
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile approach to create sensitive and selective Cu(II) sensors on carbon fiber microelectrodes†
Uma Nudurupati a,
Terdha Narlab,
David Punihaoleac and
Yangguang Ou*ac
a,
Terdha Narlab,
David Punihaoleac and
Yangguang Ou*ac
aDepartment of Chemistry, University of Vermont, USA. E-mail: you@uvm.edu
bDepartment of Pharmacology, University of Vermont, USA
cPipeline Investigator in Vermont Centre for Cardiovascular & Brain Health, USA
First published on 16th November 2023
Abstract
A facile platform derived from deposition of ethynyl linkers on carbon fiber microelectrodes has been developed for sensitive and selective sensing of Cu(II). This study is the first to demonstrate the successful anodic deposition of ethynyl linkers, specifically 1,4-diethynylbenzene, onto carbon fiber microelectrodes. Multi-scan deposition of DEB on these microelectrodes resulted in an increased sensitivity and selectivity towards Cu(II) that persists amidst other divalent interferents and displays sustained performance over four days while stored at room temperature. This method can be extended to other ethynyl terminal moieties, thereby creating a versatile chemical platform that will enable improved sensitivity and selectivity for a new frontier of biomarker measurement.
Introduction
Metal ions such as copper(II) (Cu(II)) are imperative for normal brain health and function, including as cofactors for enzymes, signaling molecules, factors for neurite growth, modulators of vesicular trafficking, and regulators of signaling receptors.1 Glutamate-induced activation of N-methyl-D-aspartate (NMDA)-receptors is hypothesized to trigger the translocation of copper-bound transporters to the synapse (on millisecond timescale).1 This results in free Cu(II) being released into the synaptic cleft, the nanometre-sized gap between two communicating neurons. In one hypothesis, this Cu(II) release prevents the over-excitability of the NMDA receptors from excessive glutamate,1 a phenomenon that is toxic to the neurons and causes cell death. High glutamate release, NMDA activation, and resulting excitotoxicity are hypothesized to be one of the significant causes of neurodegeneration in disorders such as Alzheimer's disease (AD)2 (reviewed in Parameshwaran et al.3).One of the hallmarks of AD is the aggregation of amyloid-β into senile plaques. Studies have indicated that Cu(II) is among the primary cations found inside senile plaques.4 Interestingly, some studies suggest that Cu(II) binding to amyloid-β makes it more prone to aggregation and increases the production of reactive oxygen species,5,6 thereby leading to cognitive decline and neurodegeneration. It has even been suggested that amyloid-β is increased to combat toxic levels of metal ions such as Cu(II).7–10
There is thus still an ongoing debate on whether Cu(II) is neuroprotective (preventing excitotoxicity) or deleterious (stabilizing amyloid aggregates) in the context of neurodegeneration. The role of Cu(II) in AD is generally controversial, as some studies observed a copper deficiency in AD patients11 while others show a copper overload12,13 (also reviewed in Ejaz et al.14 and Bagheri et al.4). Moreover, studies have revealed different Cu content in different regions of the brain.15,16 This is important because the hippocampus and cortex, for example, are more susceptible to plaque formation.17
Copper quantitation in biological samples is commonly accomplished by separations and spectroscopic techniques with high sensitivity. Separation methods like high-performance liquid chromatography (HPLC) and inductively coupled plasma mass spectrometry are championed for their multiplexing capabilities and are widely utilized in analysing clinical samples for copper.18–22 However, these methods are restricted to measuring total copper concentration (free Cu and those bound to proteins) and require multiple sample preparation steps (including sampling, extraction, digestion, and dilution) and longer analysis time. On the other hand, while colorimetric assays23,24 are cost-effective and have a more rapid readout than HPLC,25 they are limited by analyte–chromogen interaction kinetics.26 Fluorescent probes have faster response times and have been deployed in cells27–30 but rely on complicated fluorophore design and synthesis. Additionally, fluorescence assays are limited in their time resolution as they require incubation for 30 min–1 h for the fluorophore to localize appropriately. Probe-free methods like surface-enhanced Raman spectroscopy has also been applied to quantify Cu(II) in aqueous medium,31,32 but its relatively longer data acquisition time prevents its applicability to study shorter time-scale phenomena.
Many of these studies above quantify total Cu content, i.e., bound and free Cu together, rather than free Cu(II) alone. The majority of Cu(II) is usually in the bound form and coordinated to biomolecules (i.e., protein).33 While total Cu(II) may not change much or as rapidly, free Cu(II) can in response to different stimuli (i.e., depolarization, immune response, etc.) and can readily participate in events such as coordination with amyloid-β, production of reactive oxygen species, protect against excitotoxicity, etc.1,34–37 Thus, making measurements of free Cu(II) dynamics in a spatiotemporally resolved manner in vivo is essential to elucidate the role of Cu(II) in the etiology of AD and other neurodegenerative diseases.
Fast scan cyclic voltammetry (FSCV) at carbon-fiber microelectrodes (CFMs) has been gaining traction as a technique with high spatiotemporal resolution capable of in situ measurements. It typically operates with 100 ms time resolution and can measure from 10 s–100 s synaptic clefts simultaneously.38 One of the limitations of this method, however, is selectivity. Researchers have leveraged different approaches to improve selectivity for quantitation of different analytes (reviewed in Ou et al.39). Recent research have demonstrated that FSCV at CFMs is sensitive to metal ion reduction, including for Cu(II).40–44 To improve selectivity for Cu(II) sensing, Hashemi and colleagues grafted Cu(II)-selective ionophores onto the CFM surface.45 This methodology yielded a highly selective Cu(II) sensor while maintaining good sensitivity. The limitation, however, is that it involved a three-step synthetic approach, including electrochemical deposition of diazonium moiety, protection of unreacted hydroxyl groups with silyl ethers, and ionophore grafting via Click chemistry.
Here we detail a facile single-step method that generates a sensitive and selective Cu(II) sensor on CFMs for utilization with FSCV. We perform anodic deposition of 1,4-diethynylbenzene (DEB), a symmetric compound with two ethynyl moieties, directly onto CFMs. The primary inspiration for this work is from Geiger and colleagues,46–48 who demonstrated that ethynyl functional groups can be anodically deposited on glassy carbon electrodes with complete surface coverage (and even multi-layer coverage) in a controlled manner. We demonstrate here that we can anodically deposit ethynyl-containing compound like DEB onto CFM surface in a similar fashion. Moreover, the covalent attachment of DEB improves both sensitivity and selectivity of CFMs to Cu(II). This approach bypasses the need for a protection step and an ionophore step and reproducibly generates Cu(II)-selective biosensors in a more facile fashion. The long-term goal is to use these Cu(II)-selective biosensors to measure Cu(II) levels in cell and rodent models of neurodegeneration to elucidate the role of Cu(II) levels in the etiology of AD and other neurodegenerative disorders.
Experimental
Chemicals and solution
The salts used in the study were analytical grade, purchased from Krackeler Scientific (Albany, NY). We prepared all the salt solutions (0.1 M KCl, 11.5 μM CuCl2 solutions, mixed ion solutions (each ion being 1× equivalent to CuCl2 concentration)) by dissolving the appropriate amount of salt(s) in nanopure water (Milli-Q IQ7000, MilliporeSigma, St. Louis, MO). Phosphate buffered saline (PBS) tablets were purchased from Fisher Bioreagents (Waltham, MA, USA). 1× PBS (pH 7.40) was prepared by dissolving five PBS tablets in 1.0 L nanopure water (Milli-Q IQ7000, MilliporeSigma, St. Louis, MO). We prepared a stock solution of 1.79 ± 0.03 mM DEB (Krackeler Scientific, Albany, NY) in 0.1 ± 0.001 M [NBu4][PF6] (Krackeler Scientific, Albany, NY) in dry dichloromethane (DCM, Fisher Scientific, Waltham, MA). DCM was dried using molecular sieves method described by Geiger and colleagues.49 The organic electrolyte did not need to be dried.Electrode fabrication
We used a vacuum pump (Gast, Benton Harbor, MI) to aspirate a single 7 μm thick carbon fiber (GoodFellow, Pittsburgh, PA, USA) into a 0.5 mm inner diameter (1 mm outer diameter) borosilicate glass capillary (A-M Systems, Inc., Carlsberg, WA, USA). Then the borosilicate capillaries were inserted into a vertical puller (PC-100 or PE-22, Narishige Group, Japan), resulting in two electrodes with a tapered seal insulating the carbon fiber. The exposed carbon tip was manually cut to between 130 μm to 160 μm under a CX43 microscope with EP50 camera (Evident, formerly Olympus, Tokyo, Japan). All the results in this study are reported as current densities, where the measured current was normalized to the geometric surface area of the electrode, reported in unit of nA μm−2. We completed the electrode fabrication by attaching a silver epoxy paint (GC Electronics, Rockford, IL, USA) coated nichrome wire into the distal end of the fiber-filled capillary and sealing the nichrome wire using heat-sealed polyolefin shrink tubing (Mouser, Mansfield, TX, USA) at the electrode-nichrome wire interface.Electrochemical deposition of 1,4-diethynylbenzene on carbon fiber microelectrodes
The flask containing DEB, [NBu4][PF6], and DCM was sealed with a rubber septum, covered with aluminium foil during a coating experiment, and flushed with nitrogen for 10 minutes. We prepared a fresh solution each day of the experiment, and for coating each electrode, the required amount was withdrawn from the stock using a glass syringe. Because any water can quench the radical reaction, all electrodes must be air dried before use. DCM must be dried well.We placed a CFM of the desired length along with a pseudo-Ag/AgCl electrode in an amber, septum-capped 20 mL vial (Fisher Scientific, Waltham, MA) containing approximately 10 mL of the above stock. We flushed the entire vial for 10 min with nitrogen or until we observed condensation outside the vial. We then applied the waveform from −1 V to +1.9 V at a scan rate of 100 mV s−1 using the Wavedriver 100 Potentiostat (Pine Research Instrumentation, Durham, NC). We performed these waveform applications either once (which we will denote later on as “1 scan” or “single scan”), twice (2-scan), or thrice (3-scan). The cyclic voltammograms were collected and processed on the Aftermath software.
Fast scan cyclic voltammetry
We performed FSCV using a custom-built integration unit that includes a Dagan Chem-Clamp (Dagan Corporation, Minneapolis, MN, USA), National Instrument data acquisition cards (National Instrument, Austin, TX), Dagan-compatible headstage (Pine Research Instrumentation, Durham, NC), and WCCV 4.0 software (Knowmad Technologies LLC, Tucson, AZ, USA). All experiments were performed in a Faraday cage (AutoMate, Berkeley, CA, USA) with a two-electrode system comprised of a pseudo-Ag/AgCl reference (made in-house) and the CFM working electrode. We cycled the electrodes in each solution for 10 min prior to any data acquisition. The waveform we used for Cu(II) measurements was developed by Hashemi and colleagues (−1 V to 0.8 V at 400 V s−1 scan rate and a resting at 0 V, 10 Hz).45 All data was processed in Microsoft Excel and GraphPad Prism 9. Error bars and uncertainties are all reported as SEM.In vitro testing of Cu(II) using fast scan cyclic voltammetry
We equilibrated the CFM at 60 Hz and 10 Hz for 10 min each respectively with the applied waveform before the measurement. We performed FSCV in a beaker containing 0.1 M KCl with a stir bar rotating at 350 RPM. For calibration curve, we tested 0.115–11.5 μM concentration range. We added the analyte to the beaker at approximately 4 s after beginning data acquisition, with a total acquisition time of 30 s for each FSCV data file. For analysis, we exported all plots (the current vs. time curves and cyclic voltammograms) and converted the ordinate to current densities by dividing the current obtained by the geometric surface area. We performed linear regression using GraphPad Prism 9 on the current density vs. time plot for each measurement. The y-intercept of this regression line obtained was used as the average current density for that experimental condition.Electrochemical impedance spectroscopy (EIS)
We conducted EIS measurements for the bare and modified electrodes in 1× PBS buffer using the WaveDriver 100 Bipoteniostat/Galvanostat (Pine Research Instrumentation, Durham, NC). We applied the sinusoidal waveform of 5 mV amplitude from 100 kHz to 1 Hz on the Aftermath software (Pine Research Instrumentation, Durham, NC) and collected data using the Aftermath software. Equivalent circuit modelling was also performed in this software.Scanning electron microscopy (SEM)
We cut the CFMs leaving only the visible carbon fiber tip, which was then scatter coated with 60%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40% Pt:Au prior to imaging on SEM using Zeiss Sigma 300 VP Field-Emission Scanning Electron Microscope (Zeiss, Oberkochen, Germany). We utilized an excitation energy of 10 keV and adjusted the working distance to ensure that striations of the carbon fibres were in focus.
:40% Pt:Au prior to imaging on SEM using Zeiss Sigma 300 VP Field-Emission Scanning Electron Microscope (Zeiss, Oberkochen, Germany). We utilized an excitation energy of 10 keV and adjusted the working distance to ensure that striations of the carbon fibres were in focus.
Fast scan controlled adsorption voltammetry (FSCAV)
We performed FSCAV measurements per the methods developed in the studies by Heien, Hashemi, and colleagues.43 For data analysis, we manually integrated the Cu(II) reduction peak on the WCCV 4.0 software, and performed a linearized Langmuir fit to obtain the Kads (discussed below).Results and discussion
Anodic deposition of 1,4-diethynylbenzene is feasible on carbon fiber microelectrodes
Anodic deposition of DEB onto CFMs displayed a characteristic peak for generation of the ethynyl radical, as seen in Fig. 1. This characteristic peak appeared at a different potential for different electrodes, as seen in Fig. 1, but generally appeared in the potential range of 1.5–1.7 V. This is comparable to observations previously reported by Geiger and colleagues for glassy carbon electrodes.46,47,50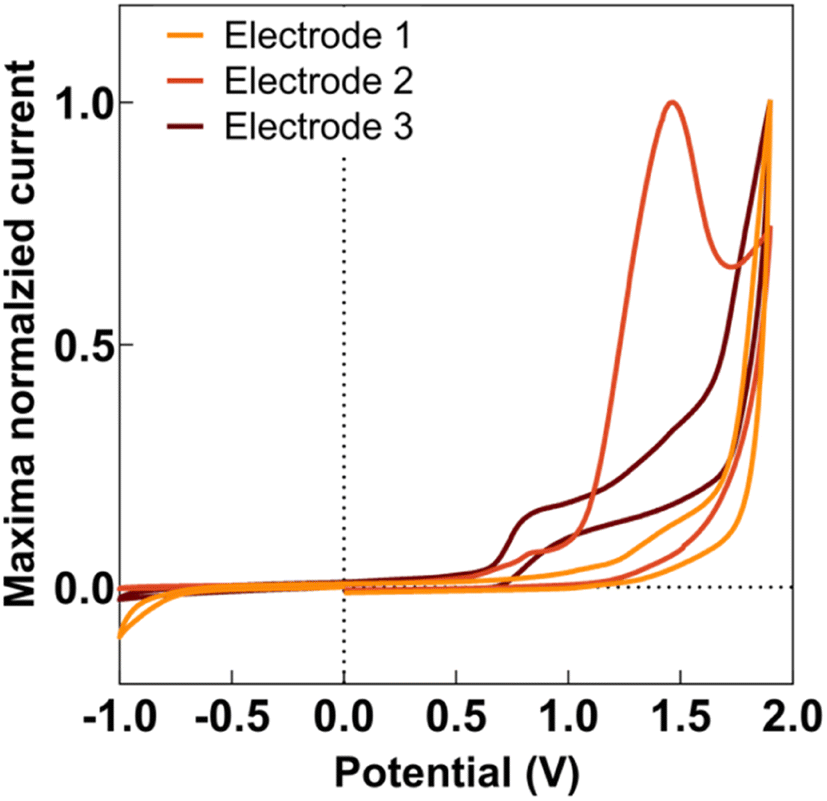 | ||
| Fig. 1 Representative cyclic voltammogram for the first scan of anodic electrodeposition of DEB on 3 different CFMs. The y-axis is normalized current density to maximum value for each data set. The distinctive peak for electrode #2 (orange) at ∼1.50 indicates the formation of an ethynyl radical. This potential shifts from electrode to electrode but generally shows up between 1.5 and 1.7 V. | ||
Geiger and colleagues previously reported successful deposition of multiple monolayers when they applied multiple scans of the deposition waveform.48 In this paper, we refer to each application of an entire waveform for deposition as a ‘scan.’ Thus, when we applied the waveform thrice, we refer to it as three scans. Geiger and colleagues observed the polymerization of ethynyl terminal molecules during multi-scan depositions, characterized by an increase in current at the potential at which ethynyl radical formation is observed.51 In contrast, as seen in Fig. S1,† we observed a decrease in the current at the ethynyl radical potential for subsequent scans, suggesting the absence of polymerization. This was advantageous as we did not want the DEB to self-polymerize before attaching to the electrode. Instead, our data suggests that the DEB formed covalent bonds to the carbon surface. With each scan, there were less surface area on the CFM for the DEB to attach, thereby reducing ethynyl radical formation and resulting current.
Multi-scan DEB deposition resulted in elevated sensitivity and improved selectivity of CFM to Cu(II)
We performed FSCV on bare and DEB-modified CFMs and discovered that the multi-scan DEB modified CFM possessed increased sensitivity to Cu(II) reduction compared to an unmodified electrode. As seen in Fig. 2a, the current density observed for the Cu(II) reduction peak (−0.7 V vs. pseudo-Ag/AgCl) increased for the same Cu(II) concentration as the number of scans of DEB deposition increased. The highest number of scans we tested was three, which yielded three-fold higher signal for Cu(II) than a bare electrode. We performed FSCV on the same bare and DEB (3 scans)-modified electrodes over 6 days and found that the enhanced sensitivity for DEB-modified electrodes (green, Fig. 2b) remained stable for up to 4 days when stored under room-temperature. After day 4, the 3-scan coated DEB electrode observed a significant drop in signal (p = 0.0069 comparing day 3 and 5, p = 0.0203 comparing day 3 and day 6). There were no statistical differences between days 1–4 for DEB-coated electrodes. There were no statical differences between days 1–6 for bare electrodes.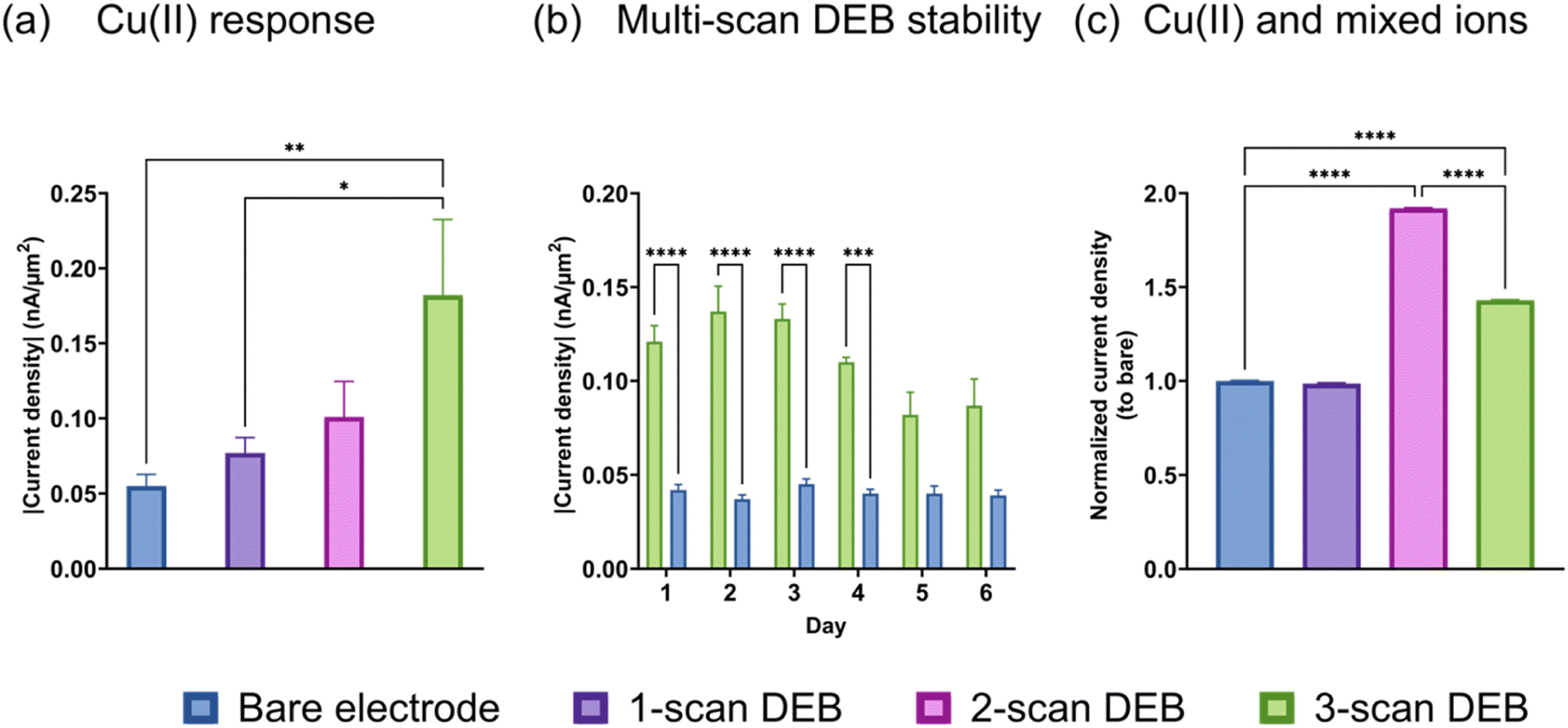 | ||
| Fig. 2 (a) Current density in response of bare (blue) and DEB-modified electrodes (1-scan DEB (purple), 2-scan DEB (pink), 3-scan DEB (green)) to injection of 11.5 μM Cu(II); solvent was 0.1 M KCl. (b) Current density response of bare and 3-scan DEB-modified electrodes to injection of 11.5 μM Cu(II) over 6 days after coating. The electrodes were stored at room-temperature in a box during this period. (c) Response of electrodes to Cu(II) in the presence of other divalent ion interferences (Mg(II),Ca(II),Zn(II)). The y-axis was calculated as the ratio of current density of modified electrode to that of the bare electrode. *p < 0.05 **p < 0.01 ***p < 0.0005 ****p < 0.0001. | ||
Next, we monitored the response of the bare and modified electrodes to Cu(II) in the presence of equal molar interfering divalent species (ions (Mg(II), Zn(II) Ca(II))). This means that the total concentration of the other metal ions is three times as high as Cu(II) in the solution. We compared the relative current density (the ratio of current density of the electrode of interest to that of the bare electrode) to quantitatively evaluate whether the presence of interfering species affected the elevated signal. The signal from Cu(II) reduction in mixed metal solution using a single-scan DEB-coated electrode (Fig. 2c, purple) was not statistically different than that for bare (p = 0.0836). For 2-scan DEB-coated electrodes, its elevated sensitivity to Cu(II) remained unchanged by the presence of other divalent ions Fig. 2c, pink. For 3-scan DEB, the presence of the other metal ions did attenuate the signal of the modified electrode. However, the 3-scan DEB-coated electrodes (green in Fig. 2c) still displayed higher sensitivity compared to that for bare (p < 0.0001). When we further monitored the response of the electrodes to the mixture of divalent metal ions without Cu(II), we discovered some interesting results. The bare electrode yielded anodic current densities (Fig. S3†) for mixtures of metal ions, with broad peaks. In the presence of equal molar Cu(II) and the same mixture of Mg(II), Zn(II), and Ca(II), the anodic current was supplanted by a cathodic current. Note that the total concentration of other divalent metal ions in these mixtures were 3-fold that of Cu(II). DEB-modified (3 scans) electrodes yielded cathodic currents for the mixed metals as well as mixed metals plus Cu(II). The signal of the modified electrodes for mixed metals with Cu(II) was 17.4 (±0.1)-fold higher than that for mixed metals alone. This increase was higher than that observed for bare electrode (Fig. S3†). These results suggest that perhaps the DEB coating increase electroplating or preconcentration of other divalent metals on the CFM surface. As indicated by the change of cathodic to anodic currents, this increase was greatest for Cu(II), yielding the highest sensitivity for it over other divalent ions. Combined with data from Fig. 2c, these results suggest that the attenuation of Cu(II) signal by the other metal ions can be attributed to the presence of other ions shielding Cu(II) and/or due to some faradaic interactions between other ions and DEB-coating. Nevertheless, even with a 3-fold higher molar equivalence of other divalent ions, the sensitivity of DEB-modified CFM for Cu(II) remained elevated.
We also tested to see if the elevated signal for 3-scan DEB coated CFMs persisted at different concentrations. Indeed, as seen in Fig. 3, the 3-scan DEB coated CFMs displayed significantly higher signal at all concentrations tested. The bare CFM had a sensitivity of 4.1 ± 0.8 pA μM−1 μm−2. For 3-scan DEB coated CFMs, the sensitivity is 11.4 ± 0.8 pA μM−1 μm−2, which is a 3-fold increase. The bare electrodes had a zero intercept (5 ± 5 pA μm−2) but the coated CFMs had a nonzero intercept (16 ± 5 pA μm−2).
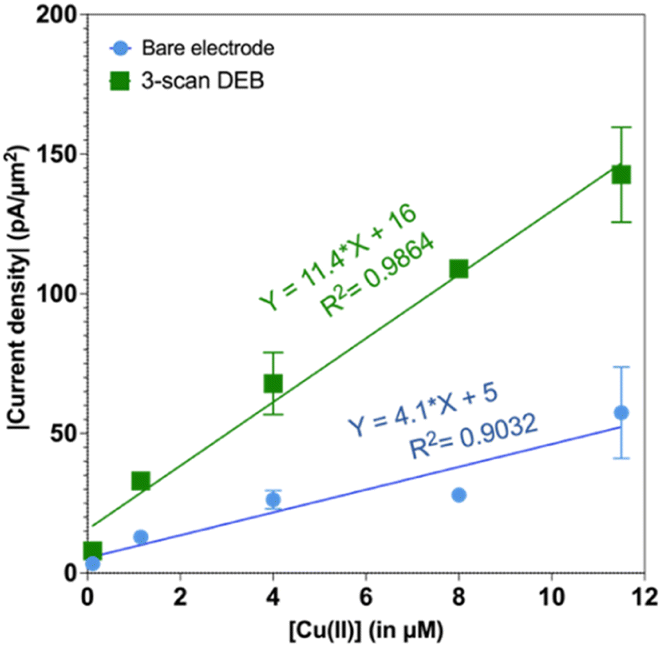 | ||
| Fig. 3 Plot of absolute value of current density as a function of concentration of Cu(II). Responses from bare electrodes is in blue and the responses from 3-scan DEB coated CFMs are in green. | ||
Investigating increased sensitivity of 3-scan electrodes
Next, we investigated the mechanism by which multi-scan DEB coating afforded the modified electrodes their increased sensitivity to Cu(II). We probed this via a few different approaches.First, we imaged the surface morphology using scanning electron microscopy. As seen in Fig. 4b, the bare electrode displayed a highly striated surface characteristic of carbon fiber in agreement with those reported by others.52–54 The single scan electrodeposition of DEB yielded a film-like smooth layer (Fig. 4c) over the striated features, which were no longer visible. The 3-scan DEB coating yielded more apparent striations compared to the single scan DEB coating but less than the bare. It also yielded small island-like features of varying sizes (Fig. 4d). This finding was surprising and points to the formation of an uneven surface for the 3-scan DEB deposition on the CFMs.
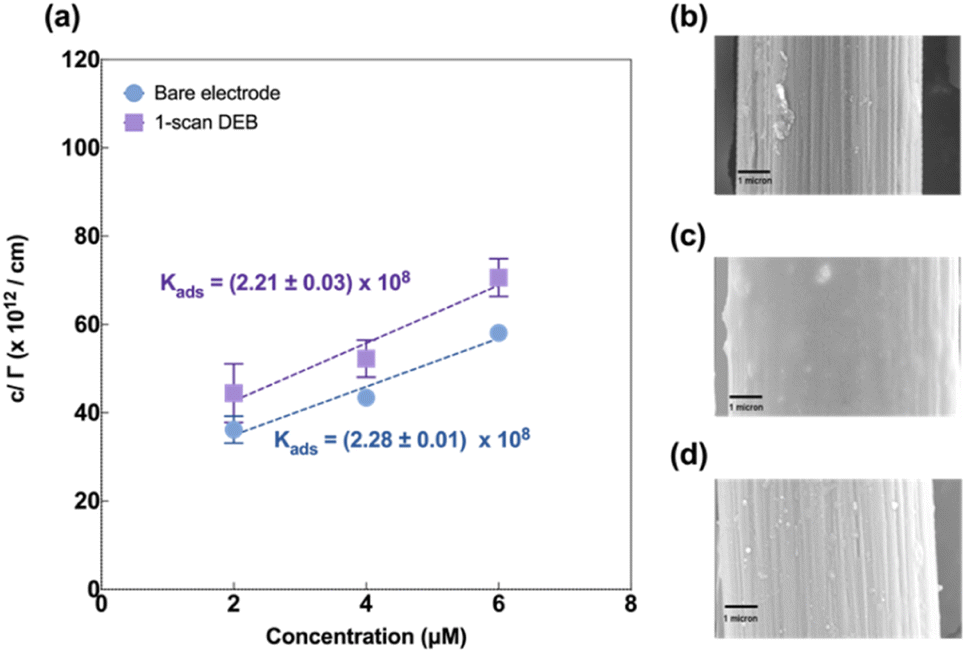 | ||
| Fig. 4 (a) Linearized adsorption isotherm for a bare or unmodified electrode (blue) and 1-scan DEB modified electrode (purple) scanning electron micrographs of (b) an unmodified carbon fiber electrode (c) 1-scan DEB modified electrode (d) 3-scan DEB modified electrode. Scale bar = 1 μm. | ||
Next, we evaluated Cu(II) adsorption behaviour on both modified and unmodified electrodes using FSCAV (please see ESI).† For an unmodified CFM, Cu(II) forms a monolayer on the electrode surface that follows the Langmuir adsorption isotherm (Fig. 4a, blue), in agreement with those reported by others.55 Cu(II) adsorption on DEB-modified CFM (1 scan) was found to follow Langmuir adsorption isotherm (Fig. 4a, purple) as well. The adsorption coefficient between DEB-modified electrode and Cu(II) (Kads = (2.21 ± 0.03) × 108) for 1-scan DEB-modified electrode was comparable to that of a bare CFM at the 95% confidence level (Kads = (2.28 ± 0.01) × 108 (p = 0.0913)).
Three-scan DEB deposition modified CFM on the other hand, could not be fitted with a Langmuir adsorption model (Fig. S2†). This suggests that this modification yielded a surface with greater heterogeneity, on which Cu(II) coverage could not form a monolayer. This agreed with the SEM results, which showed the island-like features for the 3-scan DEB-deposited electrodes.
To evaluate the impact of DEB modification on the CFM electrical conductivity, EIS was performed. Theoretically, modifications that increase the conductivity of the surface would yield lower impedance response in EIS across the applied perturbation frequencies.56,57 As seen in Fig. 5a and b, respectively, neither the single-scan electrodeposition of DEB on CFMs nor the multi-scan DEB coated electrodes yielded statistically different impedance than that of an unmodified electrode across perturbation frequencies. Such similarity indicates that unmodified electrodes and single scan DEB coating modified CFM are similar in surface electrical conductivity. Additionally, this is further evidence that self-polymerization was not observed during our electrodeposition experiments of DEB, as self-polymerization would yield a highly conductive polymer and is expected to decrease impedance response across perturbation frequencies.
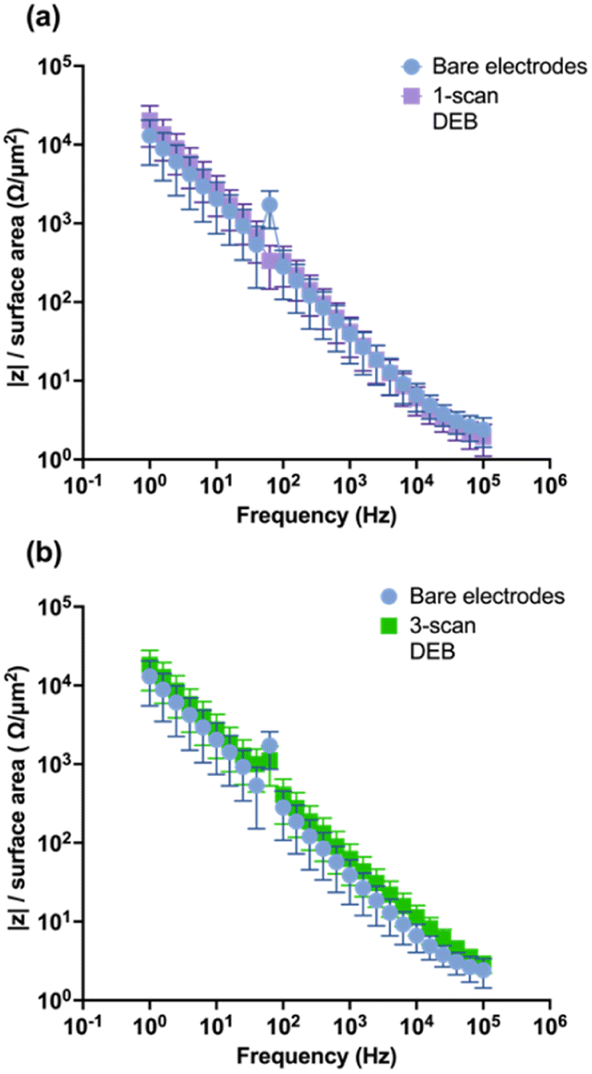 | ||
| Fig. 5 Bode plot containing surface area normalized impedance vs. frequency of sinusoidal perturbation for (a) 1-scan DEB modified electrodes (purple) and an unmodified electrode (blue) (b) 3-scan DEB modified electrode (green) and bare electrode (blue). | ||
The EIS data were fitted with the equivalent circuit model proposed by Sombers and colleagues58 comprising of two resistors and two constant phase elements. Table 1 lists the fitted parameters for the 3-scan DEB electrodes and unmodified electrodes. These terms include Q1 (double layer capacitance), R1 (CFM surface and solution resistance combined), Q2 (capacitance arising from the seal of the CFM), and R2 (shunt). The parameters α1 and α2 are modifiers for the constant phase element, which is used to model nonideal capacitors. For instance, an ideal capacitor would have a modifier α equal to 1. One-scan DEB coated electrodes displayed a marginal decrease of 11% in α1. Three-scan DEB deposition decreased α1 by 60% compared to an unmodified electrode. These results suggest that increasing scans of DEB increased the nonideal capacitor nature of the CFM. Nonideal behavior can arise from a few different phenomena, one of which is increased surface roughness.58 This makes sense for our system as 3-scan DEB had the greatest deviation from ideal capacitor behavior and has the greatest surface roughness in the form of island-like structures (Fig. 4d).
| Parameter | Electrode type | |||
|---|---|---|---|---|
| Bare | 1-scan DEB coating | 3-scan DEB coating | ||
| Q1 | nF cm−2 Sα−1 | 2.3 ± 0.7 | 1.6 ± 1.3 | 1.2 ± 0.4 |
| α1 | 0.85 ± 0.04 | 0.8 ± 0.2 | 0.3 ± 0.4 | |
| Q2 | nF cm−2 Sα−1 | 1.1 ± 1.1 | 0.7 ± 0.6 | 0.8 ± 0.3 |
| α2 | 0.6 ± 0.3 | 0.96 ± 0.04 | 0.4 ± 2.6 | |
| R1 | kΩ | 5.8 ± 0.6 | 4.0 ± 3.4 | 2.7 ± 1.8 |
| R2 | kΩ | 22.0 ± 27.1 | 187.0 ± 165.3 | 126.5 ± 86.7 |
Another phenomenon that can contribute to nonideal capacitor behavior is electrolyte absorption into the polymeric coating.59 We speculate that this could also be occurring with our 3-scan DEB-coated system. This electrolyte absorption could also explain another feature of the equivalent circuit modelling results (Table 1). We observed that 3-scan DEB-modified electrode had a significant decrease (by almost half) in the double layer capacitance compared to the bare electrode. If electrolytes were absorbing into the polymeric layer, it is possible that the overall permittivity could be reduced due to reduced polarizability, resulting in decrease in the observed double layer capacitance. The shunt capacitance (R2) also increased for both 1-scan and 3-scan DEB coated compared to that of bare. More studies will be needed to understand these complex phenomena and verify these hypotheses.
Conclusions
Successful anodic deposition of ethynyl terminal 1,4-diethynylbenzene on carbon fiber microelectrodes has been demonstrated for the first time. Single scan deposition of DEB resulted in a smooth film layer on the CFM surface that resulted in similar characteristics as that of an unmodified electrode, including adsorption coefficient to Cu(II) and net impedance across perturbation frequencies in EIS. One-scan DEB coated electrodes also had similar sensitivity towards Cu(II) in an FSCV measurement as that of an unmodified electrode. Three-scan deposition of DEB resulted in island-like structures on the CFM surface, a rough surface that prevented monolayer coverage by Cu(II), and a three-fold higher sensitivity towards Cu(II) compared to the bare CFM. This elevated sensitivity persisted even in the presence of other divalent interfering species (Zn(II), Mg(II), Ca(II)) and for up to four days when stored at room temperature. Characterization experiments revealed that 3-scan DEB modified electrodes had reduced double layer capacitance and increased nonideal capacitor behavior compared to bare electrodes.Author contributions
YO contributed to the conceptualization, acquiring funding, project administration, formal analysis, visualization of data, and writing, review, and editing process. UN contributed to the methodology development, formal analyses, conducting investigations and analyses, preparing the original draft of the manuscript (and review and editing), and the visualization of the data. TN contributed to methodology development, validation and analyses for standard curve and stability studies. DP assisted in formal analyses of the adsorption data, including assessing the appropriate mathematical model, and reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Prof. William Geiger for helpful conversations and Anna Sellon for helping with preliminary tangential experiments in the conceptualization phase of this work. The SEM utilized in this study was purchased with the support of the award number 1828371 from the National Science Foundation. The authors thank Michele von Turkovich and Kenneth Shepherd Jr. for their support with the SEM instrument. Funding for this work was provided by the University of Vermont (Y. O., D. P., U. N., and T. N.) and the National Institutes of Health, P20 GM1350007 through the Vermont Centre for Cardiovascular and Brain Health pilot grant award (Y. O., D. P., and U. N.).References
- N. D'Ambrosi and L. Rossi, Neurochem. Int., 2015, 90, 36–45 CrossRef PubMed.
- T. Harkany, I. Abrahám, W. Timmerman, G. Laskay, B. Tóth, M. Sasvári, C. Kónya, J. B. Sebens, J. Korf, C. Nyakas, M. Zarándi, K. Soós, B. Penke and P. G. Luiten, Eur. J. Neurosci., 2000, 12, 2735–2745 CrossRef CAS PubMed.
- K. Parameshwaran, M. Dhanasekaran and V. Suppiramaniam, Exp. Neurol., 2008, 210, 7–13 CrossRef CAS PubMed.
- S. Bagheri, R. Squitti, T. Haertlé, M. Siotto and A. A. Saboury, Front. Aging Neurosci., 2018, 9, 446 CrossRef PubMed.
- G. z. Eskici and P. H. Axelsen, Biochemistry, 2012, 51, 6289–6311 CrossRef CAS PubMed.
- X. Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, R. E. Tanzi and A. I. Bush, JBIC, J. Biol. Inorg. Chem., 2004, 9, 954–960 CrossRef CAS PubMed.
- H. G. Lee, R. J. Castellani, X. Zhu, G. Perry and M. A. Smith, Int. J. Exp. Pathol., 2005, 86, 133–138 CrossRef CAS PubMed.
- F. Cavaleri, Med. Hypotheses, 2015, 84, 460–469 CrossRef PubMed.
- K. P. Kepp, Prog. Neurobiol., 2016, 143, 36–60 CrossRef CAS PubMed.
- H. Hua, L. Münter, A. Harmeier, O. Georgiev, G. Multhaup and W. Schaffner, Biol. Chem., 2011, 392, 919–926 CrossRef CAS PubMed.
- C. Exley, E. House, A. Polwart and M. M. Esiri, J. Alzheimer's Dis., 2012, 31, 725–730 CAS.
- D. Kaden, A. I. Bush, R. Danzeisen, T. A. Bayer and G. Multhaup, Int. J. Alzheimer's Dis., 2011, 2011 Search PubMed.
- R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. McLean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y. S. Kim, X. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters and A. I. Bush, Neuron, 2001, 30, 665–676 CrossRef CAS.
- H. W. Ejaz, W. Wang and M. Lang, Int. J. Mol. Sci., 2020, 21, 7660 CrossRef CAS PubMed.
- E. Zieminska, A. Ruszczynska, J. Augustyniak, B. Toczylowska and J. W. Lazarewicz, Front. Mol. Neurosci., 2021, 14, 656740 CrossRef CAS PubMed.
- A. Pal and R. Prasad, Indian J. Clin. Biochem., 2016, 31, 93–98 CrossRef CAS PubMed.
- A. Pignataro and S. Middei, Neural Plast., 2017, 2017, 5281829 Search PubMed.
- J. S. Becker, M. V. Zoriy, C. Pickhardt, N. Palomero-Gallagher and K. Zilles, Anal. Chem., 2005, 77, 3208–3216 CrossRef CAS PubMed.
- S. Kubo, H. Fukuda, M. Ebara, N. Ikota, H. Saisho, H. Nakagawa, T. Ozawa, M. Yukawa, K. Kato, T. Satoh, T. Watayo and H. Sakurai, Biol. Pharm. Bull., 2005, 28, 1137–1141 CrossRef CAS PubMed.
- M. M. Quarles CD Jr, B. Michalke, H. Zischka, U. Karst and P. Sullivan, Metallomics, 2020, 12, 1348–1355 CrossRef.
- A. T. Townsend, K. A. Miller, S. McLean and S. Aldous, J. Anal. At. Spectrom., 1998, 13, 1213–1219 RSC.
- J. S. Becker, A. Matusch, C. Depboylu, J. Dobrowolska and M. V. Zoriy, Anal. Chem., 2007, 79, 6074–6080 CrossRef CAS PubMed.
- A. Abe, S. Yamashita and A. Noma, Clin. Chem., 1989, 35, 552–554 CrossRef CAS.
- H. H. Deng, G. W. Li, A. L. Liu, W. Chen, X. H. Lin and X. H. Xia, Microchim. Acta, 2014, 181, 911–916 CrossRef CAS.
- J. Arnaud, P. Chappuis, R. Zawislak, O. Houot, M. C. Jaudon, F. Bienvenu and F. Bureau, Clin. Biochem., 1993, 26, 43–49 CrossRef CAS PubMed.
- B. R. Gangapuram, R. Bandi, R. Dadigala, G. M. Kotu and V. Guttena, J. Cluster Sci., 2017, 28, 2873–2890 CrossRef CAS.
- L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 10–11 CrossRef CAS PubMed.
- Y. Niu, T. Ding, J. Liu, G. Zhang, L. Tong, X. Cheng, Y. Yang, Z. Chen and B. Tang, Talanta, 2021, 223 Search PubMed.
- C. Cheng, R. Zhang, J. Wang, Y. Zhang, C. Wen, Y. Tan and M. Yang, Analyst, 2020, 145, 797–804 RSC.
- Y. Fu, C. Ding, A. Zhu, Z. Deng, Y. Tian and M. Jin, Anal. Chem., 2013, 85, 11936–11943 CrossRef CAS PubMed.
- M. Y. Hsieh and P. J. Huang, RSC Adv., 2021, 12, 921–928 RSC.
- N. Ly, C. Seo and S.-W. Joo, Sensors, 2016, 16, 1785 CrossRef PubMed.
- G. Gromadzka, B. Tarnacka, A. Flaga and A. Adamczyk, Int. J. Mol. Sci., 2020, 21, 9259 CrossRef CAS PubMed.
- V. W.-S. Hung, H. Masoom and K. Kerman, J. Electroanal. Chem., 2012, 681, 89–95 CrossRef CAS.
- M. Del Barrio, V. Borghesani, C. Hureau and P. Faller, in Biometals in Neurodegenerative Diseases, Elsevier, 2017, pp. 265–281 Search PubMed.
- L. E. Cassagnes, V. Hervé, F. Nepveu, C. Hureau, P. Faller and F. Collin, Angew. Chem., 2013, 125, 11316–11319 CrossRef.
- V. Balland, C. Hureau and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17113–17118 CrossRef CAS PubMed.
- E. C. Dankoski and R. M. Wightman, Front. Integr. Neurosci., 2013, 7, 44 CAS.
- Y. Ou, A. M. Buchanan, C. E. Witt and P. Hashemi, Anal. Methods, 2019, 11, 2738–2755 RSC.
- J. Holmes, P. Pathirathna and P. Hashemi, TrAC, Trends Anal. Chem., 2019, 111, 206–219 CrossRef CAS.
- A. N. Perry, M. T. Cryan and A. E. Ross, Anal. Bioanal. Chem., 2021, 413, 6727–6735 CrossRef CAS PubMed.
- T. Siriwardhane, Y. Ou, P. Pathirathna and P. Hashemi, Anal. Chem., 2018, 90, 11917–11924 CrossRef CAS PubMed.
- P. Pathirathna, S. Samaranayake, C. W. Atcherley, K. L. Parent, M. L. Heien, S. P. McElmurry and P. Hashemi, Analyst, 2014, 139, 4673–4680 RSC.
- N. Manring, M. M. N. Ahmed, J. L. Smeltz and P. Pathirathna, Anal. Bioanal. Chem., 2023, 1–8 Search PubMed.
- Y. Yang, A. A. Ibrahim, P. Hashemi and J. L. Stockdill, Anal. Chem., 2016, 88, 6962–6966 CrossRef CAS PubMed.
- M. V. Sheridan, K. Lam and W. E. Geiger, Angew. Chem., 2013, 125, 13135–13138 CrossRef.
- M. V. Sheridan, K. Lam and W. E. Geiger, J. Am. Chem. Soc., 2013, 135, 2939–2942 CrossRef CAS PubMed.
- M. V. Sheridan, K. Lam, R. Waterman and W. E. Geiger, ChemElectroChem, 2019, 6, 5880–5887 CrossRef CAS.
- M. V. Sheridan, K. Lam, M. Sharafi, S. T. Schneebeli and W. E. Geiger, Langmuir, 2016, 32, 1645–1657 CrossRef CAS PubMed.
- P. Gamm, M. V. Sheridan, S. J. Van Wyck, A. Meindl, M. O. Senge and W. E. Geiger, Langmuir, 2019, 36, 96–108 CrossRef PubMed.
- M. V. Sheridan, K. Lam and W. E. Geiger, Angew. Chem., Int. Ed., 2013, 52, 12897–12900 CrossRef CAS PubMed.
- S. Tiwari, J. Bijwe and S. Panier, Wear, 2012, 274–275, 326–334 CrossRef CAS.
- P. Pathirathna, Y. Yang, K. Forzley, S. P. McElmurry and P. Hashemi, Anal. Chem., 2012, 84, 6298–6302 CrossRef CAS PubMed.
- Y. Li, A. L. Keller, M. T. Cryan and A. E. Ross, ACS Meas. Sci. Au, 2021, 2(2), 96–105 CrossRef PubMed.
- P. Pathirathna, T. Siriwardhane, S. P. McElmurry, S. L. Morgan and P. Hashemi, Analyst, 2016, 141, 6432–6437 RSC.
- C. L. Weaver, H. Li, X. Luo and X. T. Cui, J. Mater. Chem. B, 2014, 2, 5209–5219 RSC.
- E. Hernández-Balaguera, H. Vara and J. L. Polo, J. Electroanal. Chem., 2016, 775, 251–257 CrossRef.
- C. J. Meunier, J. D. Denison, G. S. McCarty and L. A. Sombers, Langmuir, 2020, 36, 4214–4223 CrossRef CAS PubMed.
- A. Trentin, A. Pakseresht, A. Duran, Y. Castro and D. Galusek, Polymers, 2022, 14, 2306 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05119f |
| This journal is © The Royal Society of Chemistry 2023 |