, mFold modeling, and NUPACK web servers to predict the potential of G-quadruplex formation, the most thermodynamically stable free energy (ΔG), and the secondary structure of aptamers, respectively. Among these 211 aptamers, 50 aptamers had at least four guanine-rich regions to form G-quadruplex tetrahelical structures, including 16 and 34 aptamers predicted by NUPACK to have hairpin and linear conformations, respectively (ESI Table 2†). The distribution of QGRS vs. ΔG of the potential hairpin (colored diamond) and linear (uncolored circle) aptamers is shown in Fig. 2B. Several aptamers, including Apt-1, Apt-3, Apt-8, and Apt-10, exhibited hairpin loop structure predicted by NUPACK (Fig. 2C), were further tested. Using the OVA257–264-H-2Kb pentamer in the pull-down assay, we found that the Apt-1 and Apt-10 molecules, but not Apt-3 and Apt-8, increased as a function of increasing the input of the aptamers (Fig. 2D). The dissociation constant (Kd) values of Apt-1 and Apt-10 were 136.2 nM and 155.1 nM, respectively, which are lower than most wild-type TCRs,29,30 indicating that these two aptamers have sufficient binding capability to their targets under physiological conditions.
097, 62494, 43661, and 28987 aptamer sequences in samples #1, #2, #3, and #4, respectively. Some aptamer sequences were shared in more than one sequencing sample. For example, while 109988 aptamers only appeared in sample #1, 7520 aptamers simultaneously appeared in both samples #1 and #2; 41 aptamers simultaneously appeared in samples #1, #2, and #3; and two aptamers appeared in all four sequencing samples (Fig. 2A and ESI Table 1†). We focused on the aptamers that simultaneously appeared in more than three samples, including sample #4 and two or more other samples, assuming that candidate aptamers can sustain prior selections until the last cycle of amplification. Consequently, a total of 211 aptamers (including 125 aptamers in samples #1, #2, #4; 28 aptamers in samples #1, #3, #4; 56 aptamers in samples #2, #3, #4; and 2 aptamers in sample #1, #2, #3, #4) were selected for further analysis. We then used the quadruplex forming G-rich sequences (QGRS) mapper, mFold modeling, and NUPACK web servers to predict the potential of G-quadruplex formation, the most thermodynamically stable free energy (ΔG), and the secondary structure of aptamers, respectively. Among these 211 aptamers, 50 aptamers had at least four guanine-rich regions to form G-quadruplex tetrahelical structures, including 16 and 34 aptamers predicted by NUPACK to have hairpin and linear conformations, respectively (ESI Table 2†). The distribution of QGRS vs. ΔG of the potential hairpin (colored diamond) and linear (uncolored circle) aptamers is shown in Fig. 2B. Several aptamers, including Apt-1, Apt-3, Apt-8, and Apt-10, exhibited hairpin loop structure predicted by NUPACK (Fig. 2C), were further tested. Using the OVA257–264-H-2Kb pentamer in the pull-down assay, we found that the Apt-1 and Apt-10 molecules, but not Apt-3 and Apt-8, increased as a function of increasing the input of the aptamers (Fig. 2D). The dissociation constant (Kd) values of Apt-1 and Apt-10 were 136.2 nM and 155.1 nM, respectively, which are lower than most wild-type TCRs,29,30 indicating that these two aptamers have sufficient binding capability to their targets under physiological conditions.
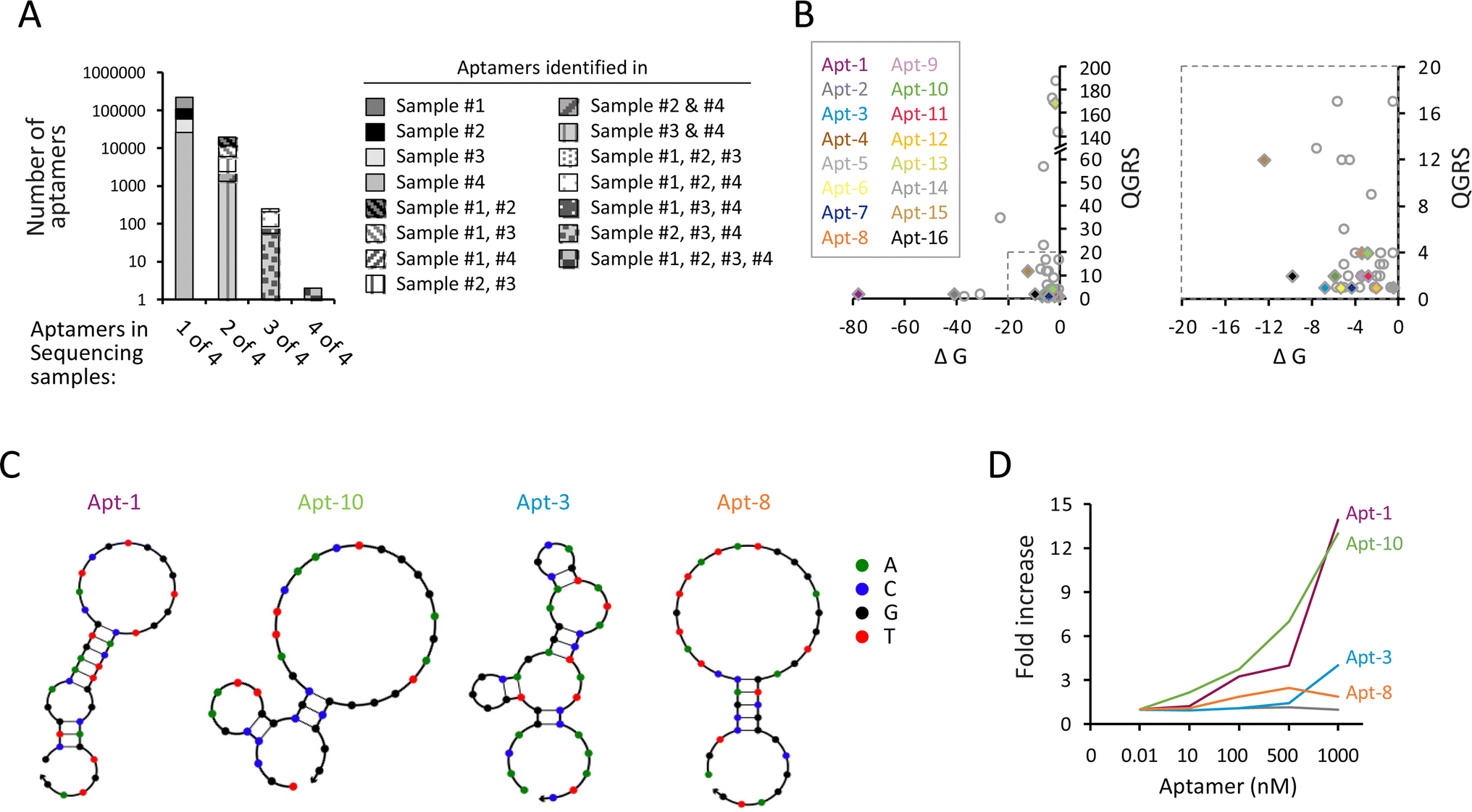 | ||
| Fig. 2 Systemic consecutive selection with minimal PCR amplification enables the identification of aptamers with binding specificity to OVA257–264-H-2Kb pentamers. (A) The number of aptamers identified in one (sample #1, #2, #3, or #4), two (sample #1/2, #1/3, #1/4, #2/3, #2/4, or #3/4), three (sample #1/2/3, #1/2/4, or #2/3/4), or all (sample #1/2/3/4) of four sequencing samples. (B) Distribution of hairpin (colored diamond) and linear (uncolored circle) aptamers with QGRS vs. ΔG ranging from 0 to 200 and 0 to −80 (left) or from 0 to 20 and 0 to −20 (right). (C) Secondary structures of candidate aptamers predicted by NUPACK. (D) qPCR-based binding assay (n = 3) showing the fold increase of Apt-1, Apt-3, Apt-8, and Apt-10 pulled down with 0.01 nM OVA257–264-H-2Kb pentamer. | ||
Conventional SELEX underwent more rounds of selection and amplification produced more PCR by-products
To illustrate the merits of the modified method, we also used OVA257–264-H-2Kb pentamers, MTCQ1 cells, and SA-beads to perform a conventional SELEX for comparison. Briefly, the aptamer library was first incubated with the OVA257–264-H-2Kb pentamers. After removing unbound aptamers, pentamer-bound aptamers were amplified by PCR and then refolded for subsequent incubation with MTCQ1 cells as the first negative selection. The MTCQ1-unbound aptamers were retrieved, PCR amplified, and refolded for the next round of SELEX. Beginning from the 9th round, a second negative selection using SA-beads was incorporated into each round of SELEX. The refolded aptamers underwent cell-based negative selection were incubated with SA-beads, and the bead-unbound aptamers were retrieved, PCR amplified, and refolded for the next round of SELEX (Fig. 3A). To determine the appropriate number of PCR cycles for aptamer amplification without producing artificial by-products, the aptamers after each selection were tentatively amplified by running 4, 6, 8, or 10 PCR cycles. We found that the initial PCR amplification of samples in round #1 resulted in a smear-like pattern of PCR products, which was likely caused by the high template concentration used in PCR reaction.31 The smear-like artifacts were notably diminished following the subsequent negative selection, indicating the removal of a substantial number of unspecific aptamers (Fig. 3B). Meanwhile, we found that more PCR cycles in each round of amplification often led to more ladder-type artifacts, which became increasingly prevalent as the SELEX process continued. For example, amplifying pentamer-bound aptamers from round #5 with 10 PCR cycles resulted in the formation of ladder-type artifacts that was observed with only 8 PCR amplification cycles of samples from round #8. These ladder-type artifacts, which resemble those documented in several previous studies, have been sequenced and verified as PCR by-products caused by primer–primer or primer–product hybridization.10,13 Accordingly, we selected the PCR cycles that showed minimal by-product on polyacrylamide gel for the subsequent aptamer amplification, i.e., 8 PCR cycles for round #2 to #7, and 6 PCR cycles for round #1 and #8 to #12. In addition, to further minimize the accumulation of the undesired by-product, a single correctly sized band corresponding to 80 base pairs on the polyacrylamide gel was excised and purified before proceeding to each selection of the entire SELEX.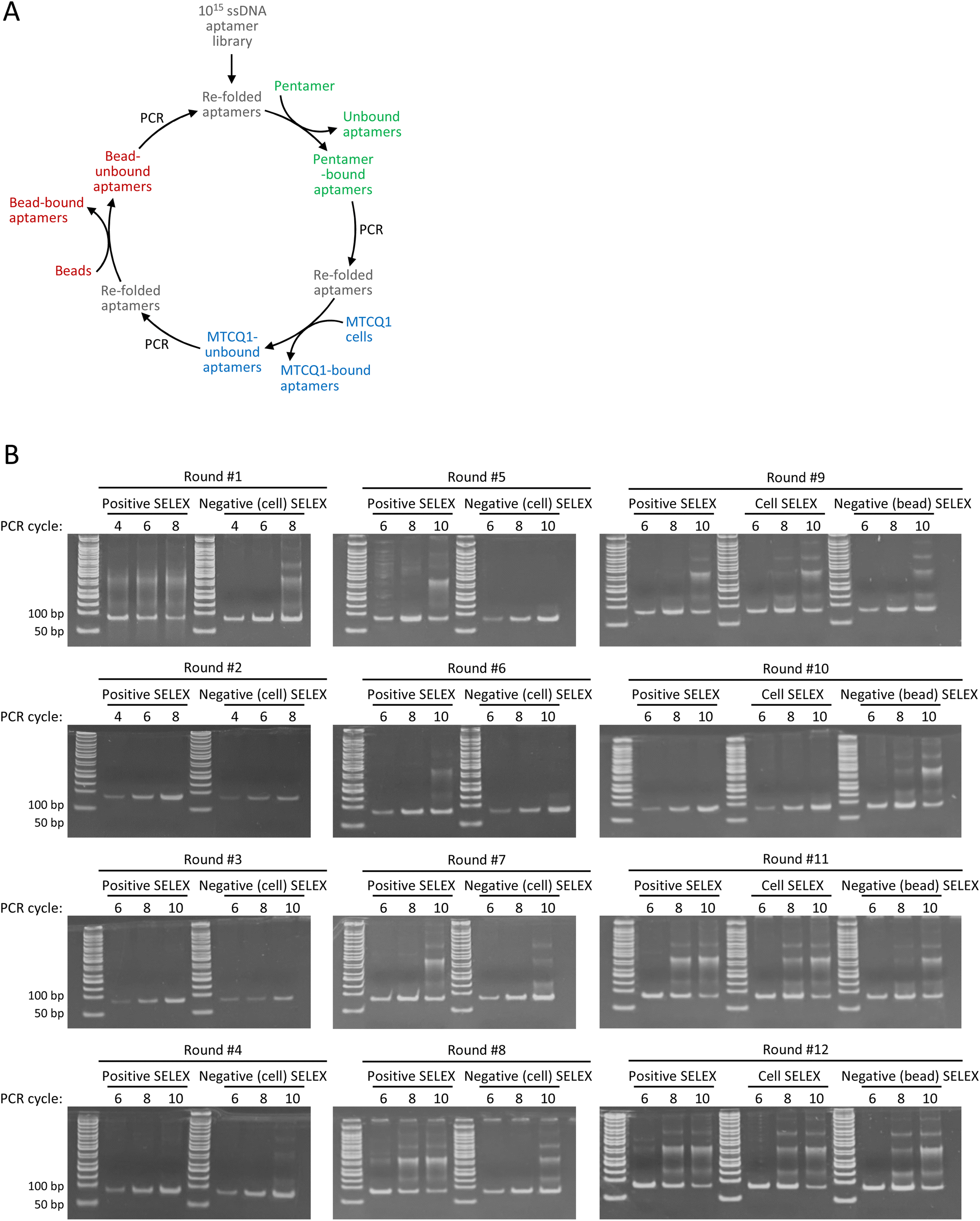 | ||
| Fig. 3 PCR by-product formation increased as more rounds of conventional SELEX were performed. (A) Scheme of conventional SELEX consisting of a positive selection (using OVA257–264-H-2Kb pentamers) and two negative selections (using MTCQ1 cells and SA-beads) per round. (B) Polyacrylamide gel analysis of the PCR products of each round of conventional SELEX. | ||
Conventional SELEX tends to enrich the aptamers that contain fractions of primer sequences
To identify aptamers that were enriched through conventional SELEX, the PCR products obtained from rounds #4, #6, #7, #8, #9, #10, #11, and #12 were subjected to high-throughput sequencing. The aptamer sequencing data was loaded into AptaSUITE software for preprocessing.32,33 We observed a gradual decrease in the number of unique aptamer sequences with each successive round of SELEX, implying a trend of aptamer enrichment (Fig. 4A). The aptamer sequences of the last round (round #12) were clustered into 42 families, while the most abundant cluster contained more than 2000000 reads. The top 150 enriched aptamers based on read counts of the last round of SELEX were selected for analysis. As shown in Fig. 4B, we plotted each of these top 150 aptamers as a horizontal bar, with the length of the bar corresponding to its cluster size in read counts and the most enriched aptamer positioned at the top of the graph. Remarkably, the sequences of the majority of these aptamers (indicated in gray) contained segments of primer sequences, with the exception of two aptamers (highlighted in purple and green). Interestingly, the sequence of these two aptamers coincides with the sequences of Apt-1 and Apt-10 that we previously identified using consecutive selection with minimal PCR amplification. Importantly, these two aptamers were not highly enriched compared to other aptamers (Fig. 4C), likely because these aptamers were outcompeted during PCR by aptamers that had sequences at least partially complementary to the primer sequences with a low binding affinity.
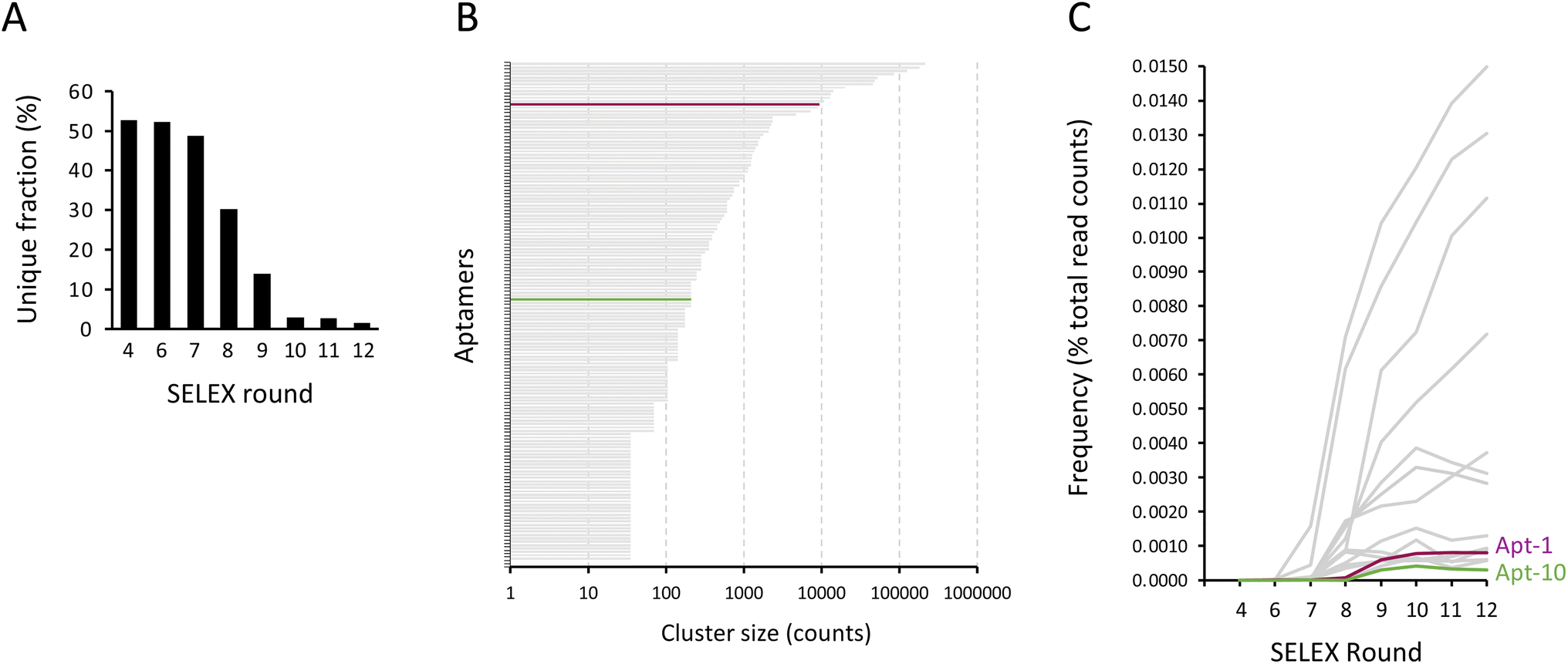 | ||
| Fig. 4 Conventional SELEX often results in the enrichment of aptamers with fractional primer sequences. (A) The fractions of the unique sequence in round #4 and #5 to #12 of the conventional SELEX. (B) The cluster size of the top 150 enriched aptamers based on read counts from the last round of the conventional SELEX. Each horizontal bar represents an aptamer. The aptamer containing various fractions of primer sequence was colored gray. Aptamers with sequences identical to Apt-1 and Apt-10 were colored purple and green, respectively. (C) The frequency (estimated by the percentage of total read counts) of the top 150 enriched aptamers (in gray), as well as Apt-1 (in purple) and Apt-10 (in green) during selections. | ||
In addition, we further analyzed the distributions of the top 150 enriched aptamers in these 8 sequencing samples (round #4, #6, #7, #8, #9, #10, #11, and #12), as we did for the modified SELEX analysis, showing that 14 aptamers were identified in all 8 sequencing samples; 5 aptamers were identified in 6 out of 8 sequencing samples; 30 aptamers were identified in 5 out of 8 sequencing samples; 34 aptamers were identified in 4 out of 8 sequencing samples; 16 aptamers were identified in 3 out of 8 sequencing samples; 10 aptamers were identified in 2 out of 8 sequencing samples; and 41 aptamers were identified in one of 8 sequencing samples (ESI Fig. 1†). Notably, the aptamer sequence that coincided with Apt-1 was found in only 6 of 8 sequencing samples (round #6, #7, #8, #9, #10, #11, and #12), and the aptamer sequence that coincided with Apt-10 was found in only 5 of 8 sequencing samples (round #9, #10, #11, and #12). These findings suggest that the aptamers identified in multiple selection rounds of conventional SELEX may not necessarily be the aptamers that survived the selection processes, but rather the aptamers that were enriched due to PCR bias.
Aptamers have the potential to target cancer cells expressing specific peptide–MHC complexes
To further examine the ability of Apt-1 and Apt-10 to recognize MTCQ1 cells expressing OVA257–264-H-2Kb MHC complexes, we established MTCQ1 cells that stably produce ovalbumin model antigen (named MTCQ1-OVA) by lentiviral transduction. This validation is important because Apt-1 and Apt-10 were identified by OVA257–264-H-2Kb pentamer-based selection, but not by cell-based SELEX. As shown in Fig. 5A, the signal intensity of flow cytometric analysis using OVA257–264-H-2Kb-specific antibody was significantly higher in MTCQ1-OVA cells than that in wild-type MTCQ1 cells, supporting the MHC presentation of ovalbumin model antigen in MTCQ1-OVA cells. Using these cells with FITC-conjugated aptamers, we showed that Apt-1 and Apt-10 selectively bound to MTCQ1-OVA cells, whereas Apt-3 and Apt-8 did not exhibit binding specificity to wild-type MTCQ1 or MTCQ1-OVA cells. Consistently, high immunofluorescence was observed only in MTCQ1-OVA cells, but not in wild-type MTCQ1 cells, incubated with fluorophore-conjugated Apt-1 and Apt-10 (ESI Fig. 2†). To further demonstrate the in vivo efficacy of Apt-1 and Apt-10 in recognizing cells expressing OVA257–264-H-2Kb MHC complexes, we used biotin-conjugated Apt-1, Apt-10, Apt-3, and Apt-8 to perform immunohistochemistry on tumors derived from inoculation of wild-type MTCQ1 or MTCQ1-OVA cells in the back of nude mice. We showed that positive immunointensity was only observed in tissue sections of MTCQ1-OVA tumors stained with biotin-conjugated Apt-1 and Apt-10; neither the MTCQ1-OVA tumor stained with Apt-3 and Apt-8 nor the MTCQ1 tumor stained with Apt-1, Apt-10, Apt-3, or Apt-8 showed immunopositivity (Fig. 5B).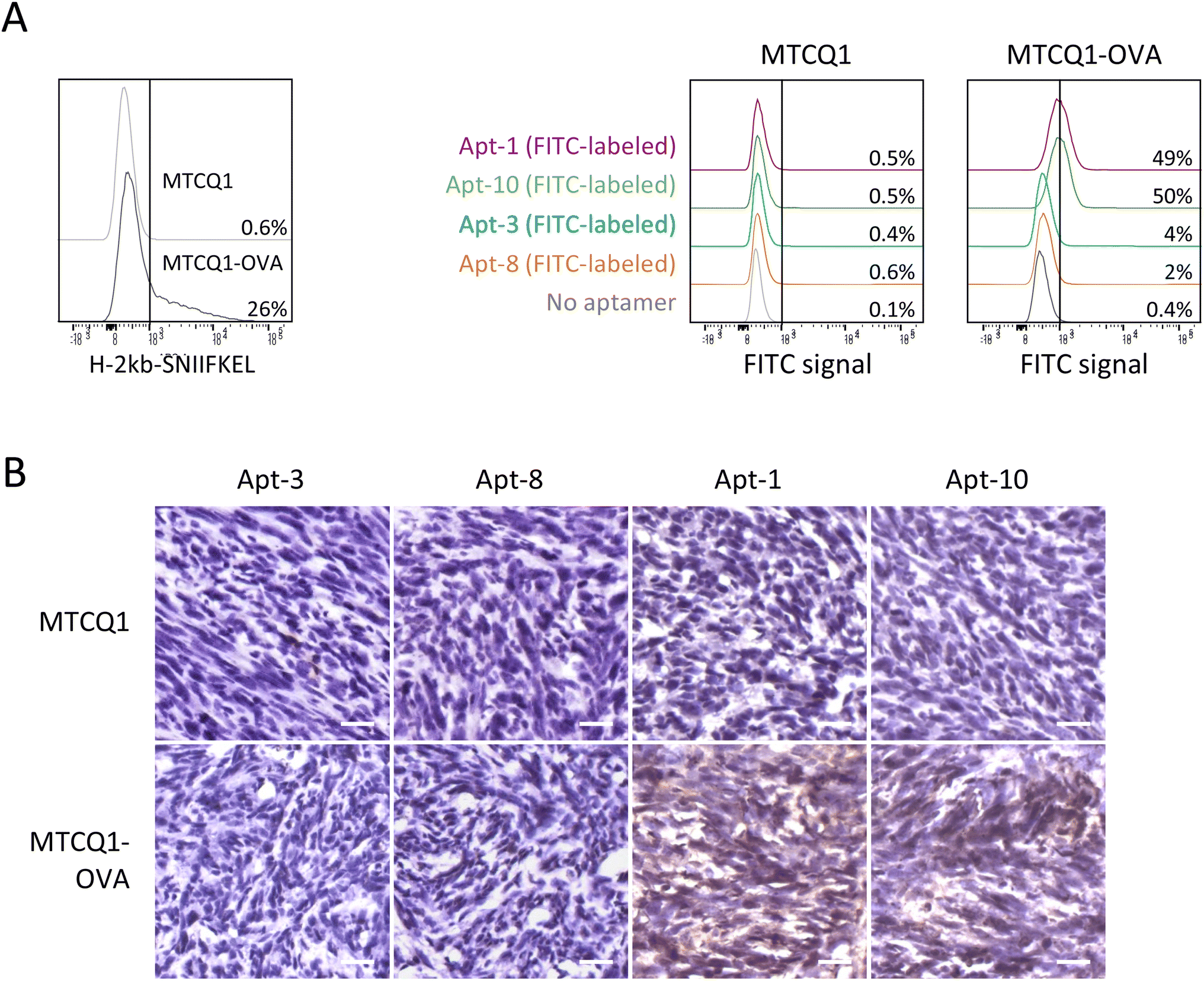 | ||
| Fig. 5 Apt-1 and Apt-10 specifically target cancer cells presenting OVA model antigen. (A) Flow cytometry analysis of MTCQ1 and MTCQ1-OVA cells stained with the OVA257–264-H-2Kb specific antibody, as well as the FITC-conjugated Apt-1, Apt-3, Apt-8, and Apt-10. The percentage of cells that were bound by specific aptamers was shown in the overlaid histograms. (B) Tumor xenografts derived from inoculation of wild-type MTCQ1 or MTCQ1-OVA cells in the back of nude mice were immunostained with biotin-conjugated Apt-1, Apt-10, Apt-3, and Apt-8. (200× magnification, scale bar 100 μm). | ||
Discussion
Aptamers are valuable tools in the fields of therapeutics and diagnostics due to their ability to bind to a wide range of molecules. However, the conventional SELEX, which includes PCR in each selection cycle, can suffer from the undesired pairing of primers and random sequences, potentially biasing sequence convergence and misleading the enrichment of aptamers.10 To address this issue, our strategy takes a different approach by reducing the number of PCR cycles and utilizing PCR primarily to replenish the aptamer pool. Additionally, we adopt a distinct strategy to identify candidate aptamers by selecting those that can be detected in multiple sequencing samples, rather than relying on the sequence enrichment in read counts in each sample. These strategies enable us to efficiently refine the selection from a vast pool of aptamers. Subsequently, we identified two aptamers with high affinity to our target molecule. Notably, these two aptamers were also among the top 150 enriched aptamers obtained through conventional SELEX, despite not being highly enriched based on read counts analysis, thus highlighting the benefit of our strategy to efficiently identify candidate aptamers. Importantly, these two aptamers exhibited higher affinity to target molecules compared to most of highly enriched aptamers in conventional SELEX. Taken together, our approach holds great potential to improve the aptamer selection process.As mentioned previously, the potential for generating detrimental PCR by-products using a random DNA library as a template is one of the major obstacles to successful identification of candidate aptamers through SELEX. Several studies have proposed alternative primer/template designs and PCR techniques.34–37 Among these modifications, emulsion PCR (ePCR) stands out as a noteworthy approach. Indeed, by enclosing only a few aptamers in each droplet, ePCR not only reduces the possible hybridization of partially or fully complementary aptamers for by-product formation, but also preserves the GC-rich aptamers that might otherwise be outcompeted by other aptamers during PCR. However, to effectively minimize by-products and bias, optimizing the ePCR reaction condition is needed, and even with optimization, there have been reports of emerging PCR by-products.38–40 Furthermore, the excessive preservation of rare aptamers with weak binding affinity may expand the pool of candidate aptamers, potentially complicating the identification of target aptamers. In addition to ePCR, the non-SELEX method, which involves sequential selections without amplification steps, represents another approach designed to address the PCR-related challenge of conventional SELEX.14,41 While non-SELEX techniques have been successful in quickly isolating candidate aptamers,42,43 these methods typically require specialized equipment, such as capillary electrophoresis or electrochemical sensors.14,44 As a result, non-SELEX techniques hardly work with cell-SELEX where cells are used as the target molecules. Moreover, high-throughput sequencing of samples derived from non-SELEX methods only generates a list of aptamers without information on their cycle-to-cycle dynamics, making the identification of target aptamers particularly challenging.
Notably, the number of consecutive selections appropriate for this approach needs to be discussed. Besides a complex formula that has been proposed for estimating the number of non-amplification cycles,15 we have developed a relatively simple rationale to address this issue. Consecutive selections gradually deplete the concentration of aptamers, and we observed that approximately 10 cycles of PCR can replenish the aptamer pool that has been depleted by 4 to 5 consecutive selections, without significant by-product accumulation. Certainly, higher PCR cycles are required for more consecutive selections to replenish the aptamer pool. Unfortunately, this can lead to some by-product formation, even at extremely low template concentrations, consistent with a previous study showing that by-product formation is less associated with the initial amount of aptamer molecules in PCR mixture.36 Nonetheless, the optimal number of consecutive selections and PCR cycles can vary depending on the type of target molecule and the design of primers and templates. Therefore, a pre-test to determine these parameters is necessary if this method is to be used for other target molecules.
To date, aptamers have been shown to exhibit binding affinity (ranging from 10−12 to 10−6 M) to a wide variety of molecules, including proteins, small molecules, and metal ions,45–49 except for the peptide–MHC complex. In fact, to our knowledge, only one relevant study reported the identification of a DNA aptamer against tumor-specific antigen peptide that can be presented by several MHC molecules.50 The peptide–MHC complex is a cell surface molecule; therefore, both protein-SELEX and cell-SELEX, which use synthetic peptide–MHC molecules and cells harboring the exact antigen and the MHC molecule as target proteins, respectively, can be employed for aptamer selection and offer distinct advantages. Protein-SELEX utilizes well-defined and purified target proteins, allowing for the isolation of aptamers that specifically bind to the protein of interest.51,52 This is particularly important because obtaining cells that exhibit a single peptide–MHC complex for cell-SELEX is essentially impossible. Additionally, the use of synthetic peptide–MHC molecules tagged with magnetic beads in protein-SELEX simplifies the process compared to the time-consuming retrieval of aptamers from cells in cell-SELEX.53,54 Moreover, aptamers that are incubated with cells during positive selection may be internalized,55 posing further challenges for aptamer purification. On the other hand, cell-SELEX provides the advantage of preserving the natural conformation and appropriate post-translational modifications of proteins exhibited on the cell membrane, enabling the identification of biologically relevant aptamers that can be directly applicable to in vivo settings.56,57 In this study, we employed pentamers, a multimer technology that authentically recapitulates the presentation of peptide–MHC complexes on the cell surface,58 as the target molecules for the selection of aptamers. Furthermore, negative selection using cells that express a repertoire of normally presented peptides but not the antigen peptide allowed for the counter-selection of aptamers binding to MHC molecules presenting non-target peptides. By combining the strengths of both protein-SELEX and cell-SELEX, we successfully identified aptamers with the ability to recognize cells displaying specific peptide–MHC complexes. Given that cancer cells may express tumor-specific antigens presented by MHC complexes, this approach holds the potential to generate aptamers that serve as probes for precisely targeting tumor cells based on their specific peptide–MHC complexes.59
In conclusion, aptamers are undeniably an appealing substitute for antibodies in the realm of biomedicine. However, several technical aspects of the selection process hinder the success rate of aptamer identification. Consequently, multiple variations to improve key steps of SELEX, such as library design, target usage, and PCR modification, have been proposed during the past decades. Building upon this effort, we tailored the method by performing consecutive selection with minimal PCR amplification that is repurposed to replenish the aptamer pool and selecting aptamers that emerged across multiple selection rounds regardless of read counts. Importantly, we showed that traditional SELEX tends to bias the enrichment and that candidate aptamers are easily missed in the traditional SELEX process. We believe that these modifications, based on the current understanding of SELEX, will facilitate the identification of aptamers, making aptamers a superior alternative to various antibodies.
Methods
Single-stranded DNA library
All oligonucleotides, including the single-stranded DNA (ssDNA) library and primer, were synthesized by IDT (Integrated DNA Technologies, IA, ISA). The aptamer library was composed of 80 nucleotide-long ssDNAs, consisting of 40 bp random sequences flanked by primer-binding sequences. The primers used to amplify ssDNA were 5′-ACG CTC GGA TGC CAC TAC AG-3′ (forward), 5′-GTC ACC AGC ACG TCC ATG AG-3′ (reverse), and 5′ Biotin-GTC ACC AGC ACG TCC ATG AG-3′ (biotin-conjugated reverse).Cell culture and construction of stable OVA-expressing cell lines
Mouse 4NQO-induced tongue cancer cell lines (MTCQ1) were established and obtained from Dr Kuo-Wei Chang, National Yang Ming Chiao Tung University.60,61 Cells were cultured in Dulbecco's modified Eagle medium (DMEM, Corning) supplemented with 10% fetal bovine serum (Gibco-BRL) and penicillin-streptomycin (Invitrogen) in a 100 mm culture plate (Corning) incubated in the 5% CO2 incubator at 37 °C regularly. Cells were passaged three times a week when cells had reached 80–90% confluences. The MTCQ1 cancer cell lines were transduced with a lentiviral vector encoding the OVA model antigen. After transduction, cells were selected with the puromycin-containing medium. The expression of the OVA 257–264 (SIINFEKL) peptide by H-2Kb was validated using the APC-conjugated OVA 257–264 (SIINFEKL) peptide bound to H-2Kb monoclonal antibodies (1:125) (Invitrogen).
Pentamer-based selection
The modified SELEX begins with pentamer-based selection. The aptamer pool was prepared by dissolving 40 μg ssDNAs library in 300 μl binding buffer (2.5 mM MgCl2, 0.02% Tween-20 in PBS), which was then heated at 95 °C for 10 minutes and snap-cooled on ice for 10 minutes, followed by a slow return to room temperature for 1 hour for aptamer folding. To perform pentamer-based selection, a total amount of 2.4 μg of biotinylated OVA257–264-H-2Kb pentamer (Cat.no. F093-1A-G, Proimmune) was incubated with 2 μl of Streptavidin Mag Sepharose (SA-beads, Cat. no.04123003, GE Healthcare Bioscience, Pittsburgh, PA, USA) at room temperature. The mixture was gently mixed with the rotary shaker (Intelli Mixer RM-2S, ELMI, Riga, Latvia) for 30 minutes in binding buffer, and the OVA257–264-H-2Kb pentamer bound SA beads were collected using the DynaMag-2 magnetic strip (Cat. no. 123-21D, Life Technologies, USA). The folded ssDNA library in binding buffer was then incubated with an OVA257–264-H-2Kb pentamer/SA-beads mixture in 1:500 molar ratios at room temperature, and the mixture was mixed with the rotary shaker for 1 hour. The wash step was then performed five times using wash buffer (1 mM MgCl2, 40 mM HEPES, 4 mM KCl, 2.5 mM CaCl2, 140 mM NaCl, 0.02% Tween-20 in PBS) to remove unbound ssDNAs from the pentamers. Subsequently, the pentamer–bead complexes were heat-denatured, and the ssDNAs that remained bound to the pentamers were resuspended in 200 μl of binding buffer. For aptamer refolding, resuspended ssDNAs were heated at 95 °C for 10 minutes, centrifuged at 4 °C, 13000×g for 5 minutes, and then incubated on ice for 10 minutes, followed by a slow return to room temperature for the next round of selection. After five rounds of pentamer-based selection, the retained amount of target-bound ssDNA was reduced to approximately 20 ng. To replenish the pool of aptamers for subsequent selection cycles, a total of 20 ng of selected ssDNAs (equivalent to approximately 2 × 1011 copies of aptamers as determined by the amount and the length of amplicons62) that remained bound to pentamers after five consecutive selections was amplified by running 10 PCR cycles. The amplification was carried out using high-fidelity polymerase (Cat. no. PR1000-HF-S, Accuris, Edison, NJ, USA), along with un-conjugated forward primer and biotin-conjugated reverse primers, on an ABI 2720-PCR machine (Cat. no. 4359659, ABI, Carlsbad, California, USA). Approximately 200 ng of DNA, estimated to be 2 × 1012 copies of aptamers, were obtained from the final products of 10 PCR cycles. Because running PCR for more than 10 cycles produced higher quantity of aptamers but also more PCR by-products, PCR amplification was limited to 10 cycles. The PCR product was then heat denatured and incubated with SA-beads to remove the reverse strands. The remaining forward strands were then heated and refolded for subsequent cell-based SELEX.
Cell-based selection
The ssDNAs that were obtained following pentamer-based selection were refolded in the binding buffer as described above. To perform cell-based selection, wild-type MTCQ1 cells were first harvested and resuspended with 100 μl binding buffer in a 1.5 ml tube. Subsequently, a total amount of 200 ng of refolded aptamers that was obtained after PCR amplification of pentamer-selected ssDNAs was added to the tube. Cell suspension and ssDNAs were incubated and gently mixed with the rotary shaker at 4 °C for 1 hour. Cell-unbound ssDNAs in the supernatant were collected and refolded for subsequent beads-based negative selection. Four rounds of cell-based selection left approximately 20 ng of ssDNAs that remained unbound to the MTCQ1 cells. The aptamer pool was replenished by PCR amplification. To obtain an appropriate number of aptamers for the subsequent selection process, we tested different amplification cycles. We found that running PCR with less than 12 cycles could yield PCR end products in the range of 100 to 200 ng without significant accumulation of PCR by-products. Therefore, to simplify the PCR process throughout the modified SELEX, we performed PCR with 10 amplification cycles, as we did for PCR after five rounds of pentamer-based selection.Bead-based selection
A total of 200 ng of ssDNAs that were not bound to MTCQ1 cells after four consecutive selections was refolded and incubated with 20 μl of SA-beads at room temperature with the rotary shaker for 1 hour in the binding buffer. SA-beads-unbound ssDNA (supernatant) were collected by a magnetic strip. After five consecutive bead-based negative selections, approximately 20 ng of ssDNAs was obtained and amplified with 10 PCR cycles as above.PCR and polyacrylamide gel electrophoresis
To amplify aptamers during modified SELEX and conventional SELEX, we tested a variety of PCR conditions, including various target protein/primer/aptamer ratios, binding buffer and wash steps, as well as using different high-fidelity DNA polymerases, which were summarized in ESI Table 4.† The PCR conditions that were used in our study are summarized in ESI Table 5.† Briefly, PCR was performed with Accuris high-fidelity polymerase on an ABI-2720 PCR machine. The total volume of the PCR reaction mixture for each tube is 25 μl. DNA concentration was measured with the Qubit Fluorimeter 3.0 (Cat. no. Q33216, Invitrogen, California, USA) and NanoDrop™ One/OneC Microvolume UV-vis Spectrophotometer (Cat. no.701-058112, Thermo Fisher Scientific, Waltham, Massachusetts, USA). The size and purity of the PCR products were validated on a 10% polyacrylamide gel with 5× TBE buffer (Cat. no. BL0280-1000, Genestar Biotechnologies, TAIWAN). A total of 12 μl of samples and 3 μl of 50 bp DNA ladder RTU (Cat. no. DM012-R500, GeneDireX Inc., TAIWAN) were loaded onto lanes and electrophorized at 100 V for 1 hour. After electrophoresis, 1× SYBR® Safe DNA Gel stain (Cat. no. S33102, Invitrogen, Mount Waverley, Australia) was added, and the bands were observed under the BLook™ LED transilluminator (Cat. no. BK001, GeneDireX Inc., TAIWAN).High throughput sequencing and analysis
After the selection process, target-bound ssDNA was amplified using barcoded primers and then purified by gel extraction and the MinElute PCR Purification Kit (Cat. no. 28004, Qiagen, Hilden, Germany). The barcoded PCR products were subjected to MiSeq 150 (Illumina, San Diego, California, USA) high-throughput sequencing. The secondary structure and ΔG of candidate aptamers, including Apt-1, Apt-3, Apt-8, and Apt-10, were predicted by NUPACK and Mfold web software.qPCR-based binding assay
qPCR-based binding affinity experiments were performed on QuantStudio™ 3 Real-Time PCR Systems (Cat. no. A28567, ThermoFisher Scientific, Massachusetts, USA) and analyzed using the QuantStudio™ Design and Analysis software v1.5.2. For these binding experiments, 96-well plates were used. Candidate aptamers, including Apt-1, Apt-3, Apt-8, and Apt-10, were 2-fold serially diluted in the binding buffer from 1000 nM to 0.01 nM. The diluted aptamers were then incubated with 2 μg of OVA257–264-H-2Kb pentamer at room temperature for 1 hour. After the binding process, the amounts of aptamers were determined. The Kd values of the candidate aptamers were calculated using the equation: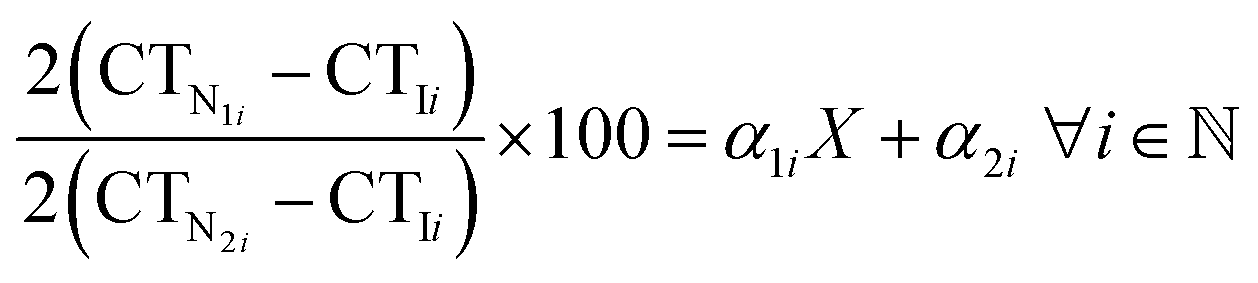
Where X represents the Kd value, CTI is the CT value of candidate aptamers at the minimum concentration, CTN1 is the CT value of candidate aptamers at 0.1–100 nM, CTN2 is the CT value of candidate aptamers at the maximum concentration, and the α1 and α2 is the coefficient of regression curve.
Flow cytometry analysis
For experiments validating the binding capacity of candidate aptamers to OVA257–264-H-2Kb pentamers, MTCQ1-WT and MTCQ1-OVA cells were harvested by trypsinization and washed with PBS. Each sample was divided into 2 aliquots for different staining patterns: (i) biotinylated Apt-1, Apt-3, Apt-8, Apt-10; and (ii) OVA257–264-H-2Kb antibody. For staining pattern (i), cells were incubated with 200 nM biotinylated candidate aptamers at room temperature for 1 hour and subsequently with FITC-conjugated streptavidin (1:500) (Cat. no. 405202, BioLegend, San Diego, California, USA) at 4 °C for 30 minutes. For staining pattern (ii), cells were incubated with biotin-conjugated OVA257-264 (SIINFEKL) peptide bound to H-2Kb monoclonal antibodies (1:125) (Cat. no. 17-5743-80, Invitrogen) at 4 °C for 1 h and subsequently with FITC-conjugated streptavidin (1:500) at 4 °C for 30 minutes. After staining, all cells were washed twice with PBS by centrifuged at 4 °C, 500×g for 3 minutes, resuspended in PBS containing 1% BSA, and then subjected to flow cytometry. All samples were analyzed using the BD FACSCanto™ II Flow cytometer System (BD Biosciences, USA). Data were analyzed using Flowjo™ v10 software (BD Biosciences, USA).
Confocal image
After washing with PBS, MTCQ1-WT, and MTCQ1-OVA cells were fixed with 4% PFA for 10 minutes and then incubated with 200 nM biotinylated Apt-1 and Apt-10 at room temperature for 30 minutes followed by staining with the 1:500 secondary antibody Streptavidin Alexa-594 (Cat. no.405240, BioLegend, USA) at 4 °C for 30 minutes. Nuclei were stained with 300 nM DAPI at 4 °C for 15 minutes. The images were acquired using the confocal laser scanning microscope (LSM 880, Carl Zeiss GmbH, Jena, Germany). DAPI was excited at 405 nm and Streptavidin Alexa 594 filter (filter set 10; 590 nm excitation, 618 nm emission) was used to determine the immunofluorescence signal. Camera exposure time was set to 100 microseconds and the 63 × 1.4 oil DIC objective lens was used. We quantified the immunofluorescence signals of the acquired images using ImageJ software (https://imagej.nih.gov/ij/download.html). The quantification of mean immunofluorescence was expressed as an arbitrary unit (AU).
Murine syngeneic tumor models and immunohistochemical staining
BALB/C nude mice were inoculated subcutaneously with 5 × 106 MTCQ1-WT or MTCQ1-OVA cells, respectively. When the long axis of the tumors reached 5 mm, mice were sacrificed. The tumors were harvested, fixed with 4% paraformaldehyde (PFA), and paraffin-embedded for tissue section. After the antigens were retrieved with a tris-based antigen unmasking solution (#H-3301, Vector Laboratories) for 1 min, a BLOXALL® Endogenous Blocking Solution (#SP-6000, Vector Laboratories) was used to inactivate endogenous peroxidase, pseudoperoxidase and alkaline phosphatase. Next, the streptavidin/biotin blocking kit (#SP-2002, Vector Laboratories) was used to block all endogenous biotin, biotin receptors, and streptavidin binding sites. The sections were then blocked in PBS with 2% rabbit serum for 30 min at room temperature and incubated with 200 nM biotinylated Apt-1, Apt-3, Apt-8, and Apt-10 at room temperature for 1 hour and then the secondary antibody Streptavidin-Alexa 594 (1:500) was used to stain at 4 °C for 30 minutes. Detections were made with a VECTASTAIN® Elite ABC-HRP kit (#PK-6100, Vector Laboratories) for 30 min at room temperature. Sections were developed using the ImmPACT® DAB substrate (#SK-4105, Vector Laboratories) and counterstaining with hematoxylin.
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Taipei Veterans General Hospital and approved by the Animal Ethics Committee of Taipei Veterans General Hospital (Protocol code no.2021-008).
AptaSUITE
The bioinformatics software AptaSUITE was used to find candidate aptamers for further verifications and post-processing in vitro and in vivo. The cluster of candidate aptamers was analyzed based on their similarity and the elucidation of shared motifs in the primary and secondary structures.32,33Data availability
All data supporting the finding of this study are available from the corresponding author on reasonable request.Author contributions
K. H. and Y. S. designed the experiments. Y. L. and B. C. performed the experiments. Y. C., Y. K., L. W., C. H., K. H, J. H., and K. H. analyzed the data. Y. L., Z. L, and K. H. prepared the manuscript. All authors discussed the results and reviewed the manuscript.Conflicts of interest
The authors declare that they have no conflicts of interest relating to the subject matter or materials discussed in this article.Acknowledgements
The authors thank Drs Bo-Tsang Huang and Pan-Chyr Yang for critical review and valuable advice of this study. This work was supported by the National Science and Technology Council, Taiwan, grant number 110-2314-B-075-022-MY3, 109-2314-B-075-006, and 109-2314-B-303-007-MY3.References
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- A. D. Gelinas, D. R. Davies and N. Janjic, Curr. Opin. Struct. Biol., 2016, 36, 122–132 CrossRef CAS PubMed.
- O. Kikin, L. D'Antonio and P. S. Bagga, Nucleic Acids Res., 2006, 34, W676–W682 CrossRef CAS PubMed.
- G. Mayer, Angew Chem. Int. Ed. Engl., 2009, 48, 2672–2689 CrossRef CAS PubMed.
- P. Dhar, R. M. Samarasinghe and S. Shigdar, Int. J. Mol. Sci., 2020, 21 Search PubMed.
- J. Zhou and J. Rossi, Nat. Rev. Drug Discovery, 2017, 16, 181–202 CrossRef CAS PubMed.
- C. A. Dougherty, W. Cai and H. Hong, Curr. Top. Med. Chem., 2015, 15, 1138–1152 CrossRef CAS PubMed.
- K. Sefah, D. Shangguan, X. Xiong, M. B. O'Donoghue and W. Tan, Nat. Protoc., 2010, 5, 1169–1185 CrossRef CAS PubMed.
- T. Wang, C. Chen, L. M. Larcher, R. A. Barrero and R. N. Veedu, Biotechnol. Adv., 2019, 37, 28–50 CrossRef CAS PubMed.
- Z. Zhuo, Y. Yu, M. Wang, J. Li, Z. Zhang, J. Liu, X. Wu, A. Lu, G. Zhang and B. Zhang, Int. J. Mol. Sci., 2017, 18(10), 2142 CrossRef PubMed.
- M. Famulok and G. Mayer, Chem. Biol., 2014, 21, 1055–1058 CrossRef CAS PubMed.
- F. Tolle, J. Wilke, J. Wengel and G. Mayer, PLoS One, 2014, 9, e114693 CrossRef PubMed.
- M. Berezovski, M. Musheev, A. Drabovich and S. N. Krylov, J. Am. Chem. Soc., 2006, 128, 1410–1411 CrossRef CAS PubMed.
- K. Szeto, D. R. Latulippe, A. Ozer, J. M. Pagano, B. S. White, D. Shalloway, J. T. Lis and H. G. Craighead, PLoS One, 2013, 8, e82667 CrossRef PubMed.
- M. F. Princiotta, D. Finzi, S. B. Qian, J. Gibbs, S. Schuchmann, F. Buttgereit, J. R. Bennink and J. W. Yewdell, Immunity, 2003, 18, 343–354 CrossRef CAS PubMed.
- P. Kourilsky, G. Chaouat, C. Rabourdin-Combe and J. M. Claverie, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 3400–3404 CrossRef CAS PubMed.
- E. Baulu, C. Gardet, N. Chuvin and S. Depil, Sci. Adv., 2023, 9, eadf3700 CrossRef CAS PubMed.
- P. S. Andersen, A. Stryhn, B. E. Hansen, L. Fugger, J. Engberg and S. Buus, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1820–1824 CrossRef CAS PubMed.
- V. P. Wittman, D. Woodburn, T. Nguyen, F. A. Neethling, S. Wright and J. A. Weidanz, J. Immunol., 2006, 177, 4187–4195 CrossRef CAS PubMed.
- M. Epel, I. Carmi, S. Soueid-Baumgarten, S. Oh, T. Bera, I. Pastan, J. Berzofsky and Y. Reiter, Eur. J. Immunol., 2008, 38, 1706–1720 CrossRef CAS PubMed.
- E. Klechevsky, M. Gallegos, G. Denkberg, K. Palucka, J. Banchereau, C. Cohen and Y. Reiter, Cancer Res., 2008, 68, 6360–6367 CrossRef CAS PubMed.
- R. Bhattacharya, Y. Xu, M. A. Rahman, P. O. Couraud, I. A. Romero, B. B. Weksler, J. A. Weidanz and U. Bickel, J. Cell. Physiol., 2010, 225, 664–672 CrossRef CAS PubMed.
- A. Sergeeva, G. Alatrash, H. He, K. Ruisaard, S. Lu, J. Wygant, B. W. McIntyre, Q. Ma, D. Li, L. St John, K. Clise-Dwyer and J. J. Molldrem, Blood, 2011, 117, 4262–4272 CrossRef CAS PubMed.
- K. Kingwell, Nat. Rev. Drug Discovery, 2022, 21, 321–323 CrossRef CAS PubMed.
- E. Dolgin, Nat. Biotechnol., 2022, 40, 441–444 CrossRef CAS PubMed.
- C. Augsberger, G. Hanel, W. Xu, V. Pulko, L. J. Hanisch, A. Augustin, J. Challier, K. Hunt, B. Vick, P. E. Rovatti, C. Krupka, M. Rothe, A. Schonle, J. Sam, E. Lezan, A. Ducret, D. Ortiz-Franyuti, A. C. Walz, J. Benz, A. Bujotzek, F. S. Lichtenegger, C. Gassner, A. Carpy, V. Lyamichev, J. Patel, N. Konstandin, A. Tunger, M. Schmitz, M. von Bergwelt-Baildon, K. Spiekermann, L. Vago, I. Jeremias, E. Marrer-Berger, P. Umana, C. Klein and M. Subklewe, Blood, 2021, 138, 2655–2669 CrossRef CAS PubMed.
- X. Yang, S. Xie, X. Yang, J. C. Cueva, X. Hou, Z. Tang, H. Yao, F. Mo, S. Yin, A. Liu and X. Lu, Theranostics, 2019, 9, 7792–7806 CrossRef CAS PubMed.
- J. D. Stone, A. S. Chervin and D. M. Kranz, Immunology, 2009, 126, 165–176 CrossRef CAS PubMed.
- M. M. Davis, J. J. Boniface, Z. Reich, D. Lyons, J. Hampl, B. Arden and Y. Chien, Annu. Rev. Immunol., 1998, 16, 523–544 CrossRef CAS PubMed.
- K. H. Roux, PCR Methods Appl., 1995, 4, S185–S194 CrossRef CAS PubMed.
- J. Hoinka and T. M. Przytycka, Methods Mol. Biol., 2023, 2570, 73–83 CrossRef PubMed.
- J. Hoinka, R. Backofen and T. M. Przytycka, Mol. Ther.–Nucleic Acids, 2018, 11, 515–517 CrossRef CAS PubMed.
- K. Shao, W. Ding, F. Wang, H. Li, D. Ma and H. Wang, PLoS One, 2011, 6, e24910 CrossRef CAS PubMed.
- K. W. Plaxco and H. T. Soh, Trends Biotechnol., 2011, 29, 1–5 CrossRef CAS PubMed.
- M. U. Musheev and S. N. Krylov, Anal. Chim. Acta, 2006, 564, 91–96 CrossRef CAS PubMed.
- R. Williams, S. G. Peisajovich, O. J. Miller, S. Magdassi, D. S. Tawfik and A. D. Griffiths, Nat. Methods, 2006, 3, 545–550 CrossRef CAS PubMed.
- S. Poolsup, E. Zaripov, N. Huttmann, Z. Minic, P. V. Artyushenko, I. A. Shchugoreva, F. N. Tomilin, A. S. Kichkailo and M. V. Berezovski, Mol. Ther.–Nucleic Acids, 2023, 31, 731–743 CrossRef CAS PubMed.
- M. Kohlberger and G. Gadermaier, Biotechnol. Appl. Biochem., 2022, 69, 1771–1792 CrossRef CAS PubMed.
- N. Komarova and A. Kuznetsov, Molecules, 2019, 24(19), 3598 CrossRef PubMed.
- M. V. Berezovski, M. U. Musheev, A. P. Drabovich, J. V. Jitkova and S. N. Krylov, Nat. Protoc., 2006, 1, 1359–1369 CrossRef CAS PubMed.
- J. Ashley, K. Ji and S. F. Li, Electrophoresis, 2012, 33, 2783–2789 CrossRef CAS PubMed.
- J. Tok, J. Lai, T. Leung and S. F. Li, Electrophoresis, 2010, 31, 2055–2062 CrossRef CAS PubMed.
- A. Kushwaha, Y. Takamura, K. Nishigaki and M. Biyani, Sci. Rep., 2019, 9, 6642 CrossRef PubMed.
- S. Tombelli, M. Minunni and M. Mascini, Biosens. Bioelectron., 2005, 20, 2424–2434 CrossRef CAS PubMed.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS PubMed.
- L. Zhang, Z. Zeng, J. Li, L. Huang, Z. Huo, K. Wang and J. Zhang, Sensors, 2018, 18(9), 2907 CrossRef PubMed.
- K. P. Williams, X. H. Liu, T. N. Schumacher, H. Y. Lin, D. A. Ausiello, P. S. Kim and D. P. Bartel, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 11285–11290 CrossRef CAS PubMed.
- A. Geiger, P. Burgstaller, H. von der Eltz, A. Roeder and M. Famulok, Nucleic Acids Res., 1996, 24, 1029–1036 CrossRef CAS PubMed.
- C. Y. Wang, B. L. Lin and C. H. Chen, Int. J. Cancer, 2016, 138, 918–926 CrossRef CAS PubMed.
- J. F. Comar, F. Suzuki-Kemmelmeier, J. Constantin and A. Bracht, J. Biomed. Sci., 2010, 17, 1 CrossRef PubMed.
- P. Bayat, R. Nosrati, M. Alibolandi, H. Rafatpanah, K. Abnous, M. Khedri and M. Ramezani, Biochimie, 2018, 154, 132–155 CrossRef CAS PubMed.
- W. H. Thiel and P. H. Giangrande, Methods, 2016, 103, 180–187 CrossRef CAS PubMed.
- K. Rahimizadeh, H. AlShamaileh, M. Fratini, M. Chakravarthy, M. Stephen, S. Shigdar and R. N. Veedu, Molecules, 2017, 22(12), 2070 CrossRef PubMed.
- Z. Xiao, D. Shangguan, Z. Cao, X. Fang and W. Tan, Chemistry, 2008, 14, 1769–1775 CrossRef CAS PubMed.
- Aptamers Selected by Cell-SELEX for Theranostics, ed. X. Fang and W. Tan, Springer, 2015, DOI:10.1007/978-3-662-46226-3.
- S. Catuogno and C. L. Esposito, Biomedicines, 2017, 5(3), 49 CrossRef PubMed.
- J. Chang, Mol. Cells, 2021, 44, 328–334 CrossRef CAS PubMed.
- I. Trenevska, D. Li and A. H. Banham, Front. Immunol., 2017, 8, 1001 CrossRef PubMed.
- Y. F. Chen, K. W. Chang, I. T. Yang, H. F. Tu and S. C. Lin, Oral Oncol., 2019, 95, 194–201 CrossRef CAS PubMed.
- Y. F. Chen, C. J. Liu, L. H. Lin, C. H. Chou, L. Y. Yeh, S. C. Lin and K. W. Chang, BMC Cancer, 2019, 19, 281 CrossRef PubMed.
- E. Prediger, Calculations: Converting from nanograms to copy number, https://sg.idtdna.com/pages/education/decoded/article/calculations-converting-from-nanograms-to-copy-number).
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04686a |
| ‡ These two first authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |