
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphorylcholine-grafted graphene oxide loaded with irinotecan for potential oncology therapy
Jia Cuia,
Ziyi Zhanga,
Han Zhonga and
Tao Zhang *ab
*ab
aCollege of Engineering and Applied Sciences, Nanjing University, Nanjing 210023, China. E-mail: ztnj@nju.edu.cn
bWuxi Xishan NJU Institute of Applied Biotechnology, Wuxi, 214105, China
First published on 2nd October 2023
Abstract
2-Methacryloyloxyethyl phosphorylcholine (MPC) zwitterions were modified onto self-made graphene oxide (GO) through the atom transfer radical polymerization method. The chemical structures of the products were verified using Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy, nuclear magnetic resonance spectroscopy (NMR), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), etc. It was found that the modified GO (GO-PCn) is well dispersed in water with an average hydrodynamic diameter of about 170 nm. By utilizing the 2D planar structure of this modified graphene, the irinotecan@GO-PCn composite can be loaded with about 20% of irinotecan via π–π stacking interaction and exhibit pH-sensitive drug release performance, releasing faster in the acidic environment. The in vitro cytotoxicity assessments confirmed that GO-PCn composed of phosphorylcholine moiety represented low cytotoxicity and acted as a certain effect on reducing the acute toxicity of irinotecan, which established a foundation for further studies of the system in oncology therapy.
1. Introduction
Graphene is a planar polycyclic aromatic hydrocarbon atomic crystal formed by interconnecting sp2 carbon atoms.1–3 Its two-dimensional planes tend to interact with other molecules through intermolecular forces and π–π stacking, thus forming a variety of hybrid materials, of which loading drugs is a typical class of potential applications.4–7 Meanwhile, the stable structure makes graphene potentially compatible with carbon-based lifeforms, and previous studies have proved that graphene presented relatively low biological toxicity compared to single carbon nanotubes.8–10 Particularly, well-designed modifications on graphene,11–13 especially on the defective structures at the edges of graphene oxide (GO) sheets prepared by wet chemical methods, can greatly improve the biocompatibility and reduce the potential biotoxicity of graphene, which may play an unexpectedly good role in the delivery of drugs, especially chemotherapeutic agents for tumors. Dai14 et al. first prepared graphene oxide with aminated polyethylene glycol successfully, the obtained product exhibited good water solubility and biocompatibility. And then the model drug SN-38 was successfully loaded on the graphene surface by the principle of π–π stacking, thus opening the way for graphene as a drug carrier. In the last decades, graphene and GO have emerged as important members with unique properties among the many widely studied drug-delivery nanomaterials.15,16In our previous research, based on 2-methacryloyloxyethyl phosphorylcholine (MPC), we conducted a series of studies on carbon nanomaterials and found that hydrophilic phosphorylcholine can be constructed on carbon nanotubes (CNT)17 and GO18 with structural defects by simple grafting or atom transfer radical polymerization (ATRP) to form a pseudo-cellular membrane structure similar to mammalian cell membrane bilayers, thereby improving their biocompatibility.19 Meanwhile, the hydrophilicity of phosphorylcholine moiety also reverses the problem that carbon nanomaterials are difficult to study and apply in living systems due to hydrophobicity, and gives these carbon materials good dispersion and stability in aqueous systems, thus making them more favorable to be used in the delivery of antitumor drugs.
In this paper, a novel complex was prepared by complexing irinotecan with phosphorylcholine-modified graphene oxide (irinotecan@GO-PCn) via π–π stacking. Irinotecan, a semi-synthetic, water-soluble camptothecin derivative, is a DNA topoisomerase I inhibitor that has effects in the S and G2 phases of cells.20 However, clinical side effects of irinotecan, such as delayed diarrhea and neutropenia, are also caused by its active metabolite SN-38.21,22 Therefore, how to effectively utilize irinotecan is a concern in the development of clinical therapeutic regimens. The development of new drug carriers for irinotecan with reduced toxicity but not reduced efficacy is an idea that deserves attention, and this paper aims to attempt to use phosphorylcholine-modified GO as a drug delivery carrier for irinotecan and to make a preliminary assessment of the potential of this composite system in tumor therapy by studying its interactions with cells. Based on the adequate characterizations of the complex, the cytotoxicity of the carrier and the preliminary in vitro antitumor effects of the complex were evaluated using L929 and HepG2 cell lines to lay the foundation for further studies. Although there are some reports on the formation of complexes between graphene and irinotecan,23–27 for phosphorylcholine-modified graphene oxide, the formation and the study on the complexes with irinotecan have not been reported.
2. Materials and methods
2.1 Materials and instruments
The graphite powder with a purity > 99.85% was purchased from Shanghai Huayuan Chemical Co., Ltd., and 2-methacryloyloxyethyl phosphorylcholine (MPC) with a purity > 97% was purchased from Nanjing Joy-Nature Institute of Technology. 2,2-Bipyridine with purity > 99.0% was purchased from TCI (Shanghai) Chemicals Co., Ltd. Thionyl chloride, hydrogen peroxide (H2O2, 30%), concentrated sulfuric acid (H2SO4), hydrochloric acid (HCl), potassium permanganate (KMnO4), ethanol amine (EA), triethylamine (TEA), cuprous bromide (CuBr), 2-bromoisobutyryl bromide, tetrahydrofuran (THF), methanol and other reagents required for synthesis are all analytical reagent, purchased from Sinopharm Chemical Reagent Co., Ltd. and used without more purification. Irinotecan hydrochloride (purity not less than 98%) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. and stored under −20 °C, and only the freshly prepared solution was used.The Fourier transform infrared spectra (FTIR) were recorded on a PE-GX spectrometer (PerkinElmer, Boston, MA, USA) at room temperature on KBr pellets with a sample concentration of ∼1% from 4000 to 400 cm−1 with a resolution of 4 cm−1. A LabRAM Aramis Raman Spectrometer (LabRAM HORIBA Jobin Yvon, Edison, NJ, USA) excited by the 532 nm coherent line of a Nd:YAG laser was used to determine the carbon structure of the samples at room temperature, The samples were prepared by casting suspensions onto the silicon substrates and drying at 60 °C for 24 h. Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker ARX 500 NMR spectrometer (Bruker, Karlsruhe, Germany) using D2O and DMSO-d6 as the solvent. X-ray diffraction (XRD) measurements were performed on a Rigaku ULTIMA-3 setup with a Mar 345 image plate as the detector, the Cu Kα as the source (wavelength of 0.1542 nm), the recorded region of 2θ = 5° to 40°, and a scanning speed of 2° min−1. X-ray photoelectron spectra (XPS) of samples were recorded on a VG Scientific ESCA Lab MK-II spectrometer (West Sussex, England) equipped with a monochromatic Mg-Kα X-ray source. XPS Peak software (v 4.1) was used to analyze and deconvolute the XPS peaks, and the peak deconvolutions were performed using fixed 80% Lorentzian–Gaussian components for C 1s and 0% for O 1s after a Shirley background subtraction. The thermogravimetric analysis (TGA) was conducted on a Netzsch STA409PC thermogravimetric analyzer (Selb, Germany) in an N2 atmosphere with a heating rate of 20 °C min−1 from room temperature to 500 °C. Both particle size and particle charge (zeta potential) were measured by a Malvern Zetasizer Nano ZS in an aqueous solution at room temperature and every measurement was repeated three times.
The L929 mouse fibroblast cells and HepG2 human hepatocellular carcinoma cells are both obtained from Zhongyuan Co., Ltd. (Beijing, China), which redistributes cell lines that initially come from ATCC (Manassas, VA, USA). The cells were stored in a −80 °C freezer and resuscitated before use. Dulbecco's modified Eagle's medium (DMEM, with 4.5 g/L-glucose and L-glutamine) was purchased from Thermo Fisher Scientific (China) Co. Ltd. (Beijing, China). Fetal bovine serum (FBS) was obtained from Hangzhou Sijiqing Biological Engineering Materials Co., Ltd. (Hangzhou, China) and was heated at 56 °C for 30 minutes and then frozen at −20 °C before use. 0.25% trypsin–EDTA (1×) with phenol red and penicillin/streptomycin (100×) were purchased from Life Technologies Co. (Shanghai, China). Cell Counting Kit-8 (CCK-8), with chemical name 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfonic acid benzene)-2H-tetrazolium monosodium salt, were purchased from Dojindo Molecular Technologies (Shanghai) Co. Ltd. (Shanghai, China). The disposable plastic culture flasks from Corning were used for all cell experiments; while 24-well and 96-well cell culture plates were purchased from Greiner AG. The cells used in the cytotoxicity evaluations were cultivated in a CO2 incubator (Heracell model 150i; Thermo Scientific, USA), and the evaluations were carried out with an RT-6000 micro-plate reader (Rayto Ltd., Shenzhen, China) at a wavelength of 450 nm.
2.2 Preparation of modifications of graphene oxide
GO was prepared by the modified Hummers' method and GO-PCn was synthesized by ATRP as shown in Fig. 1. The processes include chloride acetylation, hydroxylation of GO, preparation of ATRP precursors, and ATRP of MPC moiety. In detail, 100 mg of GO and 50 mL of SOCl2 were mixed and ultrasonically dispersed for 3 h in a dry flask and then heated to reflux for 24 h. SOCl2 was then removed by spin evaporation, 100 mL of EA was added dropwise under the cooling of cold water and ultrasonic dispersion, and then the reaction was continued with magnetic stirring for 24 h. The intermediate product was precipitated by adding acetone and separated by centrifugation, then washed with ethanol, centrifuged, and dried under vacuum after three iterations. The hydroxylated intermediate product GO-EA was obtained.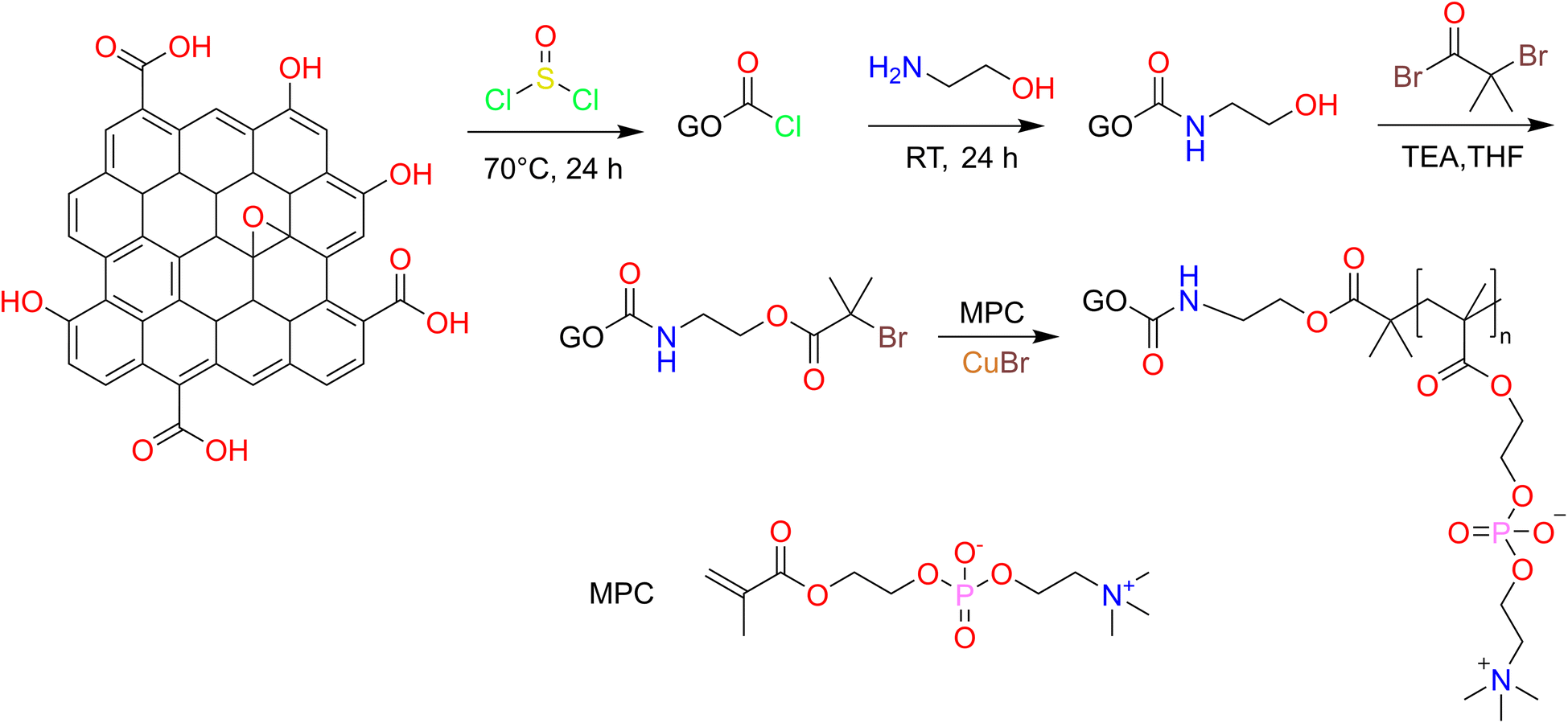 | ||
| Fig. 1 Synthesis route of GO-PCn by atom transfer radical polymerization method. | ||
The above GO-EA was transferred to a 250 mL two-necked flask containing 50 mL of THF and 10 mL of TEA, stirred magnetically for 30 min at room temperature, then 100 mg of 2-bromoisobutyryl bromide was added and stirred for 30 h. After the reaction, 100 mL of pure water was added, centrifuged and the products were washed repeatedly with ethyl ether 3 times, dried under vacuum, and then re-dispersed ultrasonically in 50 mL methanol and 1 g MPC was added, and high purity N2 was pumped for 10 min to remove the air. Then 40 mg of CuBr and 100 mg of bipyridine were rapidly added and the reaction was stirred magnetically for 24 h at room temperature in an N2 atmosphere then the acetone was added to terminate the reaction. The crude products were obtained by centrifugation and washing with acetone several times. After dialysis for 3 days and lyophilization, the GO-PCn was obtained.
2.3 Loading and release of irinotecan on phosphorylcholine grafted hydrophilic graphene oxide
The GO-PCn dispersion was prepared by sonication in pure water to a concentration of 50 μg mL−1 and set aside. In addition, 300 μg mL−1 of irinotecan hydrochloride aqueous solution was prepared and diluted into a series of concentrations (150, 100, 50, 40, 30, 20, 10, 8, 6, 4, and 2 μg mL−1). 12 parts of 5 mL GO-PCn dispersions were added to different concentrations of 5 mL irinotecan solution, mixed well by sonication, and incubated at room temperature for 24 h. The supernatants were then centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and the solids were washed with 1 mL of pure water each time and combined with the supernatants, and the black solids at the bottom were collected, lyophilized and stored in a −20 °C refrigerator. The contents of irinotecan in the supernatants were detected with high-performance liquid chromatography (HPLC), and the drug loading rate (DL) and drug loading efficiency (EE) on GO-PCn were calculated. The HPLC was conducted with a C18 reversed-phase column (filled with 5 μm silica gel in a 4.6 × 250 mm column) with a mobile phase of methanol–acetonitrile–PBS (55:5:45) at a flow rate of 1 mL min−1 and the injection volume was set to 10 μL. The mass of irinotecan in each specimen was calculated according to the standard curve of the calibration, and the DL and EE were calculated according to the following formula.
000 rpm and the solids were washed with 1 mL of pure water each time and combined with the supernatants, and the black solids at the bottom were collected, lyophilized and stored in a −20 °C refrigerator. The contents of irinotecan in the supernatants were detected with high-performance liquid chromatography (HPLC), and the drug loading rate (DL) and drug loading efficiency (EE) on GO-PCn were calculated. The HPLC was conducted with a C18 reversed-phase column (filled with 5 μm silica gel in a 4.6 × 250 mm column) with a mobile phase of methanol–acetonitrile–PBS (55:5:45) at a flow rate of 1 mL min−1 and the injection volume was set to 10 μL. The mass of irinotecan in each specimen was calculated according to the standard curve of the calibration, and the DL and EE were calculated according to the following formula.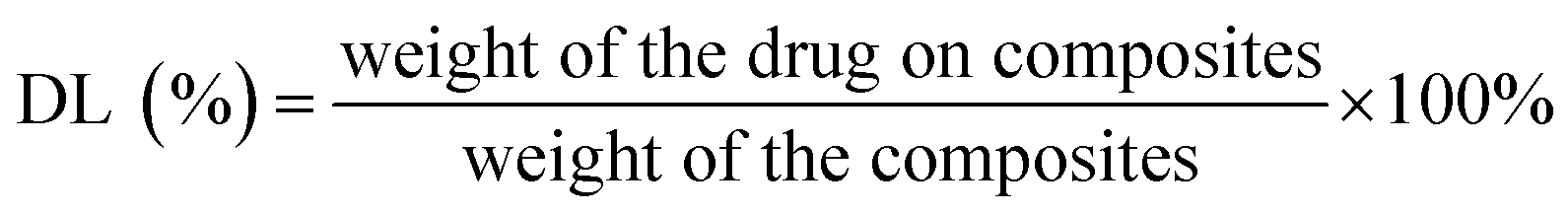
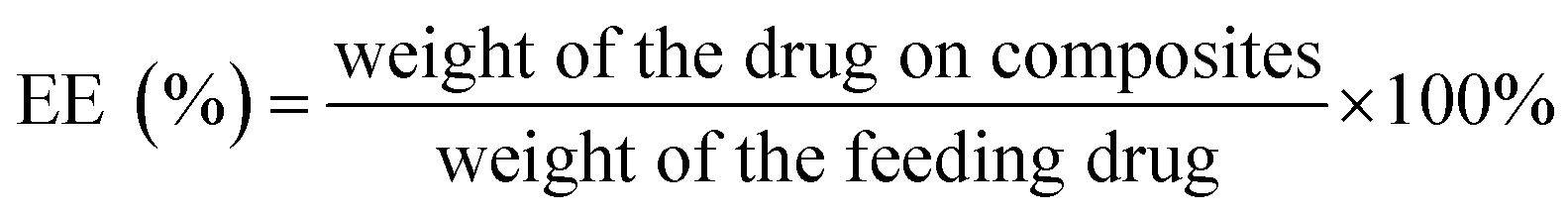
18 samples of 2 mg of the above-mentioned irinotecan@GO-PCn were accurately weighed and divided into 3 groups of 6 samples each under acidic, neutral, and basic conditions, dispersed in 2 mL of PBS with pH 5.0, 7.4, and 9.0, respectively, packed into dialysis bags with a cut-off molecular weight of 3000, submerged in glass vials containing 3 mL of PBS with the same pH value, and incubated at 37 °C with continue shaking. At every established time (0.5, 1, 1.5, 2, 4, 8, 12, 24, 36, 48, 60, and 72 h), 1 mL of PBS was pipetted from each bottle and 1 mL of PBS was supplemented. The amount of irinotecan was measured under the aforementioned HPLC conditions and the drug release curves were plotted.
2.4 The method of cell culture and cytotoxicity assay
L929 cells and HepG2 cells were recovered and incubated in DMEM medium containing 15% fetal bovine serum (FBS) and 1% antibiotics in a humidified environment at a constant temperature of 37 °C in a 5% CO2 atmosphere until reach the density of 1 × 105 cells mL−1, and then inoculated into 96-well cell culture plates at 100 μL per well and then continue incubated for 24 hours to allow the cells to adhere sufficiently to the wall and then stained.Autoclaved GO and GO-PCn were dispersed in DMEM at a concentration of 1 mg mL−1 sonicated for 30 min and diluted sequentially to 5 groups of 25, 50, 75, 100, and 150 μg mL−1, respectively. Similarly, DMEM solutions of irinotecan at a concentration of 1 mg mL−1 were prepared and diluted to 10, 25, 50, 75, 100, 150, and 200 μg mL−1, respectively, and additional DMEM dispersions of irinotecan@GO-PCn at 5 mg mL−1 were prepared and diluted to 50, 125, 250, 375, 500, 750, and 1000 μg mL−1. Based on the results of drug loading rates on GO-PCn, such concentrations were set so that the actual amount of irinotecan in the loading system was comparable to the free irinotecan group in the cytotoxicity assay. A control group was also set up with DMEM only. The upper layer of medium in the 96-well plate was pipetted off and 100 μL of each of the above solutions was added, and the number of parallel samples was set to 6 and continued to culture the cells after sampling for 24 h.
After 24 h, the medium in each well of the plate was pipetted off, washed once with PBS, and 100 μL of pre-prepared 10% CCK-8 medium solution was added in each well and continued to incubate for 4 h. The optical density (OD) value of the solution at a wavelength of 450 nm was measured with a microplate reader, and the cell viability was calculated according to the following formula.
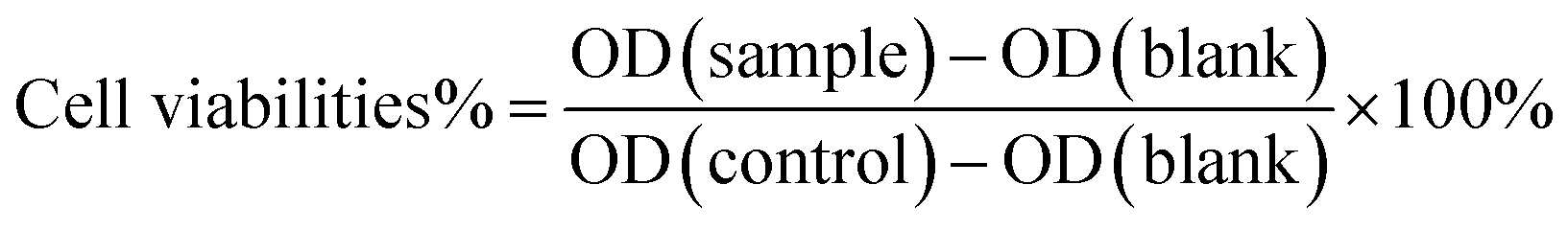
The obtained data were analyzed for statistical differences using the standard Student's t-test. p < 0.05 was considered a statistically significant difference.
3. Results and discussion
3.1 Characterization of samples
Fig. 2A shows the FTIR spectra of graphite powder, GO, and GO-PCn complexes. Among them, the IR spectrum of graphite powder is nearly a straight line, but after oxidation, various oxygen-containing groups presented on the GO sheets, such as the carbonyl groups with stretching vibration absorption peaks at 1717 and 1075 cm−1 and the hydroxyl group with absorption peak appearing at 3500 cm−1. In the GO-PCn spectrum, the absorption peak appearing at 3430 cm−1 is the absorption peak of N–H bonds and 969 cm−1 is the absorption peak of tertiary amine groups. The peak appearing at 1237 cm−1 should be attributed to the absorption of the synergistic interactions of phosphates and tertiary amine groups; the absorption peak at 1480 cm−1 is the vibrational absorption of P–O-alkyl and amino groups. These changes indicate that the phosphorylcholine moieties were successfully grafted on the GO sheets. In a comparison of the IR spectral profiles of irinotecan, GO-PCn, and irinotecan@GO-PCn, it could be seen that the absorption peaks at 1452 and 1566 cm−1 appearing in the spectra of irinotecan@GO-PCn should be attributed to the vibrational absorption peaks of the benzene ring backbone. The absorption peaks of methyl or methylene at 2966 cm−1 were significantly enhanced compared with those before the drug loading. 1128 cm−1 showed the absorption peaks of C–O bonds and 1712 cm−1 showed the vibrational absorption peaks of carbonyl groups, which are the characteristic groups of irinotecan molecule, confirming the successful loading of irinotecan on GO-PCn.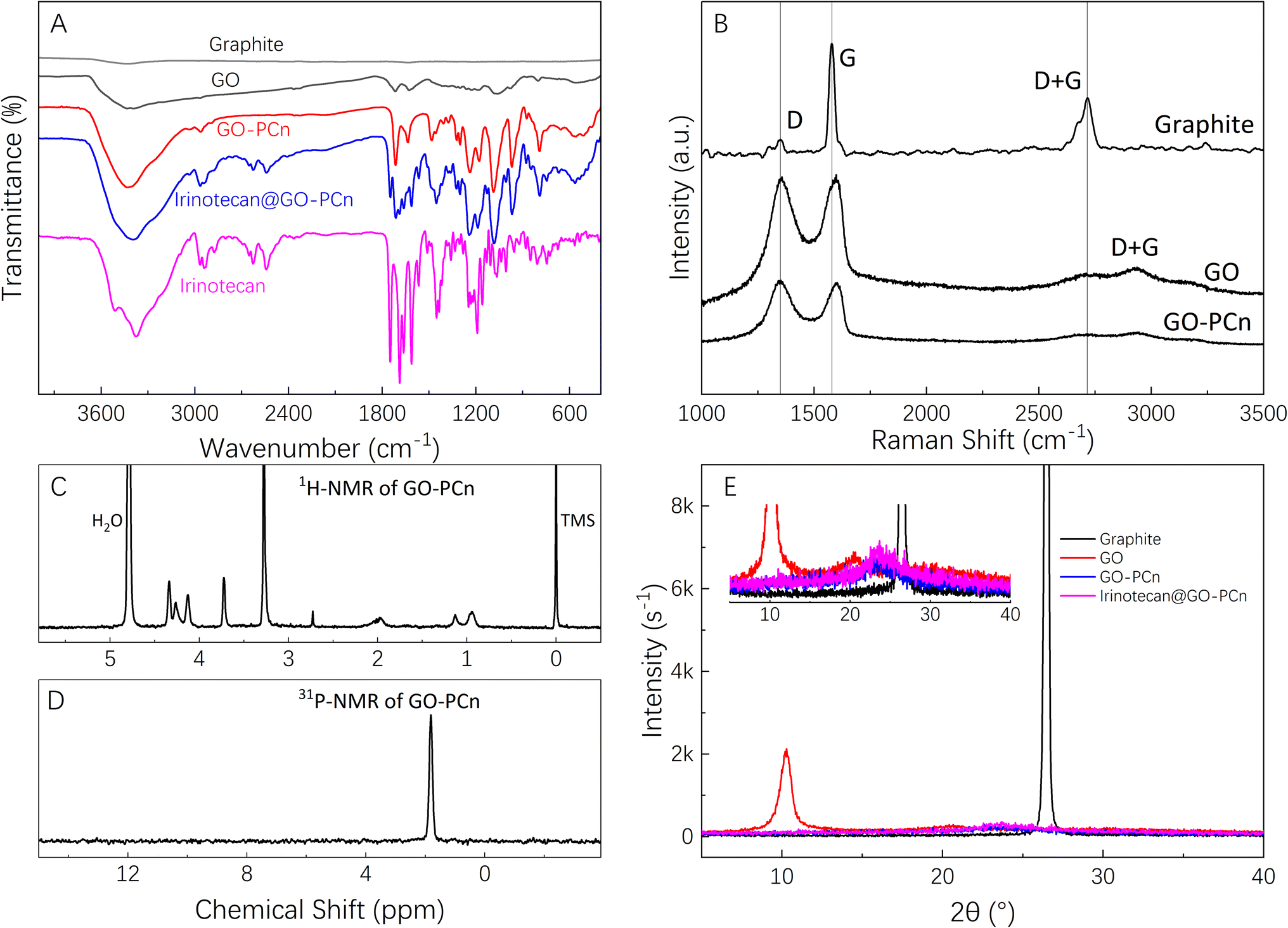 | ||
| Fig. 2 Chemical characterizations, where (A) FTIR spectra of graphite, GO, GO-PCn, irinotecan@GO-PCn, and free irinotecan; (B) Raman spectra excited at 532 nm of graphite, GO, and GO-PCn; (C) 1H-NMR in D2O, (D) 31P-NMR in d6-DMSO of GO-PCn; and (E) XRD patterns of graphite, GO, GO-PCn, and irinotecan@GO-PCn. | ||
The Raman spectra of graphite powder, GO, and GO-PCn were shown in Fig. 2B. Focus on the curve of graphite, the D-band located at 1350 cm−1 was relatively weak while the G-band located at 1580 cm−1 appeared sharp and high intensity. The D + G band located at 2700 cm−1 is an important basis for characterizing the state of graphite lamellae stacking. However, after being oxidized, the intensity of the D-band of GO increased significantly and became a sharp and strong peak while the intensity of the D + G band decreased significantly, indicating that the lamellar stacking structure of graphite was broken and split into two-dimensional GO flakes. In the Raman spectra of GO-PCn, the intensity of the D band is also significantly enhanced relative to the graphite powder, and the D + G band becomes fainter and shows a broad peak, indicating that the stacking of graphite lamellae in GO-PCn powder is further weakened and shows much of a single layer or less-layer stacking. These results presented typical of the changes in Raman spectra of GO and chemically modified GO and are consistent with the previous reports.18,28,29 Focus on the Raman spectra of graphite and graphene derivatives, the D band, the G band, and the octave-frequency band combination of the two bands (D + G) are the distinguishing bands of the Raman spectra of graphite, GO, and their derivates. The D band typically emerges at 1350 cm−1 and is activated by defects most of the time. It is caused by the symmetric stretching vibration of the radial breathing mode of the sp2 carbon atoms in the aromatic ring. The G band is most visible at about 1580 cm−1, and it is caused by the stretching vibration of the inter-atoms of the sp2 carbon atoms, which corresponds to the vibration of the E2g optical phonon in the Brillouin zone. The accumulative bands of the D and G bands tend to overlap to form the D + G band, which usually occurs near 2680–2700 cm−1 and is induced by the double resonance leaps of two phonons in carbon atoms with inverse momentum, which is a reflection of graphitic carbon atom interlayer stacking. Typically, an increase in the defects of the graphite lamellae is implied by the graphite being oxidized and chemically exfoliated, resulting in the formation of more defects and the formation of more sp3 hybridization, which provides reaction sites for the subsequent chemical modification of graphene oxide. The strength of the G band steadily diminishes during this process, representing the increasing exfoliation of the graphite lamellae. Simultaneously, the reduction in the strength of the D + G bands as the width of the half-band increases indicates that the GO produced is still multilayer graphene. If the graphene is single-layer, this band exhibits a single Lorentzian band shape with a higher intensity than the G band.30–32
Fig. 2C and D shows the 1H-NMR and 31P-NMR of GO-PCn. The peak at chemical shift of 4.70 ppm is the resonance absorption peak of D2O, while the peak at 3.36 ppm is the resonance absorption peak of the hydrogen atom located in the quaternary methyl group; the peaks at 4.18 ppm, 3.66 ppm, and 1.82 ppm are the resonance absorption peaks of the hydrogen atom of the methylene group. In the 31P-NMR spectra, there is one and only one resonance absorption peak coming from P in the phosphate structure. As for GO-PCn, only the phosphorylcholine structure contains the element of phosphorus. The presence of the phosphorus element shown in the 31P-NMR is convincing evidence for the presence of the phosphorylcholine moiety, confirming that the modification of GO with MPC yields the expected products.
The XRD patterns of graphite powder, GO, GO-PCn, and the complexes after loading with irinotecan were shown in Fig. 2E. With the modification of graphite, the interlayer expansion took place due to the introduction of various chemical groups, and thus the peak position of GO in the XRD pattern shifted to a small angle direction relative to the graphite powder. As shown in the figure, the graphite powder presented an intense and extremely sharp absorption peak near 2θ = 26°, and its layer spacing was calculated from Bragg's law to be about 0.337 nm. While in the GO, the position of this peak is shifted to 2θ = 10.3° with layer spacing about 0.86 nm. After modification on GO, almost no sharp peaks appeared on the XRD spectra of GO-PCn, indicating that the stacking structure in GO was further disrupted and single-layer or less-layer stacking appears as the main state of lamellar stacking, even the lyophilization process did not produce more stacking. Again, no significant multilayer stacking structure was observed on the XRD spectra of irinotecan@GO-PCn, indicating that the drug-loading does not affect the structures of two-dimensional planar without stacking of the already obtained GO-PCn.
Fig. 3 shows the XPS spectrum of GO and GO-PCn. Among them, Fig. 3A is the survey scan of GO, which shows that only two peaks, C 1s, and O 1s appear, corresponding to the elements of carbon and oxygen in GO, respectively. The intensive scanning for C 1s of GO shown in Fig. 3B indicated that the carbon in GO is mainly connected with other carbon atoms in C–C bonds with a binding energy of 285 eV, and also carbons in C–O or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds connected to O exist with binding energies of 287 eV and 288 eV, respectively. The intensive scanning for O 1s of GO shown in Fig. 3C, proved that all the oxygen atoms in GO are attached to carbon atoms.
O bonds connected to O exist with binding energies of 287 eV and 288 eV, respectively. The intensive scanning for O 1s of GO shown in Fig. 3C, proved that all the oxygen atoms in GO are attached to carbon atoms.
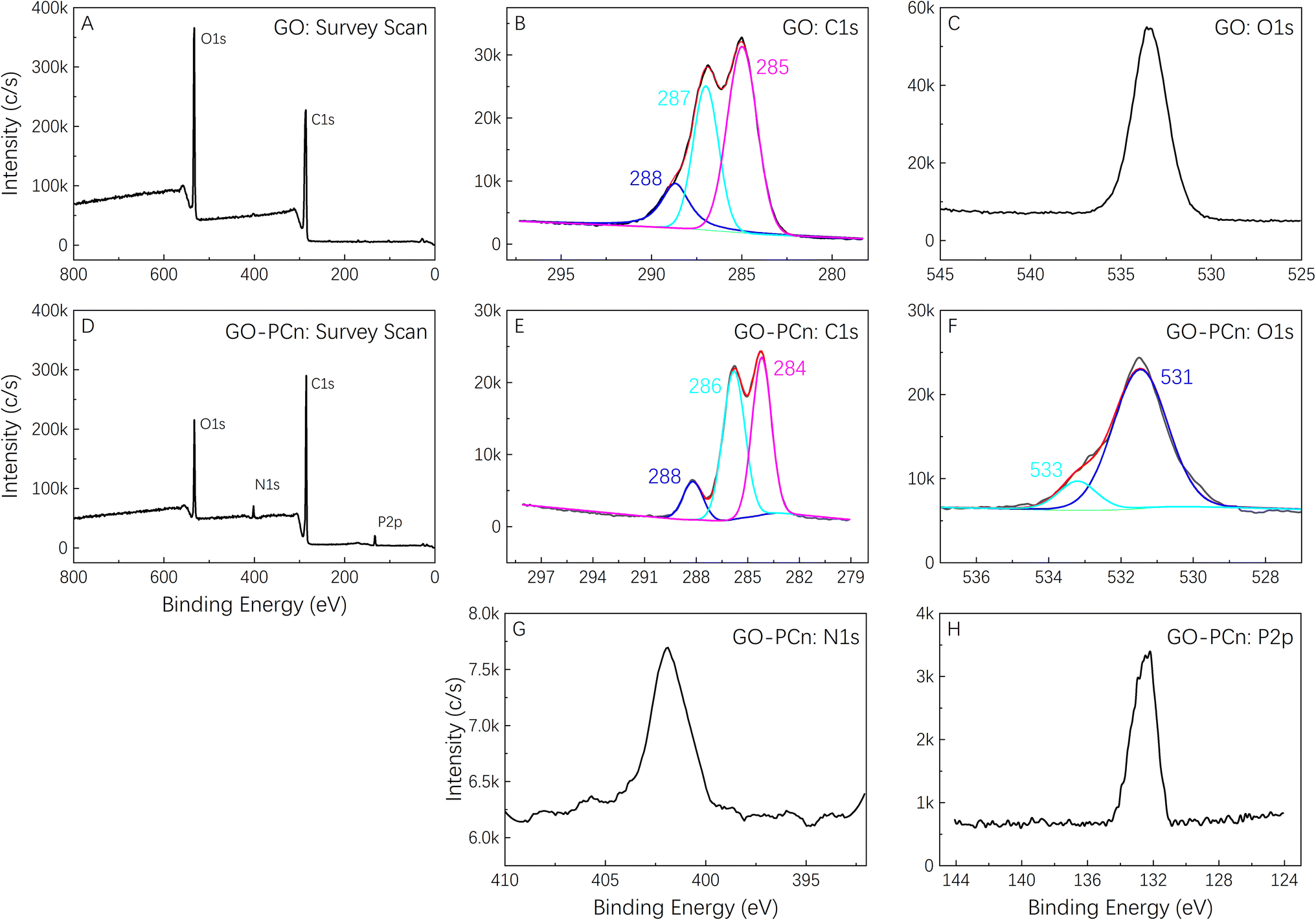 | ||
| Fig. 3 XPS Spectra of GO and GO-PCn. (A) Survey scan of GO; (B) C 1s and (C) O 1s intensive scan of GO; (D) survey scan of GO-PCn; (E) C 1s, (F) O 1s, (G) N 1s, and (H) P 2p intensive scan of GO-PCn, respectively. | ||
As for the XPS spectrum of GO-PCn, Fig. 3D showed the results of survey scanning results, indicating that besides the elements of C and O, the elements of N and P also appeared. Both elements came from the phosphorylcholine moieties introduced during the chemical modification, which also confirmed the successful modification of GO with the phosphorylcholine structure. In the intensive C 1s scanning curve shown in Fig. 3E, the binding energy of different carbon atoms in the C–C, C–O, and CO bonds still clearly appeared but slightly shifted relative to that of GO. The intensive scan spectra of O 1s in Fig. 3F showed a shoulder peak at 533 eV in addition to the oxygen that was originally present in the form of oxides. Although the attribution of this peak was still questionable, it was hypothesized that it might be related to the introduction of the phosphorylcholine structure that made an extra P–O bond in GO-PCn. Fig. 3G and H showed the intensive scans of N 1s and P 2p respectively, revealed that the peak at 402 eV corresponds to nitrogen in the quaternary amine structure, while the peak at 132 eV should be attributed to phosphorus, both of which indicated the connection of phosphorylcholine moiety on GO.
Fig. 4A showed the TGA curves of GO, GO-PCn, and the sample of irinotecan@GO-PCn produced by adequate drug feeding (irinotecan/GO-PCn = 3). The mass loss of each sample below 120 °C mainly came from the evaporation of adsorbed water. As the temperature increased to 220 °C, the oxygen-containing groups on the GO sheets gradually decomposed and the mass of the sample decreased rapidly. When the temperature reached 260 °C, the mass loss of GO slowed down significantly, and it was speculated that the mass loss until 500 °C was due to the decomposition of the carbon skeleton of the GO sheets, with a final remaining mass of about 79%. In the same temperature range, GO-PCn exhibited a similar thermal decomposition process, but with a relatively slight thermal weight loss ranging from 230 °C to 260 °C, after which it exhibited a continuous mass loss until 500 °C. This phenomenon was supposed to be caused by the gradual decomposition of the PCn structure attached to the GO sheets, with the final mass losing about 25%. Been sufficiently loaded with irinotecan, the thermal decomposition of irinotecan would be the main manifestation of the mass loss of irinotecan@GO-PCn sample, with an overall weight loss of 45% when heating up to 500 °C, and the 20% difference compared to GO-PCn suggested a 20% weight ratio of irinotecan was loaded on GO-PCn.
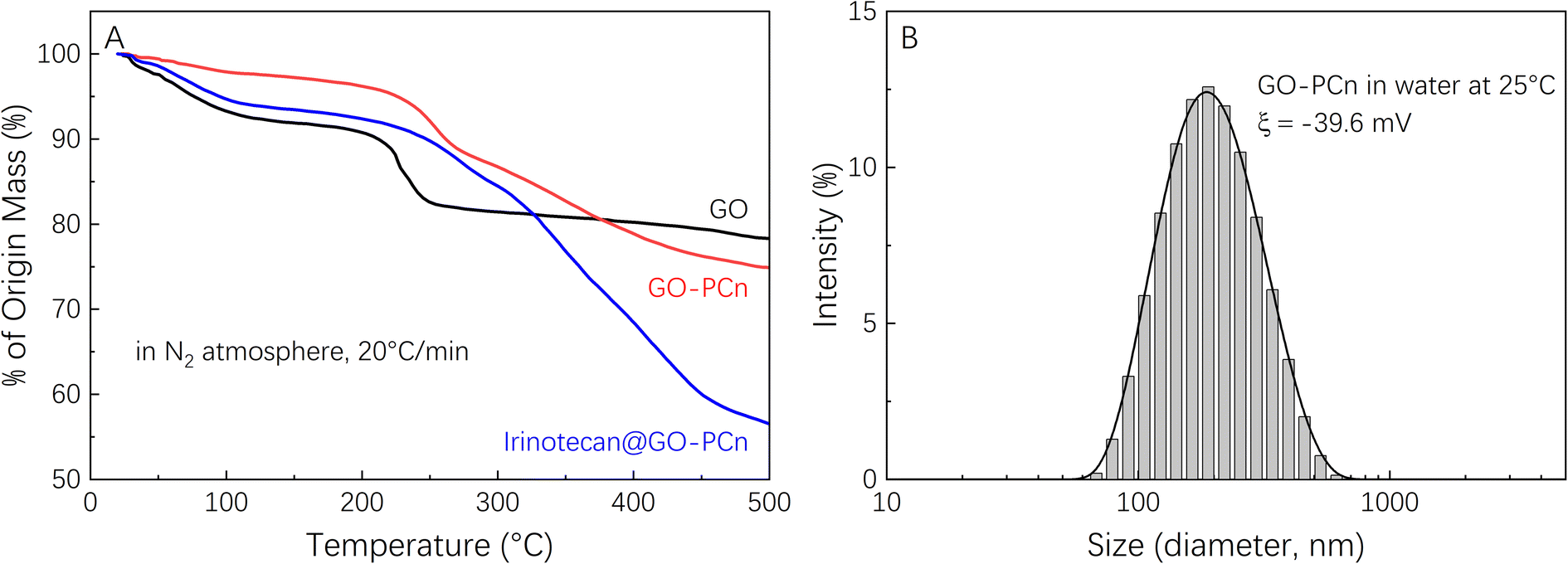 | ||
| Fig. 4 (A) Thermogravimetry analysis curves of GO, GO-PCn, and irinotecan@GO-PCn in N2 atmosphere with a heating rate of 20 °C min−1; and (B) dynamic light scattering of GO-PCn in water at room temperature. | ||
Fig. 4B exhibited the particle size distribution of GO-PCn dispersed in pure water measured by DLS and indicated that the hydrated particle size of GO-PCn ranges from 70 nm to 450 nm, and the average particle size was 174 nm. The results suggested that GO-PCn can dispersed in water wells and possess the basic possibility for biomedical research applications.
3.2 Loading and releasing of irinotecan
Fig. 5A demonstrates the drug loading rate and drug loading efficiency of GO-PCn as a drug carrier for irinotecan. Different amounts of irinotecan and GO-PCn with a series of irinotecan/GO-PCn ratios were mixed and allowed to be fully absorbed in aqueous environment through π–π stacking, irinotecan thus attached to the surface of GO sheets to form a complex for drug loading. It was found that the drug loading rate of GO-PCn increased gradually as the ratio of irinotecan to GO-PCn increased. When the ratio reached 1:1, the ratio of GO-PCn loading irinotecan was close to saturation, and the drug loading rate was about 18%. During this process, the drug loading efficiency gradually decreased, and the drug loading efficiency at close to saturation loading rate also decreased to about 16%. Continuing to increase the irinotecan/GO-PCn ratio did not significantly increase the amount of drug loaded on GO-PCn, and the drug loading rate on GO-PCn was about 20% when the sufficient loading condition of irinotecan/GO-PCn = 3 was reached, which is consistent with the 20% derived from the previous TGA analysis. This percentage of drug-loaded complex samples was also used for subsequent drug release and cytotoxicity studies.
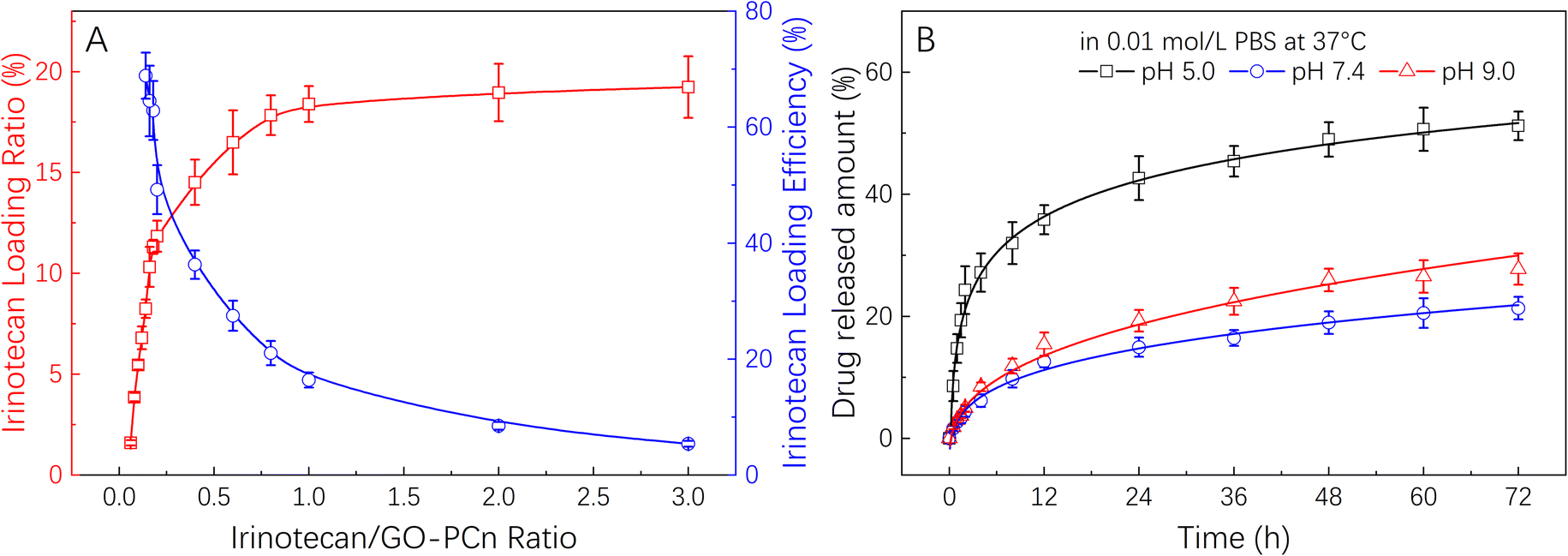 | ||
| Fig. 5 (A) Irinotecan loading on GO-PCn with different drug-carrier ratios; and (B) the release behavior of irinotecan from GO-PCn in 0.01 M PBS with different pH values at 37 °C. | ||
The drug release properties of irinotecan@GO-PCn were examined in PBS at 3 different pH values and the results of Fig. 5B were obtained. The irinotecan@GO-PCn complex exhibited pH-sensitive drug release properties. At pH 5.0, the release rate of irinotecan was significantly faster than that in pH 7.4 and pH 9.0 environments. By the release time of 72 h, about 50% of irinotecan had been released under acidic conditions, compared to only about 20% of irinotecan released under neutral and alkaline conditions. At the same time, the release rate of irinotecan was slightly higher under alkaline conditions than under neutral conditions, which was particularly evident after 12 h. The difference in release in different pH environments suggested that the loading between irinotecan and GO-PCn was mainly achieved through π–π stacking interaction, but the alkaline character of irinotecan itself makes it easy to form water-soluble salts in an acid environment, and thus faster release occurred. Moreover, uncertain interactions between irinotecan and the phospholipid and quaternary ammonium salt moieties in the GO-PCn were also supposed to work, and such charge-related interactions tend to be pH sensitive, thus also resulting in different release rates under different pH conditions. Based on the available data, we performed a kinetic analysis of the release of irinotecan from the complex under various pH settings using the Higuchi model.33,34 It was discovered that the release of irinotecan may better suit the Higuchi model in a neutral pH 7.4 environment, with the adjusted R-square of the fitted curve reaching 0.975 and the time for the release of half the amount of the drug (t1/2) being around 170 h. However, in acidic or alkaline conditions, irinotecan release deviated significantly from the Higuchi model, particularly in acidic conditions, where the corrected R-squared was only 0.906 and the t1/2 was approximately 53 hours. This was due to the drug's rapid dissolution in an acidic environment. But overall, the faster drug release in an acidic environment happens to correspond to the slightly acidic microenvironment of normal solid tumor tissues, potentially allowing this approach to release drugs more quickly in tumors and facilitate anticancer effects.
The samples of the irinotecan@GO-PCn complex were stored in a dark-protected refrigerator at −18 °C for three months before being used again for the drug release assay. Similar results to Fig. 5B were obtained, showing that the released drug did not differ from the standard irinotecan on the HPLC effusion curve, indicating that the compound is stable under these storage conditions. More research is required to confirm whether it is still stable in additional storage circumstances, such as a humid environment or room temperature.
3.3 In vitro cytotoxicity assessments
Before additional biological application investigations, the safety and biological impacts of the irinotecan@GO-PCn system should be investigated. Prior to cytotoxicity testing, hemocompatibility studies demonstrated that GO-PCn did not cause coagulation and had a hemolysis rate of less than 1%, which is consistent with earlier results for modified GO published in the literature.24,27 On this basis, we conducted a series of cytotoxicity tests on mouse fibroblast L929 and human hepatocellular carcinoma cell line HepG2 cells.Fig. 6 demonstrated the cell viability variations of both L929 and HepG2 cells to different dosages of GO and GO-PCn after 24 h exposure. Overall, it seems that neither GO nor GO-PCn exhibited significant cytotoxicity to both cells during the 24 hour exposure. Even for GO, which exhibited the greatest difference in its effect on HepG2, the cell viability was maintained at a dosage of 150 μg mL−1 for 24 h of exposure at approximately 67%. This result suggested that for both cells, GO and its derivatives have a relatively low cytotoxicity. Furthermore, no significant cytotoxic differences were exhibited between GO and GO-PCn at exposure doses below 75 μg mL−1 for both cells. Only when the dosage was further increased, GO represented slight cytotoxicity, exhibiting a statistically significant difference compared to GO-PCn, and this difference tended to widen with increasing dosage. This suggested that the introduction of phosphorylcholine moiety was beneficial in reducing the cytotoxic effect of GO. The results might be related to two factors: on the one hand, phosphorylcholine itself is the main structural feature of a phospholipid, one of the components of cell membrane, and the attachment of phosphorylcholine moiety on GO lamellae makes GO acquire cell membrane-like features, which reduced the intense reaction of cells in contact with GO-PCn, thus plays a role in improving cytocompatibility similar to cell membrane-like structures. On the other hand, the water-soluble characteristics of the phosphorylcholine structure brought significant hydrophilic changes to GO, making it easier for GO to form a hydrated construction, thus reducing the effect of hydrophobic aromatic rings on cells and the cleavage effect of the 2D planar structure of GO on cell membranes,35,36 thus reduced cytotoxicity.
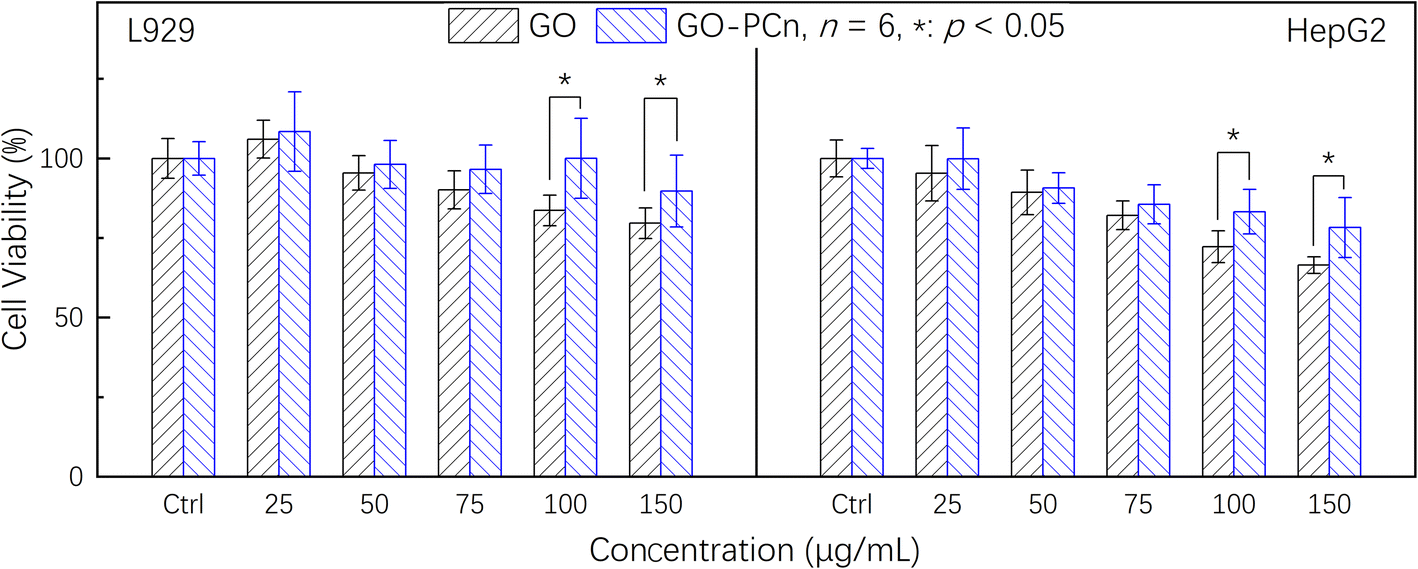 | ||
| Fig. 6 Cytotoxicity assays of GO and GO-PCn against L929 and HepG2 cells for 24 h in 5% CO2 atmosphere at 37 °C. | ||
To compare the effect of the drug-containing complexes on tumor cells and to assess their effect on normal cells, we further compared the cytotoxic effects of GO-PCn, irinotecan@GO-PCn and irinotecan on L929 and HepG2, and the results are summarized in Fig. 7. It should be noted that the staining dosage of irinotecan@GO-PCn group was set to be consistent with the dosage of free irinotecan-based on the loading rate assay, and the mass of the corresponding drug carrier GO-PCn was about 5 times the mass of the drug (20% loading rate). While the dosage of the GO-PCn group was numerically consistent with that of the free irinotecan group, the actual GO-PCn dose was only 1/5 of that of the irinotecan@GO-PCn group compared with that of the irinotecan group.
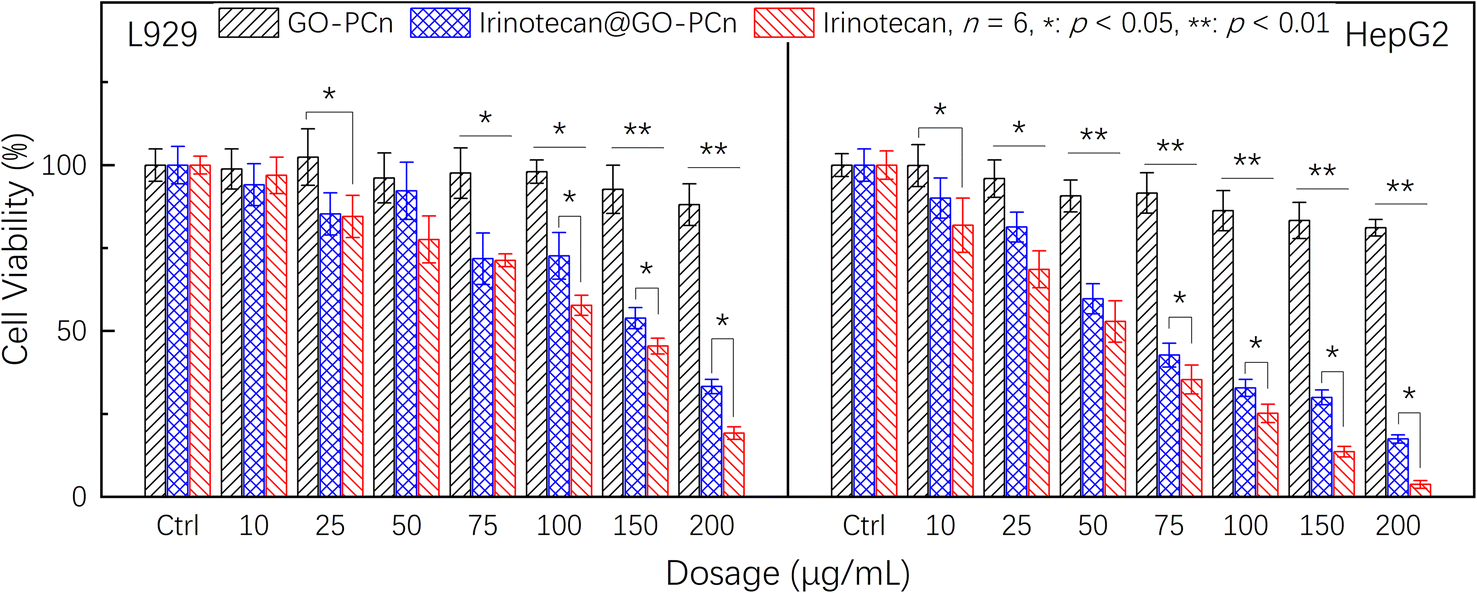 | ||
| Fig. 7 Cell viability of L929 and HepG2 cells treated with different dosages of GO-PCn, free irinotecan, and irinotecan@GO-PCn for 24 h in 5% CO2 atmosphere at 37 °C. | ||
As with the previous findings, the GO-PCn group exhibited low cytotoxicity in the dosage range of 20–200 μg mL−1 both for L929 and HepG2. The chemotherapeutic agent irinotecan, on the other hand, exhibited significant cytotoxicity, especially for HepG2, which already showed statistically significant differences in cytotoxicity at a dosage of 10 μg mL−1, and less than 5% of the cells remained viable when the dose reached 200 μg mL−1. Comparatively, L929 cells still had about 20% cell viability at this dosage, indicating that irinotecan has a more potent killing effect on tumor cells.
Compared to free irinotecan, irinotecan@GO-PCn exhibited relatively low cytotoxicity at almost all dosage levels, regardless of the cell type. In particular, at the high dosage levels, there was a statistically significant difference in the 24 hour acute cytotoxicity of both samples for both cells. Based on the current cytotoxicity data, the half-maximum inhibitory concentrations (IC50) of irinotecan@GO-PCn for L929 and HepG2 cells were estimated to be around 160 μg mL−1 and 65 μg mL−1, respectively. While for free irinotecan, the IC50 was 130 μg mL−1 and 53 μg mL−1, respectively. The reason for this difference may be related to the slow release of the irinotecan from irinotecan@GO-PCn, where only a portion of the drug-loaded on GO-PCn was released at the same drug dosage over 24 h. The actual drug dosage that interacted with the cells was relatively low and thus showed reduced cytotoxicity. This might be regarded as a certain attenuating effect of the drug complex with a carrier. The slow release of irinotecan may exhibit an inhibitory effect on tumor cell growth over a longer period.
Comparing the cytotoxicity of the GO-PCn group and the irinotecan@GO-PCn group, a significant difference in cytotoxicity was demonstrated at a slightly increased dosage. Although this is a difference due to the presence of irinotecan, it cannot be excluded they differ due to the significant difference in dosage itself due to the loading rate. The previous study confirmed that GO-PCn represented a low cytotoxicity, but the dosage of GO-PCn in the irinotecan@GO-PCn group in Fig. 7 was 5 times higher than in the neat GO-PCn group compared to the lower dosage demonstrated in Fig. 6, which still introduced some additional cytotoxicity. After all, it has been proved earlier that GO-PCn still exhibited some degree of cytotoxic effects at high dosages, but a more accurate answer still depends on more studies.
4. Conclusions
Based on the ATRP method, zwitterions containing phosphorylcholine structures were successfully grafted on GO. It was confirmed that the hydrophilic phosphorylcholine structure rendered GO better hydrophilicity and could be stably and well dispersed in an aqueous environment. The introduction of phosphorylcholine as a pseudo-cell membrane reduced the cytotoxicity of GO. Based on the π–π stacking action, the chemotherapeutic drug irinotecan was successfully loaded on GO-PCn with a drug loading rate of about 20% while the release was sensitive to the pH value and had a faster release rate in an acidic environment. In vitro studies proved that the irinotecan@GO-PCn complex had lower cytotoxicity compared to free irinotecan, and the controlled release of irinotecan also established the basis for the sustained antitumor effect of the system, which accumulated basic data for further systematic investigation and in vivo studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of the National Key R&D Program of China (2022YFA01203002), and the National Natural Science Foundation of China (81671792). The works are also partly supported by the Fundamental Research Funds for the Central Universities, MOE Key Laboratory of High-Performance Polymer Materials & Technology (Nanjing University, Grant No. 02051438002).References
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- S. Khan and M. K. Hossain, in Nanoparticle-Based Polymer Composites, Elsevier, 2022, pp. 15–54, DOI:10.1016/b978-0-12-824272-8.00009-9.
- L. Feng and Z. Liu, Nanomedicine, 2011, 6, 317–324 CrossRef CAS PubMed.
- K. Yang, L. Feng, X. Shi and Z. Liu, Chem. Soc. Rev., 2013, 42, 530–547 RSC.
- D. Placha and J. Jampilek, Nanomaterials, 2019, 9, 1758 CrossRef CAS PubMed.
- H. Huang, W. Feng and Y. Chen, Chem. Soc. Rev., 2021, 50, 11381–11485 RSC.
- Y. Zhang, S. F. Ali, E. Dervishi, Y. Xu, Z. Li, D. Casciano and A. S. Biris, ACS Nano, 2010, 4, 3181–3186 CrossRef CAS PubMed.
- Y. Chang, S. T. Yang, J. H. Liu, E. Dong, Y. Wang, A. Cao, Y. Liu and H. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef CAS PubMed.
- O. Akhavan, E. Ghaderi and A. Akhavan, Biomaterials, 2012, 33, 8017–8025 CrossRef CAS PubMed.
- A. Hirsch, J. M. Englert and F. Hauke, Acc. Chem. Res., 2013, 46, 87–96 CrossRef CAS PubMed.
- N. Lu, L. Wang, M. Lv, Z. Tang and C. Fan, Nano Res., 2019, 12, 247–264 CrossRef CAS PubMed.
- X. Zhang, C. Gong, O. U. Akakuru, Z. Su, A. Wu and G. Wei, Chem. Soc. Rev., 2019, 48, 5564–5595 RSC.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- M. I. Khan, M. I. Hossain, M. K. Hossain, M. H. K. Rubel, K. M. Hossain, A. Mahfuz and M. I. Anik, ACS Appl. Bio Mater., 2022, 5, 971–1012 CrossRef CAS PubMed.
- S. Hossen, M. K. Hossain, M. K. Basher, M. N. H. Mia, M. T. Rahman and M. J. Uddin, J. Adv. Res., 2019, 15, 1–18 CrossRef CAS PubMed.
- D. Chassoux and J. C. Salomon, Int. J. Cancer, 1975, 16, 515–525 CrossRef CAS PubMed.
- Y. Liu, Y. Zhang, T. Zhang, Y. J. Jiang and X. F. Liu, Carbon, 2014, 71, 166–175 CrossRef CAS.
- Q. Wu, L. Yin, X. Li, M. Tang, T. Zhang and D. Wang, Nanoscale, 2013, 5, 9934–9943 RSC.
- L. Marcus, S. J. Lemery, S. Khasar, E. Wearne, W. S. Helms, W. Yuan, K. He, X. Cao, J. Yu, H. Zhao, Y. Wang, O. Stephens, E. Englund, R. Agarwal, P. Keegan and R. Pazdur, Clin. Cancer Res., 2017, 23, 2924–2927 CrossRef CAS PubMed.
- C. Blandizzi, B. De Paolis, R. Colucci, G. Lazzeri, F. Baschiera and M. Del Tacca, Br. J. Pharmacol., 2001, 132, 73–84 CrossRef CAS PubMed.
- K. Fujita, Y. Kubota, H. Ishida and Y. Sasaki, World J. Gastroenterol., 2015, 21, 12234–12248 CrossRef CAS PubMed.
- T. H. Tran, H. T. Nguyen, T. T. Pham, J. Y. Choi, H. G. Choi, C. S. Yong and J. O. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 28647–28655 CrossRef CAS PubMed.
- C. Fiorica, N. Mauro, G. Pitarresi, C. Scialabba, F. S. Palumbo and G. Giammona, Biomacromolecules, 2017, 18, 1010–1018 CrossRef CAS PubMed.
- N. Karki, H. Tiwari, M. Pal, A. Chaurasia, R. Bal, P. Joshi and N. G. Sahoo, Colloids Surf., B, 2018, 169, 265–272 CrossRef CAS PubMed.
- B. Hatamluyi, Z. Es'haghi, F. M. Zahed and M. Darroudi, Sens. Actuators, B, 2019, 286, 540–549 CrossRef CAS.
- C. C. Chuang, Y. H. Lan, Y. J. Lu, Y. L. Weng and J. P. Chen, Biomater. Sci., 2022, 10, 3201–3222 RSC.
- Y. Qin, C. Y. Wang, Y. Jiang, T. Liu, J. Y. Yang, R. Lin and T. Zhang, RSC Adv., 2017, 7, 41675–41685 RSC.
- Y. Liu, H. Zhong, Y. Qin, Y. Zhang, X. F. Liu and T. Zhang, RSC Adv., 2016, 6, 30184–30193 RSC.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
- J. B. Wu, X. Zhang, M. Ijas, W. P. Han, X. F. Qiao, X. L. Li, D. S. Jiang, A. C. Ferrari and P. H. Tan, Nat. Commun., 2014, 5, 5309 CrossRef CAS PubMed.
- A. A. King, B. R. Davies, N. Noorbehesht, P. Newman, T. L. Church, A. T. Harris, J. M. Razal and A. I. Minett, Sci. Rep., 2016, 6, 19491 CrossRef CAS PubMed.
- J. Siepmann and N. A. Peppas, Int. J. Pharm., 2011, 418, 6–12 CrossRef CAS PubMed.
- D. R. Paul, Int. J. Pharm., 2011, 418, 13–17 CrossRef CAS PubMed.
- F. Xiaoli, C. Qiyue, G. Weihong, Z. Yaqing, H. Chen, W. Junrong and S. Longquan, Arch. Toxicol., 2020, 94, 1915–1939 CrossRef PubMed.
- K. Tadyszak, J. K. Wychowaniec and J. Litowczenko, Nanomaterials, 2018, 8, 944 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |