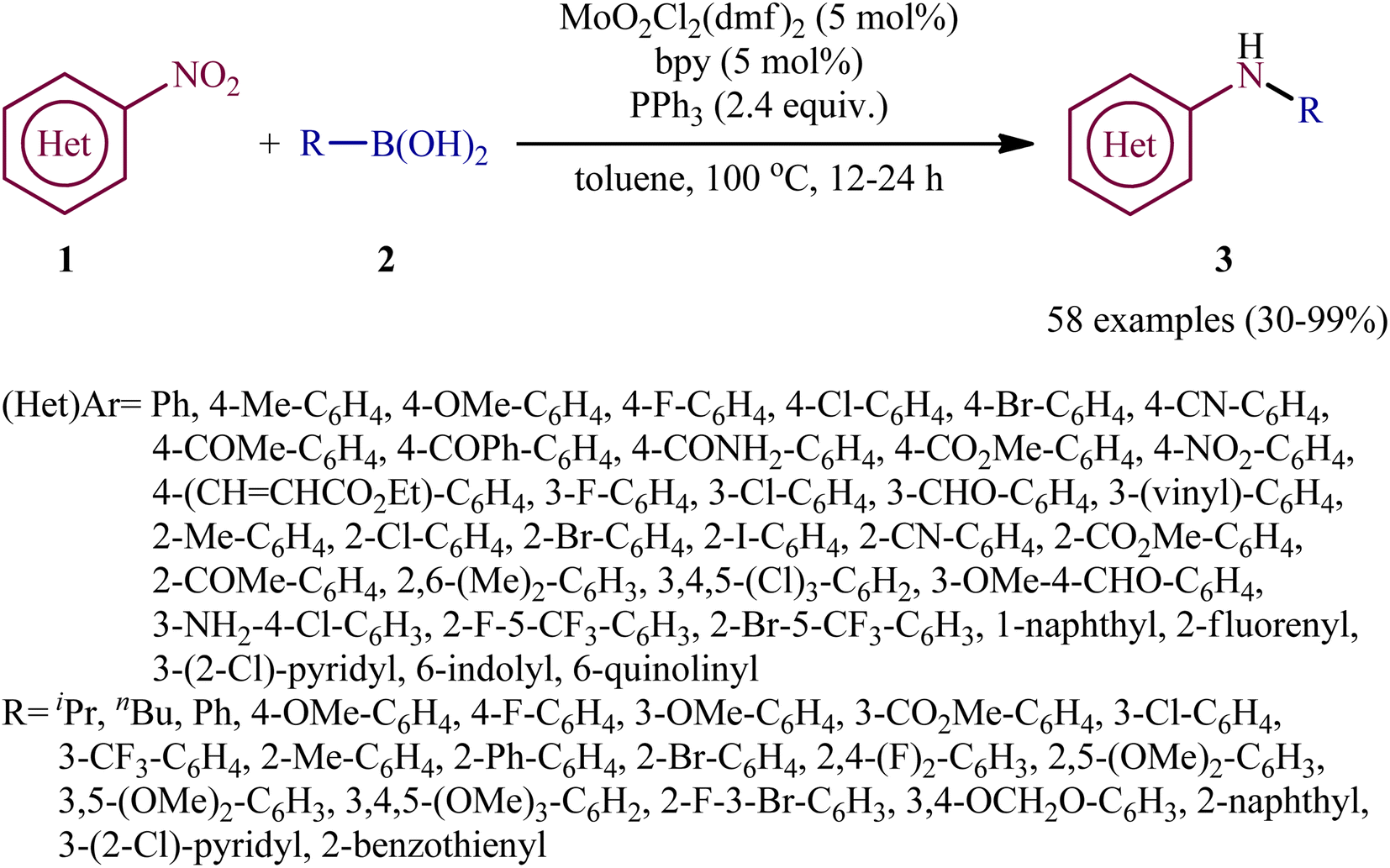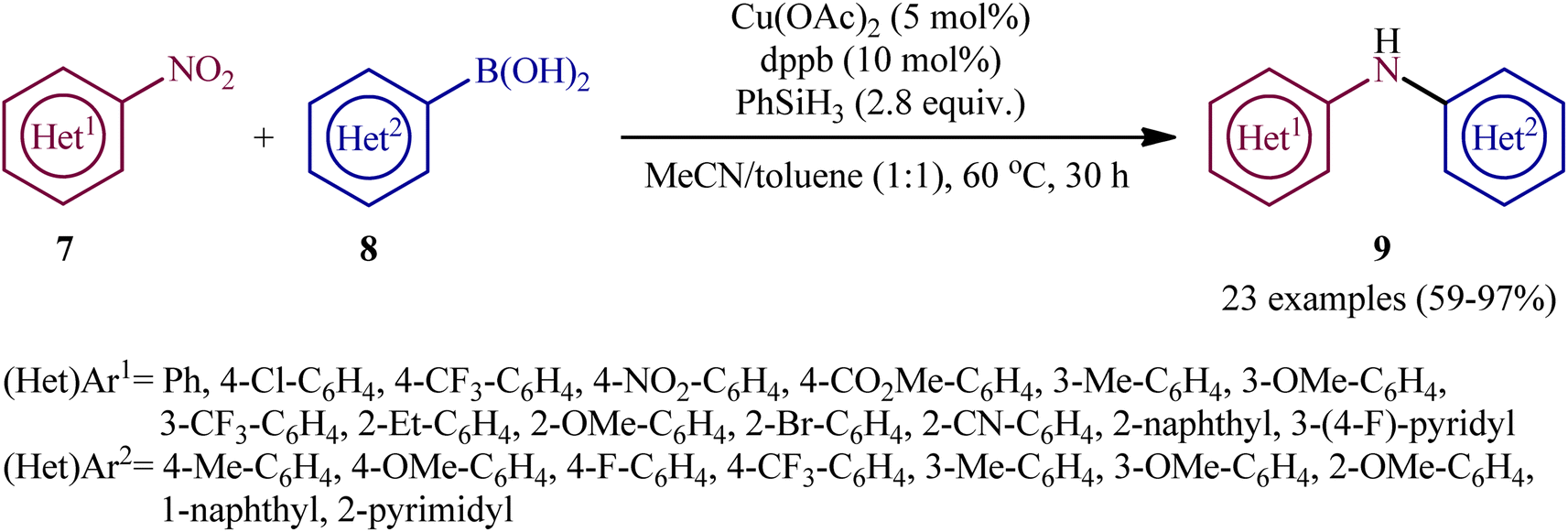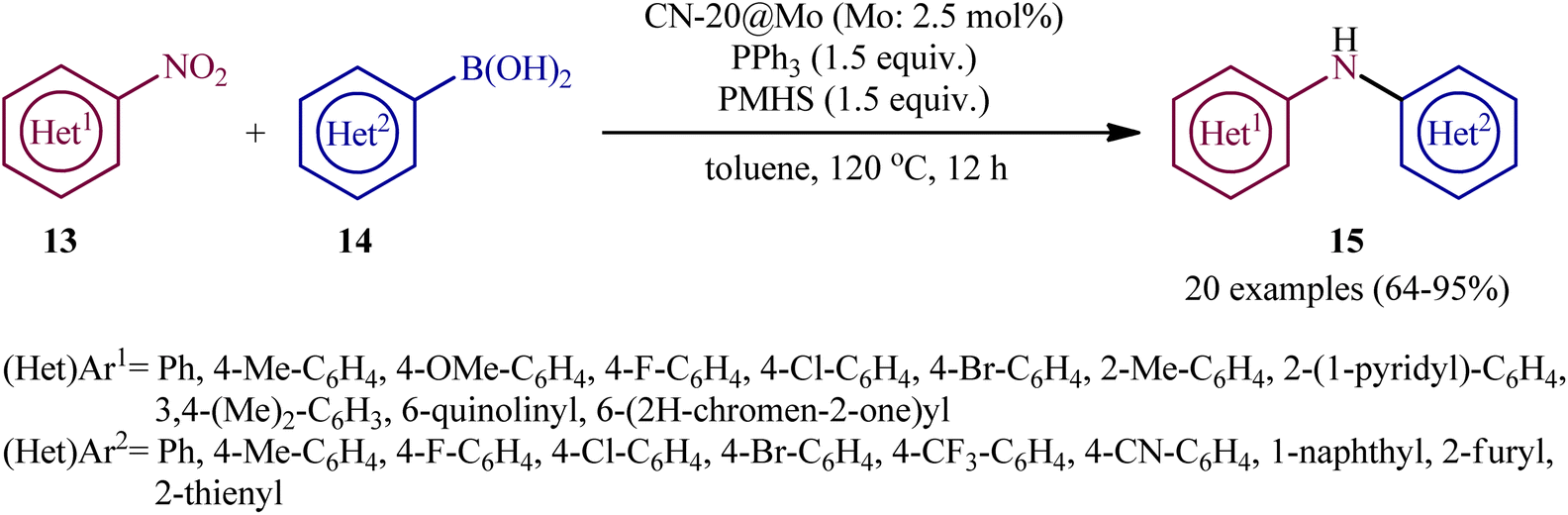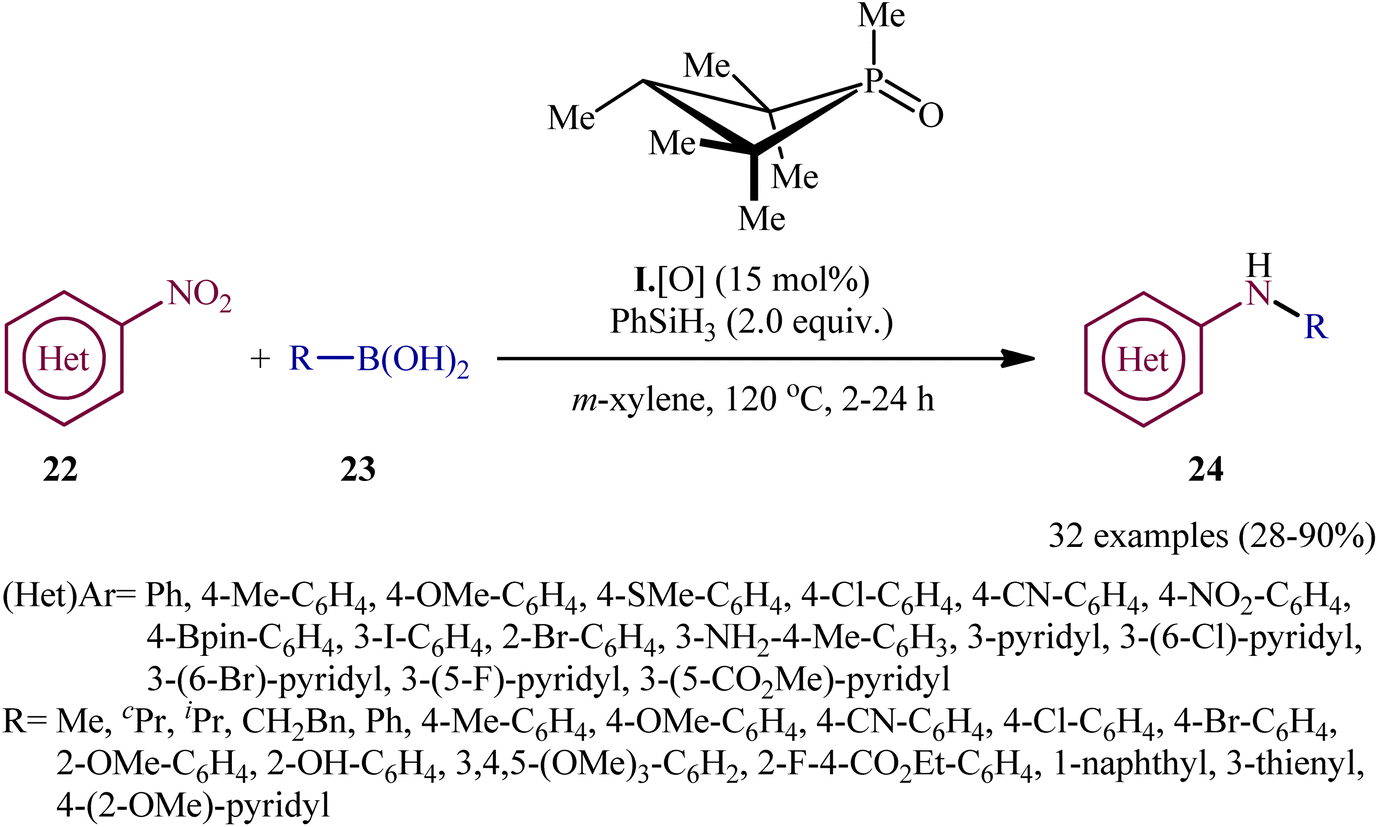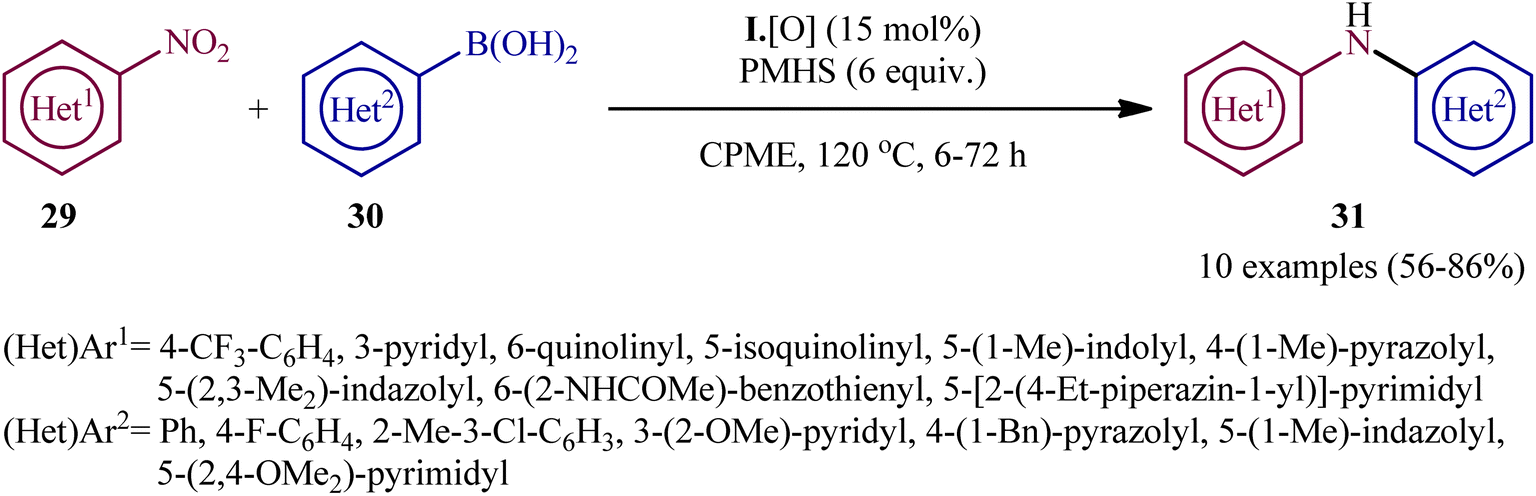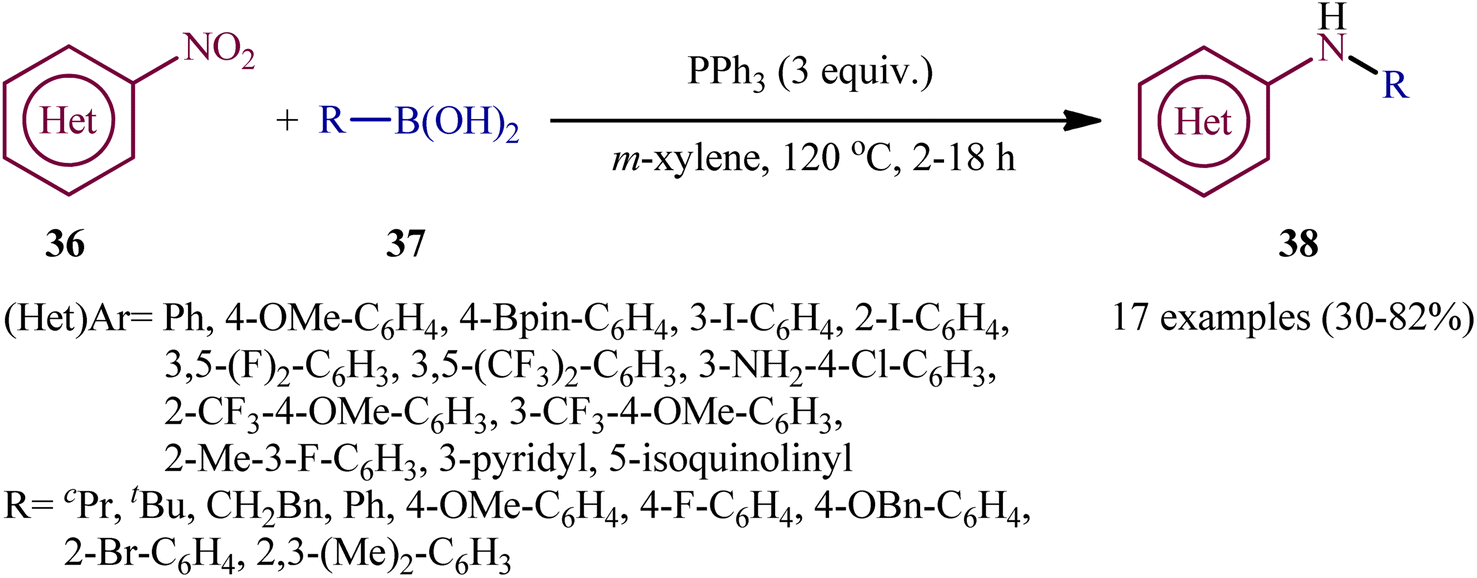DOI:
10.1039/D3RA05100E
(Review Article)
RSC Adv., 2023,
13, 33390-33402
Reductive coupling of nitro compounds with boronic acid derivatives: an overview
Received
27th July 2023
, Accepted 16th October 2023
First published on 13th November 2023
Abstract
The purpose of this review is to summarize the current literature on reductive C–N coupling of nitro compounds and boronic acids, with special emphasis on the mechanistic features of the reactions. The metal-catalyzed reactions are discussed first. This is followed by electro-synthesis and organophosphorus-catalyzed reactions. Finally, the available examples of catalyst-free reactions will be covered at the end of this review.
1. Introduction
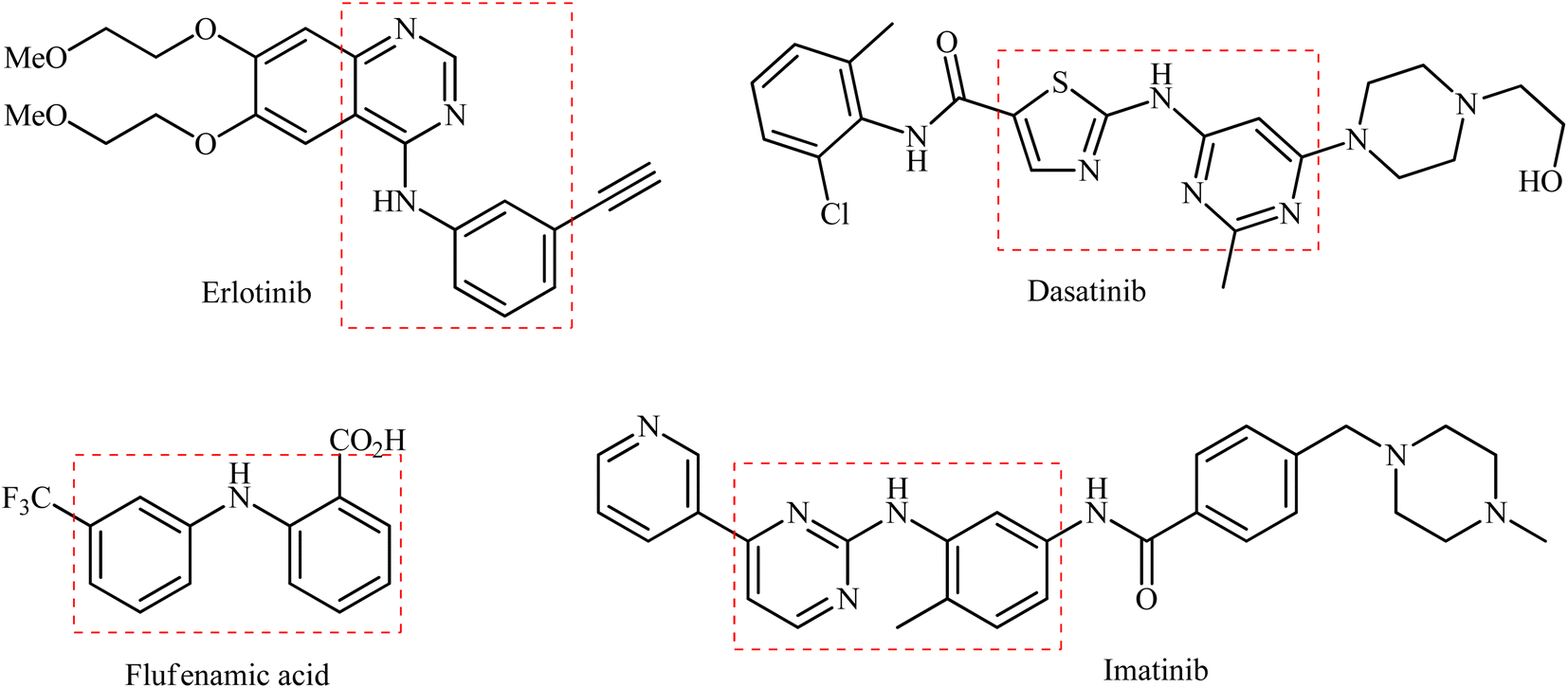
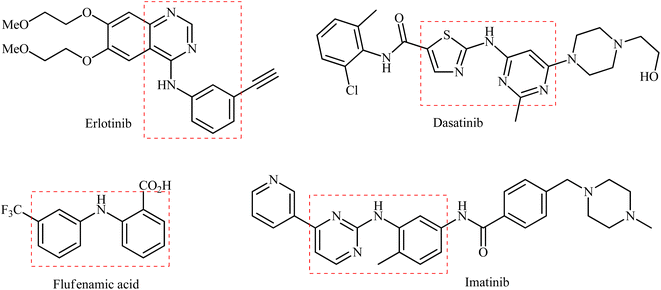
Secondary (hetero)arylamines are common structural motifs encountered in the architecture of numerous natural products,1 pharmaceutical agents,2 and agrochemicals (Scheme 1).3 Due to their wide importance in diverse fields, over the years, a number of synthetic approaches have been developed for their preparation. The most general transformations include direct alkylation of (hetero)aromatic amines with alkyl halides,4 Buchwald–Hartwig,5 Ullmann,6 and Chan–Lam–Evans7 and cross-coupling reactions. Without exception, all these strategies utilize free amines as the nitrogen source. As it is well-known, the main method for the preparation of primary amines, especially for anilines, is stoichiometric reduction of the corresponding nitro compounds.8 Indeed, a recent study on the most-used reactions in medicinal chemistry found reduction of nitroarenes to anilines was the most-used reduction.9 Therefore, without question, the development of efficient and practical approaches for the synthesis of titled compounds directly from the corresponding nitro compounds is extremely desirable in terms of saving energy and time.10
 |
| | Scheme 1 Selected examples of pharmaceutically important (hetero)arylamine derivatives. | |
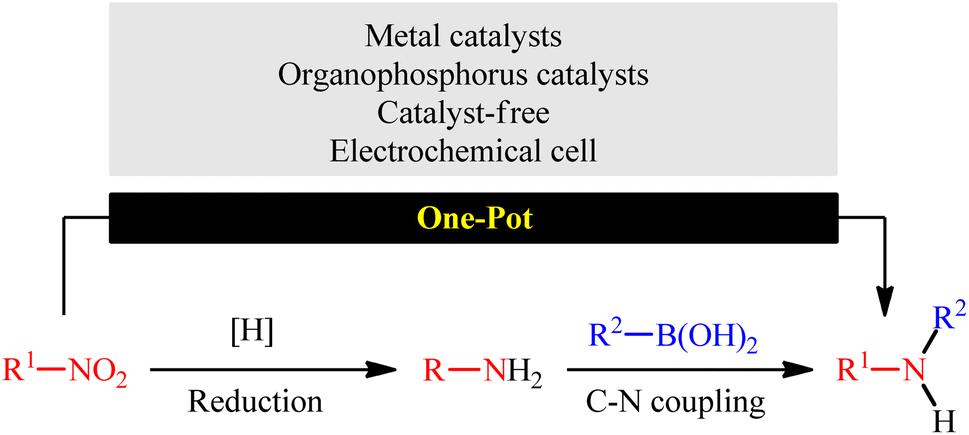
In this context, recently, reductive C–N coupling of abundant and readily available nitro compounds with stable and non-toxic boronic acid derivatives has attracted more and more attention as a general and straightforward route for the construction of secondary amines (Fig. 1), which beside high step economy, using non-toxic and easily accessible starting materials, high functional group tolerance, simplicity as well as avoiding the pre-preparation of amine substrates can be considered as the main advantages of this synthetic strategy over conventional methodologies.
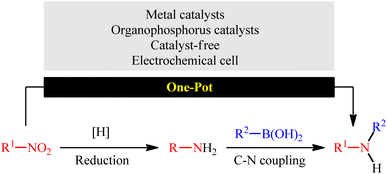 |
| | Fig. 1 Direct reductive coupling of nitro compounds with boronic acids. | |
Despite the significant progress that has been achieved in this attractive research topic over the past five years, a comprehensive review has not appeared on this hot and fast-growing arena in the literature thus far. As a continuation of our previous works on modern cross-coupling reaction,11 herein, we will try to provide an updated overview of the most important advances and developments of the reductive C–N coupling of nitro compounds with boronic acids with special emphasis on the mechanistic features of the reactions. For clarity, the review was structured based on the type catalysts (i.e., metal-catalyzed, electro-catalyzed, organophosphorus-catalyzed, catalyst-free reactions).
2. Metal-catalyzed reactions
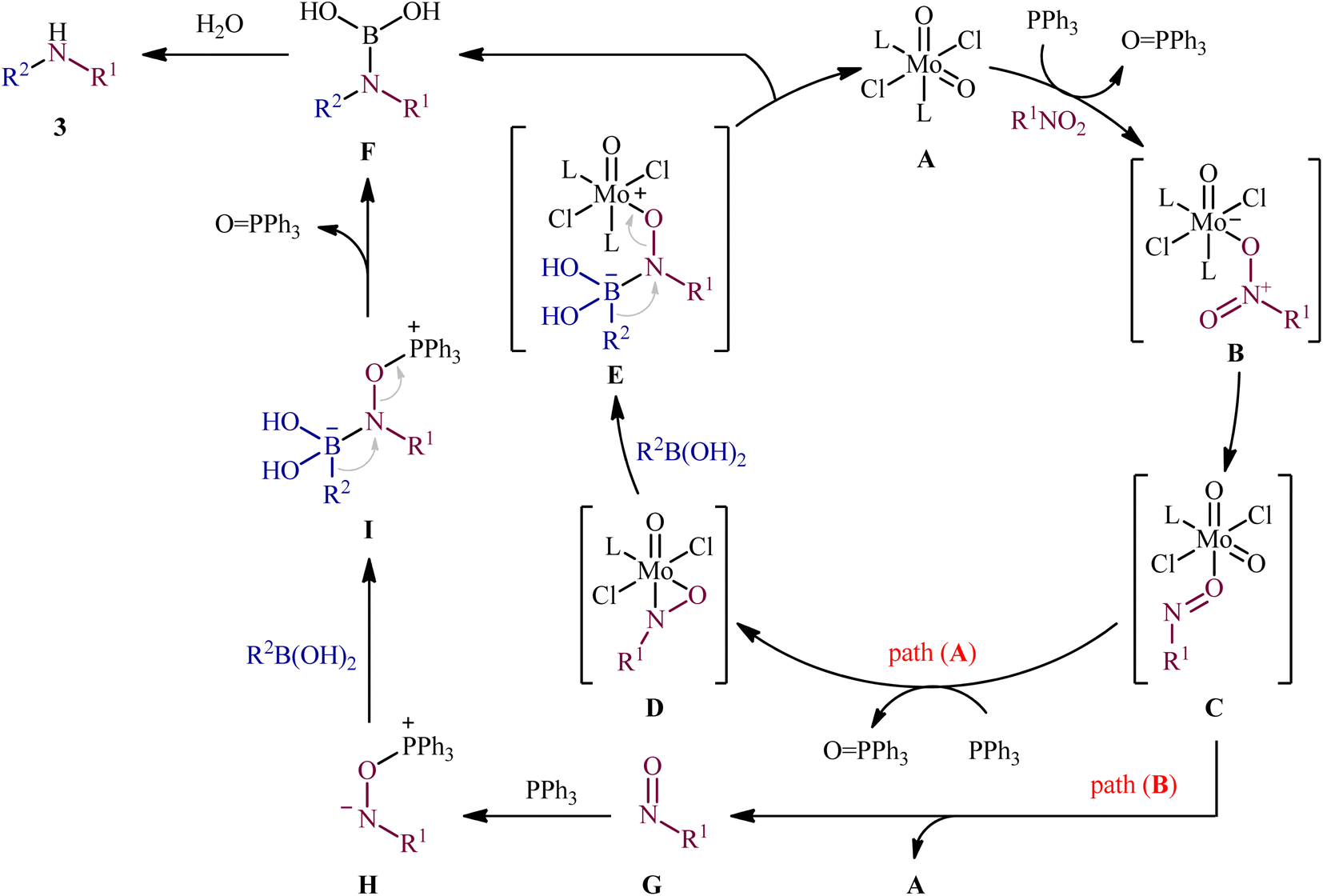
Reductive C–N coupling of nitro compounds and boronic acid derivatives with the assistance of a metal-catalyst is probably the area that has experienced the most growth in the field in the last few years. In 2019, Pantiga, Sanz, and co-workers demonstrated for the first time the usefulness of metal catalysts for reductive amination of boronic acids with nitro compounds.12 Thus, in the presence of 5 mol% of MoO2Cl2(dmf)2, as a catalyst and 2,2′-bipyridine (bpy) as a ligand, in combination with 2.4 equiv. of PPh3 as a reducing agent in toluene at 100 °C, the reaction of nitro(hetero)arenes 1 with various (hetero)aryl and alkyl boronic acids 2 furnished the corresponding secondary amines 3 in moderate to quantitative yields, ranging from 30% to 99% (Scheme 2). The reaction tolerated a wide panel of important functional groups (e.g., F, Cl, Br, I, CF3, CN, NH2, COMe, CO2Me, CONH2, CHO), which might provide potential opportunities for further manipulation of amine products. Particularly remarkable is that the reaction exhibited extremely high degree of site-selectivity, in which amination is exclusively took place over the nitro group when free amine is also present without the need for protecting group chemistry. It is worthwhile to note that reductive arylation of aliphatic nitro compounds including nitromethane, nitroethane, 2-nitropropane, 1-nitrohexane, and ethyl 2-nitroacetate was also achieved with use of similar reaction conditions. However, they were slightly sluggish to participate in this protocol and required microwave (MW) irradiation to achieve full conversion (7 examples, 51–72%). Unfortunately, no comment was made by the authors regarding the relationship between structure of starting materials and their associated catalytic activity as well as regarding the recovery and reusability of the catalyst. According to the authors proposed mechanism (Scheme 3), this C–N coupling reaction might start with the generation of Mo species B via the reduction of dioxomolybdenum(VI) catalyst A with PPh3 to a MoIV species followed by coordination to the nitro compound 1. Subsequently, reoxidation of the Mo center by the coordinated nitro compound gives intermediate C bearing a nitroso ligand, which after the second deoxygenation with PPh3 and subsequent reduction of the nitroso ligand and reoxidation of the metal center converts to molybdooxaziridine (or η2-nitroso) species D (Scheme 3, path A). Next, the attack of the N atom of species D to the boronic acid 2 leading to the formation of nitrenoid intermediate E, which undergoes a rearrangement to yield aminoboronic acid F. In another possibility (Scheme 3, path b), intermediate C releases a free nitroso compound G and regenerates catalyst A. Subsequently, the former reacts with another molecule of PPh3 to afford the reduced adduct H, which after interaction with the boronic acid 2 delivers nitrenoid borate intermediate I. Later, 1,2-migration of a nucleophilic R2 group to adjacent N center and spontaneous release of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3 leads to the formation the same aminoboronic acid F. Finally, hydrolysis of this intermediate F affords the observed amine 3.
PPh3 leads to the formation the same aminoboronic acid F. Finally, hydrolysis of this intermediate F affords the observed amine 3.
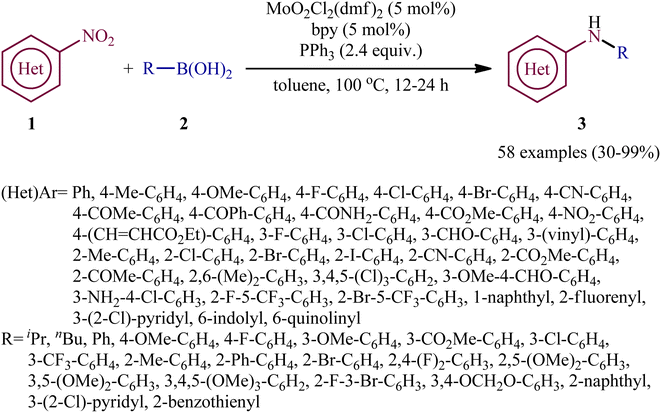 |
| | Scheme 2 Mo-catalyzed reductive C–N coupling of nitro(hetero)arenes 1 with boronic acids 2. | |
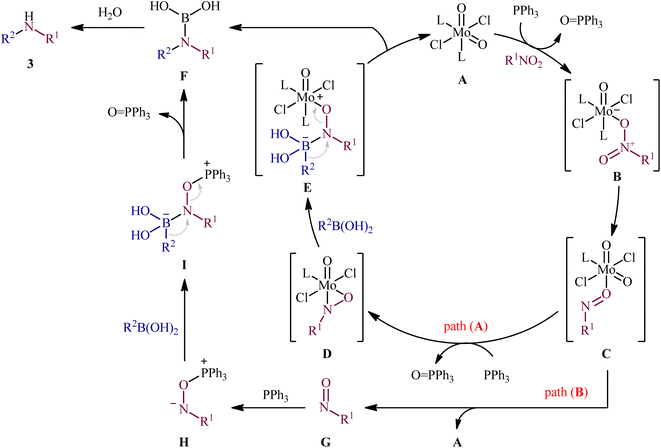 |
| | Scheme 3 Mechanism that accounts for the formation of secondary amines 3. | |
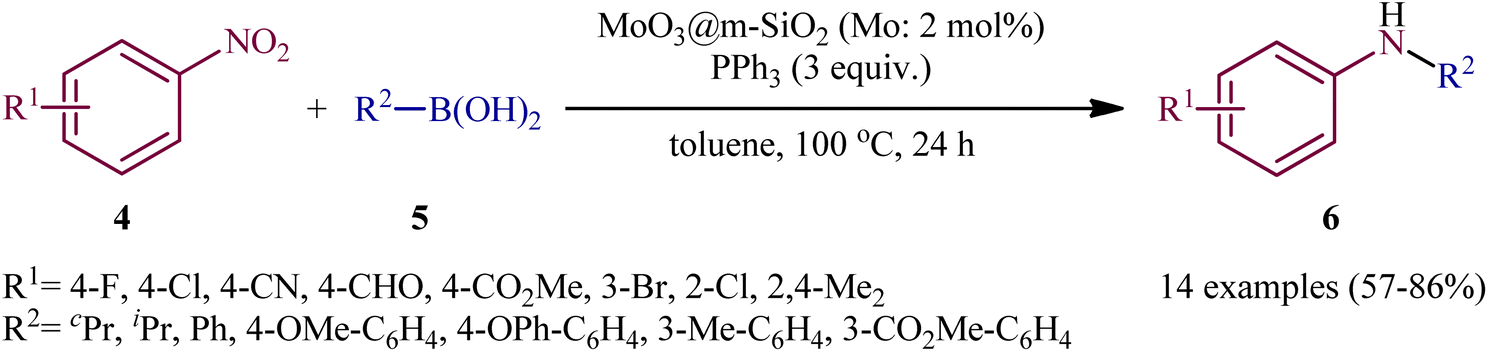
Two years later, Song and colleagues developed this chemistry using a novel heterogeneous catalyst composed of nanoporous MoO3 confined in mesoporous silica (MoO3@m-SiO2).13 Thus, with the assistance of only 2 mol% MoO3@m-SiO2 (based on Mo atoms) and 3 equivalent PPh3, a variety of nitroarenes 4 reacted with aryl/alkyl boronic acids 5 under an inert atmosphere to afford the target N-arylamine products 6 in good to high yields within 24 h (Scheme 4). Noteworthy, recycling tests indicated that the catalyst could be reused at least for five consecutive trials without tangible decrease in its catalytic activity. A comparison of the catalytic performance of MoO3@m-SiO2 and commercial MoO3 NPs in the reaction of 1-fluoro-4-nitrobenzene with phenylboronic acid revealed that the former was more efficient under the same condition. The yield of coupling amines increased from 17% for commercial catalyst to 83% for the newly developed catalyst, while the turnover number (TON), increased from 16 to 42.
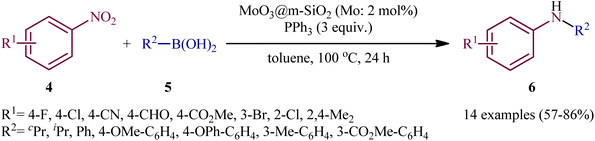 |
| | Scheme 4 Reductive coupling of nitroarenes 4 with aryl/alkyl boronic acids 5 catalyzed by MoO3@m-SiO2. | |
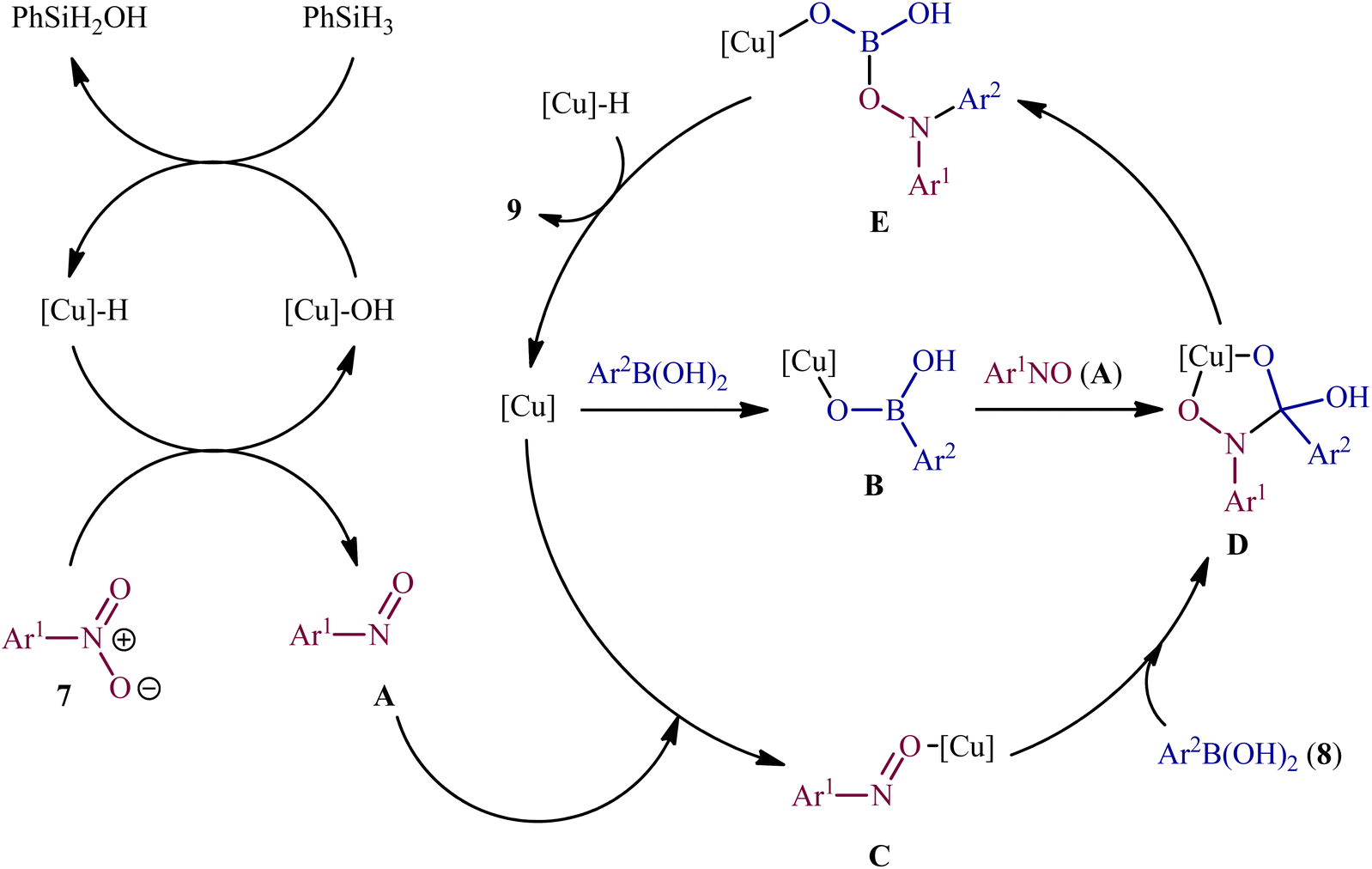
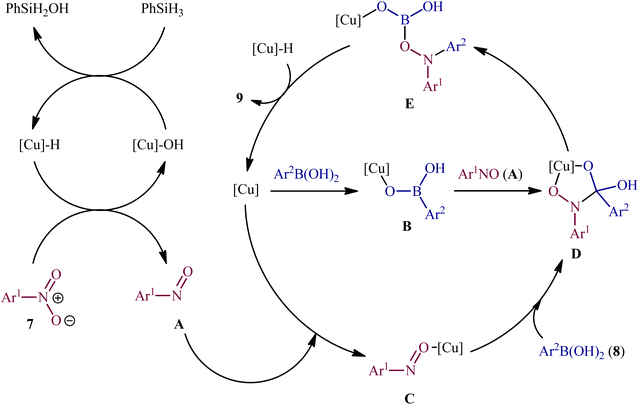
Concurrently, Driver's research group has identified easily available Cu(OAc)2 as a cheap and non-toxic catalyst for conversion of nitro(hetero)arenes 7 and (hetero)aryl boronic acids 8 into the corresponding di(hetero)arylamines 9 under relatively mild conditions.14 In this report, 23 amines were synthesized in good to almost quantitative yields by means of 5 mol% of Cu(OAc)2 and 1,4-bis(diphenylphosphino)butane (dppb) in a binary solvent MeCN/toluene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in the presence of phenyl silane (PhSiH3) as the stoichiometric terminal reductant (Scheme 5). In the investigation of the scope of this transformation, it was found that the electron-deficient nitroarenes were provided better results than electron-rich ones, whereas the electronic character of the substituent attached to the aromatic ring periphery of aryl boronic acids had no effect on the facility of this reaction. On the other hand, the steric hindrance effects of substituents on the aryl boronic acids are very strong, as the presence of one ortho-ethyl group prevented the product formation. Unfortunately, the applicability of neither aliphatic nitro compounds nor any alkyl boronic acids as starting materials was investigated in this study. After a series of mechanistic investigations, it was confirmed that this C–N bond forming reaction most likely proceeds via a nitrosoarene intermediate A (Scheme 6), and that copper is required to catalyze both the deoxygenation of the nitroarene to afford nitrosoarene A and C–N bond formation of the nitrosoarene with the boronic acid.
:1) in the presence of phenyl silane (PhSiH3) as the stoichiometric terminal reductant (Scheme 5). In the investigation of the scope of this transformation, it was found that the electron-deficient nitroarenes were provided better results than electron-rich ones, whereas the electronic character of the substituent attached to the aromatic ring periphery of aryl boronic acids had no effect on the facility of this reaction. On the other hand, the steric hindrance effects of substituents on the aryl boronic acids are very strong, as the presence of one ortho-ethyl group prevented the product formation. Unfortunately, the applicability of neither aliphatic nitro compounds nor any alkyl boronic acids as starting materials was investigated in this study. After a series of mechanistic investigations, it was confirmed that this C–N bond forming reaction most likely proceeds via a nitrosoarene intermediate A (Scheme 6), and that copper is required to catalyze both the deoxygenation of the nitroarene to afford nitrosoarene A and C–N bond formation of the nitrosoarene with the boronic acid.
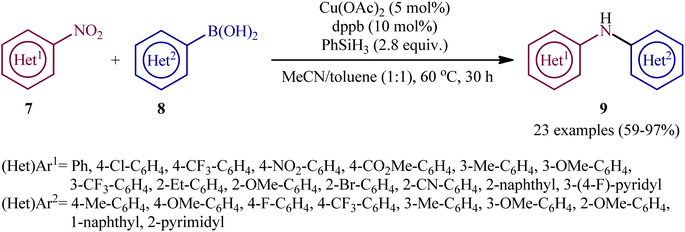 |
| | Scheme 5 Cu-catalyzed reductive coupling of nitro(hetero)arenes 7 and (hetero)aryl boronic acids 8. | |
 |
| | Scheme 6 Mechanistic proposal for the formation of di(hetero)arylamines 9. | |
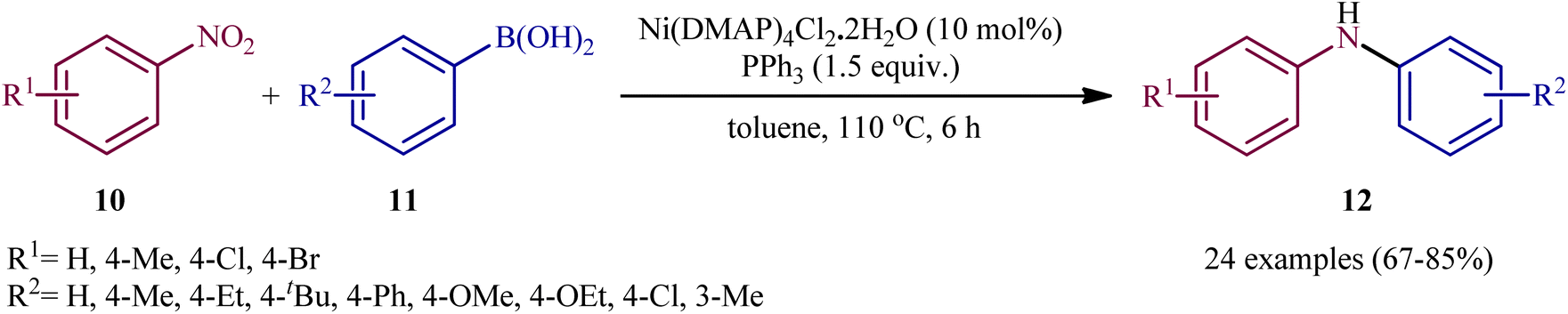
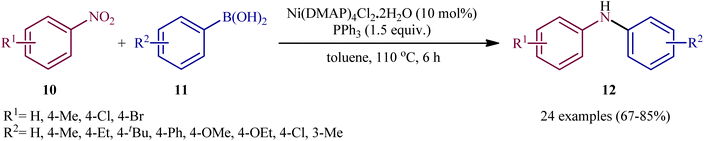
Very recently, Talukdar, Gogoi, and Phukan studied the scope of the Ni-catalyzed fashion of this reaction.15 Thus, a novel octahedral Ni(DMAP)4Cl2·2H2O complex was prepared by simply treating NiCl2·6H2O with DMAP at room temperature and applied as an efficient catalyst for the synthesis of diarylamines 12 through reductive coupling of corresponding nitroarenes 10 with aryl boronic acids 11 in the presence of PPh3 in refluxing toluene (Scheme 7). These authors demonstrated significant scope of the aryl boronic acid reagent, but limited scope of the nitroarene substrate; however, the yields were good. It should be mentioned that other nickel catalysts such as NiCl2·6H2O and NiSO4·6H2O, could also promote this transformation; albeit, at lower efficiencies. Employing their catalyst, the authors have also presented the synthesis of a library of diaryl sulfides through C–S coupling of aryl boronic acid and thiols. The system was also amenable to the Chan–Lam coupling of various boronic acids with different amines as well as azides, providing N-aryl amine products in good yields.
 |
| | Scheme 7 Ni-catalyzed synthesis of diarylamines 12 from nitroarenes 10 with aryl boronic acids 11. | |
Drawing inspiration from these works, quite recently, Chen, Lu, and co-workers designed and synthesized a novel zeolitic imidazolate framework (ZIF)-derived CN-20@Mo catalyst by a simple and scalable strategy through the addition of a solution of 2-methylimidazole and aniline in deionized water to an as-prepared solution of Zn(NO3)2·6H2O and Na2MoO4·2H2O in water followed by stirring the resulted mixture at room temperature and subsequent centrifugation and calcination.16 The nanocomposite system was applied as an efficient catalyst for the synthesis of a panel of 20 di(hetero)arylamines 15 through reductive coupling of the corresponding nitro(hetero)arenes 13 with (hetero)aryl boronic acids 14 in the presence of the combination of PPh3 and polymethylhydrosiloxane (PMHS) as reducing system in refluxing toluene (Scheme 8). Unfortunately, 1-nitropropane and cyclohexylboronic acid did not take part in this coupling reaction and therefore no other alkyl nitro compounds and alkyl boronic acid substrates were examined in the protocol. Nevertheless, the catalyst exhibited good recycling properties on at least six consecutive reuses without significant decline deactivation in its catalytic performance.
 |
| | Scheme 8 CN-20@Mo-catalyzed reductive C–N coupling of nitro(hetero)arenes 13 and (hetero)aryl boronic acids 14. | |
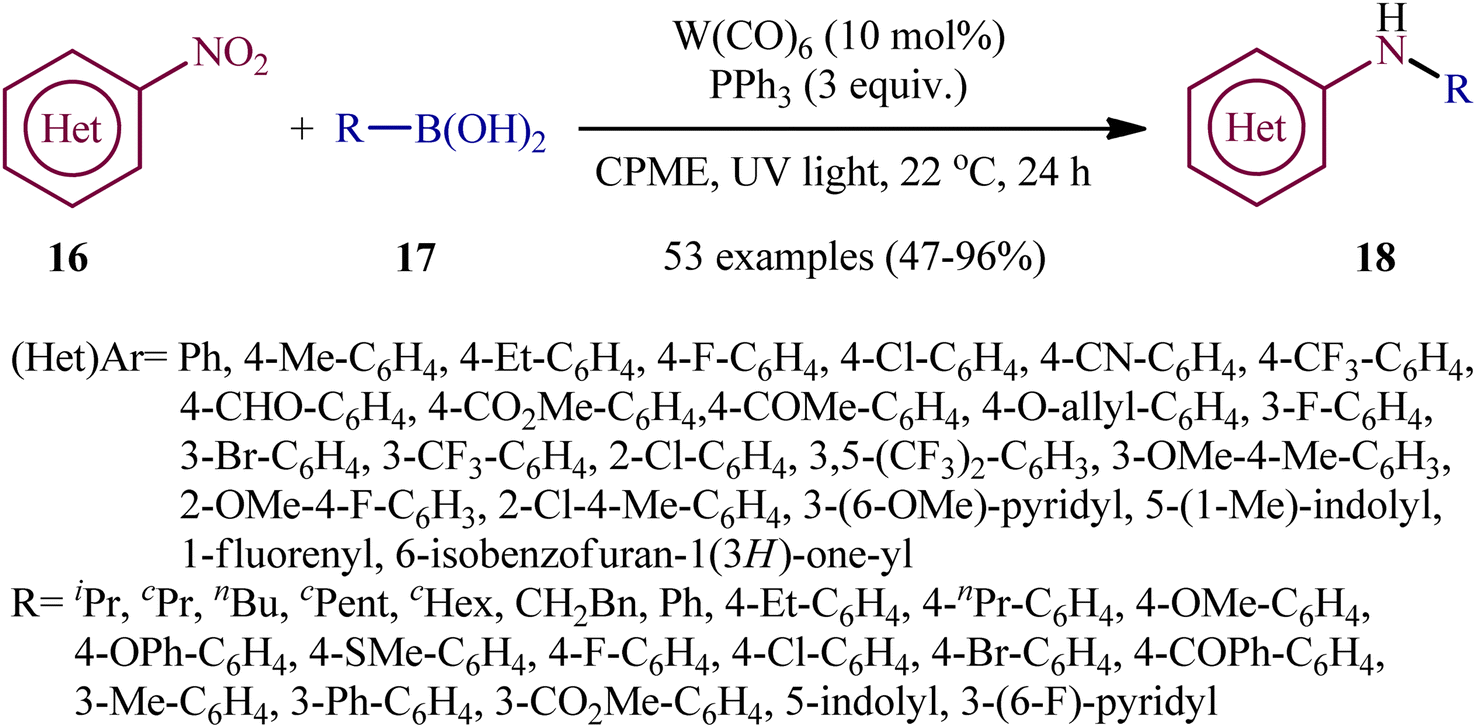
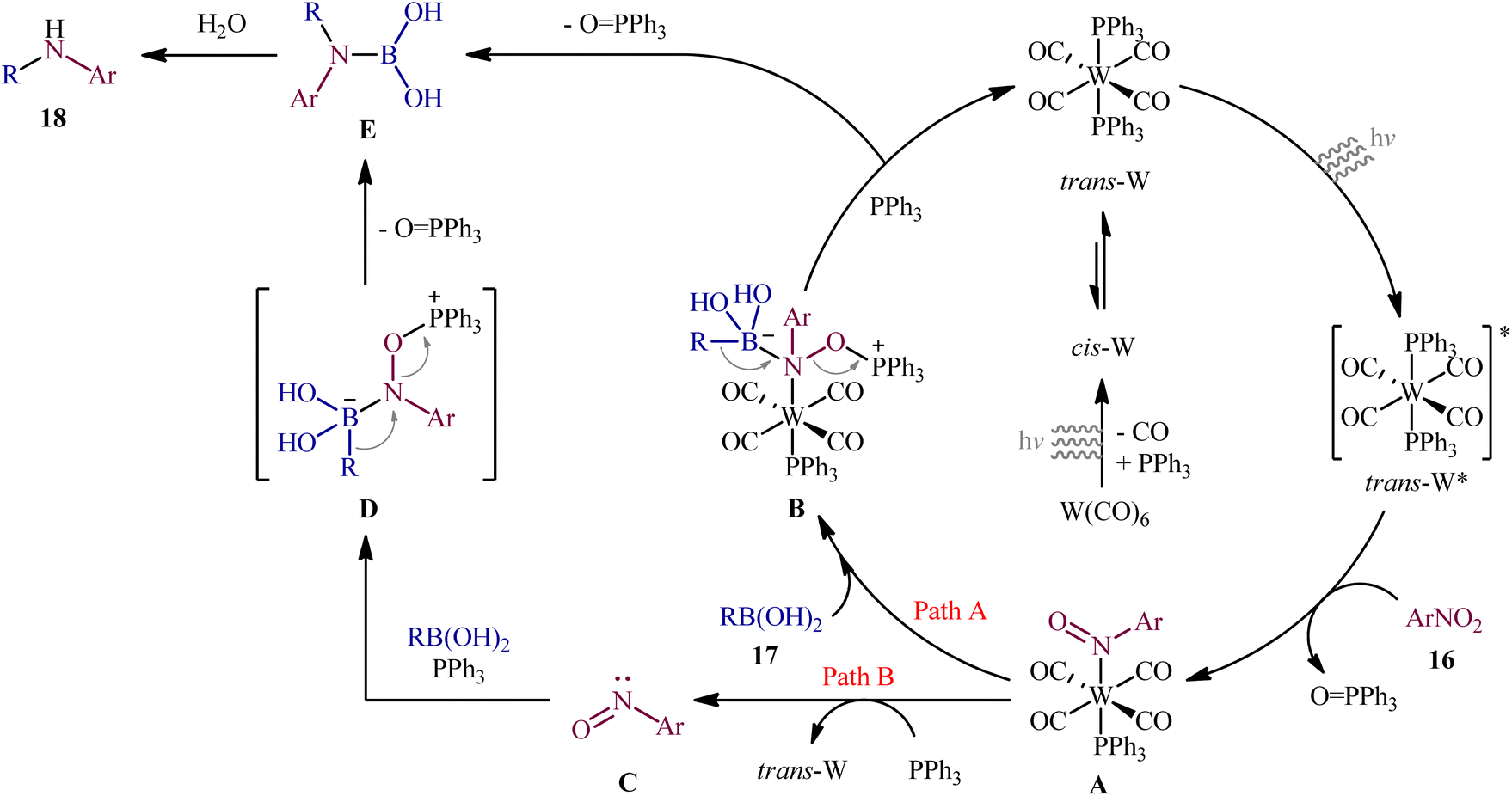
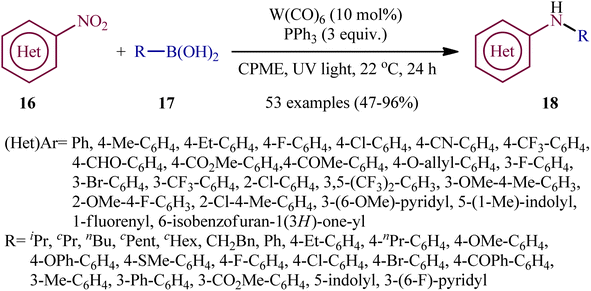
Recently, in an attractive contribution in this field, Song et al. disclosed an interesting visible-light induced deoxygenative cross-coupling of nitro compounds 16 and boronic acids 17 using readily available W(CO)6 as a precatalyst and PPh3 as a reductant.17 The reactions proceed under UV light irradiation at 22 °C and providing the secondary (hetero)arylamines 18 in moderate to good yields, ranging from 47% to 96% yield (Scheme 9). The method showed a broad substrate scope including both aliphatic and aromatic boronic acids and various nitro(hetero)arenes. However, the applicability of nitroalkanes as starting materials was not investigated in this study. To gain mechanistic insights, several preliminary experiments were performed. No products were obtained in the absence of light irradiation or catalyst. The possibility of a radical pathway was neglected since the reaction with BHT (butylated hydroxytoluene; radical scavengers) did not prevent the product formation. The author speculated that this transformation is initiated by the deoxygenation of nitro compounds by a trans-[W(CO)4(PPh3)2] complex, which forms in situ via ligand replacement, as shown in Scheme 10.
 |
| | Scheme 9 Visible-light induced, tungsten-catalyzed amination of boronic acids 17 with nitro(hetero)arenes 16. | |
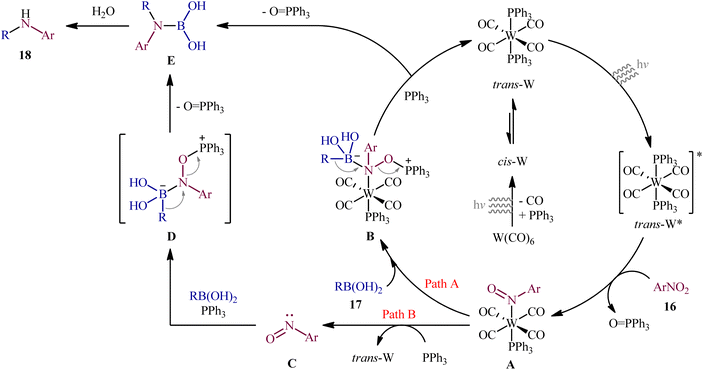 |
| | Scheme 10 Mechanistic proposal for the reaction in Scheme 9. | |
3. Electrochemical reactions
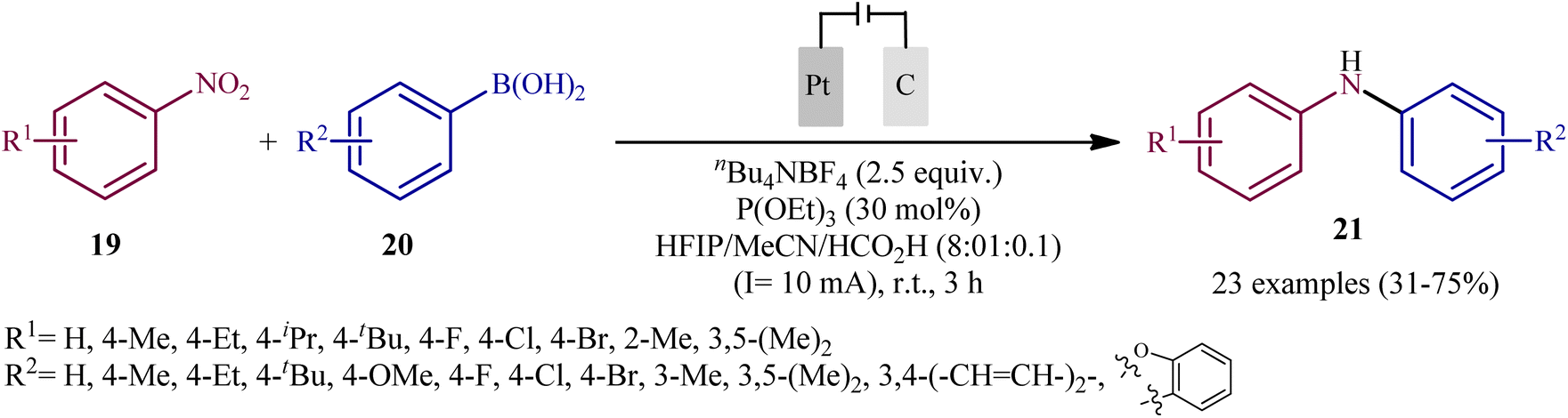
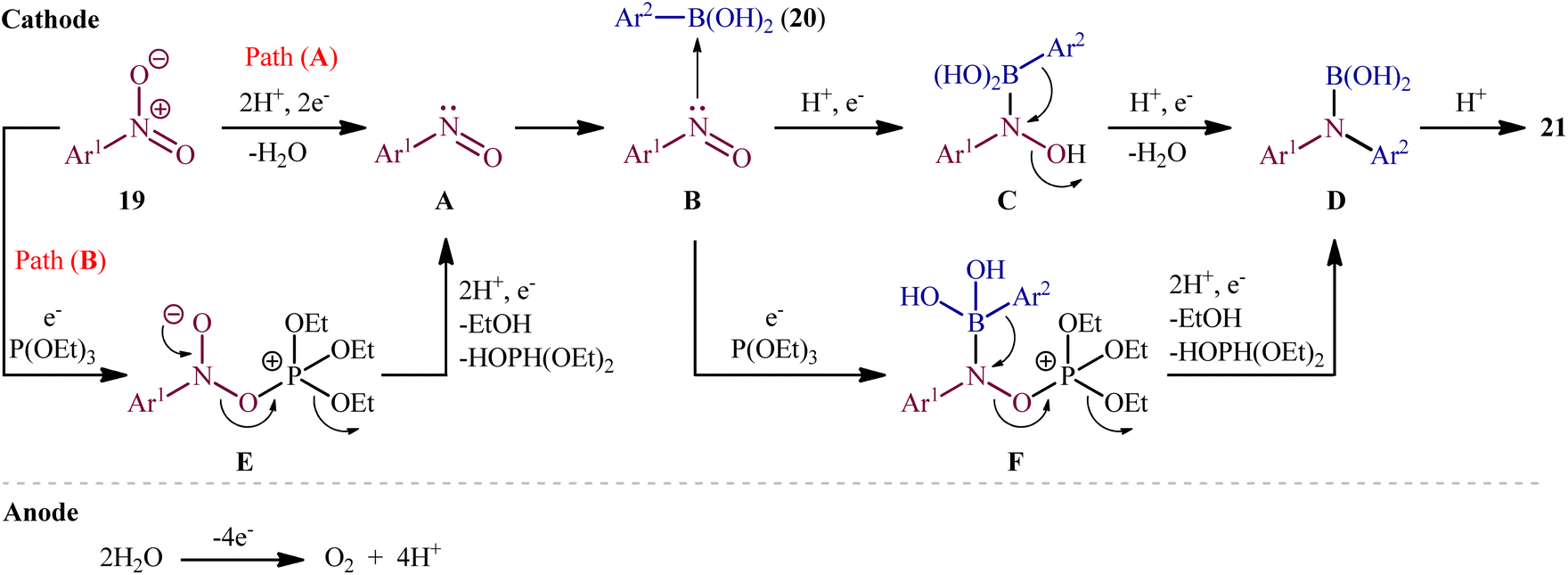
In 2021, Yi and Lei along with their co-workers investigated the possibility of synthesizing diarylamines by electrochemical reductive arylation of nitroarenes with aryl boronic acids.18 The electrolysis was performed with a platinum plate anode and a carbon cloth cathode in an undivided cell, with hexafluoroisopropanol (HFIP)/MeCN/HCO2H as the mixed solvent containing nBu4NBr as the supporting electrolyte and triethyl phosphite [P(OEt)3] as a reducing agent under a constant current of 10 mA (Scheme 11). The transformation was suitable for several nitroarenes 19 and a variety of electron rich- and deficient-substituted aryl boronic acids 20 and the desired diarylamines 21 were obtained in fair to good yields. However, when methoxy- or trifluoromethyl-substituted nitroarenes were employed under the optimal reaction conditions, only corresponding amino compounds were obtained. Furthermore, the process is not viable for gram-scale due to the drastic reduction in the yield (from 75% in the 0.2 mmol scale, to 60% in the 5.0 mmol scale). The main features of this method are the mild reaction conditions and carried out in a short reaction time. It is interesting to mention that under the standard reaction conditions, replacing nitroarenes by either anilines or 1,2-diaryldiazenes stopped the reaction completely. Since this electrosynthesis was doable with or without presence of a reducing agent, although the best results were observed in the presence of P(OEt)3, two possible mechanisms were proposed by the authors for this transformation (Scheme 12). In pathway A, nitroarene 19 is reduced to the nitroso compound A by two-electron uptake from the cathode and losing a water molecule with the help of acid. Next, with the coordination of the nitroso intermediate A with arylboronic acid 20 and receiving one electron from the cathode, intermediate C is generated. This intermediate is then converted to diarylboramidic acid D via aryl migration and another dehydration process. Finally, through the protonation of intermediate D, the expected diarylamine 21 is formed. On the other hand, in pathway B through the nucleophilic attack of a lone-pair electron of P(OEt)3 on the oxygen atom of the nitro group of nitroarene 19, followed by the acceptance of one electron from the cathode, the reactive species E is generated, which after receiving one electron from the cathode and losing a EtOH and HOP(OEt)2 with the help of acid, transformed to the nitroso compound A. It has been suggested that both pathways may be operative at the same time. To the best of our knowledge, this is the first and only example reported on the electrochemical reductive arylation of nitroarenes with arylboronic acids till date.
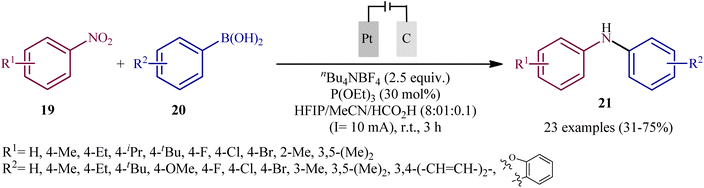 |
| | Scheme 11 Yi–Lei's synthesis of diarylamines 21. | |
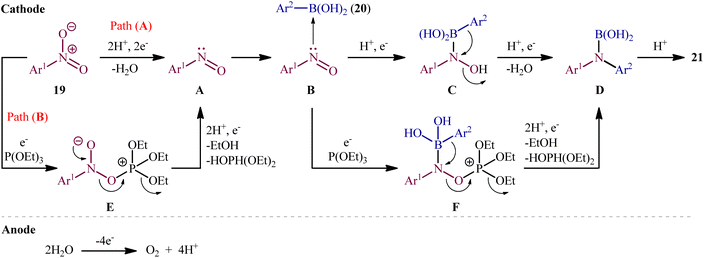 |
| | Scheme 12 Plausible mechanism for the reaction in Scheme 11. | |
4. Organophosphorus-catalyzed reactions
In recent years, organophosphorus compounds have been extensively used as catalysts in a variety of synthetic reactions and in drug development.19 Particularly, chiral organophosphorus catalysts play an extremely important role in the field of asymmetric catalysis.20 The first report on the utilization of phosphorus-based organocatalysis in the deoxygenative C–N cross-coupling between nitro compounds with boronic acid derivatives was published by Luzung and Radosevich along with their co-workers in 2018,21 who showed that the treatment of functionalized (hetero)aromatic nitro compounds 22 with various alkyl, aryl, as well as heteroaryl boronic acids 23 in the presence of a catalytic amount of 1,2,2,3,4,4-hexamethylphosphetane (I·[O]), a small-ring phosphacycle, and over-stoichiometric amounts of PhSiH3 as a reducing agent in m-xylene, resulted in the formation of the corresponding secondary aryl amines 24 in moderate to excellent (Scheme 13). Remarkably, it was found that the reaction is stereospecific with respect to Csp3–N bond formation, when using the stereochemical probe molecules as substrate. The authors, also nicely exemplified the complementarity of their methodology for C–N coupling with respect to existing transition metal strategies (i.e., Buchwald–Hartwig and Chan–Lam couplings) utilizing N-(3-bromo-5-nitrophenyl)acetamide as the reactant. The results indicated that whereas C–N coupling under Cu-mediated or Pd-catalyzed methods permitted chemoselective functionalization at the anilide or aryl bromide positions, respectively, catalytic arylamination by the newly developed organophosphorus-catalyzed coupling approach resulted in selective functionalization at the nitro moiety. Unfortunately, the applicability of aliphatic nitro compounds as starting materials was not investigated in this study.
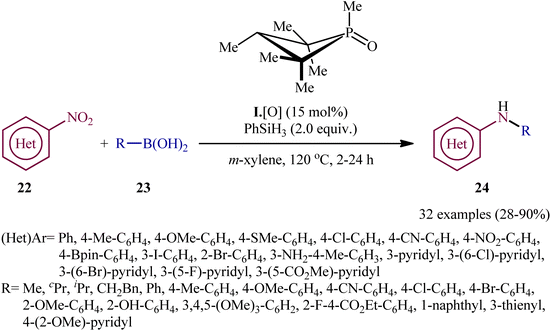 |
| | Scheme 13 Luzung–Radosevich's synthesis of secondary aryl amines 24. | |
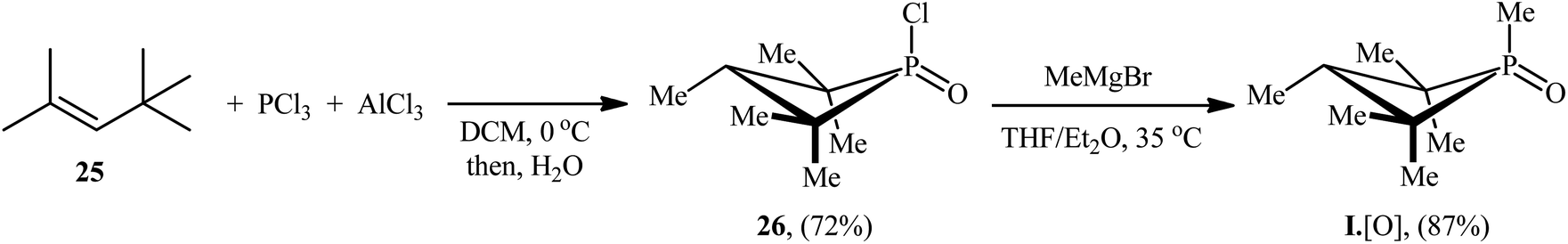
The synthetic route for the preparation of I·[O] is shown in Scheme 14.22 The key step in this synthesis was to prepare the intermediate phosphetane oxide 26 via treatment of 2,4,4-trimethyl-2-pentene 25 with phosphorus trichloride (PCl3) in the presence of aluminum chloride.
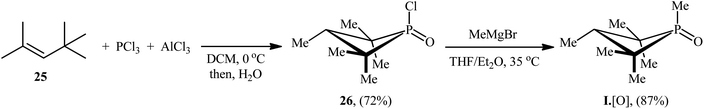 |
| | Scheme 14 Synthesis of I·[O] developed by Radosevich. | |
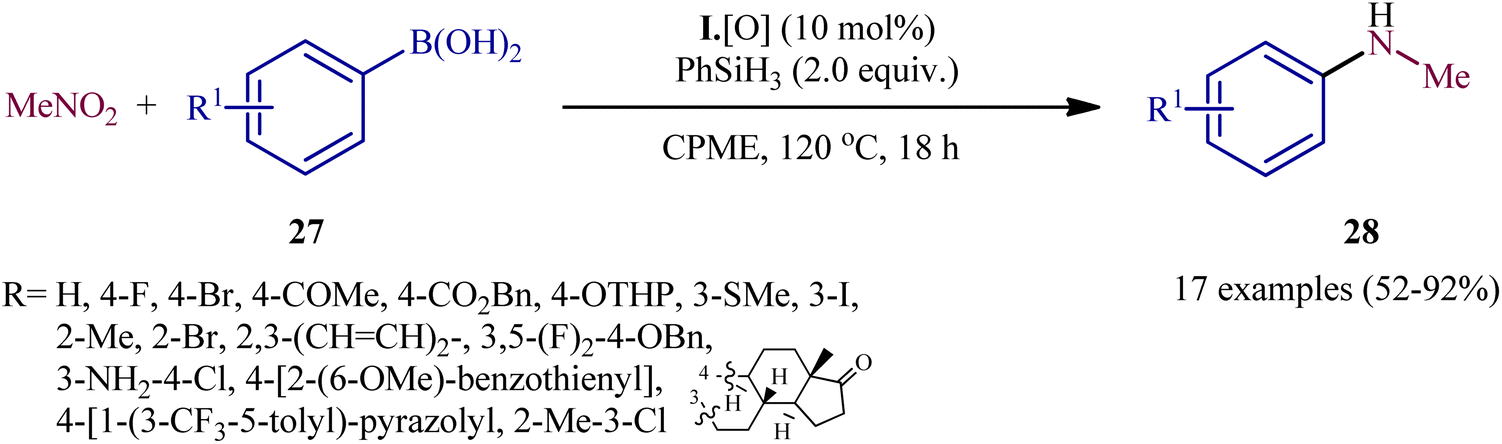
In a subsequent extension of the substrate scope of the methodology, it was shown that various functionalized aryl boronic acids 27 could be treated with 3 equiv. of nitromethane (MeNO2) under the same catalytic system in refluxing cyclopentyl methyl ether (CPME) to give the corresponding N-methylanilines 28 in moderate to excellent yields (Scheme 15).23 The system was also amenable to the methylamination reaction of aryl boronic acids with the nitromethane isotopologue (viz. 2H3C–NO2, H313C–NO2, H3C–15NO2), providing isotopically labeled N-methylaniline products in high yields. It should be mentioned that apart from aryl boronic acids, aryl boronic esters were also compatible with this methylamination scenario. In order to further value the applicability of the method, the authors elegantly synthesized triflubazam (an anxiolytic drug) from commercially available 4,4,5,5-tetramethyl-2-(2-nitro-5-(trifluoromethyl)phenyl)-1,3,2-dioxaborolane through a two-pot, three step procedure involving two PIII/PV-catalyzed deoxygenative C–N cross-coupling steps.
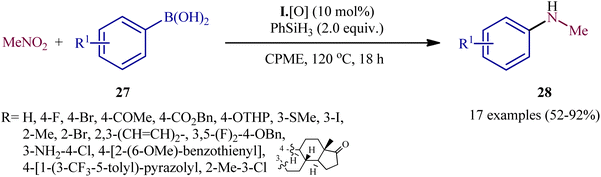 |
| | Scheme 15 P(III)/P(V)-catalyzed methylamination of arylboronic acids 27 with MeNO2. | |
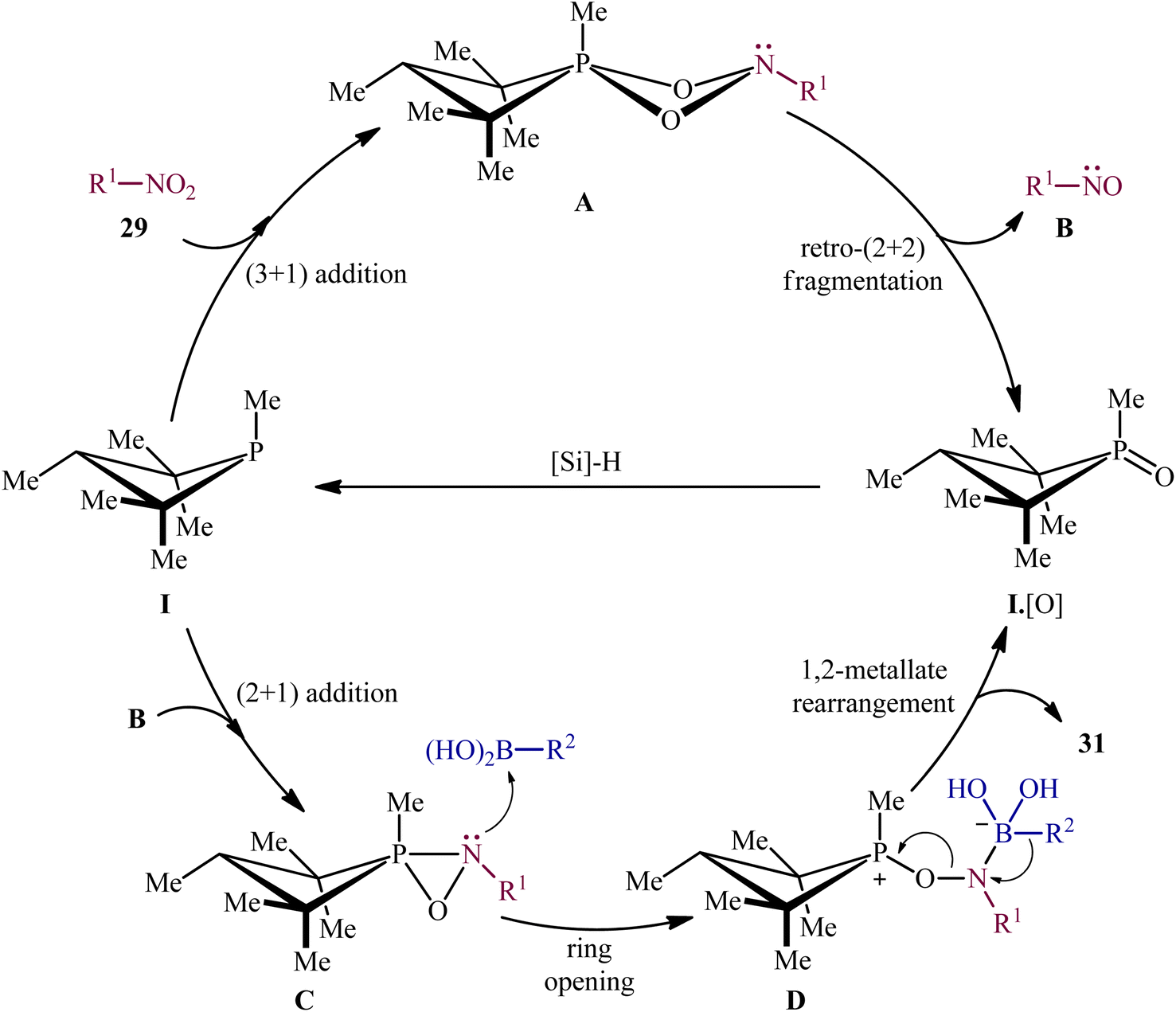
Subsequently, based on the above catalyst, this research group developed an improved catalytic system by replacing PhSiH3 with PMHS and performing the process in CPME, which resulted in a broader substrate scope and significantly improved yields.24 Thus, with the modified “second-generation” conditions, a library of heteroarylnitro substrates 27 engaged effortlessly with various heteroaryl boronic acids 30 to give the corresponding di(hetero)arylamines 31 in good yields (Scheme 16). In this report, several control experiments, such as DFT calculation and spectroscopic studies, were conducted for the insight of the reaction mechanism, which indicated a two-stage deoxygenation sequence for this reductive C–N coupling (Scheme 17).24,25 The first cycle starts with the [3 + 1] cheletropic addition of nitro compound 29 to phosphetane I leading to the formation of cycloadduct A, which undergoes a retro-[2 + 2] fragmentation to furnish the corresponding nitroso compound B and phosphetane oxide I·[O]. Finally, the later rapidly converts to I through reduction by the hydrosilane. In the second cycle, a [2 + 1] addition between the nitroso compound B and phosphetane I affords oxazaphosphirane C, which then reacts with boronic acid 30 to furnish betaine intermediate D. Finally, 1,2-metallate rearrangement of intermediate D yields the expected product 31.
 |
| | Scheme 16 P(III)/P(V)-catalyzed reductive C–N coupling of nitro(hetero)arenes 29 and (hetero)aryl boronic acids 30 using PMHS as the reductant. | |
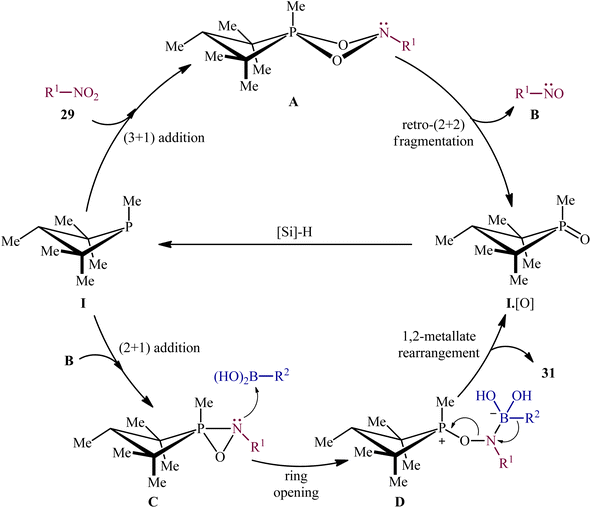 |
| | Scheme 17 Mechanistic proposal for the P(III)/P(V)-catalyzed reductive C–N coupling of nitro compounds 29 and boronic acids 30. | |
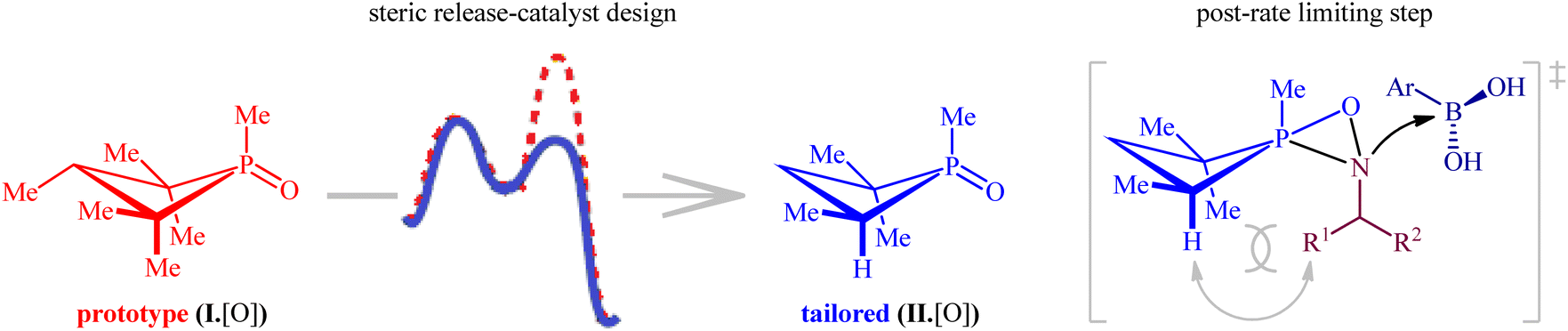
Later, this innovative research group undertook in-depth experimental kinetics and computational studies to elucidate the barriers to the compatibility of their methodology with nitroalkanes as coupling partners.26 The results indicated that a steric perturbation on the phosphorus catalyst I·[O] is a key factor that disrupts the desired reaction pathway when nitroalkanes were used as the substrates (Fig. 2). Therefore, a small peripheral alteration to the catalyst dramatically improves the efficiency of reductive coupling of nitroalkanes with boronic acids.
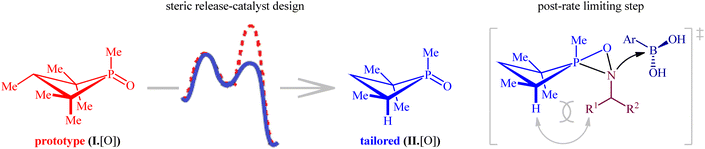 |
| | Fig. 2 Steric considerations enable P(III)/P(V)-catalyzed reductive C–N coupling of nitroalkanes and boronic acids. | |
Very recently, this creative research team disclosed that treatment of readily available 2-nitropropane with various (hetero)aryl boronic acids in the present of a catalytic amount of sterically reduced phosphetane II·[O] resulted in good yields of corresponding primary anilines through the Nef decomposition of 2-nitropropane to form the nitroxyl intermediate HNO followed by II·[O]-catalyzed reaction with boronic acids.27
5. Catalyst-free reactions
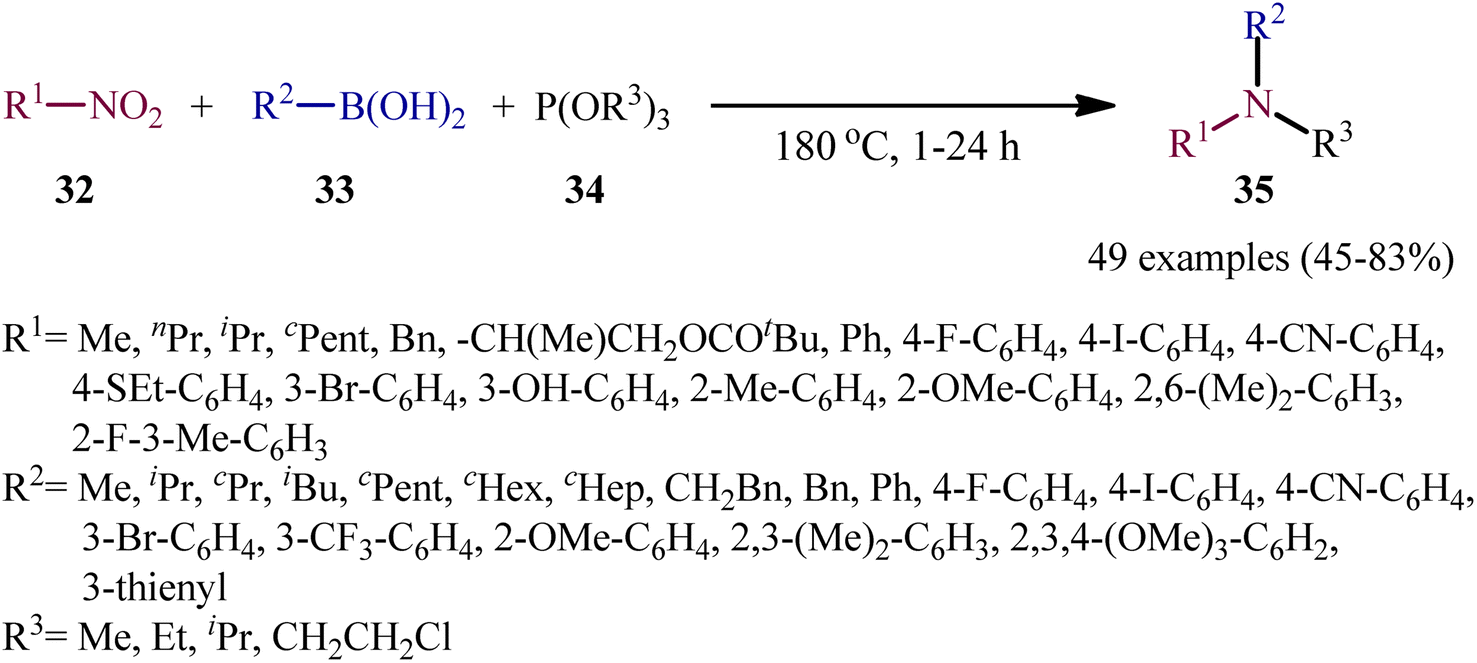
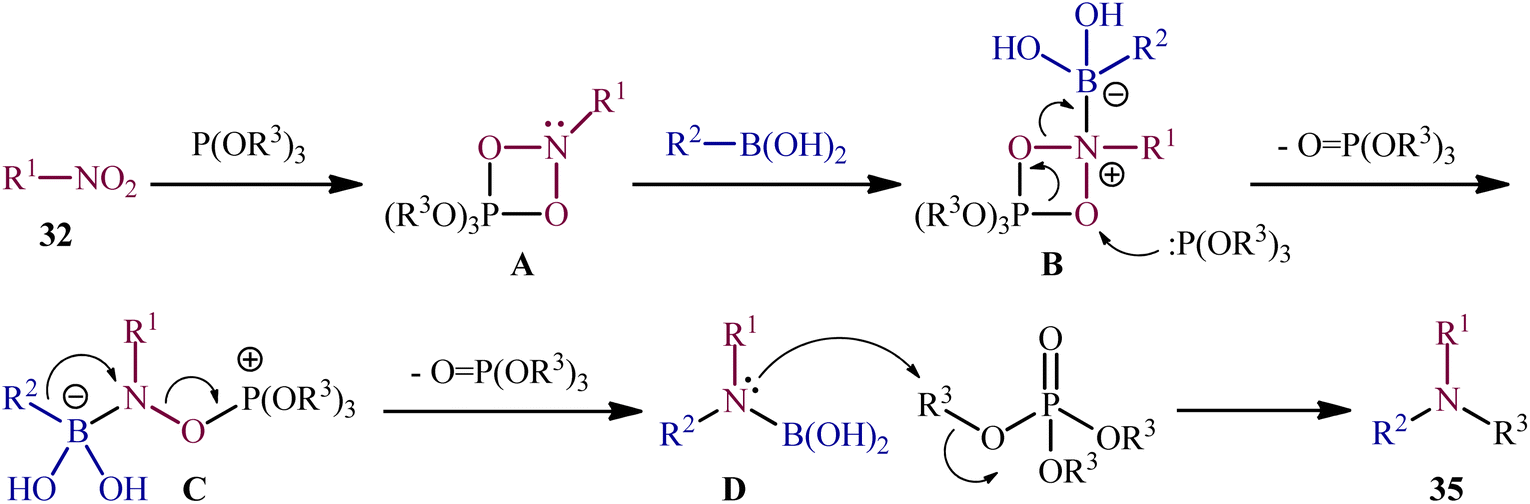
As a continuation of their previous work on the synthesis of di(hetero)arylamines through the transition-metal-free cross-coupling between nitrosoarenes and boronic acids in the presence of a trivalent phosphorus species,28 in 2020, Roscales and Csáky reported a beautiful protocol for the synthesis of tertiary aromatic amines 35 through the three-component reaction between nitro compounds 32, boronic acids 33 and trialkyl phosphites 34 under catalyst- and additive-free conditions (Scheme 18).29 Interestingly, in this reaction trialkyl phosphites played multiple roles; the substrate, solvent, and mediator. The transformation was very general and functional-group tolerant, compatible with a wide variety of nitro compounds, including aromatic (electron-rich and -poor), aliphatic (primary and secondary), and benzylic nitro compounds. In additions, the scope of boronic acids that underwent reaction was broad enough to include alkyl, aryl, and heteroaryl boronic acid derivatives. Additionally, various functionalized phosphites (primary and secondary, including halogen functionalization) were well tolerated by this method. The broad synthetic applicability of this three-component reaction was further demonstrated by performing reductive engaging of the corresponding α-nitro esters with aryl boronic acids and trialkyl phosphites to give biologically important α-amino esters. According to the authors, this reaction most likely proceeds through an azadioxaphosphetane intermediate A, as depicted in Scheme 19.
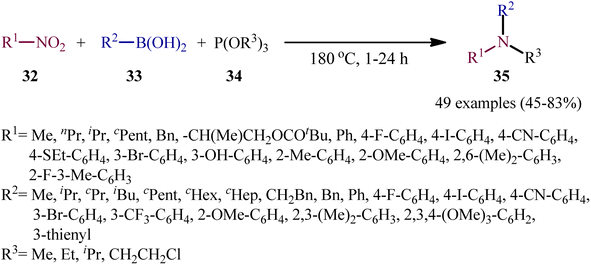 |
| | Scheme 18 Catalyst-free three-component coupling of nitro compounds 32, boronic acids 33, and trialkyl phosphites 34. | |
 |
| | Scheme 19 Possible mechanism for the formation of tertiary aromatic amines 35. | |
In a related investigation, Radosevich's research team reported the synthesis of a library of (hetero)arylamines 38 via reductive C–N coupling of corresponding nitro(hetero)arenes 36 and boronic acids 37 using PPh3 as a mediator without addition of any transition metal catalysts/additives.30 The reactions carried out in m-xylene at 120 °C, tolerated a series of sensitive functional groups (e.g., OMe, F, Cl, Br, I, Bpin, NH2), and provided the desired amine products in moderate to good yields, within hours (Scheme 20). The protocol was compatible with both aromatic and aliphatic boronic acids and various nitro(hetero)arenes. The results demonstrated that the nitroarenes bearing electron-withdrawing afforded better yields compared to electron-donating groups ones. On the other hand, aromatic boronic acids gave higher yield of coupling products than aliphatic and benzylic acids.
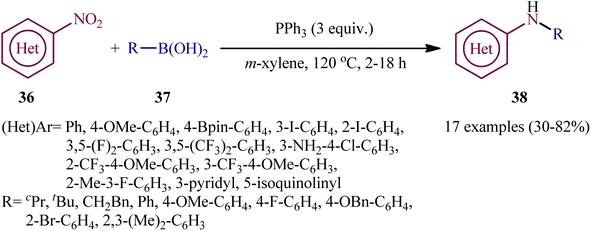 |
| | Scheme 20 PPh3-mediated reductive coupling of nitro(hetero)arenes 36 and boronic acids 37 reported by Radosevich. | |
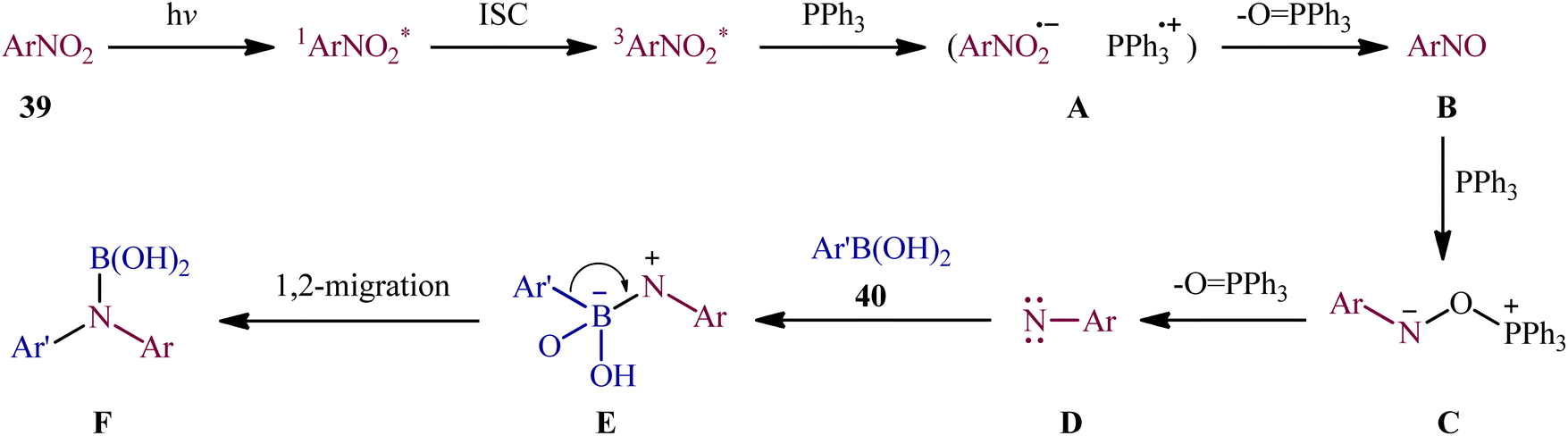
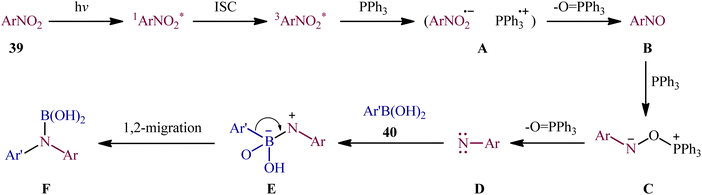
With the aim of designing a milder procedure to aryl amines through catalyst-free deoxygenative C–N coupling of nitroarenes and boronic acids, Baitalik and Jana along with their colleagues were able to demonstrate that a diverse range of N-substituted aniline derivatives 41 (52 examples) could be obtained in fair to good yields (up to 86%) from the reaction of corresponding nitro(hetero)arenes 39 with boronic acids 40 (aliphatic, aromatic and heteroaromatic) in the presence of 2 equiv. PPh3 under the irradiation of visible light at 30 °C, in the absence of any photoredox catalyst (Scheme 21).31 However, the process is not viable for gram-scale due to the noticeable reduction in the yield (from 86% in the 0.2 mmol scale, to 70% in the 4.0 mmol scale). Based on literature and the experimental results, the authors suggested a plausible mechanism for this C–N bond forming reaction as depicted in Scheme 22. The transformation may start with the singlet excited state of nitroarene via the excitation of nitroarene 39 under visible light irradiation, which after ultrafast intersystem crossing and electron transfer from PPh3 affords the nitroso compound B through the redox couple A. Subsequently, that nucleophilic addition of another molecule of PPh3 to the oxygen of the nitroso compound B leads to the formation of tetravalent phosphorus intermediate C, which after decomposition affords he nitrene intermediate D and 1 equiv. OPPh3. Thereafter, the newly formed intermediate D undergoes nucleophilic addition to the boronic acid 40 to form the boronate species E, which then undergoes a rearrangement to give the aminoboronic acid F. Finally, hydrolysis of this intermediate F affords the desired amine 41.
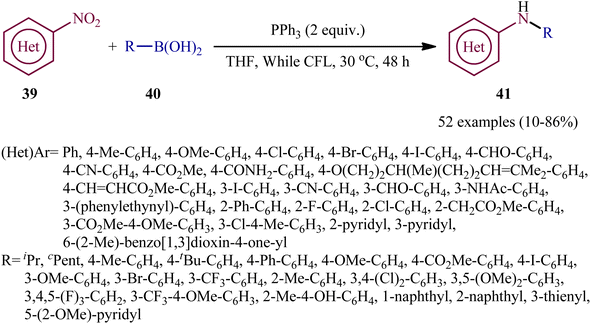 |
| | Scheme 21 Visible-light- and PPh3-mediated reductive coupling of nitro(hetero)arenes 39 with boronic acids 40. | |
 |
| | Scheme 22 The plausible mechanism for the reactions in Scheme 21. | |
Another independent light-mediated reductive amination method was published by Song–Yang and co-workers32 almost using identical conditions demonstrated by Baitalik and Jana group.33 However, in this method, the reaction was performed in toluene using an UV lamp (365 nm) as the light source. Although various nitro(hetero)arenes, and aromatic boronic acids were well tolerated under the reaction conditions, aliphatic boronic acids did not respond the reaction under standard conditions.
6. Conclusion
The direct reductive coupling of widely available nitro compounds with boronic acids has recently emerged as the reliable, powerful, and step-economical approach for the construction of biologically and synthetically important secondary amines in a one-pot manner, without the need to isolation of intermediates and use of pre-prepared amine substrates. As illustrated, this novel strategy for C–N bond formation was successfully applied for the construction of various C(sp2)–N–C(sp2) and C(sp3)–N–C(sp2) bonds. However, its applicability in the formation of C(sp3)–N–C(sp3) bonds was not investigated. Therefore, many more studies are needed to extend this chemistry to C(sp3)–N–C(sp3) bond formation. In addition, the majority of reactions covered in this review were performed at elevated temperatures. Thus, further research is still needed to development of efficient catalytic systems that allow this C–N bond forming reaction under milder conditions. Additionally, the scope of nitro compounds is largely limited to nitroarenes. Therefore, methodologies compatible with nitroalkanes should be developed.
Conflicts of interest
There are no conflicts to declare.
References
- Selected examples:
(a) G. R. Martinez, P. A. Grieco, E. Williams, K. Kanai and C. V. Srinivasan, J. Am. Chem. Soc., 1982, 104, 1436–1438 CrossRef CAS;
(b) C. C. Hughes and W. Fenical, J. Am. Chem. Soc., 2010, 132, 2528–2529 CrossRef CAS PubMed;
(c) P. N. Reddy, B. Banerjee and M. Cushma, Org. Lett., 2010, 12, 3112–3114 CrossRef CAS PubMed;
(d) K. C. Nicolaou, D. Y. K. Chen, X. Huang, T. Ling, M. Bella and S. A. Snyder, Chemistry and biology of diazonamide A: first total synthesis and confirmation of the true structure, J. Am. Chem. Soc., 2004, 126, 12888–12896 CrossRef CAS PubMed.
- Selected examples:
(a) G. Wozel and C. Blasum, Arch. Dermatol., 2014, 306, 103–124 CrossRef CAS PubMed;
(b) S. Dhillon and M. Clark, Drugs, 2014, 74, 1285–1291 CrossRef CAS PubMed;
(c) R. N. Brogden, R. C. Heel, T. M. Speight and G. S. Avery, Drugs, 1979, 18, 1–24 CrossRef CAS PubMed;
(d) D. K. Mahajan and S. N. London, Fertil. Steril., 1997, 68, 967–976 CrossRef CAS PubMed.
- Selected examples:
(a) R. Fritz, C. Lanen, V. Colas and P. Leroux, Pestic. Sci., 1997, 49, 40–46 CrossRef CAS;
(b) P. G. Bartels and C. W. Watson, Weed Sci., 1978, 26, 198–203 CrossRef CAS;
(c) H. J. Liang, Y. L. Di, J. L. Li and F. X. Zhu, Eur. J. Plant Pathol., 2015, 142, 691–699 CrossRef CAS.
- R. N. Salvatore, C. H. Yoon and K. W. Jung, Tetrahedron, 2001, 57, 7785–7812 CrossRef CAS.
- R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- H. Lin and D. Sun, Org. Prep. Proced. Int., 2013, 45, 341–394 CrossRef CAS PubMed.
- J. Q. Chen, J. H. Li and Z. B. Dong, Adv. Synth. Catal., 2020, 362, 3311–3331 CrossRef CAS.
- Z. Jiang, E. A. Mahmood, N. Z. Harofteh, A. G. Ebadi, M. Toughani and E. Vessally, Top. Curr. Chem., 2022, 380, 27 CrossRef CAS PubMed.
- B. D. Akana-Schneider and D. Weix, J. Am. Chem. Soc., 2023, 145, 16150–16159 CrossRef CAS PubMed.
-
(a) F. Ferretti, D. R. Ramadan and F. Ragaini, ChemCatChem, 2019, 11, 4450–4488 CrossRef CAS;
(b) S. Suárez-Pantiga and R. Sanz, Org. Biomol. Chem., 2021, 19, 10472–10492 RSC;
(c) J. Gui, C. M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. La Cruz and M. A. Schmidt, Science, 2015, 348, 886–891 CrossRef CAS PubMed;
(d) C. W. Cheung and X. Hu, Nat. Commun., 2016, 7, 12494 CrossRef PubMed;
(e) I. Sapountzis and P. Knochel, J. Am. Chem. Soc., 2002, 124, 9390–9391 CrossRef CAS PubMed.
- Selected examples:
(a) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 1–19 CrossRef PubMed;
(b) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 1–22 CrossRef CAS PubMed;
(c) Y. Yang, D. Zhang and E. Vessally, Top. Curr. Chem., 2020, 378, 1–32 CrossRef PubMed;
(d) Z. He, D. Wu and E. Vessally, Top. Curr. Chem., 2020, 378, 1–30 CrossRef PubMed;
(e) M. R. P. Heravi, A. Hosseinian, Z. Rahmani, A. Ebadi and E. Vessally, J. Chin. Chem. Soc., 2021, 68, 723–737 CrossRef CAS;
(f) S. Sarhandi, Z. Rahmani, R. Moghadami, M. Vali and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15, DOI:10.22034/crl.2018.85109;
(g) M. R. J. Sarvestani, N. Mert and E. Vessally, J. Chem. Lett., 2020, 1, 93–102, DOI:10.22034/jchemlett.2020.120304.
- S. Suárez-Pantiga, R. Hernández-Ruiz, C. Virumbrales, M. R. Pedrosa and R. Sanz, Angew. Chem., Int. Ed., 2019, 58, 2129–2133 CrossRef PubMed.
- F. Yang, X. Dong, Y. Shen, M. Liu, H. Zhou, X. Wang, L. Li, A. Yuan and H. Song, ChemSusChem, 2021, 14, 3413–3421 CrossRef CAS PubMed.
- X. Guan, H. Zhu and T. G. Driver, ACS Catal., 2021, 11, 12417–12422 CrossRef CAS PubMed.
- H. Talukdar, D. Gogoi and P. Phukan, Tetrahedron, 2023, 133251 CrossRef CAS.
- Y. Zhu, X. Zhang, F. Chen and G. P. Lu, Tetrahedron, 2023, 133444 CrossRef CAS.
- H. Song, Y. Shen, H. Zhou, D. Ding, F. Yang, Y. Wang, C. Xu and X. Cai, J. Org. Chem., 2022, 87, 5303–5314 CrossRef CAS PubMed.
- D. Wang, Z. Wan, H. Zhang, H. Alhumade, H. Yi and A. Lei, ChemSusChem, 2021, 14, 5399–5404 CrossRef CAS PubMed.
-
(a) C. Xie, A. J. Smaligo, X. R. Song and O. Kwon, ACS Cent. Sci., 2021, 7, 536–558 CrossRef CAS PubMed;
(b) K. Biswas, A. Das and V. Ganesh, Adv. Synth. Catal., 2021, 363, 4475–4496 CrossRef CAS.
-
(a) J. M. Brunel and G. Buono, Top. Curr. Chem., New Aspects in Phosphorus Chemistry, 2002, vol. 220, pp. 79–105 Search PubMed;
(b) O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2012, 23, 1–46 CrossRef CAS.
- T. V. Nykaza, J. C. Cooper, G. Li, N. Mahieu, A. Ramirez, M. R. Luzung and A. T. Radosevich, J. Am. Chem. Soc., 2018, 140, 15200–15205 CrossRef CAS PubMed.
- T. V. Nykaza, J. C. Cooper and A. T. Radosevich, Org. Synth., 2019, 96, 418–435 CrossRef PubMed.
- G. Li, Z. Qin and A. T. Radosevich, J. Am. Chem. Soc., 2020, 142, 16205–16210 CrossRef CAS PubMed.
- G. Li, T. V. Nykaza, J. C. Cooper, A. Ramirez, M. R. Luzung and A. T. Radosevich, J. Am. Chem. Soc., 2020, 142, 6786–6799 CrossRef CAS PubMed.
- G. Li, C. Te Grotenhuis and A. T. Radosevich, Trends Chem., 2021, 3, 72–73 CrossRef CAS PubMed.
- G. Li, Y. Kanda, S. Y. Hong and A. T. Radosevich, J. Am. Chem. Soc., 2022, 144, 8242–8248 CrossRef CAS PubMed.
- S. Y. Hong and A. T. Radosevich, J. Am. Chem. Soc., 2022, 144(20), 8902–8907 CrossRef CAS PubMed.
- S. Roscales and A. G. Csaky, Org. Lett., 2018, 20, 1667–1671 CrossRef CAS PubMed.
- S. Roscales and A. G. Csáky, Adv. Synth. Catal., 2020, 362, 111–117 CrossRef CAS.
- T. V. Nykaza, J. Yang and A. T. Radosevich, Tetrahedron, 2019, 75, 3248–3252 CrossRef CAS PubMed.
- K. Manna, T. Ganguly, S. Baitalik and R. Jana, Org. Lett., 2021, 23, 8634–8639 CrossRef CAS PubMed.
- X. Dong, Y. Yang, Y. Shen, A. Yuan, Z. Guo, H. Song and F. Yang, Green Chem., 2022, 24, 4012–4025 RSC.
- K. Manna, T. Ganguly, S. Baitalik and R. Jana, Org. Lett., 2021, 23, 8634–8639 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Seyedeh Bahareh Azimic,
Mohammed Ubaidd,
Abbas Talib Abd Alie,
Ayat Hussein Adhabf,
Mustafa Humam Samig,
Somayeh Soleimani-Amiri
b,
Seyedeh Bahareh Azimic,
Mohammed Ubaidd,
Abbas Talib Abd Alie,
Ayat Hussein Adhabf,
Mustafa Humam Samig,
Somayeh Soleimani-Amiri