
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheophylline-based hybrids as acetylcholinesterase inhibitors endowed with anti-inflammatory activity: synthesis, bioevaluation, in silico and preliminary kinetic studies†
Abdullah A. Elgazar a,
Ramadan A. El-Domanyb,
Wagdy M. Eldehnac and
Farid A. Badria*d
a,
Ramadan A. El-Domanyb,
Wagdy M. Eldehnac and
Farid A. Badria*d
aDepartment of Pharmacognosy, Faculty of Pharmacy, Kafrelsheikh University, P.O. Box 33516, Kafrelsheikh, Egypt
bDepartment of Microbiology and Immunology, Faculty of Pharmacy, Kafrelsheikh University, P.O. Box 33516, Kafrelsheikh, Egypt
cDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Kafrelsheikh University, P.O. Box 33516, Kafrelsheikh, Egypt
dDepartment of Pharmacognosy, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt. E-mail: faridbadria@gmail.com; Tel: +20-1001762927
First published on 29th August 2023
Abstract
In this study, we investigated the conjugation of theophylline with different compounds of natural origin hoping to construct new hybrids with dual activity against cholinergic and inflammatory pathways as potential agents for the treatment of Alzheimer's disease (AD). Out of 28 tested hybrids, two hybrids, acefylline–eugenol 6d and acefylline–isatin 19, were able to inhibit acetylcholinesterase (AChE) at low micromolar concentration displaying IC50 values of 1.8 and 3.3 μM, respectively, when compared to the galantamine standard AChE inhibitor. Moreover, the prepared hybrids exhibited a significant anti-inflammatory effect against lipopolysaccharide induced inflammation in RAW 264.7 and reduced nitric oxide (NO), tumor necrosis alpha (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) levels in a dose dependent manner. These hybrids demonstrated significant reductions in nitric oxide (NO), tumor necrosis alpha (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) levels in RAW 264.7 cells induced by lipopolysaccharide (LPS). The findings of this study were further explained in light of network pharmacology analysis which suggested that AChE and nitric oxide synthase were the main targets of the most active compounds. Molecular docking studies revealed their ability to bind to the heme binding site of nitric oxide synthase 3 (NOS-3) and effectively occupy the active site of AChE, interacting with both the peripheral aromatic subsite and catalytic triad. Finally, the compounds demonstrated stability in simulated gastric and intestinal environments, suggesting potential absorption into the bloodstream without significant hydrolysis. These findings highlight the possible therapeutic potential of acefylline–eugenol 6d and acefylline–isatin 19 hybrids in targeting multiple pathological mechanisms involved in AD, offering promising avenues for further development as potential treatments for this devastating disease.
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by progressive cognitive decline, memory loss, and behavioral changes. The number of individuals affected by AD is alarmingly high, with an estimated 50 million people worldwide suffering from the disease. Hence, AD is placing a significant burden on individuals, families, and healthcare systems worldwide. Understanding the underlying mechanisms and developing effective therapeutic strategies for AD is of paramount importance which would eventually lead to developing innovative therapeutic strategies that target different pathways involved in AD pathology.1,2Current therapeutic strategies for management of AD are based on the acetylcholinesterase (AChE) hypothesis which proposes that the decline in cholinergic neurotransmission contributes to the cognitive decline observed in AD as reduced levels of acetylcholine have been observed in AD patients, so Acetylcholinesterase, an enzyme responsible for acetylcholine breakdown, appears to be a promising target. As a result, cholinesterase inhibitors have been developed as a symptomatic treatment for AD, aiming to increase acetylcholine levels. However, these drugs provide only modest benefits and do not alter the underlying disease progression.3,4
Another significant theory in AD research is the amyloid hypothesis. Amyloid-beta (Aβ) peptides, derived from the amyloid precursor protein (APP), aggregate and form amyloid plaques, a hallmark of AD pathology which is linked to neurodegeneration in AD patients. This hypothesis has guided the development of therapeutic approaches targeting Aβ production or clearance.5 However, clinical trials targeting Aβ have thus far yielded disappointing results, indicating the complexity of AD pathogenesis and the need for alternative therapeutic strategies.6
Recently, the role of inflammation in AD has gained significant attention. Chronic activation of the immune cells of the central nervous system such as microglia and astrocytes, induce the release of pro-inflammatory molecules, including cytokines and reactive oxygen species. These inflammatory mediators contribute to neuronal dysfunction and promote neurodegeneration in AD.7 Targeting inflammation represents a promising approach for AD treatment, as it addresses a common pathway involved in disease progression.8
Hence, several attempts were made to modify the activity of clinically relevant agents such as tacrine, donepezil, and galantamine. This led to the development of hybrids with multiple activities against other targets involved in AD. For example, the conjugation of tacrine with ferulic acid give the produced hybrid the ability to inhibit Aβ aggregation, also beta secretase inhibition was feasible after the conjugation with chromone moiety. In case of donepezil, molecular hybridization of the benzyl piperidine moiety with chromone and coumarin scaffolds enabled these compounds to inhibit monoamineoxidase-1 (MAO-1) and increased its antioxidant activity, respectively. Also, galantamine-curcumin hybrids showed significant reduction in acetylcholinesterase and promoted the antioxidant mechanisms in vivo (Fig. 1a).9–12
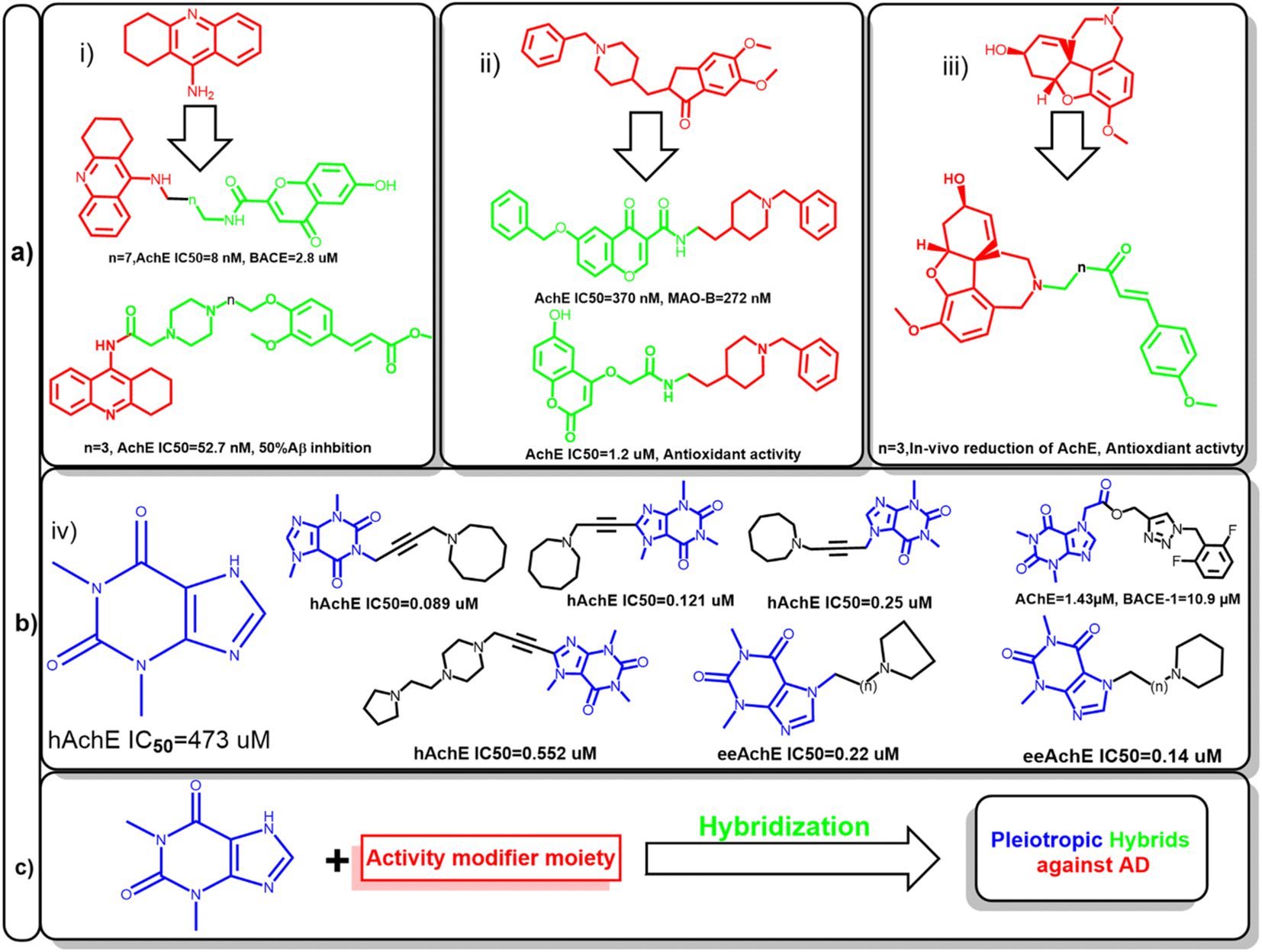 | ||
| Fig. 1 (a) Representative examples for previously reported inhibitor against acetylcholinesterase (AChE), (i) tacrine, (ii) donepezil, (iii) galantamine and their corresponding hybrids to modify their activity to be active against other enzymes involved in Alzheimer's disease such as monoamine oxidase (MAO) or betas secretase (BACE). (b) Theophylline (iv) has low affinity to AChE; new hybrids of theophylline had enhanced activity by linking with various moiety against human acetylcholinesterase (hAChE) or electric eel acetylcholinesterase (eeAchE). (c) Proposed strategy for anti-Alzheimer's agent based on xanthine hybrids, the counterpart of the conjugate would determine the activity of the prepared hybrids. | ||
Amidst the search for effective AD therapeutics, natural products have emerged as a valuable source of potential compounds since they offer a vast array of chemical diversity and biological activities. In the context of AD, natural products have exhibited neuroprotective, anti-amyloidogenic, and anti-inflammatory properties, positioning them as promising candidates for drug development. Xanthine alkaloids, such as caffeine, was linked to protection against several cardiovascular and neurodegenerative diseases.13–16
Since it can cross blood–brain barrier, it would be a privileged scaffold for targeting neurodegenerative disease. Several caffeine derivatives could be accessible through chemical modification of theophylline. Acefylline, which is theophylline-7-acetic acid was extensively studied to develop new bioactive compounds. Theophylline and its derivatives have been utilized to obtain hybrid compounds combining different pharmacophores or functional groups which was proven to improve the activity of the parent drugs (Fig. 1b).17–19
Therefore, this study aims at preparing hybrid compounds based on theophylline and other known natural products to fabricate new compounds with dual activity targeting different pathways in AD as shown in Fig. 1c. In silico approaches such as network pharmacology and molecular docking would be used to identify their potential mechanism of action. Also, the stability of the most active hybrids would be assessed.
2. Results and discussion
2.1. Chemistry
Firstly, acefylline 2 (7-acetic acid derivative of theophylline) was prepared to enhance the synthetic accessibility of theophylline in further reactions. Appearance of new signals in 13C NMR (CMR) at δ 47 and 168 ppm and in 1HNMR (PMR) at δ 5–5.5 ppm as well as melting point are among compiled evidences of the success of formation of, acefylline 2.Acefylline hybrids were formed by reactions with different natural products bearing either alcohol OH (menthol) or phenolic OH (thymol, vanillin, eugenol), through Steglich esterification20 as in hybrids 4a–d (Scheme 1) or alkylation by bromoacetate derivatives 5a–d to result in hybrids 6a–d (Scheme 2).
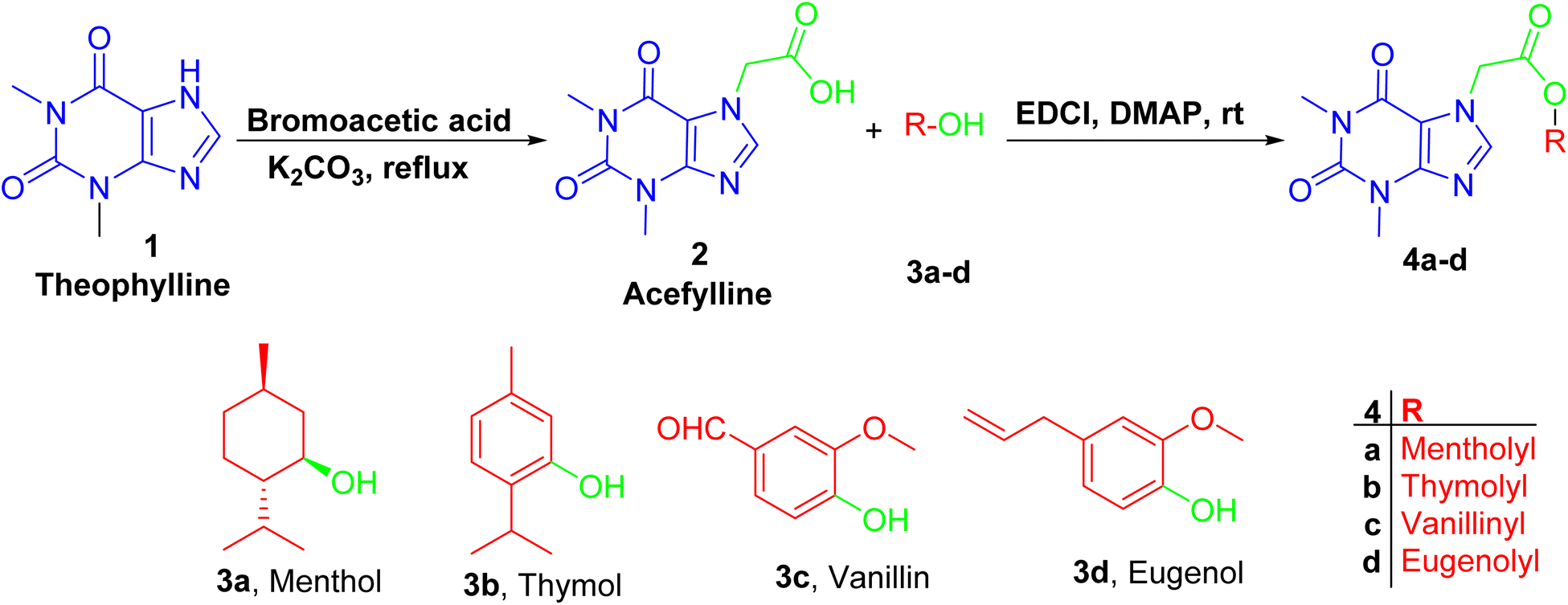 | ||
| Scheme 1 Preparation of theophylline-7-acetic acid (acefylline) followed by Steglich esterification, using EDCI as coupling agent in presence of menthol, thymol, eugenol or vanillin to give the corresponding ester. | ||
 | ||
| Scheme 2 Preparation of ester derivative 6a–d was facilitated by reaction of bromoacetyl bromide with respective phenol or alcohol followed by alkylation of acefylline with acetyl derivatives. | ||
The prepared hybrids showed characteristic spectroscopic profile including the presence of 2 signals at ranges of δ 3.2–3.4 ppm in PMR and δ 27–29 ppm in CMR assigned for methyl functionality of xanthine ring. In addition, one signal was shown at δ 5 ppm in PMR and δ 47 ppm in CMR assigned for the methylene carbon of acetic acid linker. Also, a downfield proton at ranges of δ 7.6–8 ppm in 1H NMR and a signal at δ 144 ppm in 13C NMR were observed and assigned for proton and carbon 4 of the purine ring, while the two carbonyl groups of the xanthine ring were found at δ 154 and 151 ppm.21
For hybrids 4a–d, the spectroscopic data showed consistent pattern with the reported parent compounds except the decrease in the chemical shift value of carboxylic acid of acefylline from δ 169 to 168–165 ppm confirming the formation of ester. Additionally, in case of menthol hybrid 4a, the chemical shift value of proton 1′ of menthol increased from δ 3.4 to 4.7 ppm as shown in Tables S1–S4.†
While in case of hybrids containing acetyl spacer 6a–d, acefylline 2 was reacted with bromoacetate derivatives of the corresponding alcohol or phenol. Hence, an additional signal were observed around δ 4.6 ppm (s, 2H) in PMR and δ 61 and 166 ppm in CMR corresponding to the acetyl linker as recorded in Tables S5–S8.†
The formation of 1,3,4-oxadiazole-containing hybrids 9a–d was facile through the reaction of the hydrazide derivative of acefylline with carbon disulfide in basic medium followed by alkylation with bromoacetate derivatives of the corresponding alcohol or phenol 5a–d (Scheme 3). Thus, these derivatives exhibited similar PMR behavior as their corresponding ester derivatives 6a–d except for a new signal was shown around δ 162 ppm in CMR indicating the formation of the oxadiazole ring and the chemical shift value of the methylene linker showed significant decrease from δ 60 and 47 ppm to around δ 40 and 34 ppm, respectively, as demonstrated in Tables S9–S12.†
 | ||
| Scheme 3 Preparation of 1,3,4-oxadiazole-containing acefylline hybrids 9a–d. | ||
For compounds containing 1,2,3-triazole linker 15a–c, they were successfully obtained through copper catalyzed azide alkyne cycloaddition reaction of the propargylated derivative of acefylline 14 with respective azides 10a-b and 13 that prepared from bromoacetylated derivatives 5a-b and 4-chloro-coumarin 12, respectively, as demonstrated in Scheme 4. The reaction was confirmed by the appearance of additional signals around δ 8 (1H) and 5.2 (2H) ppm corresponding to the triazole ring and the methylene bridge between the xanthine moiety and the triazole ring, respectively. Moreover, three new signals were observed in CMR corresponding to the two olefinic carbons in the triazole ring and the methylene bridge around δ 142, 122, and 60 ppm, respectively.
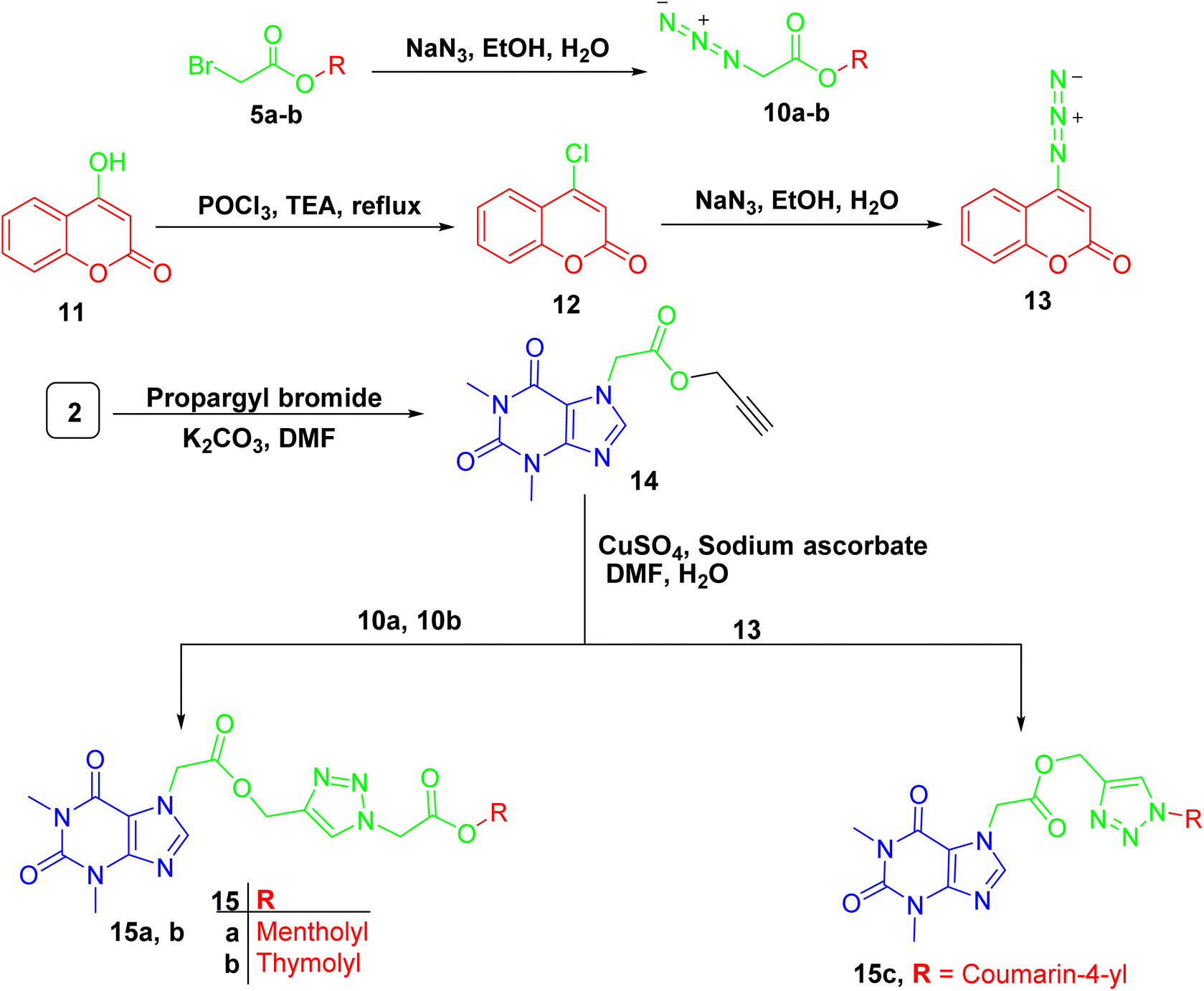 | ||
| Scheme 4 Preparation of acefylline triazole hybrids 15a–c. | ||
The other data showed no significant change in the NMR profile in comparison to that found in the parent compounds when compared to spectroscopic data in literature22 except for 4-hydroxycoumarin derivative 13 where carbon 4′ showed significant upfield shift from δ 162 to 154 ppm due to the substitution of hydroxyl group with azide group as recorded in Tables S13–S15.†
Finally, hydrazone linker-tethered derivatives 18a–l and 19 were furnished through coupling of hydrazide 7 with various aromatic aldehydes, 16a–l, and isatin 17, respectively as shown in Scheme 5. The disappearance of aldehydic proton around δ 9–10 ppm and the appearance of two new signals at δ 11 and 8 ppm corresponding to the hydrazone linker protons. Furthermore, the decrease in chemical shift of aldehydic carbon from δ 190 to 140 ppm were among the spectroscopic features to confirm the formation of acefylline-hydrazone hybrids 18a–l. Lastly, the methylene group of acefylline showed significant decrease in chemical shift from around δ 60 to 47 ppm. For acefylline–isatin hybrid 19, the hydrazone linker was observed at δ 11.36 and 143.21 ppm as explained in Tables S16–S28.†
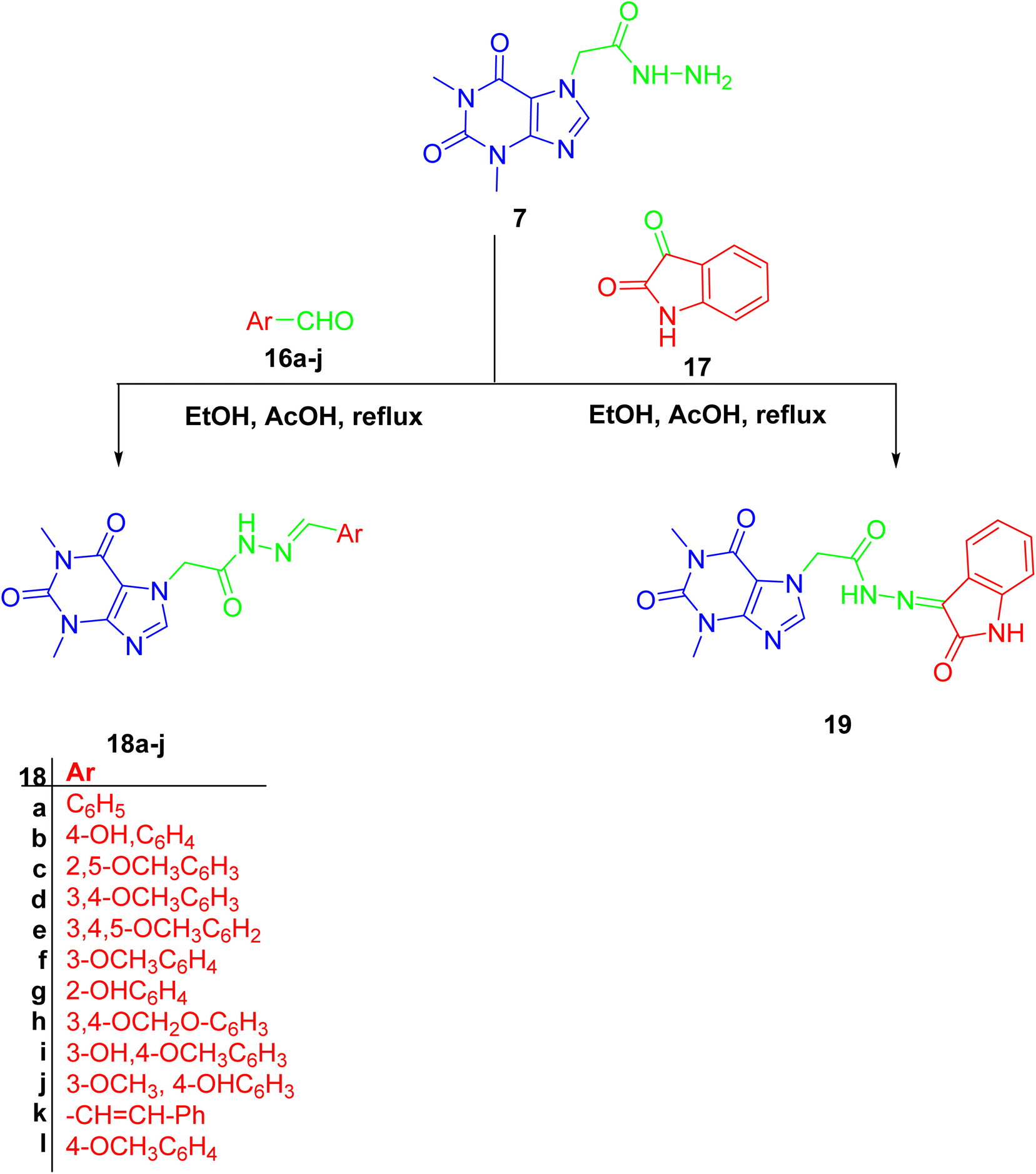 | ||
| Scheme 5 Preparation of acefylline hydrazone hybrids 18a–j and 19 through the reaction of hydrazide with corresponding aldehydes or ketone. | ||
2.2. Biological evaluation
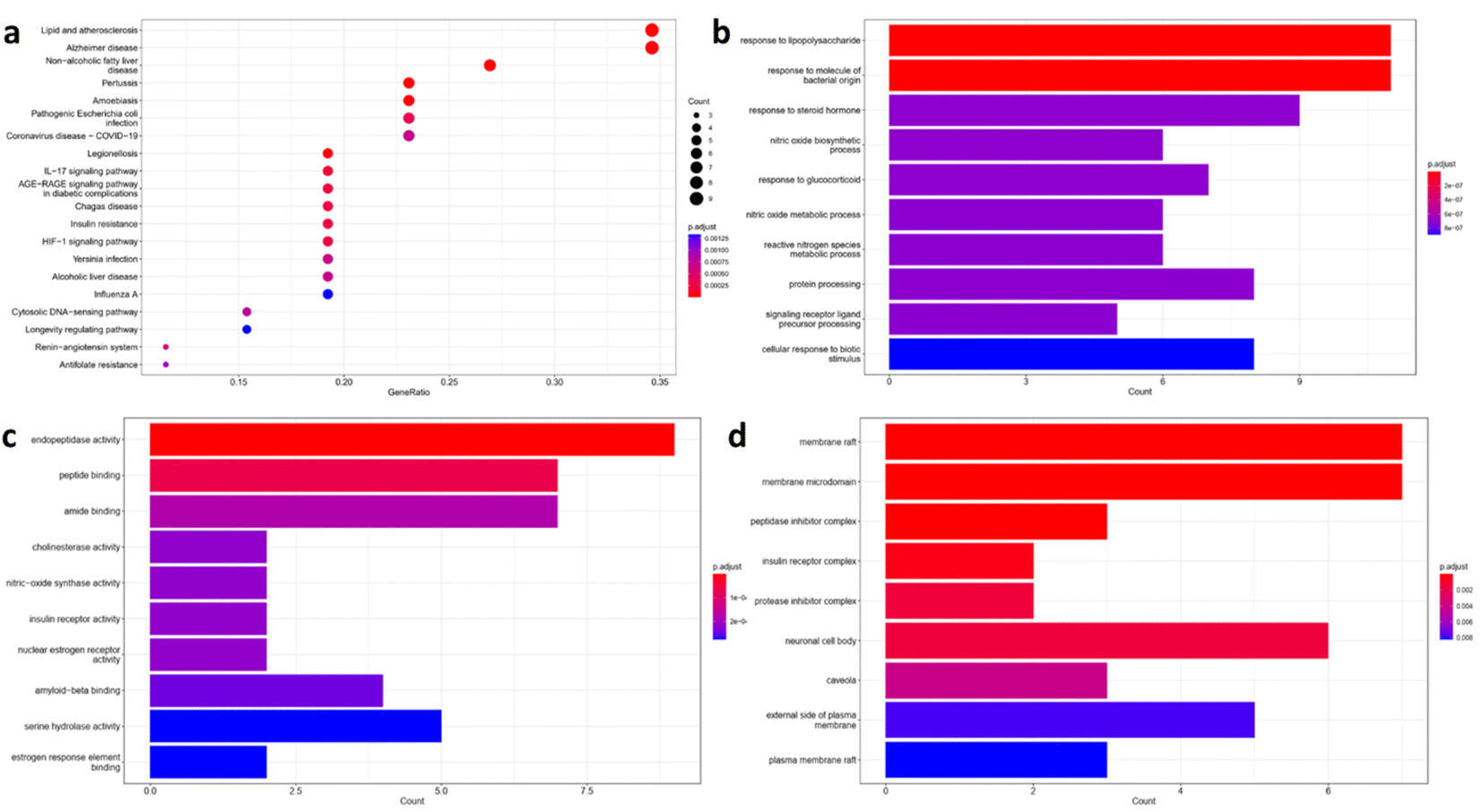 | ||
| Fig. 2 Enrichment analysis results of Alzheimer's disease-related targets in relation with acefylline hybrids significantly enriched terms in, (a) KEGG, (b) BP, (c) MF, and (d) CC. | ||
Also, gene enrichment reveals other genes involved in biological processes (BP), molecular functions (MF) and cellular component (CC) associated with the predicted targets. The top five enriched terms in BP was related to response to stimuli that trigger or suppress inflammation such as LPS, NO or glucocorticoid respectively (Fig. 2b). For MF, endopeptidase activity, peptide binding, amide binding, choline esterase activity and nitric oxide activity were among the top five enriched terms where targets such as nitric oxide synthase, beta secretase, acetylcholinesterase and butyl cholinesterase appeared as the most significant ones as depicted in Fig. 2c. Finally, the top enriched terms of CC were a reflection to what have been found in KEGG analysis where membrane rafts and membrane microdomain were found to be the most significant term with P ≥ 0.002 (Fig. 2d). Interestingly, lipid rafts were shown to play important role in the development of AD through regulation of Aβ deposition and AChE signaling.28
Protein–protein interaction (PPI) of the enriched targets was also studied, where the constructed network showed eNOS, AChE, TNF, IL-1β and GSK-3β as the most prominent targets. Still eNOS and AChE were the most prominent target with 47/68 and 40/68 interactions respectively as shown in Fig. 3a and b. AChE targeting is one of the clinical pillars for management of AD while NOS is still under investigation, nevertheless the correlation between overexpression NOS and AD is well established,29 also NOS inhibitors have shown neuroprotective activity in experimental animal.30
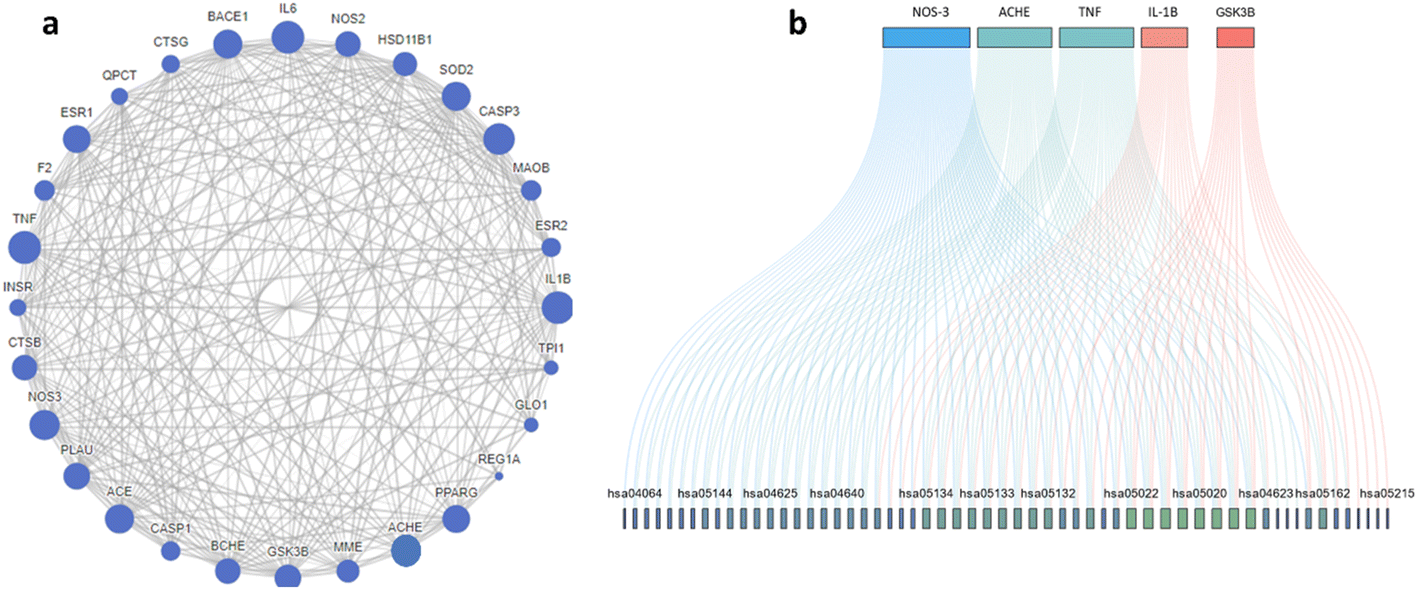 | ||
| Fig. 3 (a) Protein–protein interactions of targets predicted to be relevant to acefylline hybrid activity. (b) Top five targets included in KEGG pathways where the greater the degree of overlap of genes, the more the same pathways involved. Lines with different colors indicate different gene-pathway relationships. | ||
In summary, network pharmacology suggests that the ability of the prepared hybrids to modulate pathways correlated to AD through regulation of inflammatory response and cholinesterase interactions with lipid rafts which would consequently reduce Aβ precipitation and plaque formation.
2.3. In vitro anti-inflammatory assay of acefylline-hybrids
| Code | Viability (%) |
|---|---|
| a Data represent the mean and SD of % viability based on two independent experiment. | |
| 2 | 85 ± 1.7 |
| 4a | 91.4 ± 0.1 |
| 4b | 94 ± 0.2 |
| 4c | 90 ± 1 |
| 4d | 93 ± 1 |
| 6a | 88 ± 0.8 |
| 6b | 89 ± 1 |
| 6c | 90 ± 1.1 |
| 6d | 92 ± 1 |
| 9a | 90 ± 1.1 |
| 9b | 93 ± 1 |
| 9c | 92 ± 1 |
| 9d | 98 ± 0.6 |
| 15a | 86 ± 1.3 |
| 15b | 97 ± 0.6 |
| 15c | 88 ± 0.8 |
| 18a | 94 ± 0.2 |
| 18b | 87 ± 0.8 |
| 18c | 88 ± 0.8 |
| 18d | 90 ± 1 |
| 18e | 92 ± 1 |
| 18f | 91 ± 1 |
| 18g | 92 ± 1 |
| 18h | 93 ± 1 |
| 18i | 104.7 ± 1.2 |
| 18j | 89 ± 1 |
| 18k | 94.7 ± 1.2 |
| 18l | 93 ± 1 |
| 19 | 89.5 ± 1.1 |
| No. | Code | % inhibition |
|---|---|---|
| a L-NAME was used at 250 μM, data represent the mean and SD of % inhibition based on two independent experiment. | ||
| 1 | 2 | 58.7 ± 1.82 |
| 2 | 4a | 66.4 ± 0.79 |
| 3 | 4b | 70.7 ± 0.87 |
| 4 | 4c | 65.4 ± 0.84 |
| 5 | 4d | 86.8 ± 0.87 |
| 6 | 6a | 79.3 ± 0.85 |
| 7 | 6b | 74 ± 0.87 |
| 8 | 6c | 68.6 ± 0.92 |
| 9 | 6d | 88 ± 1 |
| 10 | 9a | 69.6 ± 0.92 |
| 11 | 9b | 92.8 ± 0.9 |
| 12 | 9c | 86.4 ± 0.86 |
| 13 | 9d | 93.2 ± 0.87 |
| 14 | 15a | 61.1 ± 0.9 |
| 15 | 15b | 68.6 ± 0.92 |
| 16 | 15c | 85 ± 0.88 |
| 17 | 18a | 65.4 ± 0.84 |
| 18 | 18b | 71.8 ± 0.9 |
| 19 | 18c | 75 ± 0.88 |
| 20 | 18d | 70.7 ± 0.87 |
| 21 | 18e | 71.8 ± 0.9 |
| 22 | 18f | 63.2 ± 0.93 |
| 23 | 18g | 66.4 ± 0.79 |
| 24 | 18h | 96.6 ± 0.87 |
| 25 | 18i | 63.2 ± 0.93 |
| 26 | 18j | 92.2 ± 0.86 |
| 27 | 18k | 65.4 ± 0.84 |
| 28 | 18l | 85.4 ± 0.87 |
| 29 | 19 | 90 ± 0.88 |
| 30 | L-NAMEa | 84.8 ± 0.9 |
| No. | Compound | Inhibition (%) |
|---|---|---|
| a Data represent the mean and SD of % inhibition based on two independent experiment. | ||
| 1 | Acefylline | 6.5 ± 1.5 |
| 2 | 4d | 61.39 ± 3.79 |
| 3 | 6d | 97 ± 3.2 |
| 4 | 9b | 61.07 ± 1.79 |
| 5 | 9c | 88.2 ± 3.7 |
| 6 | 9d | 41.33 ± 3.57 |
| 7 | 15c | 85 ± 2.8 |
| 8 | 18h | 73.6 ± 3 |
| 9 | 18j | 78 ± 3.9 |
| 10 | 18l | 55.42 ± 2.07 |
| 11 | 19 | 92.5 ± 2.4 |
It is worthy to mention that acefylline–eugenol conjugates through acetyl linker had more advantage over other conjugation strategies such as oxadiazole and direct esterification where the first achieved 97% inhibitory effect, while the later achieved only 61% and 41% inhibition against AChE. For acefylline–vanillin conjugates, hybrids containing oxadiazole linker achieved better inhibition than that observed in case of the hydrazone spacer.
In case of isatin hydrazone, it achieved the 2nd best inhibitory activity, also the coumarin triazole hybrid inhibit AChE by 85%. So, the ability of these 5 hybrids to inhibit the enzyme at different doses was studied and their IC50 was calculated. Compounds 6d and 19 demonstrated the best IC50 = 1.8 and 3.3 μM respectively which was comparable to the standard AChE inhibitor galantamine as shown in Table 4 and Fig. S1†. While other hybrids showed relatively higher IC50 but still stronger by thirty-fold stronger inhibitory effect compared to the parent drug theophylline.
| No. | Activity modifying moiety | Linker | Compound | IC50 (μM) |
|---|---|---|---|---|
| 1 | Eugenol | Acetyl | 6d | 1.8 ± 0.3 |
| 2 | Vanillin | Oxadiazole | 9c | 5.55 ± 0.33 |
| 3 | 4-Hydroxy coumarin | Triazole | 15c | 9.8 ± 0.7 |
| 4 | Vanillin | Hydrazone | 18j | 14.6 ± 0.5 |
| 5 | Isatin | Hydrazone | 19 | 3.3 ± 0.2 |
| 6 | — | — | Galantamine | 1.3 ± 0.25 |
 | ||
| Fig. 4 Effect of compounds 6d and 19 on (a) TNF-α, (b) IL-1β, and (c) IL-6 levels in RAW 264.7 treated with 1 μg ml−1 LPS in comparison to dexamethasone as standard. ****Significantly different from control group at p < 0.0001. | ||
The biological evaluation of acefylline hybrids indicated their dual activity as anti-inflammatory and acetylcholinesterase inhibitor as suggested by network pharmacology. Thymol, eugenol, coumarin and aldehydes conjugates with acefylline through acetyl, oxadiazole, triazole and hydrazone linker were observed as the most actives. Two compounds 6d which is acefylline–eugenol conjugate through acetyl linker and 19 which is acefylline conjugate through hydrazone linker achieved the best activity among the prepared hybrids.
While theophylline itself is understudied as anti-inflammatory agent it was reported to suppress TNF production at 20 μM in LPS stimulated RAW264.7 (ref. 31) On the other hand, caffeine which is the methylated analogue of theophylline is well studied. For example, caffeine and its analogue pentoxifylline were previously reported to possess anti-inflammatory activity in RAW264.7 induced by LPS but at relatively high dose 100–800 μM and 0.5–8 mM respectively.32,33 Also, caffeine attenuated neuroinflammation induced by LPS in BV2 microglial cells, where pro-inflammatory such as TNF-α and iNOS through prevention of phosphorylation of AKT and ERK leading to suppress of the downstream inflammatory cascade.34
Remarkably, the neuroprotective effect of caffeine against LPS induced neurodegeneration in mice through activation of nuclear factor erythroid 2-related factor 2 (Nrf2) and reduction of NO, TNF-α and oxidative stress.35 Moreover, it decreased number of activated microglia in hippocampus of experimental animals subjected to inflammation associated with LPS administration or aging.36
Furthermore, caffeine was found to protect against cognitive impairment in AD transgenic mice through decreasing Aβ levels and β-secretase.37 Despite caffeine is a weak inhibitor to AChE, it was found to have indirect inhibitory effect on AChE through inactivation of protein kinase A, which is correlated with enhancing the activity of AChE to hydrolyze acetylcholine.38–40 In this context, chemical derivatization of theophylline and caffeine led to the development of novel acetylcholinesterase inhibitors41–43 which support our proposal to choose xanthine alkaloid to design hybrids for management of AD.
For the counterpart moiety of hybrid 6d, eugenol is known for its ability to exert substantial anti-inflammatory and antioxidant activity in vitro and in vivo which is attributed to the modulation of several and overlapping pathways.44 So, numerous studies investigated its application in management of AD. For instance, eugenol at 0.01 mg kg−1 was able to reduce plaques induced in rats injected by Aβ which was reflected by improvement in memory of the treated rat.45 Also, it mitigated the cholinergic dysfunction in hippocampus induced by scopolamine through decreasing in AChE activity.
It also protected against neurotoxicity by decreasing glutamate levels, boosting antioxidant mechanisms through increasing SOD and catalase activity and decreasing mitochondrial reactive oxygen species (ROS) and NO production.46 In combination with psilocybin, it was able to protect against inflammation induced in mice brain by LPS where several cytokines levels such as TNF, IL-1β, IL-6 were reduced.47 This was also reproducible in other experimental model based on aluminum chloride induced neurotoxicity.48
Furthermore, eugenol was evaluated as potential pleotropic agent against AD. While it showed remarkable protection against hydrogen peroxide and Aβ toxicity to PC12 cell line, it showed weak inhibition to AChE and β-secretase.49 Still chemical derivatization has shown to improve the activity to micromolar range.50,51 Taking these studies in consideration the conjugation of acefylline and eugenol hybrid through acetyl linker could be an intuitive approach to target different pathways involved in AD. Also, since hybrid compound could be considered as prodrug, acetylcholinesterase itself would help to release its original parent compounds once it reached the action site leading to extended efficacy.52
In case of compound 19, isatin was linked to acefylline. Isatin is a remarkable molecule with plethora of activities. In the last decade, it was thoroughly investigated for its ability to modulate molecular targets associated with AD since it is one of the endogenous neuroprotector molecules.53,54 So, isatin itself was recognized as a ligand for multiple binding sites of Aβ preventing its interaction with several target and maintain it in monomer state allowing their physiological degradation.55
In Aβ-induced Human Neuroblastoma (SK-N-SH Cells), isatin thiosemicarbazone increased the survival rate from 20% to approximately 100% which was explained by their inhibitory effect against GSK-3β which is one of the molecular targets predicted by our network pharmacology analysis.56 Moreover, several isatin derivatives were designed as AChE inhibitors; interestingly, isatin–tacrine and isatin–triazole hybrids had pronounced ability to prevent Aβ aggregation.57–59
Regarding the contribution of isatin in resolving inflammation, isatin, its derivatives were able to reduce the levels of COX-2, TNF and iNOS in LPS challenged RAW 264.7 at relatively high dose (100 μM)60 but when conjugated with Nalidixic acid through amino acid linkers, the prepared hybrids were able to inhibit iNOS by 80% at 10 μM.61 Hence, the observed acetylcholinesterase and anti-inflammatory effect of compound 19 could be justified in the light of these studies.
2.4. In silico analysis
In case of NOS-3, the co-crystallized ligand was found to be near the heme-porphyrin catalytic site, and it interacted with Trp356, Met358, Glu361 through hydrogen bonding. On the other hand, hydrophobic interactions with Pro334, Val 336 Met339, and Phe353 were also identified.62 Compound 6d showed similar interaction profile where it was able to form hydrogen bonds with Cys184, Val185, Gly186, Ser226, TRP356,Met358 and Glu361 through acefylline moiety while other hydrophobic interactions such as TRP178, Leu193, Ile228, Phe353 and Phe473 were exerted through the eugenyl moiety as shown in Fig. 5a and b. This was also the case of compound 19 where the acefylline scaffold formed hydrogen bond with TRP178, Val185, Ser226, TRP356, Met 358 while isatin moiety formed hydrophobic interactions with Ala181, Arg183, Cys184, Met339 and Gly355 as shown in Fig. 5c and d.
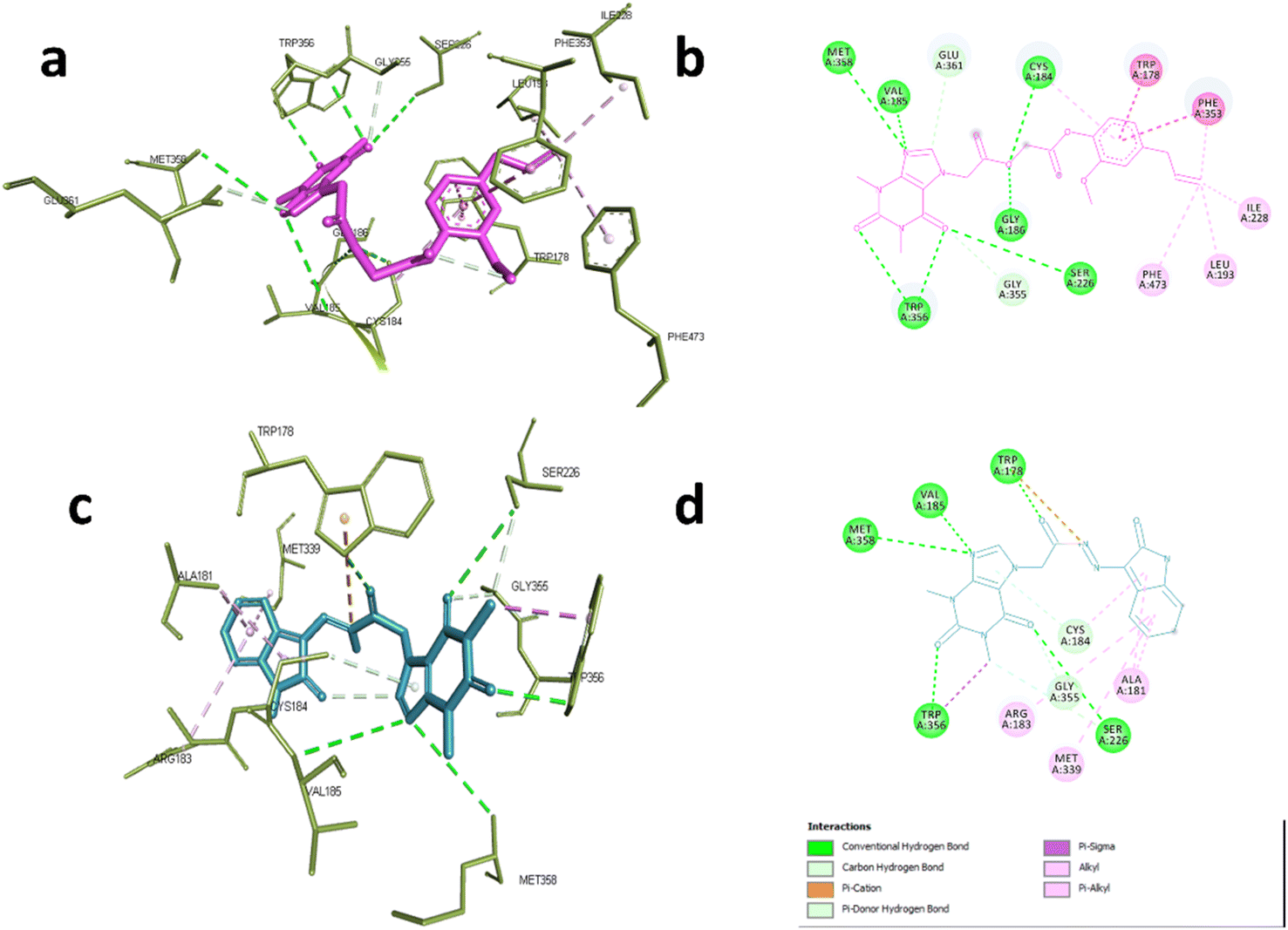 | ||
| Fig. 5 Interactions with eNOS complex (PDB:6AV7). (a) 3D and (b) 2D interactions of 6d (magenta). (c) 3D and (d) 2D interactions of 19 (cyan). | ||
AChE was one of the predicted targets for the prepared hybrids and compounds 6d and 19 also showed good inhibitory activity against this enzyme experimentally. Post docking analysis gave insights about their binding mode in comparison to known inhibitors. The co-crystallized ligand donepezil mainly exert its remarkable inhibitory activity through hydrophobic interactions with Trp86, Trp286, Tyr337, Phe338, Tyr341and one hydrogen bond with Phe295 while galantamine the standard inhibitor inhibitory effect depends on blocking the catalytic active site by interaction with Glu202, Ser203, Tyr337 through hydrogen bonding and Trp86, Gly122 through hydrophobic interactions.
These interactions were reproducible by compound 6d where it formed hydrogen bonds with Ser203, Gly120, Tyr124, Ser125, Tyr133 and Tyr 341, also hydrophobic interactions with Tyr72, Trp86, Trp286 indicating that the compound 6d occupy the full active site gorge-channel which is similar to the binding mode of donepezil rather than galantamine. Compound 19 was also able to interact with Try124, Tyr133, Ser125 and Try341 through hydrogen bonding and Trp86 and Trp286 through hydrophobic interactions as demonstrated in Fig. 6a–d.
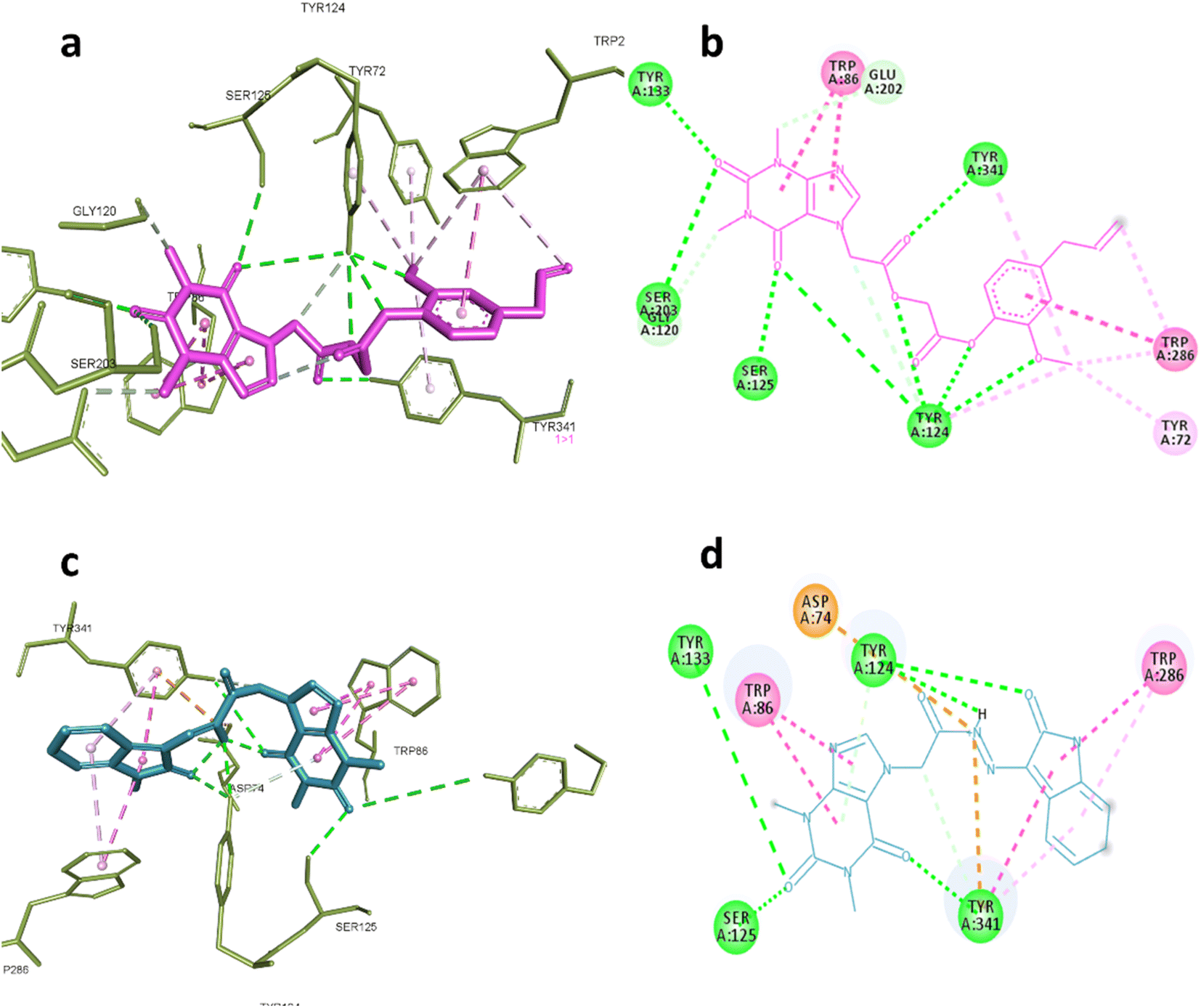 | ||
| Fig. 6 Interactions with AChE (PDB:4EY7). (a) 3D and (b) 2D interactions of 6d (magenta). (c) 3D and (d) 2D interactions of 19 (cyan). | ||
Nevertheless, the length of linker may explain the difference in the inhibitory activity of compound 6d and 19 where the acetyl linker in case of the first allowed the xanthine moiety to reach the catalytic triad and interact with Ser203, which is important to achieve good inhibitory activity.63 Overall, the analysis of binding mode of 6d and 19 showed their ability to bind to both of catalytic site and peripheral aromatic site (PAS) suggesting that their ability to neutralize the pathological role of AChE through preventing acetyl choline hydrolysis and the aggregation of Aβ by disrupting their interaction with PAS of AChE.64
2.5. Preliminary stability studies
Given the enhanced bioactivity observed in compounds 6d and 19 relative to other hybrid derivatives, our investigation centered on evaluating their stability under simulated gastric and intestinal conditions. Compound 6d demonstrated exceptional stability when exposed to 0.1 M HCl, exhibiting a half-life (t1/2) of 35 hours, while compound 19 exhibited a significantly shorter t1/2 of 1.8 hours, as shown in Fig. 7a.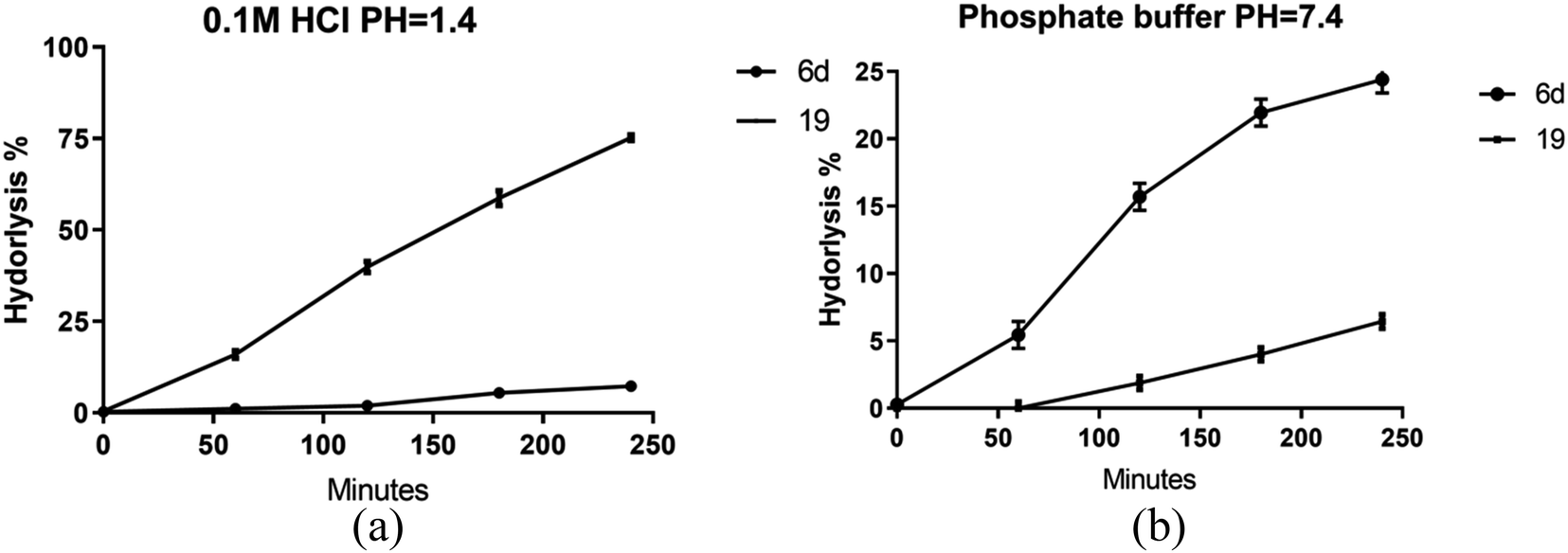 | ||
| Fig. 7 Stability of prepared hybrids at different buffers (a) pH = 1.4, (b) phosphate buffer pH = 7.4. Data presented as mean values ±SD of three independent experiments. | ||
However, a contrasting pattern emerged when the pH was adjusted to 7.4. Notably, due to its ester composition, compound 6d displayed increased susceptibility to hydrolysis, resulting in a t1/2 of 11.3 hours. Conversely, compound 19 displayed a prolonged t1/2 of 76 hours, as depicted in Fig. 7b. These findings are supported by prior investigations, emphasizing the diminished stability of hydrazones within acidic pH in comparison with basic or neutral environments. In contrast, esters assume a prodrug role in relatively alkaline pH levels, facilitating sustained release of the active substrate.
Importantly, both compounds exhibited substantive resistance to acid hydrolysis.65,66 Based on the data we gathered, it can be inferred that both compounds are not greatly affected by gastric or intestinal fluids. This suggests that they have the ability to be absorbed into the body without undergoing substantial breakdown.
3. Conclusion
This study investigated the conjugation of theophylline with different compounds of natural origin hoping to construct new hybrids with dual activity against cholinergic and inflammatory pathways as potential agents for treatment of Alzheimer's disease (AD). In this study, a series of 28 theophylline-based hybrids were synthesized and investigated against acetylcholinesterase (AChE) and inflammation. Out of 28 tested hybrids; two hybrids, acefylline–eugenol 6d and acefylline–isatin 19 were able to inhibit acetylcholinesterase (AChE) at low micromolar concentration when compared to “galantamine” a known standard AChE inhibitor. Moreover, the prepared hybrids possessed significant anti-inflammatory effect against lipopolysaccharide induced inflammation in RAW 264.7 and reduced nitric oxide (NO), tumor necrosis alpha (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) levels in dose dependent manner. The findings of this study were further explained in the light of network pharmacology analysis which suggested that AChE and nitric oxide synthase are the main targets of the most active compounds. Our molecular docking studies supported the obtained results. Finally, the two promising hybrids proved to be stable when studied in medium simulating gastric and intestinal environment suggesting their possible mode of absorption without substantial breakdown.Particularly, the hybrids of theophylline with either eugenol or isatin revealed very promising and unleashing their therapeutic potential for managing complex health disorders, e.g., Alzheimer's disease and other neurodegenerative disorders. However, further investigation of the kinetic properties of these hybrids is necessary before translating these findings into preclinical and clinical practice.
4. Material and methods
4.1. Chemistry
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol 7:3).
:methanol 7:3).4.1.3.1. 2-Isopropyl-5-methylcyclohexyl-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetate (4a). Yield 75% as semisolid, [α]25D = −52.5 (c 15 g l−1, EtOH). 1H NMR (400 MHz, CDCl3) δ ppm: 7.59 (s, 1H), 5.01 (s, 2H), 4.71 (td, J = 10.9, 4.4 Hz, 1H), 3.53 (s, 3H), 3.31 (s, 3H), 1.96 (d, J = 11.5 Hz, 1H), 1.78 (qt, J = 7.2, 3.2 Hz, 1H), 1.61 (dt, J = 12.1, 2.8 Hz, 2H), 1.36 (dtt, J = 38.3, 11.3, 3.0 Hz, 2H), 1.05–0.89 (m, 2H), 0.83 (t, J = 7.0 Hz, 7H), 0.68 (d, J = 6.9 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ ppm: 166.60, 155.16, 151.69, 148.29, 141.79, 107.20, 76.88, 47.62, 46.90, 40.62, 34.02, 31.40, 29.91, 27.93, 26.19, 23.27, 21.95, 20.76, 16.23; displayed a molecular ion at m/z 376.46 calculated for the molecular formula anal. calcd for C19H28N4O4, C, 60.62; H, 7.5; N, 14.88; found C, 60.71; H, 7.27; N, 14.95.
4.1.3.2. 2-Isopropyl-5-methylphenyl-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetate (4b). Yield 65%, 1H NMR (400 MHz, CDCl3) δ ppm: 7.64 (d, J = 6.2 Hz, 1H), 7.12 (dd, J = 8.0, 2.2 Hz, 1H), 7.00–6.93 (m, 1H), 6.80 (d, J = 2.2 Hz, 1H), 5.28 (d, J = 2.3 Hz, 2H), 3.53 (d, J = 2.2 Hz, 3H), 3.33 (d, J = 2.2 Hz, 3H), 2.98–2.86 (m, 1H), 2.22 (s, 3H), 1.09 (dd, J = 6.9, 2.2 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ ppm: 165.80, 155.19, 151.57, 148.36, 147.18, 141.61, 136.79, 136.70, 127.70, 126.54, 122.14, 107.12, 47.44, 29.84, 27.87, 26.86, 23.00, 20.73; displayed a molecular ion at m/z 370.41 calculated for the molecular formula anal. calcd for C19H22N4O4, C, 61.61; H, 5.99; N, 15.13; found C, 61.64; H, 5.42; N, 15.26.
4.1.3.3. 4-Formyl-2-methoxyphenyl-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetate (4c). Yield 25%, 1H NMR (400 MHz, CDCl3) δ ppm: 9.84 (s, 1H), 7.85 (s, 1H), 7.48–7.41 (m, 2H), 7.06 (d, J = 8.5 Hz, 1H), 3.98 (d, J = 5.1 Hz, 2H), 3.68 (s, 3H), 3.51 (d, J = 5.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ ppm: 191.11, 168.81, 156.26, 151.67, 148.99, 140.23, 127.71, 114.55, 107.03, 56.26, 50.97, 30.40, 28.63; displayed a molecular ion at m/z 372.34 calculated for the molecular formula anal. calcd for C17H16N4O6, C, 54.84; H, 4.33; N, 15.05; found C, 55.11; H, 4.89; N, 14.54.
4.1.3.4. 4-Allyl-2-methoxyphenyl-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetate (4d). Yield 75%, 1H NMR (400 MHz, CDCl3) δ ppm: 7.63 (s, 1H), 6.96 (d, J = 7.8 Hz, 1H), 6.69 (d, J = 10.4 Hz, 2H), 5.91–5.81 (m, 1H), 5.30 (s, 2H), 5.06–4.98 (m, 2H), 3.74 (s, 3H), 3.52 (s, 3H), 3.33 (s, 3H), 3.29 (d, J = 6.9 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ ppm: 165.49, 155.29, 151.70, 150.41, 148.50, 141.99, 139.76, 137.27, 136.85, 122.27, 120.78, 116.35, 112.74, 107.16, 55.85, 47.14, 40.05, 29.86, 27.96; displayed a molecular ion at m/z 384.39 calculated for the molecular formula anal. calcd for C19H20N5O5, C, 77.49; H, 10.22% found C, 77.93; H, 10.03% C, 59.37; H, 5.24; N, 14.58; found C, 59.89; H, 4.78; N, 14.26.
4.1.5.1. 2-Isopropyl-5-methylcyclohexyl-2-(2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetoxy)acetate (6a). Yield 60%, [α]25D = −67.5(c 15 g l−1, EtOH). 1H NMR (400 MHz, CDCl3) δ ppm: 7.63 (s, 1H), 5.18 (s, 2H), 4.70 (td, J = 11.0, 4.5 Hz, 1H), 4.64 (s, 2H), 3.54 (s, 3H), 3.32 (s, 3H), 1.95–1.86 (m, 1H), 1.79–1.67 (m, 1H), 1.66–1.57 (m, 2H), 1.47–1.26 (m, 2H), 1.06–0.90 (m, 2H), 0.82 (dd, J = 8.7, 6.7 Hz, 7H), 0.67 (d, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ ppm: 166.68, 166.57, 155.29, 151.64, 148.43, 141.95, 107.06, 76.21, 61.92, 47.09, 46.89, 40.67, 34.05, 31.39, 29.94, 27.96, 26.26, 23.38, 21.96, 20.69, 16.27; displayed a molecular ion at m/z 434.49 calculated for the molecular formula anal. calcd for C21H30N4O6 C, 58.05; H, 6.96; N, 12.9; found C, 58.25; H, 6.96; N, 12.6%.
4.1.5.2. 2-(2-Isopropyl-5-methylphenoxy)-2-oxoethyl-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetate (6b). Yield 45%, 1H NMR (400 MHz, CDCl3) δ ppm: 7.60 (s, 1H), 7.13 (d, J = 7.9 Hz, 1H), 6.98 (dd, J = 8.0, 1.8 Hz, 1H), 6.77–6.70 (m, 1H), 5.19 (s, 2H), 4.94 (s, 2H), 3.53 (s, 3H), 3.30 (s, 3H), 2.85 (dt, J = 13.7, 6.8 Hz, 1H), 2.23 (s, 3H), 1.09 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ ppm: 166.73, 165.92, 155.30, 151.61, 148.44, 147.04, 141.89, 136.89, 136.87, 127.77, 126.69, 122.25, 107.05, 61.71, 47.09, 29.94, 27.93, 27.06, 23.02, 20.79; displayed a molecular ion at 428.45 m/z calculated for the molecular formula anal. calcd for C21H24N4O6 C, 58.87; H, 5.65; N, 13.08; found C, 58.42; H, 6.13; N, 13.
4.1.5.3. 2-(4-Formyl-2-methoxyphenoxy)-2-oxoethyl-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetate (6c). Yield 25%, 1H NMR (400 MHz, CDCl3) δ ppm: 9.88 (s, 1H), 7.61 (d, J = 1.8 Hz, 1H), 7.42 (dt, J = 9.7, 1.6 Hz, 2H), 7.18 (dd, J = 7.9, 3.0 Hz, 1H), 5.25–5.15 (m, 2H), 4.97 (d, J = 17.3 Hz, 2H), 3.83 (d, J = 3.9 Hz, 3H), 3.53 (s, 3H), 3.30 (d, J = 2.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ ppm: 190.95, 166.61, 164.67, 155.32, 151.62, 148.55, 148.51, 143.78, 141.94, 135.66, 124.66, 123.17, 110.94, 107.06, 61.36, 56.20, 47.07, 29.91, 27.94; displayed a molecular ion at 430.37 m/z calculated for the molecular formula anal. calcd for C19H18N4O8 C, 53.03; H, 4.22; N, 13.02; found C, 53.07; H, 3.63; N, 13.59.
4.1.5.4. 2-(4-Allyl-2-methoxyphenoxy)-2-oxoethyl-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetate (6d). Yield 45%, 1H NMR (400 MHz, CDCl3) δ ppm: 7.59 (s, 1H), 6.89 (d, J = 8.0 Hz, 1H), 6.74–6.66 (m, 2H), 5.93–5.82 (m, 1H), 5.18 (s, 2H), 5.08–4.98 (m, 2H), 4.95 (s, 2H), 3.73 (s, 3H), 3.53 (s, 3H), 3.36–3.27 (m, 2H), 3.30 (s, 3H); 13C NMR (101 MHz, CDCl3) δ ppm: 166.61, 165.38, 155.31, 151.63, 150.50, 148.47, 141.95, 139.74, 137.04, 136.85, 122.19, 120.73, 116.35, 112.79, 107.06, 61.55, 55.86, 47.03, 40.07, 29.92, 27.94; displayed a molecular ion at 442.43 m/z calculated for the molecular formula anal. calcd for C21H22N4O7, C, 57.01; H, 5.01; N, 12.66; found C, 56.86; H, 4.64; N, 13.15.
A mixture of hydrazide 7 (1.26 g, 5 mM) and potassium hydroxide (0.28, 5 mM) was formed by dissolving them in absolute ethanol (50 ml), carbon disulfide (0.82 ml) was added, and reaction mixture was refluxed for 6 hours. Completion of reaction was monitored through TLC, then the reaction mixture was cooled, and excess chilled water was added, acidified with dilute hydrochloric acid (10%) and the precipitate of 1,3,4-oxadiazole derivative of acefylline 8 was formed which were filtered off under vacuum to give off white powder (m.p. 252–253 °C) (reported m.p. 254 °C).69
In a 25 ml round flask, a mixture of bromoacetate 5a–d (0.2 mM), 1,3,4-oxadiazole derivative 8 (60 mg, 0.2 mM), and potassium carbonate (50 mg, 0.2 mM) in DMF (5 ml) was stirred for 24 hours in room temperature. The reaction mixture was poured into crushed ice with stirring and extracted with ethyl acetate (3 × 25 ml). The organic layer was dried over anhydrous sodium sulphate, filtered and the solvent was removed under reduced pressure to obtain semisolid residue, which was chromatographed on silica gel column using methylene chloride: ethyl acetate gradient elution from 100% to 60% methylene chloride as eluent to give compounds (9a–d).
4.1.6.1. 2-Isopropyl-5-methylcyclohexyl-2-((5-((1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)methyl)-1,3,4-oxadiazol-2-yl)thio)acetate (9a). Yield 75%, [α]25D = −46 (c 15 g l−1, EtOH). 1H NMR (400 MHz, CDCl3) δ ppm: 7.68 (s, 1H), 5.73 (s, 2H), 4.67 (td, J = 10.9, 4.4 Hz, 1H), 3.97 (s, 2H), 3.54 (s, 3H), 3.33 (s, 3H), 1.97–1.87 (m, 1H), 1.77 (ddd, J = 11.3, 8.4, 5.7 Hz, 2H), 1.66–1.56 (m, 2H), 1.38–1.21 (m, 1H), 1.04–0.90 (m, 2H), 0.82 (dd, J = 9.0, 6.7 Hz, 7H), 0.66 (d, J = 6.9 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ ppm: 166.68, 165.19, 162.15, 155.19, 151.56, 148.66, 141.35, 106.70, 76.93, 46.89, 40.58, 40.38, 34.64, 34.05, 31.39, 29.92, 28.04, 26.18, 23.30, 21.97, 20.73, 16.21; displayed a molecular ion at 490.58 m/z calculated for the molecular formula anal. calcd for C22H30N6O5S, C, 53.86; H, 6.16; N, 17.13; S, 6.54; found C, 54.09; H, 5.62; N, 17.42; S, 6.63.
4.1.6.2. 2-Isopropyl-5-methylphenyl-2-((5-((1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)methyl)-1,3,4-oxadiazol-2-yl)thio)acetate (9b). Yield 55%, 1H NMR (400 MHz, CDCl3) δ ppm: 7.68 (s, 1H), 7.12 (d, J = 7.9 Hz, 1H), 7.01–6.94 (m, 1H), 6.78–6.73 (m, 1H), 5.73 (s, 2H), 4.24 (s, 2H), 3.53 (s, 3H), 3.33 (s, 3H), 2.89 (p, J = 6.9 Hz, 1H), 2.23 (s, 3H), 1.08 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ ppm: 166.10, 164.89, 162.40, 155.18, 151.54, 148.61, 147.58, 141.33, 136.84, 136.82, 127.73, 126.60, 122.23, 106.69, 40.43, 34.26, 29.95, 28.04, 26.98, 23.05, 20.83; displayed a molecular ion at 484.53 m/z calculated for the molecular formula anal. calcd for C22H24N6O5S, C, 54.54; H, 4.99; N, 17.35; S, 6.62; found C, 54.56; H, 5.4; N, 17.43; S, 7.04.
4.1.6.3. 4-Formyl-2-methoxyphenyl-2-((5-((1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)methyl)-1,3,4-oxadiazol-2-yl)thio)acetate (9c). Yield 15%, 1H NMR (400 MHz, CDCl3) δ ppm: 9.87 (s, 1H), 7.69 (s, 1H), 7.40 (d, J = 6.9 Hz, 2H), 7.23–7.15 (m, 1H), 5.73 (s, 2H), 4.25 (s, 2H), 3.79 (s, 3H), 3.50 (s, 3H), 3.31 (s, 3H); 13C NMR (101 MHz, CDCl3) δ ppm: 190.97, 164.99, 164.62, 162.53, 155.17, 151.60, 151.51, 148.69, 144.26, 141.44, 135.60, 124.68, 123.15, 110.89, 106.68, 56.18, 40.41, 33.88, 29.89, 28.02; displayed a molecular ion at 486.46 m/z calculated for the molecular formula anal. calcd for C20H18N6O7S, C, 49.38; H, 8.23; N, 3.69; S, 4.22; found C, 48.82; H, 7.64; N, 3.76; S, 4.35.
4.1.6.4. 4-Allyl-2-methoxyphenyl-2-((5-((1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)methyl)-1,3,4-oxadiazol-2-yl)thio)acetate (9d). Yield 25%, 1H NMR (400 MHz, CDCl3) δ ppm: 7.68 (d, J = 1.9 Hz, 1H), 6.91–6.84 (m, 1H), 6.68 (t, J = 4.6 Hz, 2H), 5.94–5.79 (m, 1H), 5.73 (d, J = 2.1 Hz, 2H), 5.07–4.98 (m, 2H), 4.26–4.19 (m, 2H), 3.69 (s, 3H), 3.58–3.47 (m, 3H), 3.35–3.27 (m, 5H); 13C NMR (101 MHz, CDCl3) δ ppm: 165.66, 164.81, 162.35, 155.18, 151.53, 150.46, 148.68, 141.40, 139.68, 137.54, 136.84, 122.11, 120.69, 116.36, 112.74, 106.69, 55.85, 40.38, 40.07, 34.00, 29.87, 28.02; displayed a molecular ion at 498.51 m/z calculated for the molecular formula anal. calcd for C22H22N6O6S. C, 53.01; H, 4.45; N, 16.86; S, 6.43; found C, 52.65; H, 4.47; N, 17.01; S, 6.82.
:1) for 3 h, then the reaction mixture was evaporated under vacuum and extracted with ethyl acetate (3 × 25 ml). The organic layer was dried over anhydrous sodium sulphate, filtered and the solvent was evaporated in vacu to obtain oily liquid residues of 10a-b which were used without further purification.On the other hands, 11 (1.3 mg, 8 mmol) was charged in phosphorus oxychloride (20 ml), then, triethylamine (1.5 ml, 12 mmol) was added drop wisely, and the mixture was refluxed for 4 h. Then the reaction mixture was distilled to give yellowish brown residue which give white needles upon crystallization from petroleum ether to give 4-chloro coumarin 12, with m.p. range of 85–87 °C (reported m.p. 84–86 °C (ref. 70)).
To a solution of 12 (540 mg, 3 mmol) in dry acetone, sodium azide (292.2 mg, 4.5 mmol) was added and stirred in room temperature for 5 h. The reaction mixture was evaporated in vacu and 50 ml water was added then extracted with ethyl acetate (3 × 25 ml). The organic layer was dried over anhydrous sodium sulphate, filtered and the solvent was evaporated in-vacu to obtain white needles of 13 with m.p. range of 160–163 °C (reported m.p. 165–167 °C (ref. 71)).
Copper sulphate pentahydrate (108.4 mg, 1.2 equivalent) and sodium ascorbate (86.12 mg, 1.2 equivalent) in water was added to a stirred solution of 14 in DMF, then an azide derivative (10a-b or 13) (100 mg, 1.1 equivalent) was added to the reaction mixture and stirred in room temperature for 48 h. The reaction mixture was poured into crushed ice with stirring and extracted with ethyl acetate (3 × 25 ml). The organic layer was dried over anhydrous sodium sulphate, filtered and the solvent was removed under reduced pressure to obtain semisolid residue, which was chromatographed on silica gel column using methylene chloride: ethyl acetate gradient elution from 100% to 60% methylene chloride as eluent to give compounds (15a–c).
4.1.8.1. 2-Isopropyl-5-methylcyclohexyl-2-(4-((2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetoxy)methyl)-1H-1,2,3-triazol-1-yl)acetate (15a). Yield 25%,[α]25D = −56.7(c 15 g l−1, EtOH). 1H NMR (400 MHz, CDCl3) δ ppm: 7.79 (s, 1H), 7.57 (s, 1H), 5.32 (s, 2H), 5.16–5.03 (m, 4H), 4.72 (td, J = 10.9, 4.4 Hz, 1H), 3.54 (s, 3H), 3.31 (d, J = 1.1 Hz, 3H), 2.00–1.89 (m, 1H), 1.72 (pd, J = 6.9, 2.5 Hz, 1H), 1.67–1.57 (m, 2H), 1.46–1.38 (m, 1H), 1.37–1.29 (m, 1H), 1.06–0.89 (m, 2H), 0.84 (t, J = 7.0 Hz, 7H), 0.68 (d, J = 6.9 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ ppm: 167.02, 165.65, 155.30, 151.62, 148.52, 141.85, 125.75, 107.19, 77.13, 59.11, 51.10, 47.41, 46.84, 40.65, 33.96, 31.38, 29.92, 27.93, 26.30, 23.29, 21.94, 20.73, 16.26; displayed a molecular at 515.57 m/z calculated for the molecular formula anal. calcd for C24H33N7O6, C, 55.91; H, 6.45; N, 19.02; found C, 55.54; H, 6.9; N, 18.81.
4.1.8.2. (1-(2-(2-Isopropyl-5-methylphenoxy)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetate (15b). Yield 15%, 1H NMR (400 MHz, CDCl3) δ ppm: 7.93 (s, 1H), 7.65 (s, 1H), 7.22 (d, J = 7.9 Hz, 1H), 7.08 (d, J = 7.9 Hz, 1H), 6.86 (s, 1H), 5.48 (s, 2H), 5.41 (s, 2H), 5.12 (s, 2H), 3.61 (s, 3H), 3.37 (s, 3H), 2.90 (p, J = 6.9 Hz, 1H), 2.32 (s, 3H), 1.17 (d, J = 6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ ppm: 167.04, 164.95, 155.26, 151.58, 148.37, 147.06, 142.39, 141.88, 137.00, 136.62, 127.99, 126.75, 125.76, 122.10, 107.11, 59.06, 53.49, 50.95, 30.00, 27.95, 27.18, 23.02, 20.82; displayed a molecular ion at 509.52 m/z calculated for the molecular formula anal. calcd for C24H27N7O6, C, 56.58; H, 5.34; N, 19.24; found C, 56.45; H, 5.58; N, 19.25.
4.1.8.3. (1-(2-Oxo-2H-chromen-4-yl)-1H-1,2,3-triazol-4-yl)methyl-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetate (15c). Yield 10%, 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.86 (s, 1H), 8.09 (s, 1H), 7.85–7.75 (m, 2H), 7.60 (d, J = 8.4 Hz, 1H), 7.45 (t, J = 7.8 Hz, 1H), 6.96 (s, 1H), 5.42 (s, 2H), 5.27 (s, 2H), 3.44 (d, J = 6.8 Hz, 3H), 3.16 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.05, 159.92, 154.98, 154.15, 151.48, 148.55, 146.30, 143.70, 142.72, 134.04, 127.35, 125.90, 125.52, 117.70, 114.78, 111.28, 106.84, 58.41, 47.62, 30.00, 27.95; displayed a molecular ion at 463.41 m/z calculated for the molecular formula anal. calcd for C24H27N7O6, C, 54.43; H, 3.7; N, 21.16; found C, 54.09; H, 3.63; N, 21.46.
4.1.9.1. E-N′-Benzylidene-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetohydrazide (18a). White powder (yield 65%); m.p. 269–270 °C (reported 272–274 °C (ref. 73)); 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.80 (s, 1H), 8.12–8.05 (m, 2H), 7.75 (d, J = 7.0 Hz, 2H), 7.47 (d, J = 6.3 Hz, 3H), 5.57 (s, 2H), 3.47 (s, 3H), 3.21 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.43, 154.95, 151.51, 148.34, 144.79, 144.13, 134.31, 130.61, 129.35, 127.41, 107.19, 47.87, 29.95, 27.91; displayed a molecular ion at 340.34 m/z calculated for the molecular formula anal. calcd for C16H16N6O3, C, 56.47; H, 4.74; N, 24.69; found C, 56.91; H, 4.33; N, 25.07.
4.1.9.2. (E)-2-(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N′-(4-hydroxybenzylidene)acetohydrazide (18b). White powder (yield 80%); m.p. 235–238 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.58 (s, 1H), 9.98 (s, 1H), 8.06 (s, 1H), 7.95 (s, 1H), 7.56 (dd, J = 9.6, 2.9 Hz, 2H), 6.87–6.79 (m, 2H), 5.51 (s, 2H), 3.46 (s, 3H), 3.21 (d, J = 1.7 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.02, 159.87, 154.92, 151.50, 148.30, 145.04, 144.12, 129.14, 125.33, 116.18, 107.18, 47.83, 29.93, 27.89; displayed a molecular ion at 356.34 m/z calculated for the molecular formula anal. calcd for C16H16N6O4, C, 53.93; H, 4.53; N, 23.58; found C, 53.91; H, 4.28; N, 24.03.
4.1.9.3. E-N′-(2,5-Dimethoxybenzylidene)-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetohydrazide (18c). White powder (yield 75%); m.p. 275–280 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.74 (s, 1H), 8.36 (s, 1H), 8.07 (d, J = 6.5 Hz, 1H), 7.40 (d, J = 3.0 Hz, 1H), 7.10–6.97 (m, 2H), 5.55 (s, 2H), 3.82 (s, 3H), 3.76 (s, 3H), 3.46 (s, 3H), 3.20 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.35, 154.90, 153.71, 152.64, 151.48, 148.30, 144.06, 140.16, 122.85, 117.76, 113.75, 109.96, 107.18, 56.66, 55.95, 47.91, 29.93, 27.89; displayed a molecular ion at 400.40 m/z calculated for the molecular formula anal. calcd for C18H20N6O5, C, 54; H, 5.04; N, 20.99; found C, 54.03; H, 5.31; N, 20.6.
4.1.9.4. E-N′-(3,4-Dimethoxybenzylidene)-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetohydrazide (18d). White powder (yield 65%); m.p. 280–282 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.68 (s, 1H), 8.08 (d, J = 3.3 Hz, 1H), 7.98 (s, 1H), 7.36 (d, J = 1.9 Hz, 1H), 7.26–7.19 (m, 1H), 7.03 (d, J = 8.3 Hz, 1H), 5.55 (s, 2H), 3.82 (d, J = 4.7 Hz, 6H), 3.46 (d, J = 3.7 Hz, 3H), 3.21 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.19, 154.95, 151.53, 151.22, 149.56, 148.36, 144.89, 144.12, 127.08, 121.89, 112.06, 109.06, 107.23, 56.07, 56.00, 47.92, 29.95, 27.90; displayed a molecular ion at 356.34 m/z calculated for the molecular formula anal. calcd for C18H20N6O5, C, 54; H, 5.04; N, 20.99; found C, 53.8; H, 5.64; N, 21.25.
4.1.9.5. (E)-2-(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N′-(3,4,5-trimethoxybenzylidene)acetohydrazide (18e). White powder (yield 75%); m.p. > 300 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.82 (d, J = 12.5 Hz, 1H), 8.09 (s, 1H), 7.98 (s, 1H), 7.05 (d, J = 13.7 Hz, 2H), 5.57 (s, 2H), 3.85 (s, 6H), 3.71 (s, 3H), 3.47 (s, 3H), 3.21 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.42, 154.94, 153.67, 151.52, 148.36, 144.64, 144.11, 139.63, 129.84, 107.22, 104.85, 104.66, 60.61, 56.43, 47.98, 29.97, 27.91; displayed a molecular ion at 430.42 m/z calculated for the molecular formula anal. calcd for C19H22N6O6, C, 53.02; H, 5.15; N, 19.53; found C, 53.26; H, 4.57; N, 19.
4.1.9.6. (E)-2-(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N′-(3-methoxybenzylidene)acetohydrazide (18f). White powder (yield 75%); m.p. 255–257 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.82 (s, 1H), 8.15 (s, 1H), 8.10 (s, 1H), 7.98 (s, 1H), 7.17–7.00 (m, 3H), 5.58 (s, 2H), 3.79 (d, J = 53.2 Hz, 6H), 3.22 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.42, 154.94, 153.67, 151.52, 148.36, 144.64, 144.11, 139.63, 129.84, 107.22, 104.85, 104.66, 56.43, 47.98, 29.97, 27.91; displayed a molecular at 370.37 m/z calculated for the molecular formula anal. calcd for C17H18N6O4, 55.13; H, 4.9; N, 22.69; found C, 55.33; H, 4.95; N, 23.1.
4.1.9.7. (E)-2-(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N′-(2-hydroxybenzylidene)acetohydrazide (18g). White powder (yield 45%); m.p. 222–225 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.71 (s, 1H), 10.11 (s, 1H), 8.37 (s, 1H), 8.07 (s, 1H), 7.77 (dd, J = 7.9, 1.7 Hz, 1H), 7.35–7.23 (m, 1H), 6.96–6.84 (m, 2H), 5.54 (s, 2H), 3.47 (s, 3H), 3.21 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.11, 156.92, 154.95, 151.51, 148.32, 144.13, 141.92, 131.86, 129.48, 126.47, 119.89 (d, J = 3.6 Hz), 116.66, 107.19, 47.84, 29.95, 27.91; displayed a molecular ion in its electron impact mass spectrum at 356.34 m/z calculated for the molecular formula anal. calcd for C16H16N6O4, 55.93; H, 4.53; N, 23.58; found C, 55.4; H, 4.47; N, 24.06.
4.1.9.8. (E)-N′-(Benzo[d][1,3]dioxol-5-ylmethylene)-2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetohydrazide (18h). Yield 25%, white powder, m.p. > 300 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.68 (s, 1H), 8.07 (s, 1H), 7.96 (s, 1H), 7.37 (s, 1H), 7.17 (d, J = 8.1 Hz, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.10 (s, 2H), 5.54 (s, 2H), 3.47 (s, 3H), 3.21 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.28, 154.93, 151.51, 149.54, 148.49, 148.33, 144.45, 144.10, 128.79, 123.75, 108.92, 107.21, 105.50, 102.05, 47.91, 29.95, 27.91; displayed a molecular ion at 384.35 m/z calculated for the molecular formula anal. calcd for C17H16N6O5, 53.12; H, 4.2; N, 21.87; found C, 53.7; H, 4.28; N, 21.33.
4.1.9.9. (E)-2-(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N′-(3-hydroxy-4-methoxybenzylidene)acetohydrazide (18i). White powder (yield 75%); m.p. 265–268 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.61 (s, 1H), 9.30 (s, 1H), 8.07 (s, 1H), 7.92 (s, 1H), 7.24 (t, J = 3.0 Hz, 1H), 7.11–7.04 (m, 1H), 6.98 (dd, J = 8.4, 4.9 Hz, 1H), 5.53 (s, 2H), 3.82 (d, J = 5.2 Hz, 3H), 3.47 (s, 3H), 3.21 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.04, 154.96, 151.52, 150.26, 148.33, 147.30, 145.05, 144.18, 127.15, 120.65, 112.61, 112.30, 107.18, 56.07, 47.78, 29.95, 27.91; displayed a molecular ion at 386.37 m/z calculated for the molecular formula anal. calcd for C17H18N6O5, C, 52.85; H, 4.7; N, 21.75; found C, 52.92; H, 5.06; N, 21.95.
4.1.9.10. (E)-2-(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N′-(4-hydroxy-3-methoxybenzylidene)acetohydrazide (18j). White powder (yield 65%); m.p. 274–276 °C (reported 278–280 °C (ref. 74)); 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.61 (s, 1H), 9.58 (s, 1H), 8.08 (d, J = 2.2 Hz, 1H), 7.94 (s, 1H), 7.35–7.26 (m, 1H), 7.11 (td, J = 8.9, 1.9 Hz, 1H), 6.84 (dd, J = 8.1, 3.4 Hz, 1H), 5.54 (s, 2H), 3.84 (s, 3H), 3.47 (s, 3H), 3.21 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.08, 154.94, 151.52, 149.39, 148.47, 148.34, 145.17, 144.13, 125.75, 121.96, 115.98, 109.87, 107.22, 56.05, 47.93, 29.96, 27.91; displayed a molecular ion at 386.37 m/z calculated for the molecular formula anal. calcd for C17H18N6O5, C, 52.85; H, 4.7; N, 21.75; found C, 53.13; H, 4.19; N, 21.46.
4.1.9.11. (E)-2-(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N′-((1E,2E)-3-Phenylallylidene)acetohydrazide (18k) C18H18N6O3. White powder (yield 45%); m.p. 240–242 °C (reported m.p. 237–239 °C (ref. 75)); 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.68 (s, 1H), 8.06 (s, 1H), 7.87 (d, J = 12.3 Hz, 1H), 7.65–7.61 (m, 3H), 7.45–7.38 (m, 2H), 7.24 (d, J = 28.9 Hz, 1H), 7.05 (dd, J = 29.9, 12.6 Hz, 1H), 5.45 (s, 2H), 3.46 (s, 3H), 3.21 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.08, 154.94, 151.50, 149.82, 148.32, 147.40, 144.18, 139.93, 136.23, 129.33, 127.62, 125.25, 107.13, 47.72, 29.95, 27.91; displayed a molecular ion at 366.38 m/z calculated for the molecular formula anal. calcd for C18H18N6O3, C, 59.01; H, 4.95; N, 22.94; found C, 58.9; H, 5.04; N, 22.38.
4.1.9.12. (E)-2-(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N′-(4-methoxybenzylidene)acetohydrazide (18l). White powder (yield 50%); m.p. 245–248 °C (reported m.p. 248–250 °C (ref. 73)); 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.7 (s, 1H), 8.1 (s, 1H), 8.0 (s, 1H), 7.7 (dd, J = 7.2, 5.1 Hz, 2H), 7.0 (dd, J = 9.0, 3.1 Hz, 2H), 5.5 (s, 2H), 3.8 (s, 3H), 3.5 (s, 3H), 3.2 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 168.1, 161.3, 154.9, 151.5, 148.3, 147.7, 144.6, 144.1, 129.0, 126.9, 114.8, 107.2, 55.8, 47.8, 29.9, 27.9; displayed a molecular ion at 370.37 m/z calculated for the molecular formula anal. calcd for C17H18N6O4, C, 55.13; H, 4.9; N, 22.69; found C, 55.34; H, 4.95; N, 23.06.
4.1.9.13. (E)-2-(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N′-(2-oxoindolin-3-ylidene)acetohydrazide (19). Yellow powder (yield 85%); m.p. 240–243 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 12.72 (s, 1H), 11.36 (s, 1H), 8.16–8.10 (m, 1H), 7.53 (dd, J = 55.5, 29.1 Hz, 2H), 7.03 (dd, J = 54.1, 28.9 Hz, 2H), 5.73 (s, 2H), 3.57 (s, 3H), 3.21 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ ppm: 162.83, 154.97, 151.50, 148.44, 144.07, 143.21, 132.48, 123.17, 121.32, 119.89, 111.78, 107.05, 47.44, 29.98, 27.92; displayed a molecular ion in its electron impact mass spectrum at 381.35 m/z calculated for the molecular formula anal. calcd for C17H15N7O4, 53.54; H, 3.96; N, 25.71; found C, 53.39; H, 3.98; N, 25.85.
4.2. Biological evaluation
The identified targets were submitted to enrichment analysis module integrated in INPUT 2.0 server where KEGG pathways analysis was preformed and most relevant BP, MF, and CC were identified to establish pathways-genes network using the most significantly enriched terms. Then protein–protein interaction network was constructed using confidence score = 0.7 as cut off and finally gene distribution analysis was performed to identify targets that are significantly correlated with pathways involved in AD.
4.3. Statistical analysis
Group comparison was conducted using GraphPad Prism version 6.01 (GraphPad Software, San Diego, CA, USA). The data were expressed as mean ± SEM and analyzed using one-way analysis of variance (one-way ANOVA). A p-value of ≤ 0.05 was considered statistically significant.4.4. In silico investigation of potential mechanism of action of acefylline hybrid
4.5. Chemical stability studies
:90 (v/v%), and the elution process was carried out at a flow rate of 1 ml min−1. To establish the calibration curve, the peak areas of acefylline hybrids 6d and 19 were plotted against the corresponding standard concentrations of acefylline hybrid in methanol solutions, ranging from 12.5 to 150 μM.Data availability
The data in this manuscript would be provided by authors upon request.Conflicts of interest
The authors declare that there is no conflict of interest, and no funding was associated with this manuscript.References
- S. A. Tatulian, Drug Discovery Today, 2022, 27, 1027–1043 CrossRef PubMed.
- A. A. Tahami Monfared, M. J. Byrnes, L. A. White and Q. Zhang, Neurol. Ther., 2022, 11, 553–569 CrossRef PubMed.
- Z.-R. Chen, J.-B. Huang, S.-L. Yang and F.-F. Hong, Molecules, 2022, 27, 1816 CrossRef CAS PubMed.
- G. D. Stanciu, A. Luca, R. N. Rusu, V. Bild, S. I. Beschea Chiriac, C. Solcan, W. Bild and D. C. Ababei, Biomolecules, 2020, 10, 40 CrossRef CAS PubMed.
- R. J. Castellani, G. Plascencia-Villa and G. Perry, Lab. Invest., 2019, 99, 958–970 CrossRef PubMed.
- K. Ceyzériat, T. Zilli, P. Millet, B. G. Frisoni, V. Garibotto and B. B. Tournier, Curr. Alzheimer Res., 2020, 17, 112–125 CrossRef PubMed.
- K. A. Walker, B. N. Ficek and R. Westbrook, ACS Chem. Neurosci., 2019, 10, 3340–3342 CrossRef CAS PubMed.
- J. Xie, L. Van Hoecke and R. E. Vandenbroucke, Front. Immunol., 2022, 12, 796867 CrossRef PubMed.
- A. Bubley, A. Erofeev, P. Gorelkin, E. Beloglazkina, A. Majouga and O. Krasnovskaya, Int. J. Mol. Sci., 2023, 24, 1717 CrossRef CAS PubMed.
- M. Singh, M. Kaur, N. Chadha and O. Silakari, Mol. Diversity, 2016, 20, 271–297 CrossRef CAS PubMed.
- N. George, M. Jawaid Akhtar, K. A. Al Balushi and S. Alam Khan, Bioorg. Chem., 2022, 127, 105941 CrossRef CAS PubMed.
- E. Mezeiova, K. Spilovska, E. Nepovimova, L. Gorecki, O. Soukup, R. Dolezal, D. Malinak, J. Janockova, D. Jun, K. Kuca and J. Korabecny, J. Enzyme Inhib. Med. Chem., 2018, 33, 583–606 CrossRef CAS PubMed.
- D. R. T. Sari and G. C. Krisnamurti, Proceeding International Conference on Religion, Science and Education, 2022, vol. 1, pp. 685–692 Search PubMed.
- L. Maia and A. De Mendonça, Eur. J. Neurol., 2002, 9, 377–382 CrossRef CAS PubMed.
- P. Londzin, M. Zamora, B. Kąkol, A. Taborek and J. Folwarczna, Nutrients, 2021, 13, 537 CrossRef CAS PubMed.
- F. M. Abdel Bar, D. M. Elimam, A. S. Mira, F. F. El-Senduny and F. A. Badria, Nat. Prod. Res., 2019, 33, 2591–2599 CrossRef CAS PubMed.
- M. Sharma, A. Sharma, V. K. Nuthakki, S. Bhatt, U. Nandi and S. B. Bharate, Drug Dev. Res., 2022, 83, 1803–1821 CrossRef CAS PubMed.
- M. Yousaf, A. F. Zahoor, S. Faiz, S. Javed and M. Irfan, J. Heterocycl. Chem., 2018, 55, 2447–2479 CrossRef CAS.
- D. V. Reshetnikov, I. D. Ivanov, D. S. Baev, T. V. Rybalova, E. S. Mozhaitsev, S. S. Patrushev, V. A. Vavilin, T. G. Tolstikova and E. E. Shults, Molecules, 2022, 27, 8787 CrossRef CAS PubMed.
- B. Neises and W. Steglich, Angew Chem. Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- X. Zhao, H. Ma, Q. Pan, H. Wang, X. Qian, P. Song, L. Zou, M. Mao, S. Xia, G. Ge and L. Yang, Drug Metab. Dispos., 2020, 48, 345–352 CrossRef CAS PubMed.
- T. Yamaji, T. Saito, K. Hayamizu, O. Yamamoto, M. Yanagisawa, N. Wasada and T. Tamura, Spectral Database for Organic Compounds, SDBS, AIST, http://sdbs.db.aist.go.jp/sdbs/cgibin/cre_index.cgi Search PubMed.
- B. Xie, X. Shi, Y. Xing and Y. Tang, Brain Behav., 2020, 10, e01601 Search PubMed.
- G. Fredman, Nat. Rev. Cardiol., 2019, 16, 259–260 CrossRef PubMed.
- I. Casserly and E. J. Topol, Lancet, 2004, 363, 1139–1146 CrossRef CAS PubMed.
- I. A. Alhadi, A. M. Al Ansari, A. F. F. AlSaleh and A. M. A. Alabbasi, BMC Endocr. Disord., 2023, 23, 34 CrossRef PubMed.
- C. Cao, J. R. Cirrito, X. Lin, L. Wang, D. K. Verges, A. Dickson, M. Mamcarz, C. Zhang, T. Mori, G. W. Arendash, D. M. Holtzman and H. Potter, J. Alzheimer's Dis., 2009, 17, 681–697 CAS.
- D. Hicks, N. Nalivaeva and A. Turner, Front. Physiol., 2012, 3, 189 CAS.
- S. M. de la Monte, J.-D. Chiche, A. von dem Bussche, S. Sanyal, S. A. Lahousse, S. P. Janssens and K. D. Bloch, Lab. Invest., 2003, 83, 287–298 CrossRef CAS PubMed.
- L. M. A. Favié, A. R. Cox, A. van den Hoogen, C. H. A. Nijboer, C. M. P. C. D. Peeters-Scholte, F. van Bel, T. C. G. Egberts, C. M. A. Rademaker and F. Groenendaal, Front. Neurol., 2018, 9, 258 CrossRef PubMed.
- W.-F. Fang, Y.-M. Chen, C.-Y. Lin, H.-L. Huang, H. Yeh, Y.-T. Chang, K.-T. Huang and M.-C. Lin, J. Inflammation, 2018, 15, 3 CrossRef PubMed.
- J.-H. Hwang, K.-J. Kim, S.-J. Ryu and B.-Y. Lee, Chem.-Biol. Interact., 2016, 248, 1–7 CrossRef CAS PubMed.
- K. Vadiei, S. D. Tucker, G. Lopcz-Berestein and K. M. Wasan, Pharmacol. Toxicol., 1996, 78, 174–180 CrossRef CAS PubMed.
- C.-H. Kang, R. G. P. T. Jayasooriya, M. G. Dilshara, Y. H. Choi, Y.-K. Jeong, N. D. Kim and G.-Y. Kim, Food Chem. Toxicol., 2012, 50, 4270–4276 CrossRef CAS PubMed.
- H. Badshah, M. Ikram, W. Ali, S. Ahmad, J. R. Hahm and M. O. Kim, Biomolecules, 2019, 9, 719 CrossRef CAS PubMed.
- H. M. Brothers, Y. Marchalant and G. L. Wenk, Neurosci. Lett., 2010, 480, 97–100 CrossRef CAS PubMed.
- G. W. Arendash, W. Schleif, K. Rezai-Zadeh, E. K. Jackson, L. C. Zacharia, J. R. Cracchiolo, D. Shippy and J. Tan, Neuroscience, 2006, 142, 941–952 CrossRef CAS PubMed.
- M. Grifman, A. Arbel, D. Ginzberg, D. Glick, S. Elgavish, B. Shaanan and H. Soreq, Mol. Brain Res., 1997, 51, 179–187 CrossRef CAS PubMed.
- M. Kolahdouzan and M. J. Hamadeh, CNS Neurosci. Ther., 2017, 23, 272–290 CrossRef PubMed.
- C. Carelli-Alinovi, S. Ficarra, A. M. Russo, E. Giunta, D. Barreca, A. Galtieri, F. Misiti and E. Tellone, Biochimie, 2016, 121, 52–59 CrossRef CAS PubMed.
- C. Fabiani, B. Biscussi, J. P. Munafó, A. P. Murray, J. Corradi and S. S. Antollini, Mol. Pharmacol., 2022, 101, 154–167 CrossRef CAS PubMed.
- D. V. Reshetnikov, I. D. Ivanov, D. S. Baev, T. V. Rybalova, E. S. Mozhaitsev, S. S. Patrushev, V. A. Vavilin, T. G. Tolstikova and E. E. Shults, Molecules, 2022, 27, 8787 CrossRef CAS PubMed.
- B. Biscussi and A. P. Murray, Chem. Proc., 2022, 12, 62 Search PubMed.
- N. Parween, A. Jabeen and B. Prasad, J. Adv. Sci. Res., 2021, 12, 8–12 CAS.
- P. Taheri, P. Yaghmaei, H. S. Tehrani and A. Ebrahim-Habibi, Neurophysiology, 2019, 51, 114–119 CrossRef CAS.
- D. Garabadu and M. Sharma, Neurotoxic. Res., 2019, 35, 848–859 CrossRef CAS PubMed.
- T. Zanikov, M. Gerasymchuk, E. Ghasemi Gojani, G. I. Robinson, S. Asghari, A. Groves, L. Haselhorst, S. Nandakumar, C. Stahl, M. Cameron, D. Li, R. Rodriguez-Juarez, A. Snelling, D. Hudson, A. Fiselier, O. Kovalchuk and I. Kovalchuk, Molecules, 2023, 28, 2624 CrossRef CAS PubMed.
- M. M. Said and M. M. A. Rabo, Arch. Ind. Hyg. Toxicol., 2017, 68, 27 CAS.
- S. Chowdhury and S. Kumar, J. Food Biochem., 2021, 45, e13571 CrossRef CAS PubMed.
- F. Topal, I. Gulcin, A. Dastan and M. Guney, Int. J. Biol. Macromol., 2017, 94, 845–851 CrossRef CAS PubMed.
- H. Genç Bilgiçli, A. Kestane, P. Taslimi, O. Karabay, A. Bytyqi-Damoni, M. Zengin and İ. Gulçin, Bioorg. Chem., 2019, 88, 102931 CrossRef PubMed.
- F.-X. Toublet, C. Lecoutey, J. Lalut, B. Hatat, A. Davis, M. Since, S. Corvaisier, T. Freret, J. Sopkova de Oliveira Santos, S. Claeysen, M. Boulouard, P. Dallemagne and C. Rochais, Molecules, 2019, 24, 2786 CrossRef PubMed.
- A. Medvedev, O. Buneeva and V. Glover, Biol.: Targets Ther., 2007, 1, 151–162 CAS.
- A. Medvedev, O. Buneeva, O. Gnedenko, V. Fedchenko, M. Medvedeva, Y. Ivanov, V. Glover and M. Sandler, in Oxidative Stress and Neuroprotection, ed. H. Parvez and P. Riederer, Springer Vienna, Vienna, 2006, DOI:10.1007/978-3-211-33328-0_11, pp. 97–103.
- A. E. Medvedev, O. A. Buneeva, A. T. Kopylov, O. V. Gnedenko, M. V. Medvedeva, S. A. Kozin, A. S. Ivanov, V. G. Zgoda and A. A. Makarov, Int. J. Mol. Sci., 2015, 16, 476–495 CrossRef PubMed.
- B. Mavroidi, A. Kaminari, D. Matiadis, D. Hadjipavlou-Litina, M. Pelecanou, A. Tzinia and M. Sagnou, Brain Sci., 2022, 12, 806 CrossRef CAS PubMed.
- E. Riazimontazer, H. Sadeghpour, H. Nadri, A. Sakhteman, T. Tüylü Küçükkılınç, R. Miri and N. Edraki, Bioorg. Chem., 2019, 89, 103006 CrossRef CAS PubMed.
- K. Bhagat, J. V. Singh, A. Sharma, A. Kaur, N. Kumar, H. K. Gulati, A. Singh, H. Singh and P. M. S. Bedi, J. Mol. Struct., 2021, 1245, 131085 CrossRef CAS.
- D. O. Ozgun, C. Yamali, H. I. Gul, P. Taslimi, I. Gulcin, T. Yanik and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2016, 31, 1498–1501 CrossRef CAS PubMed.
- M. E. Matheus, F. d. A. Violante, S. J. Garden, A. C. Pinto and P. D. Fernandes, Eur. J. Pharmacol., 2007, 556, 200–206 CrossRef CAS PubMed.
- A. M. Naglah, A. F. Ahmed, Z.-H. Wen, M. A. Al-Omar, A. E.-G. E. Amr and A. Kalmouch, J. Biomol. Struct. Dyn., 2016, 21, 498 Search PubMed.
- S. Boumezber and K. Yelekçi, J. Biomol. Struct. Dyn., 2023, 41, 3607–3629 CrossRef CAS PubMed.
- M. S. Islam, A. M. Al-Majid, M. Azam, V. P. Verma, A. Barakat, M. Haukka, A. A. Elgazar, A. Mira and F. A. Badria, ACS Omega, 2021, 6, 31539–31556 CrossRef CAS PubMed.
- M. Shahidul Islam, A. M. Al-Majid, M. Azam, V. Prakash Verma, A. Barakat, M. Haukka, L. R. Domingo, A. A. Elgazar, A. Mira and F. A. Badria, ChemistrySelect, 2021, 6, 14039–14053 CrossRef CAS.
- D. A. Sánchez, G. M. Tonetto and M. L. Ferreira, Catalysts, 2023, 13, 473 CrossRef.
- L. Matesic, J. M. Locke, K. L. Vine, M. Ranson, J. B. Bremner and D. Skropeta, Bioorg. Med. Chem., 2011, 19, 1771–1778 CrossRef CAS PubMed.
- N. Pirinccioglu and A. Williams, J. Chem. Soc., Perkin Trans. 2, 1998, 37–40 RSC.
- B. Manon and P. D. Sharma, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2009, 48, 1279–1287 Search PubMed.
- I. Shahzadi, A. F. Zahoor, A. Rasul, N. Rasool, Z. Raza, S. Faisal, B. Parveen, S. Kamal, M. Zia-ur-Rehman and F. M. Zahid, J. Heterocycl. Chem., 2020, 57, 2782–2794 CrossRef CAS.
- M. L. Rao and A. Kumar, Tetrahedron, 2014, 70, 6995–7005 CrossRef CAS.
- J.-W. Zhao, Z.-H. Wu, J.-W. Guo, M.-J. Huang, Y.-Z. You, H.-M. Liu and L.-H. Huang, Eur. J. Med. Chem., 2019, 181, 111520 CrossRef PubMed.
- R. R. Ruddarraju, A. C. Murugulla, R. Kotla, M. C. B. Tirumalasetty, R. Wudayagiri, S. Donthabakthuni, R. Maroju, K. Baburao and L. S. Parasa, Eur. J. Med. Chem., 2016, 123, 379–396 CrossRef CAS PubMed.
- J. Klosa, Arch. Pharm., 1955, 288, 114–119 CrossRef CAS PubMed.
- V. K. Gopinatha, K. Mantelingu and K. S. Rangappa, J. Chin. Chem. Soc., 2020, 67, 1453–1461 CrossRef CAS.
- A. M. Alafeefy, S. I. Alqasoumi, S. G. Abdel hamid, K. E. El-Tahir, M. Mohamed, M. E. Zain and A. S. Awaad, J. Enzyme Inhib. Med. Chem., 2014, 29, 443–448 CrossRef CAS PubMed.
- X. Wang, Y. Shen, S. Wang, S. Li, W. Zhang, X. Liu, L. Lai, J. Pei and H. Li, Nucleic Acids Res., 2017, 45, W356–W360 CrossRef CAS PubMed.
- D. Gfeller, O. Michielin and V. Zoete, Bioinformatics, 2013, 29, 3073–3079 CrossRef CAS PubMed.
- X. Li, Q. Tang, F. Meng, P. Du and W. Chen, Comput. Struct. Biotechnol. J., 2022, 20, 1345–1351 CrossRef PubMed.
- F. F. El-Senduny, A. A. Elgazar, H. A. Alwasify, A. Abed, M. Foda, S. Abouzeid, L. Lewerenz, D. Selmar and F. Badria, Planta Med., 2023, 89, 964–978 CrossRef CAS PubMed.
- D. M. Elimam, A. A. Elgazar, F. F. El-Senduny, R. A. El-Domany, F. A. Badria and W. M. Eldehna, J. Enzyme Inhib. Med. Chem., 2022, 37, 39–50 CrossRef CAS PubMed.
- L.-N. Fang, M.-Q. Mao, X.-H. Zhao, L. Yang, H. Jia and S.-Y. Xia, J. Pharm. Biomed. Anal., 2019, 174, 220–225 CrossRef CAS PubMed.
- B. V. Dhokchawle, S. J. Tauro and A. B. Bhandari, Drug Res., 2016, 66, 46–50 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04867e |
| This journal is © The Royal Society of Chemistry 2023 |