
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(vinyl alcohol)/modified porous starch gel beads for microbial preservation and reactivation: preparation, characterization and its wastewater treatment performance†
Shutao Linab,
Ruiting Changb,
Xinyu Caob,
Yongheng Zhangb,
Jiabo Chenb,
Wenchao Jiang*b and
Zhi Zhang *ab
*ab
aKey Laboratory of Three Gorges Reservoir Region's Eco-Environment, Ministry of Education, Chongqing University, Chongqing 400045, China. E-mail: zhangzhicqu@cqu.edu.cn
bCollege of Environment and Ecology, Chongqing University, Chongqing 400045, China. E-mail: wchjiang@qq.com
First published on 13th October 2023
Abstract
Poly(vinyl alcohol) (PVA)/modified porous starch (MPS) gel beads were prepared through in situ chemical cross-linking by incorporating with MPS, which was obtained by modifying porous starch (PS) with polyethyleneimine (PEI) and glutaraldehyde (GA). Addition of MPS could improve the storage modulus and the effective crosslinking density (ve) of the gel beads, and the mechanical properties were enhanced. The PVA–MPS gel beads were preserved as immobilized microbial carriers for 40 d and reactivated in wastewater. Scanning electron microscope (SEM) observations showed that the beads were highly porous and conducive for microorganism adhesion. The PVA–MPS gel beads were able to remove 97% of ammonia nitrogen and 80% of chemical oxygen demand (COD) after reactivation under all four preservation conditions. The abundance of Hydrogenophaga as denitrifying bacteria on PVA–MPS gel beads increased, with abundance of 8.44%, 5.55%, 8.90% and 9.48%, respectively. It proved that the carrier provided a partial hypoxic environment for microorganisms.
1. Introduction
Untreated wastewater is rich in ammonia nitrogen, and excess ammonia nitrogen is a major driver of eutrophication.1,2 In recent years, algal–bacterial symbiosis system (ABSS) has shown promising performance in treating a variety of wastewaters.3 ABSS utilizes the synergy of physiological functions between microalgae and bacteria for wastewater purification. Many studies demonstrated that better pollutants removal from wastewater could be achieved with ABSS than single algal or bacterial cultures.4 However, the size of microalgal cells is very small and cultures are usually quite diluted, which makes separation of microalgae and water in ABSS difficult and leads to inconsistent effluent quality. To overcome this problem, various strategies have been developed. Gel entrapment technology has received widespread attention.5,6ABSS can be applied for wastewater treatment through gel entrapment technology. Entrapment immobilization entraps microalgae and functional bacteria in the pores formed by polymer materials.7 The binding of algae and bacteria within the carrier is solid, leakage-resistant, and has limited impact on the mass transfer properties of the matrix and metabolites. In comparison to suspended cells, the immobilized cells have the advantages of high shock load resistance, high biomass density and excellent pollutant degradation. Studies have reported that microbial immobilization techniques in industrial wastewater achieved the removal of 95% of COD and 97% of NH4+–N.8 Despite the better pollutant removal performance of immobilized cells, large-scale application of immobilized cells demands high active cells with stable supply, which requires commercialization and preservation of viable cells. The gel entrapment technique is also commonly reported as a viable method for cell preservation. For example, the cells of Chlorella saccharophila that immobilized in the agar containing with algal nutrients was still culturable after 200 days storage at room temperature.9
The choice of embedding carrier is an important aspect of gel entrapment technology.10 Many natural and synthetic polymer materials, including agar, alginate, chitosan, and poly(vinyl alcohol) (PVA), have been successfully applied for entrapment of microorganism.11 Among them, PVA gel beads are non-toxic and well biocompatible, and its hydrophilic three-dimensional polymer network provides a good living environment for the survival of microorganism to treat wastewater.12 However, PVA gel beads tend to expand and agglomerate in water and exhibit strong mass transfer resistance, which reduces the microbial degradation efficiency. It has been shown that the addition of sodium alginate (Alg) can reduce the mass transfer resistance and improve the swelling of PVA gel beads.13
Addition of adsorbents to hydrogels can increase the pore size of microbial gel beads to achieve the dual effect of adsorption and immobilization.14 Porous starch (PS) is a natural macromolecule with abundant micro-sized pores and has extensive application for its excellent adsorption property and environmental benignancy.15 However, PS has low strength and unstable structure. Crosslinking of PS using glutaraldehyde (GA) can stabilize the spatial structure by forming hydrogen bonds in the starch granules. In addition, polyethyleneimine (PEI) is a cationic polyelectrolyte, and modification of PS using the modifier PEI to produce modified porous starch (MPS) would cause MPS to display a positive charge.16 Therefore, MPS can immobilize cells by adsorption and charge effects. Currently, few studies have investigated the use of PVA gel beads with addition of MPS to immobilize microorganisms for preservation and reactivation.
Therefore, in this study, MPS was first prepared and PVA–MPS gel beads immobilized with microalgae and bacteria were prepared by adding MPS to the gel. Afterwards, the PVA–MPS gel beads were preserved in different environments and reactivated in wastewater. The aim of this study was to maintain high viability of microorganisms preserved in different environments and to facilitate global transportation of microbial gel beads. The main topics of this study were (1) characterization of PVA–MPS gel beads, (2) morphological changes and microstructure of gel beads, (3) the performance of gel beads after reactivation, and (4) the diversity of microbial communities within gel beads.
2. Materials and methods
2.1 Materials
PVA (average polymerization degree 1799 ± 50, alcoholysis degree 99%), sodium alginate (Alg, viscosity 200 ± 20 mPa s), polyethyleneimine (PEI, molecular weight 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, purity 99%) and glutaraldehyde (GA, 25% in H2O) were purchased from Aladdin Co. Ltd (Shanghai, China). Porous starch (food-grade) was purchased from the local market. Boric acid (H3BO3) and calcium chloride (CaCl2) were purchased from Bodi Chemical Co. Ltd (Tianjin, China). Other chemical agents were all of analytical grade and used as received. The inoculum sludge was obtained from the JiGuanShi Municipal Wastewater Plant of Chongqing (China) and the algae (Chlorella, FACHB-1068, 1.24 × 107 cell per mL) was obtained from the Freshwater Algae Culture Collection at the Institute of Hydrobiology (China) for preparation of PVA gel beads. The details can be found in Text S1.†
000, purity 99%) and glutaraldehyde (GA, 25% in H2O) were purchased from Aladdin Co. Ltd (Shanghai, China). Porous starch (food-grade) was purchased from the local market. Boric acid (H3BO3) and calcium chloride (CaCl2) were purchased from Bodi Chemical Co. Ltd (Tianjin, China). Other chemical agents were all of analytical grade and used as received. The inoculum sludge was obtained from the JiGuanShi Municipal Wastewater Plant of Chongqing (China) and the algae (Chlorella, FACHB-1068, 1.24 × 107 cell per mL) was obtained from the Freshwater Algae Culture Collection at the Institute of Hydrobiology (China) for preparation of PVA gel beads. The details can be found in Text S1.†
2.2 Preparation of modified porous starch
The preparation procedure for the MPS materials was carried out as follows: 2.0 g of PS was dissolved in 20 mL ultrapure water and then 1.3 g of PEI was added into the PS solution. The mixture solution was mechanically stirred at 300 rpm for 6 h at room temperature, and then the reaction was continued for 2 h by slowly adding 10 mL of GA solution (5.0 wt%), Afterwards dispersion solution was centrifugated (5000 rpm) and washed with ultrapure water. The resulting solids were dried in an oven at 40 °C for 24 h, finally yielding MPS materials.2.3 Preparation of PVA–MPS gel beads
The preparation procedure for the PVA–MPS gel beads was carried out as follows: 9.0 g of PVA was dispersed homogeneously in 100 mL ultrapure water in a beaker, and then Alg (1.4 g) and MPS (0.7 g) were added into the solution at 95 °C under vigorous stirring until completely dissolving. The obtained PVA–MPS dispersion solution was cooled to room temperature, and a certain amount of algal–bacterial mixture was added. The biomass of mixture was 2 g L−1 (algal–bacterial ratio of 1:3). The mixture solution was then dropped into the saturated H3BO3/CaCl2 solution (3.0 wt%), keeping for 30 min to form spherical beads, which were transferred to Na2SO4 solution (0.5 mol L−1) and placed at 4 °C for 2 h.17 Finally, the prepared beads were soaked and washed with ultrapure water, and stored in ultrapure water at room temperature. For characterization, the PVA–PS and PVA–MPS gel bead samples with unembedded microorganisms were prepared using the same method.
2.4 Characterization of PVA–MPS gel beads
Scanning electron microscopy (SEM) (Sigma 300; ZEISS, Germany) was used to determine the morphology of PS, MPS, and PVA–MPS gel beads. The zeta potential of PS and MPS particles was measured in ultrapure water by the Zetasizer Nano (ZS90; Malvern, UK). The functional group composition of the sample was determined using a Fourier transform infrared spectrometer (FTIR) (Nicolet iS5; Thermo, USA) in the wave number range 4000–400 cm−1. The sample to be tested was heated from room temperature to 600 °C in a nitrogen environment at a heating rate of 10 °C min−1. A thermogravimetric analyzer (TG209 F1; NETZSCH, Germany) was used to generate a thermogravimetric curve of the sample to determine its thermal stability and composition. The thermogravimetric curve was differentiated to reflect the rate of weight loss. The viscoelasticity properties of PVA hydrogel were performed on Rheometer System (Mars60; Haake, Germany) with parallel plates with diameter of 20 mm and a plate-to-plate distance of 1–2 mm. Both the strain and the frequency sweep experiments were performed at room temperature. In strain sweep measurements, the shear storage modulus (G′) and loss modulus (G′′) were recorded at the strain of 0.005–10% and the frequency of 1 Hz. In the frequency sweep experiments, G′ and G′′ were measured in the linear viscoelastic regime, for frequencies ranging from 0.1 to 100 Hz, at a maximum strain, γ, of 0.1%. A pore size analyzer (ASAP2460; Micromeritics, USA) was used to determine the specific surface area, pore size, pore volume, and other physical characteristics of gel bead samples with the typical monomer ratio. The tensile properties of PVA hydrogel samples were measured with a universal testing machine (CMT6103; MTS, China). Samples with a dumbbell shape and size 200 × 20 × 4 mm3 were prepared. The tensile speed and temperature were 20 mm min−1 and 23 °C, respectively. The retention rate of gel beads without damage was used to measure the hydraulic impact property. PVA gel beads were put into the ultrapure water, stirring at room temperature with stirring speed 2000 rpm. Retention rate (%) = (Nt/N0) × 100%. Where N0 was initial number of gel beads; Nt was the number of gel beads at time t. A freeze-dryer (LGJ-10A; Yuming Instruments, China) was used to lyophilize the PVA gel beads and thus measure the swelling behavior and gel mass fraction of the gel beads. The pre-weighed dry samples of PVA gel beads were immersed into the ultrapure water at room temperature until they swelled to equilibrium. Swelling ratio (%) = (Wt − W0)/W0 × 100%. Where W0 was the dried weight of gel beads; Wt was the weight of gel beads in swollen state at time t. A certain amount of dry gel was weighed and soaked in deionized water for 72 h to remove impurities. The gel was then removed and placed in a freeze-dryer to dry to constant weight. Gel mass fraction (%) = (W1/W2) × 100%. Where W1 and W2 are, respectively, the dry weight of gel samples before and after soaking in deionized water.2.5 Preservation and reactivation experiments of PVA–MPS gel beads
The synthetic wastewater was prepared using deionized water. The carbon, nitrogen and phosphorus needed for microbial growth were provided by C6H12O6, NH4Cl and KH2PO4, respectively, while Ca2+ and Mg2+ were provided by CaCl2, MgSO4·7H2O, respectively. Other trace elements and specific compositions were shown in Table S1.† The main water quality of synthetic wastewater is: COD(C6H12O6) 213 ± 17 mg L−1, NH4+–N (NH4Cl) 35 ± 2 mg L−1, TP 4.1 ± 1.1 mg L−1, MgSO4·7H2O 75 mg L−1, CaCl2 36 mg L−1, and trace element 1 mL L−1. Collected samples were filtered using a filter with 0.45 μm pore size. The water quality were determined by standard methods (APHA, 2005). Total inorganic nitrogen (TIN) = NH4+–N + NO2−–N + NO3−–N.
2.6 Microbial community
The PVA–MPS gel bead samples were collected after the end of the 60th cycle and then sent to Institute Majorbio (Shanghai, China) for 16S rRNA sequencing. The specific primers set with barcode 338F (ACTCCTACGGGAGGCAGCAG) and 806R (GGACTACHVGGGTWTCTAAT) for bacteria were used for amplification of the hypervariable region of 16S rRNA. Purified amplicons were pooled in equimolar and paired-end sequenced on the Illumina platform (Illumina, USA). Further analysis was performed on Majorbio Cloud platform (https://cloud.majorbio.com).3. Results and discussion
3.1 Structural analysis of PVA–MPS gel beads
SEM observation showed that the particle structure of MPS was more complete, the pores formed on the surface were of uniform size, and there was less pore collapse (Fig. S2†). This is because the hydroxyl group of MPS forms a chemical bond with the aldehyde group of GA, which supports and protects the spatial structure of MPS. In addition, MPS were positively charged in the range of pH 2–10 (Fig. 1(a)), and the zeta potential of MPS was significantly increased up to +37 mV and higher in intensity than that of PS (+7 mV) due to the addition of amine groups to MPS through PEI, which gave a positive charge to its surface.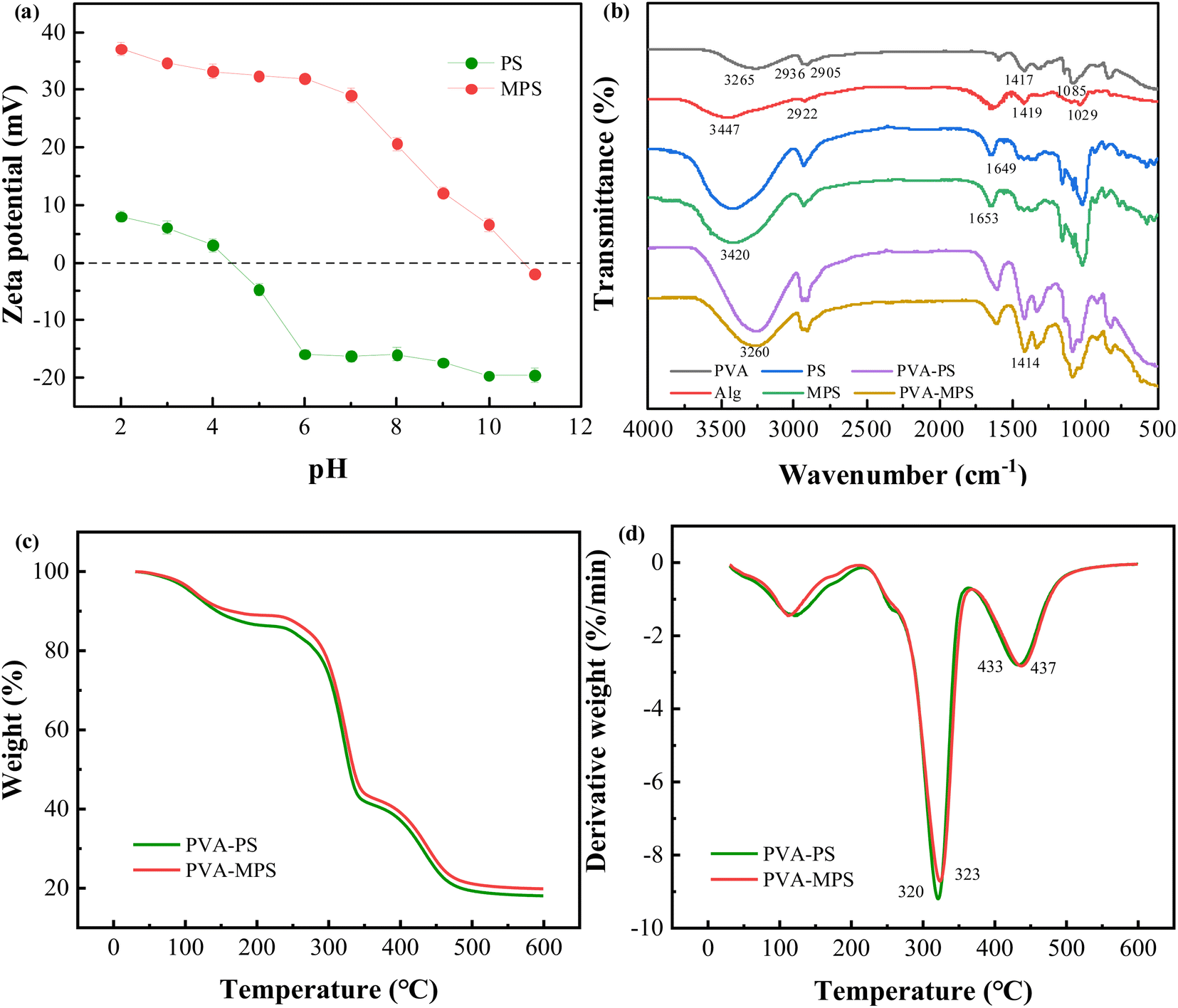 | ||
| Fig. 1 (a) Zeta potential measurements of the samples of PS and MPS, (b) FTIR spectra of PVA, Alg, PS, MPS, PVA–PS and PVA–MPS gel beads, and (c) TGA and (d) DTG curves of PVA–PS and PVA–MPS gel beads. | ||
The structural features of the constituent raw materials, PVA–PS and PVA–MPS gel beads were characterized by FTIR analysis, as shown in Fig. 1(b). For PVA sample, a strong absorption peak at 3265 cm−1 was attributed to hydroxyl (O–H) stretching vibration. The characteristic absorption peaks at 2936 and 2905 cm−1 were attributed to asymmetric and symmetric stretching vibrations of –CH2, respectively, and the absorption peaks at 1417 and 1085 cm−1 were attributed to the bending vibration of –CH2 and the stretching vibration of C–O. For Alg sample, the characteristic peaks at 3447, 2922, 1419, and 1029 cm−1 can be attributed to O–H stretching vibrations, the stretching vibrations of C–H, the stretching vibrations of –COO–, and the stretching vibrations of C–O–C, respectively. For PS and MPS samples, the two characteristic peaks are similar, indicating that MPS is made of PS and maintains the characteristics of PS through PEI modification, but the absorption peak is slightly wider near 3420 cm−1, which is due to the overlap of the characteristic peaks of N–H and O–H in MPS,18 and this is in accordance with the increase in the zeta potential value of the MPS. The 1649 cm−1 peak of the PS spectrum indicated O–H bending. The spectrum of PEI have been reported by researchers and the characteristic peak present at 1650 cm−1 for PEI was assigned to the amino group.19 Whereas, the peak at 1653 cm−1 observed in MPS with possible overlap was due to the formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, suggesting that the reaction between the PEI (primary amine groups) and the GA (aldehyde groups) formed a Schiff's base.20 For PVA–MPS gel beads sample, the characteristic peaks at 3265 cm−1 corresponding to the stretching vibration peaks of O–H of the PVA sample shifted to 3260 cm−1 and showed a broadband, indicating that O–H in PVA is crosslinked with H3BO3 through chemical bonding, and that mixing with MPS produced an overlapping of the stretching vibrations of O–H and N–H. The characteristic peaks at 1419 cm−1 corresponding to the stretching vibration peaks of –COO– on the alginate backbone shifted to 1414 cm−1. This phenomenon confirmed the formation of the ionic bonds between the Ca2+ and –COO– in Alg.21
N bonds, suggesting that the reaction between the PEI (primary amine groups) and the GA (aldehyde groups) formed a Schiff's base.20 For PVA–MPS gel beads sample, the characteristic peaks at 3265 cm−1 corresponding to the stretching vibration peaks of O–H of the PVA sample shifted to 3260 cm−1 and showed a broadband, indicating that O–H in PVA is crosslinked with H3BO3 through chemical bonding, and that mixing with MPS produced an overlapping of the stretching vibrations of O–H and N–H. The characteristic peaks at 1419 cm−1 corresponding to the stretching vibration peaks of –COO– on the alginate backbone shifted to 1414 cm−1. This phenomenon confirmed the formation of the ionic bonds between the Ca2+ and –COO– in Alg.21
We investigated the composition and thermal stability of the PVA–PS gel beads and PVA–MPS gel beads by thermogravimetric analysis. As shown in Fig. 1(c) and (d), the gel beads experienced three stages of weightlessness. First, below 215 °C the water molecules adsorbed in the material mostly evaporate; this is the first weightlessness stage.22 In this stage, the weight loss of the PVA–MPS gel beads (11.05%) was less than the PVA–PS gel beads (13.72%), which suggests that there is less bound water in the PVA–MPS gel beads, and less bound water reduces the distance of the polymer chains and may increase the intermolecular interactions of the polymer chains. The second weightlessness stage occurs between 220 and 365 °C, with peaks of 323.40 °C and 320.90 °C for PVA–MPS gel beads and PVA–PS gel beads, respectively, and is mainly caused by the fracture and decomposition of functional groups (e.g., hydroxyl groups, carboxyl groups, and ester bonds) and cationic bond.23 After 365 °C, the weight loss can mostly be attributed to the breaking of the PVA carbon chain and the Alg “egg-box” structure.24 Compared to PVA–PS gel beads, the peak of PVA–MPS gel beads was higher, and the weight residual remains above 500 °C is 19.83% for PVA–MPS gel beads which is found to be higher as compared with the weight residue of PVA–PS gel beads (18.06%), indicating that the addition of MPS improved the thermal stability of the gel beads.
3.2 The porous crosslinking network structure of PVA–MPS gel beads
Fig. 2(a) showed the strain dependence of the shear storage modulus (G′) and loss modulus (G′′) at the frequency of 1 Hz for both PVA hydrogels. Within the strain amplitudes range from 0.010 to 10.000%, the storage modulus of the two hydrogel samples is constantly greater than their loss modulus, which is evidence of typical gel networks. In addition, it can be noted that, for small strain amplitudes, G′ was independent of the strain amplitude, which indicated that the deformation imposed on the network structure was entirely reversible. The MPS hydrogel has a higher G′ and G′′ than hydrogel in the strain amplitudes stable region, which is due to the MPS loading. The higher shear storage modulus (G′) and loss modulus (G′′) in stable region is caused by the higher cross-linking degree of the MPS hydrogels. Since the MPS hydrogel has a higher cross-linking degree, the polymer network is less likely to deform under the shearing environment and shows a higher storage modulus. Moreover, the higher cross-linking degree causes a tighter structure, indicating that the friction between MPS hydrogel polymer chains is stronger, so the G′′ of magnetic hydrogel is higher than the hydrogel. At higher strain amplitudes, G′ was a decreasing function of the strain amplitude and the deformation was no longer reversible.25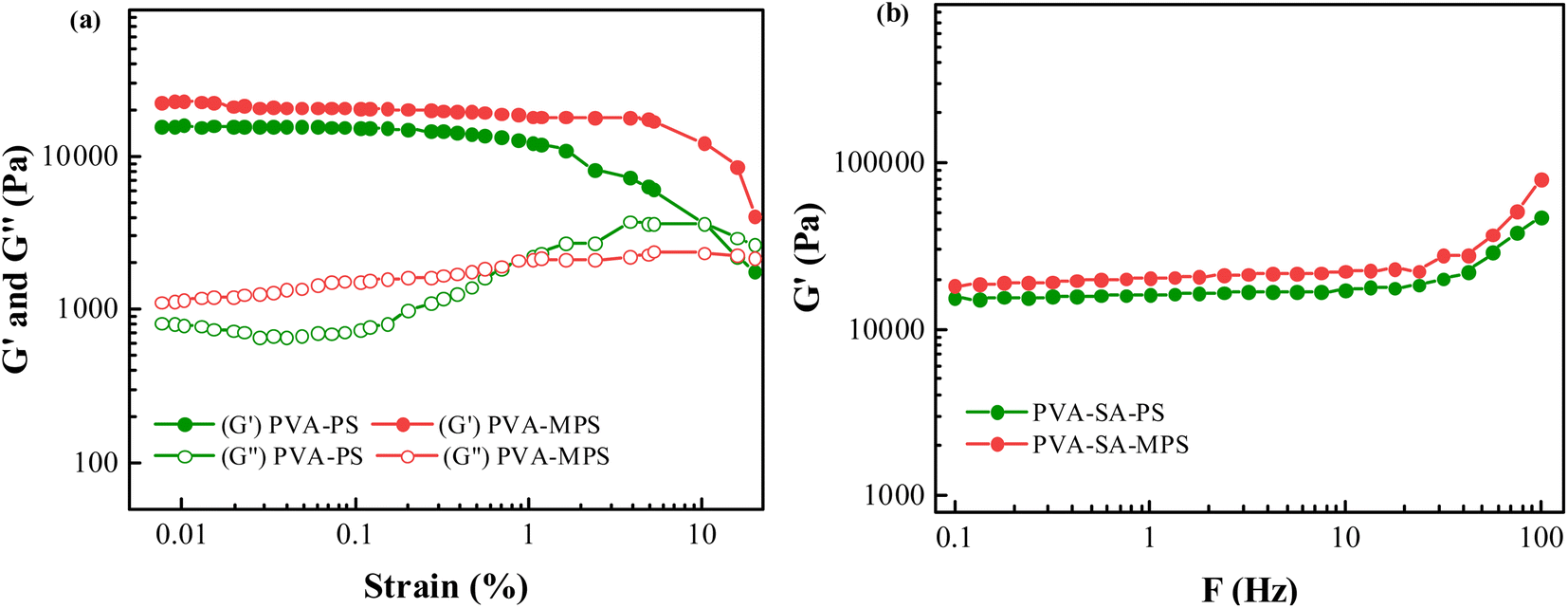 | ||
| Fig. 2 (a) Strain dependence of G′ and G′′ for PVA–PS and PVA–MPS hydrogels, and (b) Frequency dependence of G′ for PVA–PS and PVA–MPS hydrogels. | ||
The frequency dependence of G′ was plotted in Fig. 2(b) for the two hydrogels. It can be seen that G′ did not depend on the test frequency in the range between 0.1 and 100 Hz for all samples, indicating that the elastic behavior of these samples predominated over their viscous behavior, and a perfect network formed. Furthermore, a higher G′ value was observed for the samples with MPS particles, indicating that the addition of MPS had a positive effect on the gelation of PVA aqueous solution.
In the hypothesis that the hydrogel may be regarded as a classic network, the value of G′ at low strain amplitude may be related to the average number of equivalent units in a “network strand”. The effective crosslinking density of the PVA–MPS composite hydrogel was determined with the following equation based on the rubber elasticity theory:26
| G′ = RTveφ11/3φ22/3 | (1) |
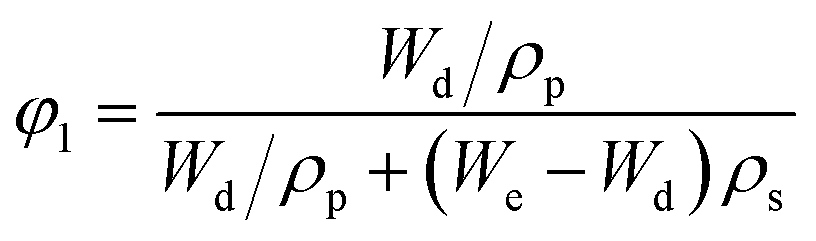 | (2) |
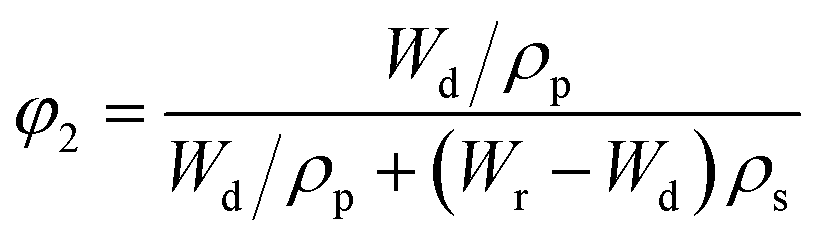 | (3) |
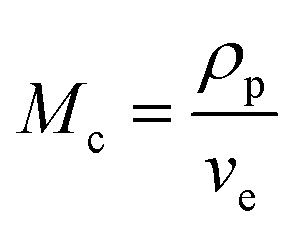 | (4) |
The value of G′, ve and Mc of PVA–PS and PVA–MPS gel beads were calculated and listed in Table 1. It could be seen that, compared with PVA hydrogel, G′ and ve of PVA–MPS gel beads increased. The existence of hydrogen bonding interaction and well dispersion MPS in PVA matrix led to the enhancement of molecular entanglement of PVA–MPS system, and the formation of network structure with PVA–MPS as a crosslinking center.
| Samples | ρp (g cm−3) | φ1 | φ2 | G′ (kPa) | ve (mol m−3) | Mc (kg mol−1) |
|---|---|---|---|---|---|---|
| PVA–PS | 1.037 | 0.1297 | 1 | 12.74 | 10.09 | 102.80 |
| PVA–MPS | 1.031 | 0.1362 | 1 | 18.55 | 14.46 | 71.32 |
The specific surface area of as-prepared gel beads was measured through N2 adsorption/desorption on the sample surface.27 Fig. S3† showed N2 adsorption and desorption isotherms for PVA–PS and PVA–MPS gel beads, and the porous characteristic parameters were listed in Table S2.† It could be seen that compared with PVA–PS gel beads, for PVA–MPS gel beads, the specific surface area, total pore volume and average pore size increased, from 4.424 m2 g−1, 0.021 cm3 g−1 and 11.790 nm to 5.413 m2 g−1, 0.040 cm3 g−1 and 12.387 nm, respectively, which was attributed to the incorporation of MPS resulted in the improvement of the dispersion of the PVA–MPS system. In addition, the increase of total pore volume can increase the microorganisms that can be accommodated in the PVA–MPS gel beads, and it has been shown that the increase of specific surface area can improve the performance of activated sludge biological nitrification and denitrification of the gel beads.28
3.3 The mechanical properties of PVA–MPS gel beads
Table 2 showed the hydraulic impact resistance of PVA–PS and PVA–MPS gel beads. It could be seen that both PVA gel beads have strong crushing strength, especially the PVA–MPS gel beads remain intact and spherical with 100% retention rate after 12 h of hydraulic impact test. These results indicate that the addition of MPS could toughen and strengthen PVA gel beads.| Sample | Time (h) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Retention rate (%) | |||||||||
| 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| PVA–PS | 100 | 100 | 100 | 100 | 100 | 100 | 94 | 82 | 76 |
| PVA–MPS | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
To quantitatively evaluate the mechanical properties of the two gel beads, tensile testing of the previous hydrogel specimens was conducted. As shown in Fig. 3 and Table 3, the elastic modulus values of the PVA–PS and PVA–MPS hydrogels were 0.21 and 0.59 MPa, respectively. Generally, the elastic modulus describes the elastic inter and intramolecular forces from the polymeric matrix that resists to deformation.29 Thus, PVA–MPS hydrogels shows a higher value of elastic modulus, resisting deformation, compared to PVA–PS hydrogels that have a lower modulus and are easily deformed. This suggests that the addition of MPS significantly improves the mechanical properties of hydrogels due to the formation of more hydrogen bonding interactions in the PVA matrix.30
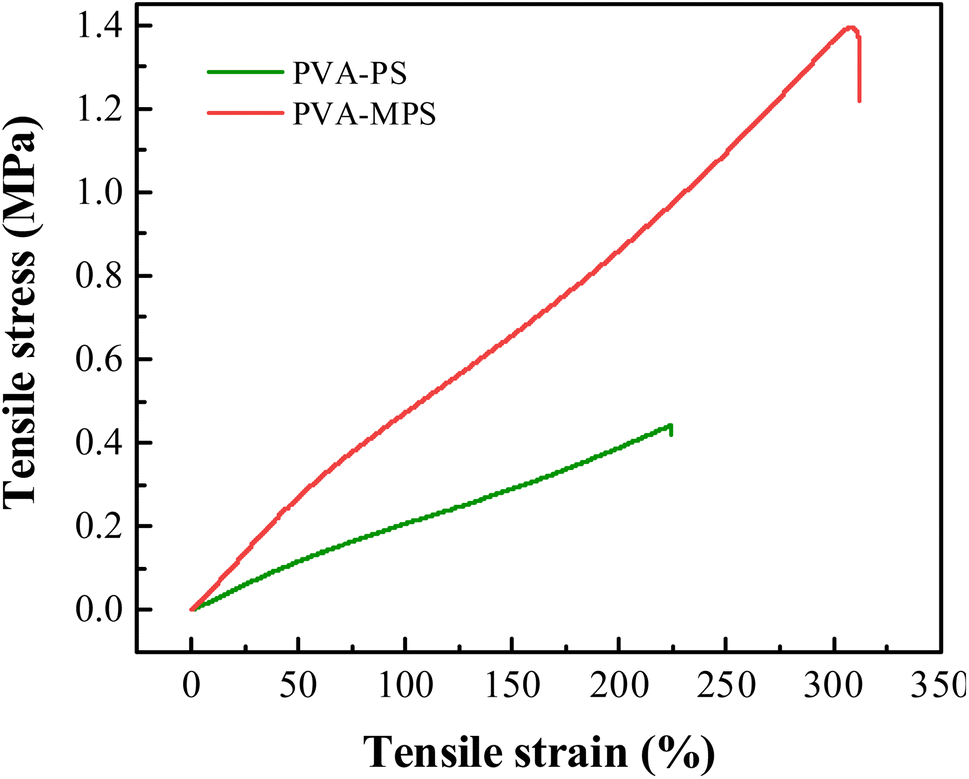 | ||
| Fig. 3 Strain–stress curves PVA–PS and PVA–MPS hydrogels. | ||
| Sample | Elastic modulus (MPa) | Tensile strength (MPa) | Elongation at break (%) |
|---|---|---|---|
| PVA–PS | 0.21 ± 0.03 | 0.43 ± 0.08 | 223.94 ± 24.2 |
| PVA–MPS | 0.59 ± 0.04 | 1.39 ± 0.13 | 312.43 ± 27.6 |
Compared to the PVA–PS hydrogel, the tensile strength of PVA–MPS hydrogel increased about 3 times from 0.43 to 1.39 MPa, and the elongation at break increased 0.4 times (from 223.94% to 312.43%). This is due to the formation of dense chemical crosslinking and a more porous structure in the PVA–MPS hydrogel after being crosslinked. Therefore, it exhibited higher tensile strength and elongation at break values.31
3.4 The swelling behavior of PVA–MPS gel beads
The swelling ratio of the two hydrogel beads as a function of swelling time was shown in Fig. 4(a). It can be seen that in the initial swelling stage, all samples of gel beads absorbed water rapidly. The swelling ratio increased dramatically with time, and reached equilibrium in about 10 min. The swelling kinetics of gel beads was analyzed by assuming that the swelling process met the first order kinetic equation:32| dQt/dt = k(Qe − Qt) | (5) |
| Qt = Qe − (Qe − Q0)/ekt | (6) |
| kt = ln[(Qe − Q0)/(Qe − Qt)] | (7) |
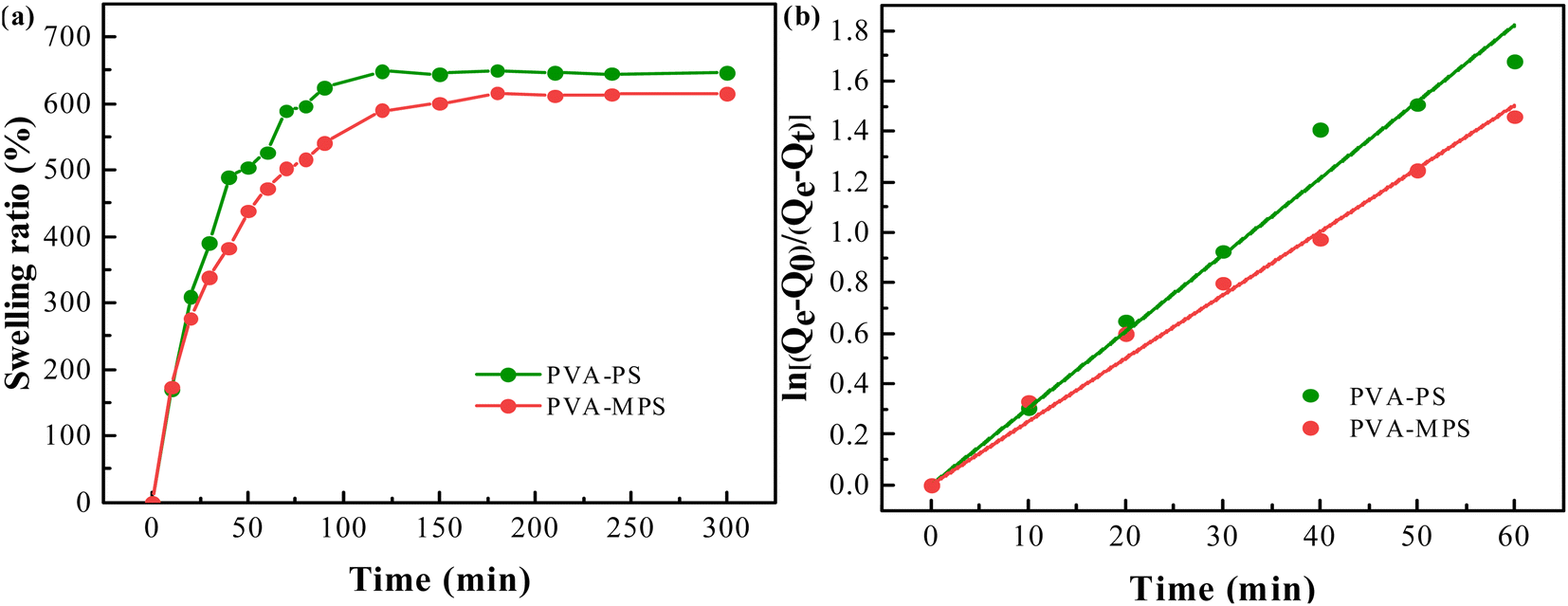 | ||
| Fig. 4 (a) The swelling ratio curve, and (b) the swelling kinetics of PVA–PS and PVA–MPS gel beads. | ||
By plotting the graph of ln[(Qe − Q0)/(Qe − Qt)] versus time t, as shown in Fig. 4(b), the swelling rate constant k can be obtained from the slope of the curve, which exhibited good linear relationship. The swelling rate constant k and equilibrium swelling rate of the two hydrogel beads were listed in Table 4. It can be seen that the swelling rate constant k and the equilibrium swelling ratio of the gel beads were reduced by the addition of MPS. Since the swelling ratio is an equilibrium between the stretching produced by the volume expansion after water molecules diffused into the polymer matrix and the elastic contraction produce by the macromolecular chains of the polymer, it is related to the crosslinking degree. A higher degree of crosslinking means a tighter structure, whose polymer chains produce a stronger contraction against the volume expanding and shows a lower swelling ratio. In this case, MPS loading causes a increased cross-linking degree of gel beads and expresses an decreased osmotic swelling pressure.33 In addition, the PVA–MPS gel beads had a higher gel mass fraction (80.34 ± 0.37) compared to the PVA–PS gel beads (76.95 ± 0.49), and the higher gel mass fraction also represented higher cross-linked structure in the PVA–MPS gel beads. The highly cross-linked network reduced water absorption by impeding the penetration of water that the PVA–MPS gel beads with higher gel mass fraction showed lower swelling ratio.
| Sample | Swelling rate constant k (h−1) | Equilibrium swelling ratio (%) |
|---|---|---|
| PVA–PS | 1.82 | 647 |
| PVA–MPS | 1.50 | 615 |
3.5 The formation mechanism of PVA–MPS gel beads
As shown in Fig. 5, PVA–MPS gel beads are interpenetrating network gels formed by the saturated H3BO3/CaCl2 solution as a crosslinking agent. PVA is chemically cross-linked by B(OH)4− in saturated H3BO3 solution to form –OB structure. The structure of –COONa on Alg interacts with Ca2+ to form the structure of –COOCaOOC, which results in the formation of an intermolecular eggshell structure of Alg. In addition, PVA and Alg are cross-linked by hydrogen bonding to form the main network structure of the double cross-linked gel beads. The surface of MPS is covered with small pores of about 1 μm in diameter, and contains active groups such as hydroxyl and amino groups. When MPS was introduced into the gel, MPS particles were dispersed in the gel and entangled in the PVA network to form PVA–MPS gel beads with MPS as the crosslinking center.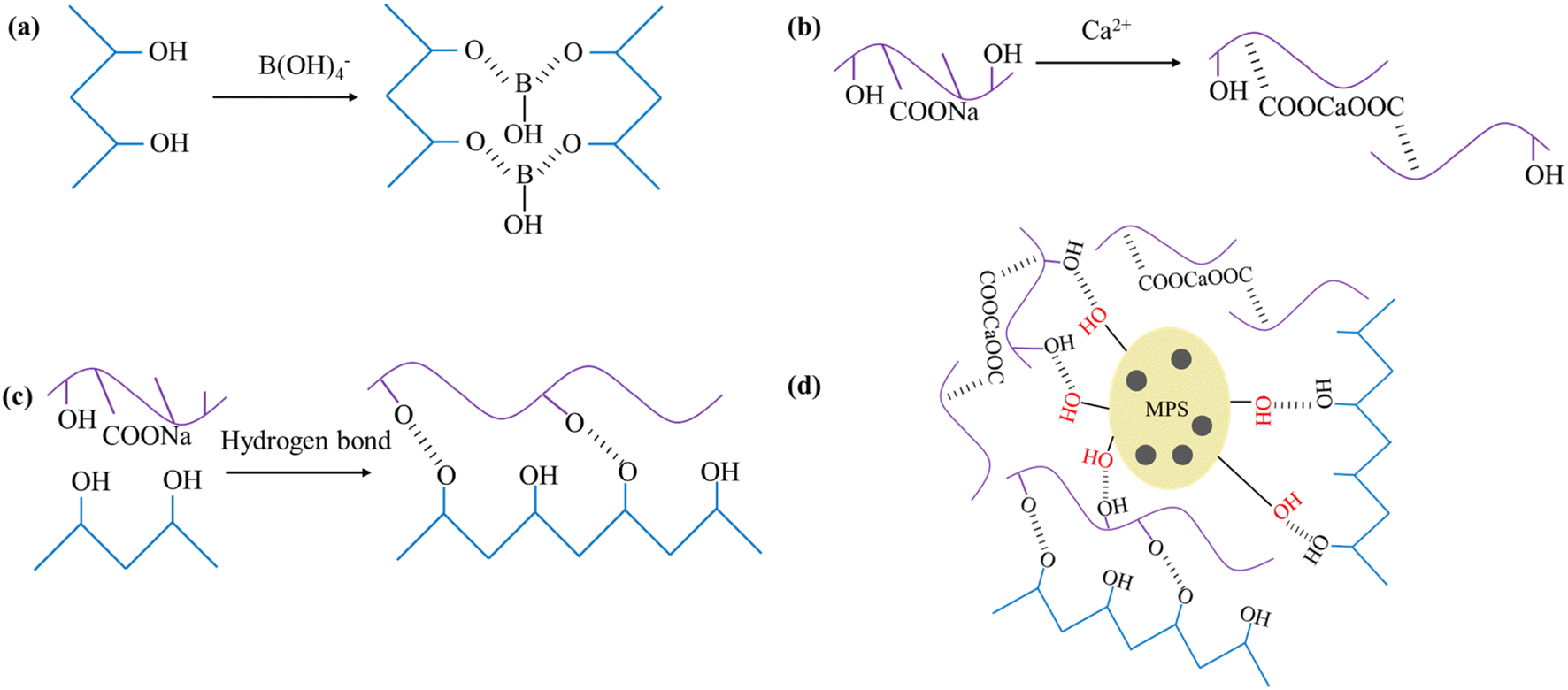 | ||
| Fig. 5 (a) PVA combined with B(OH)4−, (b) Alg chelated with Ca2+, (c) hydrogen bonding between PVA and Alg, and (d) the formation mechanism of PVA–MPS gel beads. | ||
Based on the results of the characterization of the gel beads, the good mechanical properties and lower swelling ratio enabled the PVA–MPS gel beads to maintain good shape and size in the reactor for a long period of time with good hydraulic stability. In addition, the gel beads were subjected to shock loading experiment on influent ammonia and nitrogen concentration (the details can be found in Text S2 and Fig. S4†). The nitrogen removal performance of the PVA–MPS gel beads was better than that of the PVA–PS gel beads, so the subsequent experiments were conducted using PVA–MPS gel beads for preservation and reactivation experiments.
3.6 Morphological changes in PVA–MPS gel beads before and after preservation
After 40 d of preservation, the appearance of the PVA–MPS gel beads in the four environments changed to different degrees (Fig. 6(a)–(d)). P1 and P2 were still spherical or ellipsoidal in shape and altered from emerald green(Fig. S5†) to light green in color with dense structure in both wet and dry preservation at 4 °C. P3 and P4 were spherical or ellipsoidal and changed from emerald green to black green with noticeable black spots at 25 °C. Compared to the dry-preserved P2 and P4, the beads of P1 and P3 had larger diameters, which was due to the expansion of the beads by water absorption when P1 and P3 were preserved in ultrapure water. In addition, P3 had a slight odor when the sealing bottle film was opened, which may be due to the long-term anaerobic preservation, the gel beads produce sulfate, methane and hydrogen sulfide due to endogenous respiration in the absence of oxygen, and some of these sulfide compounds were deposited on the surface of beads leading to their darkening.34 Fig. 6(e)–(h) shows the microstructure of PVA–MPS gel beads. It can be seen that the prepared gel beads had an excellent three-dimensional pore structure, and the chain structure after polymerization and crosslinking after magnification. Meanwhile, the bead cross section still maintained a relatively dense network-shaped structure, and further observation revealed that a large number of microorganisms still survived. | ||
| Fig. 6 Digital and SEM images of PVA–MPS gel beads collected from (a and e) P1, (b and f) P2, (c and g) P3, and (d and h) P4 were stored in separate environments. P1: wet storage at 4 °C; P2: dry storage at 4 °C; P3: wet storage at 25 °C; P4: dry storage at 25 °C. | ||
3.7 Removal performance of reactivated PVA–MPS gel beads
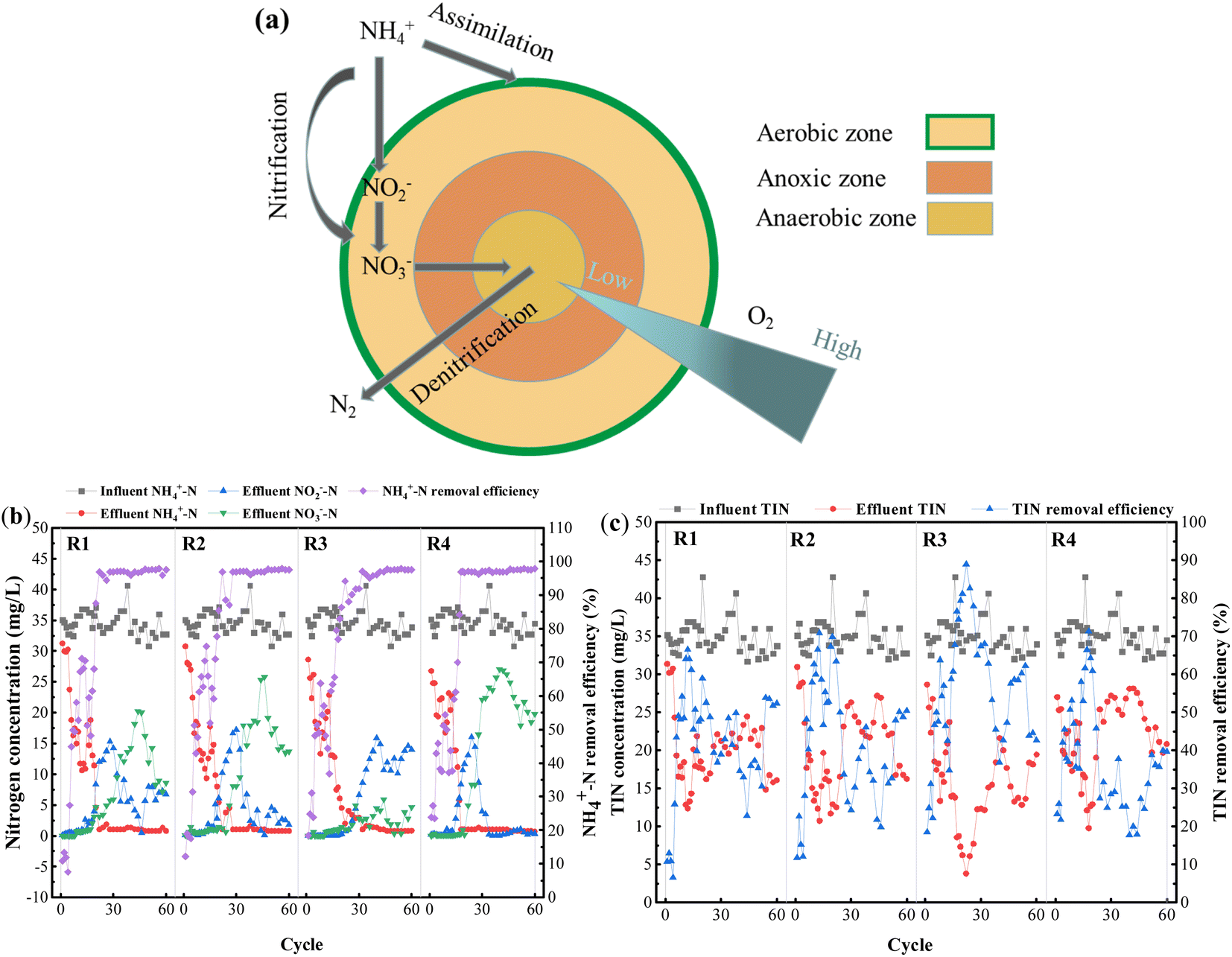 | ||
| Fig. 7 Microbial denitrification process (a), variation of each form of nitrogen in synthetic wastewater treated with PVA–MPS gel beads: nitrification and denitrification properties (b), TIN removal profile (c). R1 was inoculated with S1; R2 was inoculated with S2; R3 was inoculated with S3; R4 was inoculated with S4. | ||
The experiments were run continuously for 60 cycles. Fig. 7 shows long-term changes in various forms of nitrogen in the influent and effluent of all reactors. As shown in Fig. 7(b), stable ammonia nitrogen removal efficiencies were achieved in the 18th to 22nd cycles, basically above 90%. After 40 d of preservation under four different environmental conditions, the PVA–MPS gel beads all showed notable ammonia nitrogen removal performance, and the ammonia nitrogen removal efficiencies of each reactor at the end of 60 cycles were 97.59%, 97.59%, 97.58% and 97.91%, respectively. This is the decomposition of microbial denitrifying bacteria in the beads as well as assimilation of algae play an important role in ammonia nitrogen removal.36
As shown in Fig. 7(b), the effluent nitrite nitrogen concentrations of all reactors significantly accumulated in the 14th, 14th, 20th, and 15th cycles, and the maximum nitrite nitrogen concentrations were 15.33 mg L−1, 17.19 mg L−1, 15.84 mg L−1, and 16.07 mg L−1, respectively. This may be because PVA–MPS gel beads has a distinct spatial stratification structure that is similar to aerobic granular sludge (AGS),37 which is constrained by the diffusion of dissolved oxygen in the beads and the creation of a low dissolved oxygen environment inside them, and the competitive growth of nitrite-oxidizing bacteria (NOB) is inhibited to a certain extent due to the stronger oxygen utilization ability of ammonia-oxidizing bacteria (AOB) than NOB.38 The accumulation of effluent nitrate nitrogen concentration lagged behind that of effluent nitrite nitrogen concentration, reaching maximum values of 20.21 mg L−1, 25.76 mg L−1, 5.92 mg L−1, and 27.03 mg L−1 in cycles 44, 46, 44, and 40, respectively. This phenomenon occurred because the gel beads were stored for 40 d without oxygen, and DNB and other anaerobes proliferated, resulting in no accumulation of nitrate nitrogen occurring in the reactors during the early activation phase (1st to 20th cycles), whereas in the mid-activation phase (21st to 50th cycles), as the reactors were kept in the aeration mode, AOB and NOB gradually dominated and the number of DNB decreased, resulting in a weakening of the denitrification performance. In the late activation phase (51st to 60th cycles), with the change of aeration pattern during the cycle, the increase of anaerobic time period led to the improvement of denitrification performance and consequently the decrease of nitrate nitrogen. Among them, R3 accumulated the least amount of nitrate nitrogen, which may be attributed to the fact that at the end of preservation, R3 beads retained the least amount of NOB and more DNB.
The removal of TIN in all four reactors showed different degrees of increasing and then decreasing trends (Fig. 7(c)), and the TIN removal efficiencies reached the maximum concentrations of 66.47%, 70.83%, 88.98%, and 71.36% in the 12th, 13th, 22nd, and 18th cycles, respectively. TIN removal mainly occurred through two processes: (1) consumption by following proliferation and assimilation of microorganisms, and (2) denitrification within the gel beads. The large proliferation of microorganisms in the early activation period consumed TIN by assimilation, where the TIN removal efficiency of even cycles (light reaction cycle) was higher than that of odd cycles (dark reaction cycle), suggesting that the presence of algae is essential to increase the nitrogen removal efficiency.28 Whereas, the mid-activation phase made the denitrification of the system weaker due to the prolonged aeration of the reactors. Interestingly, the TIN removal efficiencies of R3 and R4 were higher than those of R1 and R2 at the early activation phase, which may be due to the higher preservation temperature, which resulted in higher microbial metabolism, and the more active gel beads had better TIN removal performance at the early phase. After adjusting the aeration mode, the TIN removal increased in each reactor. At the end of 60 reaction cycles, the TIN removal remained good at 52.31%, 50.43%, 42.69% and 39.65%, respectively.
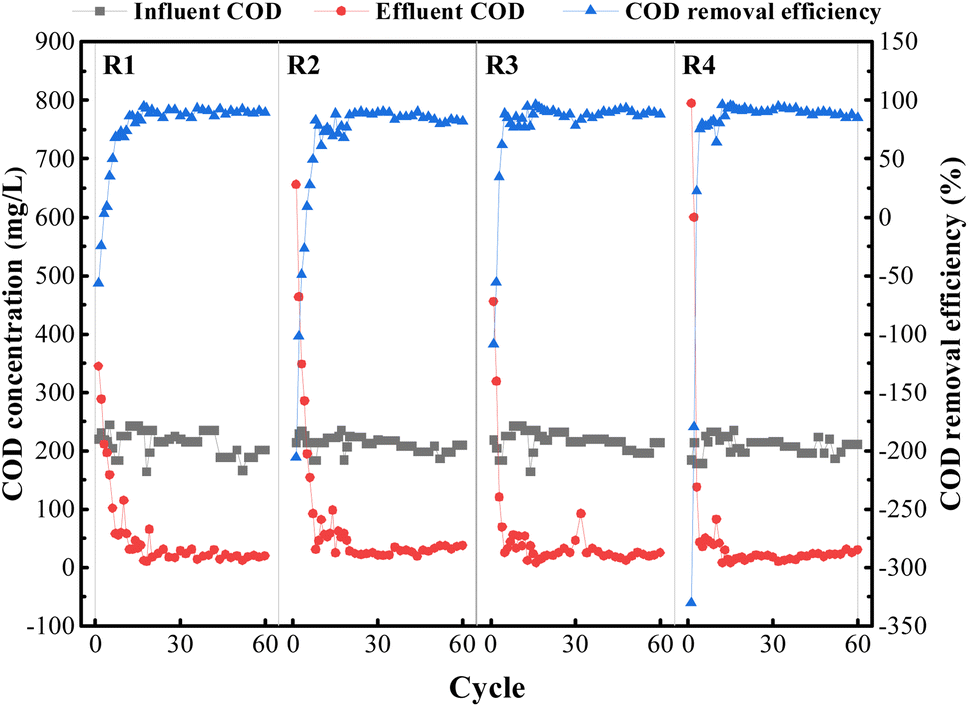 | ||
| Fig. 8 COD removal profile in four reactors, R1, R2, R3, and R4, respectively. | ||
The COD removal efficiency of each reactor basically reached about 80% after the 12th to 16th cycle. On the one hand, the gel beads could always ensure a better COD removal efficiency, indicating that the shape of the gel beads was maintained well and was not dissolved by large breakage due to aeration; on the other hand, the COD concentration of the effluent water in all the reactors was reduced to varying degrees, which indicated that the microorganisms in the gel beads effectively utilized the organic matter to realize the growth and reproduction process of microorganisms.39
3.8 Microbial properties
 | ||
| Fig. 9 SEM images of PVA–MPS gel beads collected from (a) R1, (b) R2, (c) R3, and (d) R4 were reactivated in separate reactors. R1 and R3 are the cross sections of the gel beads, R2 and R4 are the externa surfaces of the gel beads. | ||
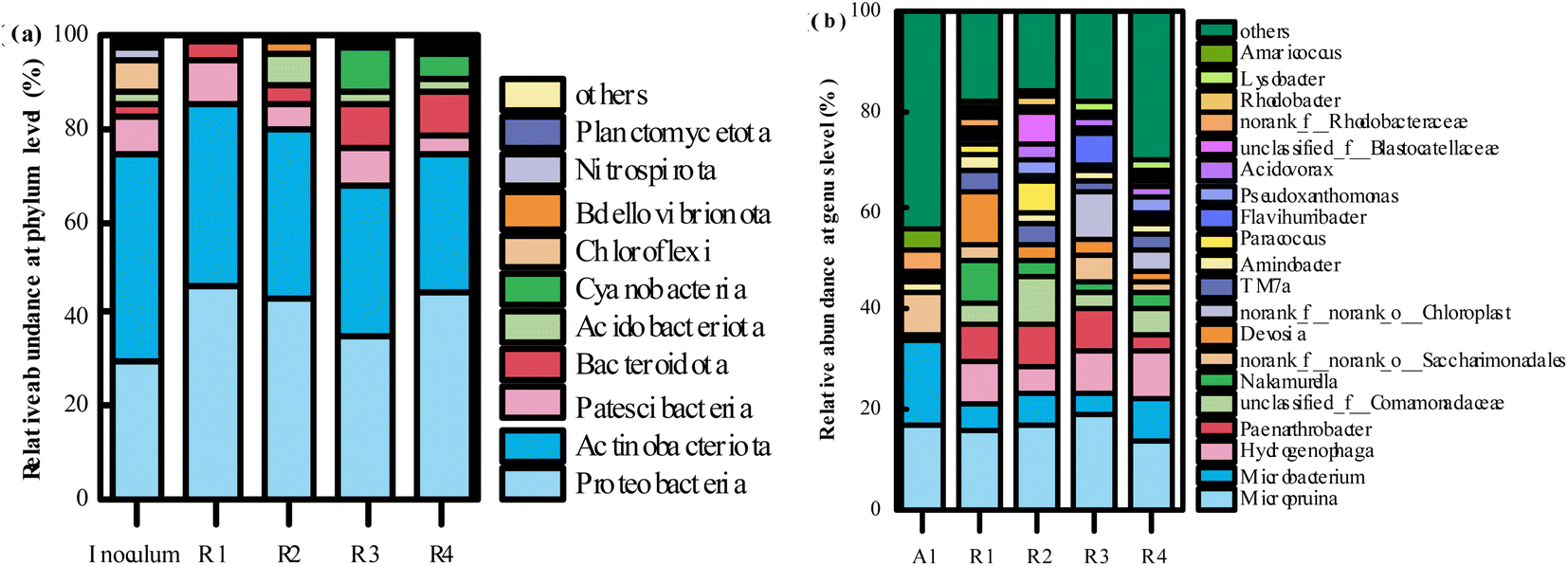 | ||
| Fig. 10 The distribution of microbial communities in the inoculum activated sludge and PVA–MPS gel beads at the phylum (a) (>0.1%) and genus (b) (top 20) level. | ||
The relative abundance of the top 20 genera at genus level is shown in Fig. 10(b). At genus level, the dominant genera in all gel beads were Micropruina (15.51%, 16.69%, 18.32%, and 13.52%, respectively); Microbacterium (5.87%, 6.14%, 4.42% and 8.34%, respectively); Hydrogenophaga (8.44%, 5.55%, 8.90%, and 9.48%, respectively). It has been shown that Micropruina is able to store large amounts of sugar polymers in the cell when glucose is used as a carbon source, thus becoming the dominant genus.43 The relative abundance of Microbacterium in all gel beads decreased significantly compared to the inoculum activated sludge (16.38%), whereas the relative abundance of Hydrogenophaga in the gel beads increased significantly compared to the inoculum activated sludge (0.06%). Hydrogenophaga is a class of aerobic partially hydrogenophilic Gram-negative bacteria that metabolize sugars by oxidative sugar metabolism with oxygen as the terminal electron acceptor, and organic pollutant degradation.44 In addition, Hydrogenophaga performed heterotrophic nitrification and aerobic denitrification and provided an essential basis for simultaneous nitrification denitrification of the reactor.45
4. Conclusions
In this paper, MPS was introduced into hydrogels to prepare PVA–MPS gel beads, and the characterization results showed that the PVA–MPS gel beads not only had good sphericity and porous network structure, but also had good rheological properties and mechanical strength, which made them an excellent microbial carrier. The PVA–MPS gel beads were preserved under four different environmental conditions and reactivated in wastewater. The results showed that the structure of PVA–MPS gel beads was well maintained during the activation process, and the microorganisms attached to the inner and outer surfaces of the beads continued to multiply and grow, and the structure of the microbial community inside the beads changed significantly. Even being preserved in different environments, the PVA–MPS gel beads still had high removal efficiencies, and the removal efficiencies of ammonia nitrogen and COD could reach more than 95% and 80%, respectively. Therefore, the strategy immobilizing microorganisms in PVA–MPS gel beads and preserving them to be a great potential for the commercialized application of algal–bacterial symbiosis on bioremediation.Author contributions
Shutao Lin: conceptualization, writing – original draft, writing – review & editing. Zhi Zhang: funding acquisition, writing – review & editing, supervision. Wenchao Jiang: visualization, supervision. Ruiting Chang: investigation. Xinyu Cao: writing – review & editing. Yongheng Zhang: writing – review & editing. Jiabo Chen: investigation.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Ministry of Housing and Urban-Rural Development of China (grant number K 2021-k-112); Technology Innovation and Application Development Project of Chongqing Municipality, China (No. CSTB2022TIAD-GPX0035).References
- W. Wang, Y. Ding, Y. Wang, X. Song, R. F. Ambrose, J. L. Ullman, B. K. Winfrey, J. Wang and J. Gong, Ecol. Eng., 2016, 94, 7–11 CrossRef.
- S. Xiang, Y. Liu, G. Zhang, R. Ruan, Y. Wang, X. Wu, H. Zheng, Q. Zhang and L. Cao, World J. Microbiol. Biotechnol., 2020, 36, 144 CrossRef CAS PubMed.
- A. Sial, B. Zhang, A. Zhang, K. Liu, S. A. Imtiaz and N. Yashir, BioEnergy Res., 2021, 14, 723–738 CrossRef.
- T.-T.-D. Nguyen, T.-T. Nguyen, Q. An Binh, X.-T. Bui, H. H. Ngo, H. N. P. Vo, K.-Y. Andrew Lin, T.-D.-H. Vo, W. Guo, C. Lin and F. Breider, Bioresour. Technol., 2020, 314, 123754 CrossRef CAS PubMed.
- M. Han, C. Zhang, F. Li and S.-H. Ho, Sci. Total Environ., 2022, 852, 158514 CrossRef CAS PubMed.
- X. Liu, M. Wang, J. Zhang, L. Wei and H. Cheng, Bioresour. Technol., 2021, 320, 124438 CrossRef CAS PubMed.
- X. Li, X. Sun, X. Zhou, Z. He, K. Lin, X. Chen and G. Jiang, Process Saf. Environ. Prot., 2022, 166, 158–165 CrossRef CAS.
- M. M. El-Sheekh, M. A. Metwally, N. Allam and H. E. Hemdan, Iran. J. Sci. Technol., Trans. A: Sci., 2020, 44, 595–604 CrossRef.
- J. Hu, H. Liu, P. Shukla, W. Lin and J. Luo, Chemosphere, 2020, 251, 126406 CrossRef CAS PubMed.
- X. Xiang, X. Yi, W. Zheng, Y. Li, C. Zhang, X. Wang, Z. Chen, M. Huang and G.-G. Ying, J. Hazard. Mater., 2023, 443, 130247 CrossRef CAS PubMed.
- Y. Yu, Y. Shu and L. Ye, Chem. Eng. J., 2018, 336, 306–314 CrossRef CAS.
- W. Yang, L. Xu, J. Su, Z. Wang and L. Zhang, Chemosphere, 2023, 326, 138460 CrossRef CAS PubMed.
- X. Lin, B. Li, M. Tian, X. Li and J. Wang, Sci. Total Environ., 2023, 900, 165599 CrossRef CAS PubMed.
- J. Wang and J. Liang, Bioresour. Technol., 2021, 326, 124778 CrossRef CAS PubMed.
- H. Wang, Q. Dai, Q. Li, J. Yang, X. Zhong, Y. Huang, A. Zhang and Z. Yan, Solid State Ionics, 2009, 180, 1429–1432 CrossRef CAS.
- X. Xie, H. Gao, X. Luo, Y. Zhang, Z. Qin and H. Ji, Adv. Compos. Hybrid Mater., 2022, 5, 2772–2786 CrossRef CAS.
- T. Takei, K. Ikeda, H. Ijima and K. Kawakami, Process Biochem., 2011, 46, 566–571 CrossRef CAS.
- P. Sankarganesh, V. Parthasarathy, A. G. Kumar, S. Ragu, M. Saraniya and N. Udayakumari, J. Sol-Gel Sci. Technol., 2022, 101, 571–578 CrossRef CAS.
- S. Wang, K. Xiao, Y. Mo, B. Yang, T. Vincent, C. Faur and E. Guibal, J. Hazard. Mater., 2020, 386, 121637 CrossRef CAS PubMed.
- X. Lin, D. T. Tran, M.-H. Song and Y.-S. Yun, J. Cleaner Prod., 2021, 328, 129545 CrossRef CAS.
- A. Yumin, D. Liguo, Y. Yi and J. Yongna, RSC Adv., 2022, 12, 11632–11639 RSC.
- Q. Liao, H. Rong, M. Zhao, H. Luo, Z. Chu and R. Wang, J. Hazard. Mater., 2022, 422, 126863 CrossRef CAS PubMed.
- X. Zhang, X. Lin, Y. He and X. Luo, Int. J. Biol. Macromol., 2019, 136, 445–459 CrossRef CAS PubMed.
- Y. Fei, Y. Li, S. Han and J. Ma, J. Colloid Interface Sci., 2016, 484, 196–204 CrossRef CAS PubMed.
- M. Chen, G. Gong, L. Zhou and F. Zhang, RSC Adv., 2017, 7, 21476–21483 RSC.
- Y. Meng and L. Ye, J. Appl. Polym. Sci., 2017, 134, 44855–44864 CrossRef.
- D. Surya Bhaskaram, R. Cheruku and G. Govindaraj, J. Mater. Sci.: Mater. Electron., 2016, 27, 10855–10863 CrossRef CAS.
- C. Xu, L. Wang, Z. Liu, G. Cai and J. Zhan, Water Sci. Technol., 2022, 85, 104–115 CrossRef CAS PubMed.
- J. Chen, Z. Yang, D. Shi, T. Zhou, D. Kaneko and M. Chen, J. Appl. Polym. Sci., 2021, 138, 49987 CrossRef CAS.
- M. Mandru, M. Bercea, L. M. Gradinaru, C. Ciobanu, M. Drobota, S. Vlad and R. Albulescu, Eur. Polym. J., 2019, 118, 137–145 CrossRef CAS.
- H. Liao, Y. Liu, Q. Wang and W. Duan, J. Appl. Polym. Sci., 2018, 135, 46402 CrossRef.
- G. Zhang and L. Ye, J. Polym. Environ., 2017, 25, 229–240 CrossRef CAS.
- L. Pang, X. Dong, C. Niu and M. Qi, J. Magn. Magn. Mater., 2020, 498, 166140 CrossRef CAS.
- D. Gao, X. Yuan and H. Liang, Water Res., 2012, 46, 3315–3322 CrossRef CAS PubMed.
- Z. Li, J. Wang, X. Chen, Z. Lei, T. Yuan, K. Shimizu, Z. Zhang and D.-J. Lee, Bioresour. Technol., 2022, 352, 127104 CrossRef CAS PubMed.
- T. Cai, S. Y. Park and Y. Li, Renewable Sustainable Energy Rev., 2013, 19, 360–369 CrossRef CAS.
- L. Tian, K. Feng, G. Qin, X. Tong, X. Feng, X. Xu and L. Zhu, J. Water Process Eng., 2022, 49, 103005 CrossRef.
- H. Bae, H. Yang, Y.-C. Chung, Y. J. Yoo and S. Lee, Bioprocess Biosyst. Eng., 2014, 37, 1115–1125 CrossRef CAS PubMed.
- G. Mujtaba, M. Rizwan and K. Lee, J. Ind. Eng. Chem., 2017, 49, 145–151 CrossRef CAS.
- V. Turon, C. Baroukh, E. Trably, E. Latrille, E. Fouilland and J.-P. Steyer, Bioresour. Technol., 2015, 175, 342–349 CrossRef CAS PubMed.
- L. Zhang, B. Long, J. Wu, Y. Cheng, B. Zhang, Y. Zeng, S. Huang and M. Zeng, Heliyon, 2019, 5, e03023 CrossRef PubMed.
- J. Fan, L. Cao, C. Gao, Y. Chen and T. C. Zhang, Water Sci. Technol., 2019, 80, 892–901 CrossRef CAS PubMed.
- L. Zhang, L. Ding, X. He, H. Ma, H. Fu, J. Wang and H. Ren, Sci. Rep., 2019, 9, 805 CrossRef PubMed.
- X. Huang, X. Yang, J. Zhu and J. Yu, Bioresour. Technol., 2020, 315, 123814 CrossRef CAS PubMed.
- S. Liu, H. Li and Y. Wang, RSC Adv., 2023, 13, 16471–16479 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05371g |
| This journal is © The Royal Society of Chemistry 2023 |