
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA useful strategy for synthesis of the disaccharide of OSW-1†
Bin Wang‡
*a,
Yan Zhang‡a and
Xiangyan He *b
*b
aDepartment of Pharmacy, People's Hospital of Xinjiang Uygur Autonomous Region, Urumqi, Xinjiang Uygur Autonomous Region, China. E-mail: binw4870@gmail.com
bScientific Research and Education Centre, People's Hospital of Xinjiang Uygur Autonomous Region, Urumqi, Xinjiang Uygur Autonomous Region, China. E-mail: xiangyan.he9@gmail.com
First published on 23rd October 2023
Abstract
A flexible, efficient, and practical synthesis route was developed to synthesize an OSW-1 disaccharide. The synthesis took 13 steps from L-arabinose and D-xylose derivatives, and the overall yield was 7.2%. The region preferentially protects various D-xylose hydroxides because the TBS group selectively reacts with this hydroxide at low concentrations due to greater activity at the C-4 hydroxyl of D-xylose. Then, high efficiency selectively protects C-2 hydroxyl and C-3 hydroxyl of D-xylose, respectively. The first high yield of glycosylation on an OSW-1 synthesis disaccharide was achieved by taking sulfide donor 4 with β-PMP anomeric L-arabinose acceptor 12. The cytotoxicity reveals that the analogy has a high IC50 for a variety of cell types. This approach should provide a versatile way to modify OSW-1's disaccharide.
Introduction
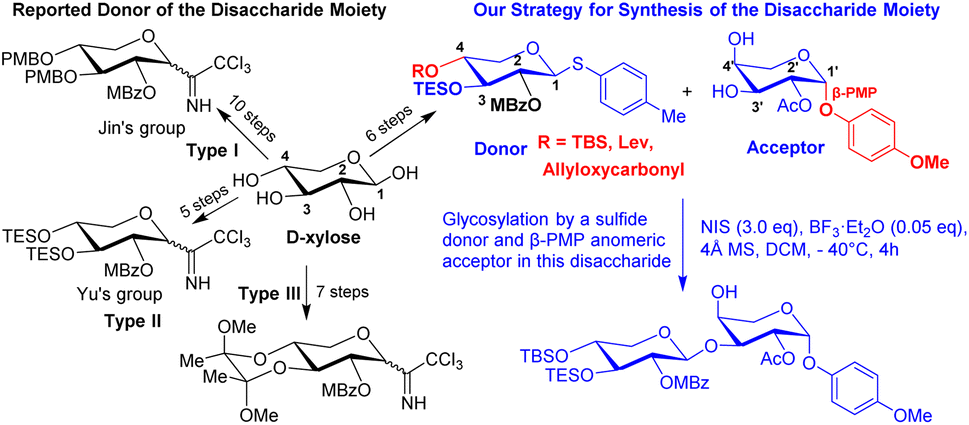
A natural saponin, OSW-1, was isolated from the bulbs of Ornithogalum saudersiae by Sashida et al. in 1992.1 Pharmacological studies showed that OSW-1 exhibits exceptionally potent antitumor activities in nanomolar concentration (IC50 0.1–0.7 nM).2a It is more potent than those used in clinical treatment as anticancer agents such as mictomycincadeiamycin and Taxol.2 Therefore, the higher potential anticancer activity and structure of OSW-1 has always attracted chemists and biologists.3The structure of OSW-1 can be separated into two parts: aglycon and disaccharide moieties. Fuchs et al. reported the first synthesis of the aglycone portion of OSW-1 in 1998.3a Yu et al. reported the first complete synthesis of OSW-1 in 1999.3b Jin et al. published another complete synthesis of OSW-1 in 2001.3c Even though many synthetic methods for the preparation of disaccharides have been published to date,3–6 Yu's method appears to be the most straightforward and practical. However, they used hydrogenation to remove the benzyl group. Three major types of donors have been reported (Scheme 1), Jin's group reported 10 steps from D-xylose to generate the type I donor.3c,d Yu's group reported the shortest synthesis from D-xylose to generate the type II donor,3b,f,6e,13a but they had to use hydrogenation to remove the benzyl group. Seven steps from D-xylose to generate type III donor by using 2,3-butanedione to selective protection of the 3,4-diol moiety of D-xylose,6b but this donor does not very commonly apply in total synthesis OSW-1 or analogies. Though these three types of donors use different protection groups, there is the same concept of using the same group to protect the 3,4-diol moiety of D-xylose. The structure–activity relationship studies of OSW-1 found that the disaccharide moiety, particularly the C-2 position of 4-methoxybenzoyl (MBz) and C-2′ position of groups acetyl (Ac) at the disaccharide unit, play a critical role. Recently, a lot of studies on site-selective functionalization of carbohydrates have been reported.7 Sakurai group reported the synthesis of OSW-1 derivatives via site-selective acylation on the xylose moiety, and they discovered an effective approach was to insert fluorescence or a biotin moiety on the xylose portion with no activity loss.8 Therefore, developing a route to simplify the selective modification of xylose and provide a flexible intermediate compound for OSW-1 or analogies modification at the end of the synthesis is extremely important.
 | ||
| Scheme 1 Synthetic plan for the disaccharide moiety. | ||
In this report, we describe a flexible, efficient, and practical synthesis of the disaccharide moiety, with a focus on shortening the xylose donor synthesis route and selectively introducing different groups at C-2 hydroxyl, C-3 hydroxyl and C-4 hydroxyl on the xylose donor. We specifically report the acceptable yield of glycosylation in this disaccharide utilizing a sulfide donor. Meanwhile, the presence of β-PMP in the anomeric position of arabinose caused C-3′ hydroxyl to be more active than C-4′ hydroxyl, which improved the yield of glycosylation (Scheme 1).
Result and discussion
The synthesis of the xylose moiety was outlined in Scheme 2 utilizing xylose as a starting material, and compound 1 was synthesized as described in the literature.3c,d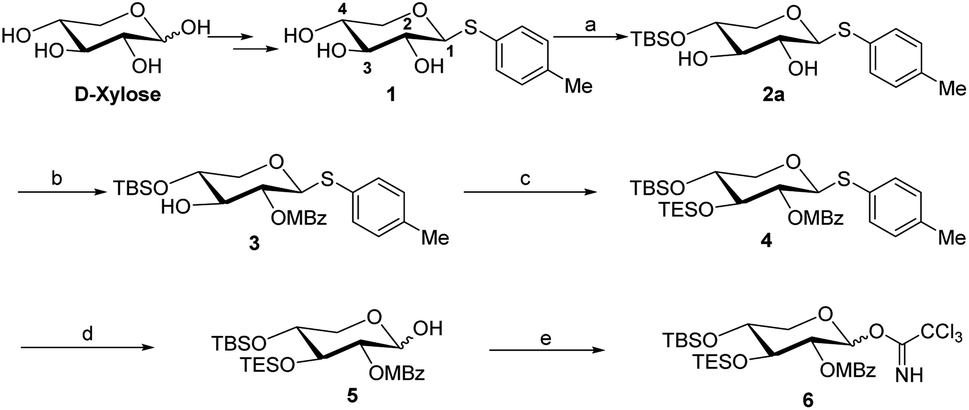 | ||
Scheme 2 Reaction condition: (a) TBSCl (1.1 eq.), imidazole, DCM (c = 0.02), rt, 85% yield; (b) MBzCl, DMAP, Et3N, DCM, room temperature, 2 h, 81% yield; (c) TESOTf, DMAP, Et3N, DMF, room temperature, 2 h, 79% yield; (d) NBS, acetone/aq. NaHCO3 (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 30 min, rt, 86% yield; (e) Cl3CCN, DBU, DCM, overnight, room temperature, 87% yield. :1), 30 min, rt, 86% yield; (e) Cl3CCN, DBU, DCM, overnight, room temperature, 87% yield. | ||
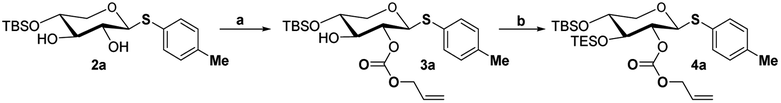
The crucial step was regioselective introducing the TBS protect group to the C-4 hydroxy position. After optimization of the reaction conditions (Table S1†), we found that a low concentration of 1 was prepared initially in dry dichloromethane, followed by slowly adding TBSCl at room temperature to afford regioselective 2a in a yield of 85%. However, we noticed that when TBSOTf replaced TBSCl or increased the concentration of compound 1, the yield of 2a was decreased, and in the meantime, we acquired a by-product that was both C-4 and C-2 hydroxyl position protected. Encouraging by successfully synthesized compound 2a, we plan to introduce another different group at the 4-hydroxyl position of xylose, after scanning several conditions (Table 1). Compound 1 could regio-selectively react with ally chloroformate using N,N-diisopropylethylamine (DIPEA) as a base to afford 2b in 70%. Meanwhile, treating compound 1 with levulinic acid and N,N′-diisopropylcarbodiimide to afford compound 2c, even though the yield was moderate (57%). This result successfully supplied an extra protection group in the xylose moiety.
|
||
|---|---|---|
| Entry | Condition | Result |
| 1 | TBSCI (1.1 eq.), imidazole DCM (c = 0.02), rt | 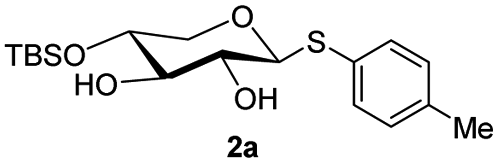 |
| 2 | Allyl chloroformate (1.1 eq.), DIPEA (c = 0.02), 0 °C | 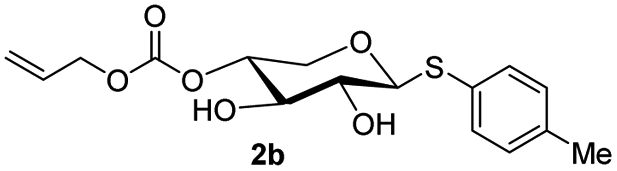 |
| 3 | Levulinic acid (1.1 eq.), DIC, Et3N DCM (c = 0.02), rt | 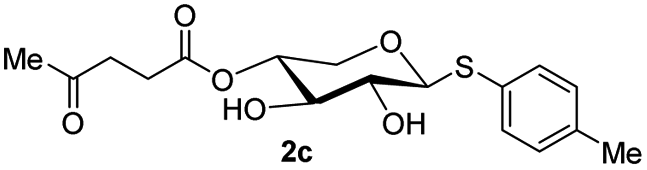 |
With compound 2a in hand, we have tried to finish the rest of the xylose part. The C-2 hydroxyl site was selectively acetylated using p-methoxybeznoyl chloride to yield 3 (Scheme 2). While compound 2a is reacted with ally chloroformate using triethylamine (Et3N) as a base, it produced the high-yield product 3a, which provides more options for disaccharide modification, as shown in ref. 9. The TES protection of the C-3 hydroxy position by TESOTf in dimethylformamide (DMF) went off without a hitch. We tried a variety of different conditions (Table S2†), such as using a different solvent (e.g., dichloromethane or tetrahydrofuran) or replacing TESOTf with TESCl as the reagent, but only the reaction employing TESOTf in DMF with DMAP yielded the best results. Finally, hemiacetal 5 was obtained by deprotecting S-Tol of glycoside 4 in the presence of N-bromosuccinimide, followed by trichloroacetimidation with trichloroacetonitrile under normal conditions to generate. After five steps from compound 1, the target Schmidt donor 6 was produced with a total yield of 39%.
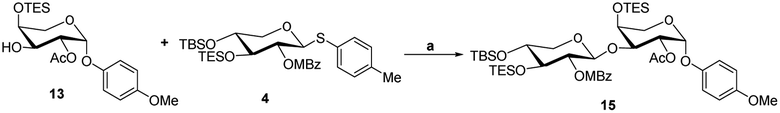
The acceptor synthesis is depicted in Scheme 3. β-PMP-glycoside 8 was made from tetra-acetyl-L-arabinose 7 utilizing BF3Et2O as a promoter, and the acetyl group was deprotected with sodium methoxide to afford 9. Protection of the cis-3′,4′-diol 9 with 2,2-dimethoxypropane resulted in 10, while protection of the C-2′ hydroxy group with acetyl anhydride resulted in 11. The 3′,4′-acetonide was deprotected by 70% acetic acid/H2O, yielding diol 12. We attempted to protect position C-4′hydroxy with TESCl reagent, but C-3′ hydroxyl protected as the major product, and only 19% of the desired compound 13. This result is different from the α-anomeric position which only protected C-4′ hydroxyl with TESCl reagent.3c,d The C-4′ hydroxyl selectivity has been observed in the previous attempts to synthesize OSW-1 analogues,6 including a detailed analysis by Pakulski and co-workers. They concluded this unusual C-4′ hydroxyl selectivity to a novel equatorial C-4′ hydroxyl of arabinose with the 1C4 conformation constrained by the intramolecular hydrogen bonding between C-3′ hydroxyl and the cis-anomeric oxygen atom of arabinoside. We assume that the presence of β-PMP in the anomeric position causes C-3′ hydroxyl more active than C-4′ hydroxyl, which is consistent with Pakulski's report.6c,d The structure of compound 13 was confirmed by 2D NMR after being treated with chloroacetyl chloride (as shown in ESI†). With compounds 6 and 13 in hand, we focused on glycosylation. After optimization of the reaction conditions (Table S3†), we found that the glycosylation of 13 with 6 as glycosyl donor and 0.1 eq. BF3·Et2O as a promoter, the reaction gave 15 with a yield of 78%. This result confirmed that glycosylation occurred in the presence of BF3·Et2O, but the yield of compound 13 is insufficient. Meanwhile, we realized that utilizing 12 as the acceptor directly should be the better option. After five processes, the necessary acceptor 12 was prepared with a total yield of 40%.
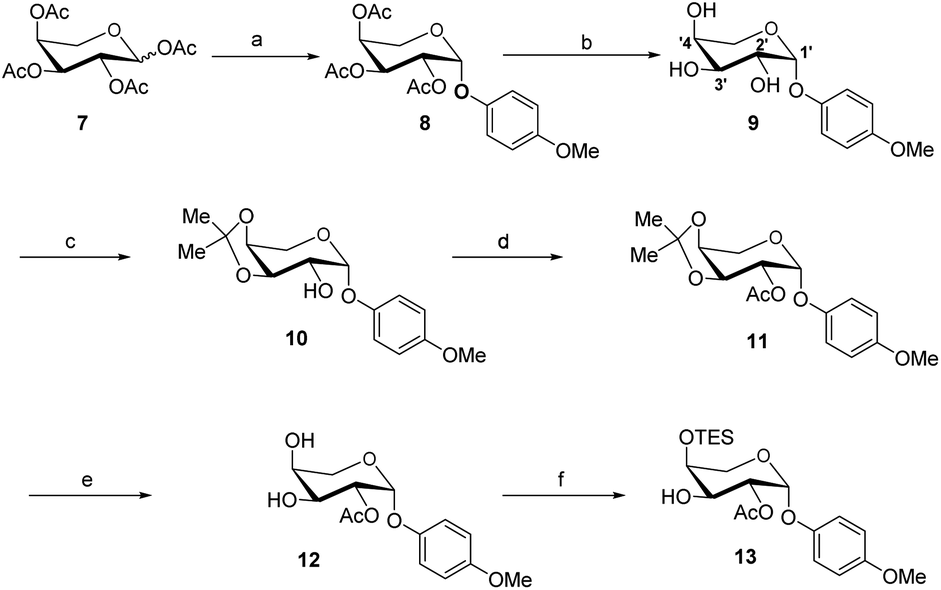 | ||
| Scheme 3 Reaction condition: (a) PMP-OH, BF3·Et2O, DCM, 0–25 °C. 3 days, 57% yield; (b) MeONa, MeOH, 95% yield; (c) CAS, 2,2-dimethoxypropane, DMF, 50 °C, 90% yield; (d) Ac2O, pyridine, 94% yield; (e) 70% AcOH, 86% yield; (f) TESCl, Et3N, DCM, −50 °C, 25% yield. | ||
In the presence of BF3·Et2O, glycosylation of 12 with 6 as the glycosyl donor resulted in an inseparable mixture of disaccharides 13 and 14 (Scheme 4). The protection of the mixture compound with TESCl using Et3N as the base. The disaccharide 15 (72% yield) and 16 (20% yield) were obtained by chromatographic separation of the combination using DCM with hexane as elution.
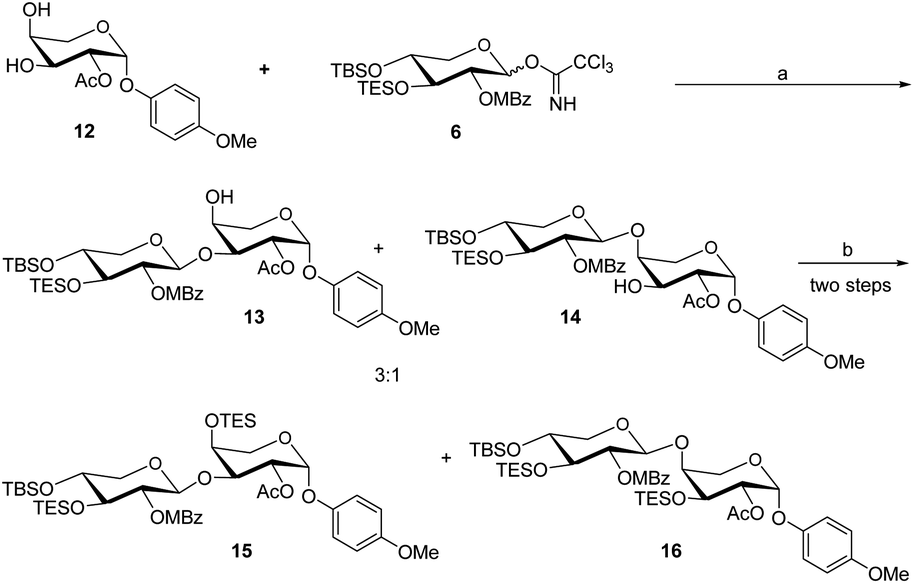 | ||
| Scheme 4 Reaction condition: (a) BF3·Et2O (0.1 eq.), 4 Å MS, DCM, −60 °C, 1.5 h; (b) TESOTf, Et3N, room temperature, DCM, 2 h. | ||
Even though we were able to properly produce disaccharide 15, this synthesis method is not very efficient. In 2008, Guo et al., attempted to glycosylate this disaccharide by a sulfide donor with α-acetyl anomeric acceptor, but were unsuccessful, despite employing N-iodosuccinimide (NIS) and triflic acid as promoters or bromine as a promoter with silver triflate.10 In 2019, Yu's group achieved the glycosylation of this disaccharide by a sulfide donor with a β-benzyl anomeric acceptor, but only 23% yield of the desired product. We thus decided to undertake the direct glycosylation of sulfide donor and β-PMP anomeric acceptor in one-pot reaction. After optimization of the glycosylation of this disaccharide by a sulfide donor with β-PMP anomeric acceptor (Table S4†), we found that utilizing NIS and BF3·Et2O as promoters, the glycosylation of 12 with 4 was successfully generated a mixture of the target 13 and by-product 14, then TES protection to generate 15 in 68% (Scheme 5). Furthermore, we confirmed β-PMP anomeric enhanced 3′-hydroxyl activity by using compound 13 as an accepter to glycosylate with donor 4 using NIS and BF3Et2O as promoters generated compound 15 in a 65% yield, as shown in ref. 11. Finally, hemiacetal 17 was obtained by deprotecting PMP of glycoside 15 in the presence of CAN, followed by trichloroacetimidation with trichloroacetonitrile under normal conditions to generate 18.
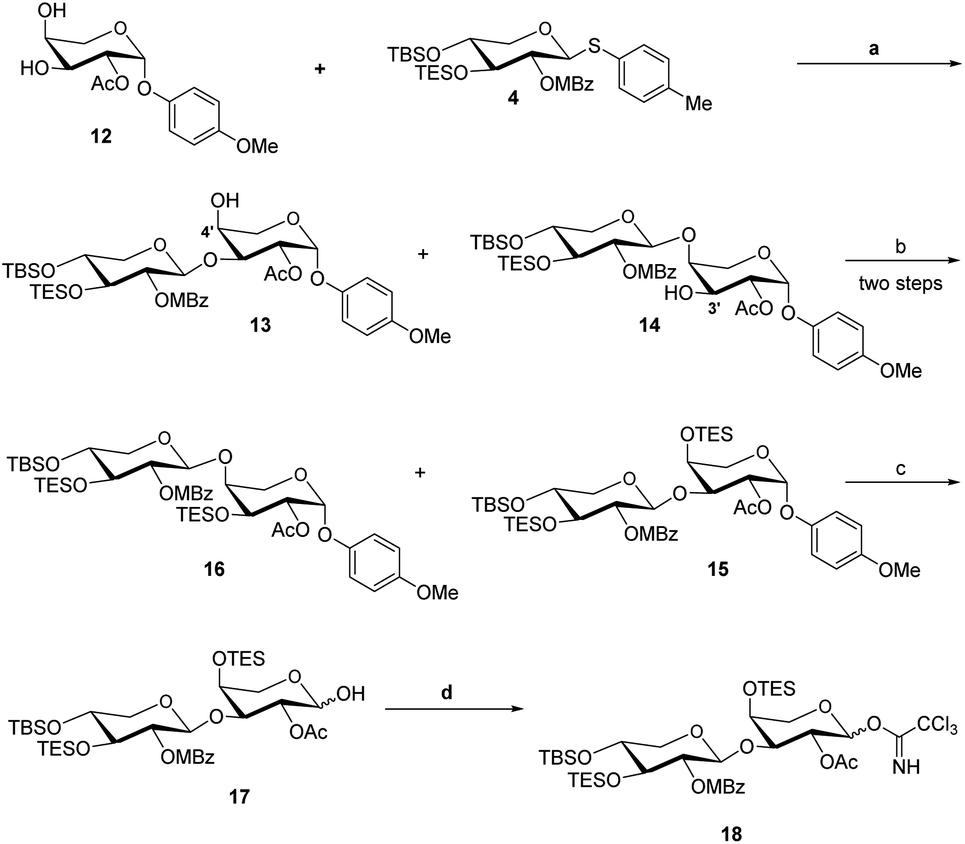 | ||
| Scheme 5 Reaction condition: (a) NIS (3.0 eq.), BF3·Et2O (0.05 eq.), 4 Å MS, DCM, −40 °C, 4 h; (b) TESOTf, Et3N, room temperature, DCM, 2 h; (c) CAN, CH3CN/H2O (4:1), −10 °C, 10 min, 75% yield; (d) Cl3CCN, DBU, DCM, overnight, room temperature, 88% yield. | ||
Sapogenin 20 was synthesized from diosgenin using a process described in the literature (Scheme 6).12 The OSW-1 analogy was synthesized with both aglycon 20 and the disaccharide donor 18 readily hand. Under typical conditions, compound 20 reacted with disaccharide donor 18 to produce 21. The OSW-1 analogue WB-6 was produced by the deprotection of 21 with Pd (MeCN)Cl2. The physical characteristics of WB-6 are the same as those reported.12b
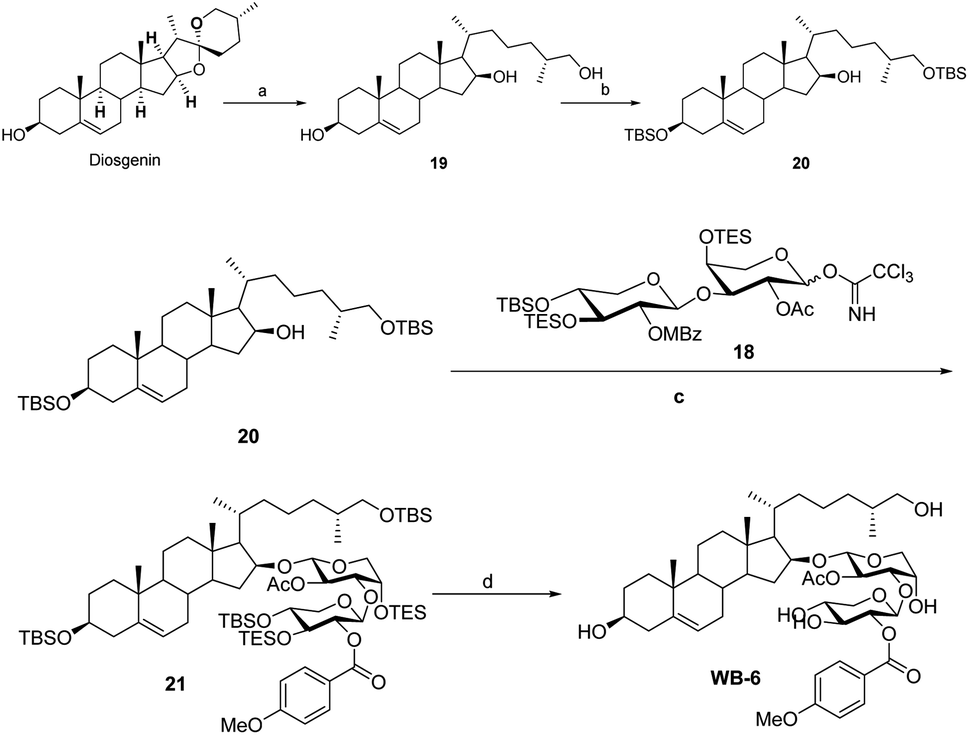 | ||
| Scheme 6 Reaction condition: a, diosgenin (1 g), Zn (150 eq.), conc. HCl (75 mL), EtOH (150 mL), reflux, 66% yield; (b) TBSCl, DBU, THF, rt 16 h, 92% yield; (c) TMSOTf, 4 Å MS, DCM, −40 °C, 3 h, 68% yield; (d) Pd (MeCN)Cl2, acetone/H2O,overnight, room temperature, 82% yield. | ||
Antitumor activities in vitro
The synthetic OSW-1 analogue WB-06 was screened against HCT-8, HeLa, MDA-MB-231, MCF7, Skov-3. HMC-3, MCF-10A cell lines by alamarBlue cytotoxicity assay. Taxol was used as a positive control; all the experiments were repeated three times. The HCT-8, HeLa, MDA-MB-231, and MCF7 cell line was selected for comparison with the literature-reported OSW-1 or OSW-1 analogy cytotoxicity data.12c,13 All of the other cell lines were screened for the first time. The results presented in Table 2 illustrate the concentrations required for 50% cell death (IC50 values) when compared to the vehicle-treated negative control. The IC50 value for WB-6 range from 18.6 nM to 110.6 nM less active to Taxol. In particular, the IC50 value for HeLa, MDA-MB-231, and MCF7 cells is comparable to the literature-reported value. WB-6 also showed valuable benefits to Skov-3 cell lines. Unfortunately, no selectivity between tumor and non-tumor cell lines (HMC-3 and MCF-10A) was observed for WB-6.| IC50 (nM) | |||||||
|---|---|---|---|---|---|---|---|
| HCT-50 | HeLa | MDA-MB-231 | MCF-7 | SKOV-3 | HMC-3M | MCF-10A | |
| WB-06 | 95.6 | 18.6 | 20.5 | 16.3 | 22.1 | 65.1 | 56.3 |
| Taxol | 57.8 | 2.1 | 5.5 | 12.1 | 9.8 | — | — |
The in vitro cytotoxicity against HCT-8 (colon carcinoma cell line), HeLa (cervical cancer cell line), MDA-MB-231 (Human Breast cancer cell line), MCF7 (breast cancer cell line), Skov-3 (ovarian cancer cell line), HMC-3 (human microglia cell line), MCF-10A (non-tumorigenic epithelial cell line) cell lines were evaluated by the standard MTT assay.
Conclusions
Finally, we have developed a flexible synthesis strategy for OSW-1 disaccharide, which was synthesized in 13 steps with an overall yield of 7.2% from xylose derivate 1 and arabinose derivate 7. A new and effective process for preparing disaccharide building blocks has been created. Donor 4 was achieved by protecting the C-2, C-3, and C-4 hydroxyl positions independently, allowing for easy modification. The xylose donor moiety is useful for studying the structure–activity relationship of OSW-1 and discovering new anticancer treatments. Specifically, the first high yield of glycosylation on an OSW-1 synthesis disaccharide was achieved by sulfide donor 4 with β-PMP anomeric L-arabinose acceptor 12. The cell analysis shows that analogy has higher anti-tumor potential. This strategy should provide a versatile way to modify OSW-1's disaccharide. Further application of this disaccharide to generate OSW-1 and analogies is in progress in our laboratory.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the National Natural Science Foundation of China (grant no. 81660725, 82060782), Urumqi science and technology talent, Tianshan Innovation Team of Xinjiang Uygur Autonomous Region (no. 202D14011). Tianshang Cedar plan (no. 2018XS21), Development Program, Specialized Research on Modernization of TCM (no. 2018YFC1708006 and 2018 YFC1708003).Notes and references
- S. Kubo, Y. Mimaki, M. Terao, Y. Sashida, T. Nikaido and T. Ohmoto, Phytochemistry, 1992, 31, 3969–3973 CrossRef CAS.
- (a) Y. Mimaki, M. Kuroda, A. Kameyama, Y. Sashida, T. Hirano, K. Oka, R. Maekawa, T. Wada, K. Sugita and J. A. Buetler, Bioorg. Med. Chem. Lett., 1997, 7, 633–636 CrossRef CAS; (b) M. Kuroda, F. Hasegawa, A. Yokosuka, Y. Mimaki and Y. Sashida, Chem. Nat. Prod., 2001, 43, 371–376 CrossRef PubMed; (c) Y. Tang, N. Ji, J. Duan and W. Tao, Chem. Rev., 2013, 113, 5480–5514 CrossRef CAS PubMed.
- (a) C. Guo and P. L. Fuchs, Tetrahedron Lett., 1998, 39, 1099–1102 CrossRef CAS; (b) S. Deng, B. Yu, Y. Lou and Y. Hui, J. Org. Chem., 1999, 64, 202–208 CrossRef CAS PubMed; (c) W. Yu and Z. Jin, J. Am. Chem. Soc., 2001, 123, 3369–3370 CrossRef CAS PubMed; (d) W. Yu and Z. Jin, J. Am. Chem. Soc., 2002, 124, 6576–6583 CrossRef CAS PubMed; (e) J. W. Morzycki and A. Wojtkielewicz, Carbohydr. Res., 2002, 337, 1269–1274 CrossRef CAS PubMed; (f) B. Shi, P. Tang, X. Hu, J. O. Liu and B. Yu, J. Org. Chem., 2005, 70, 10354–10367 CrossRef CAS PubMed.
- (a) M. Tsubuki, S. Matsuo and T. Honda, Tetrahedron Lett., 2008, 49, 229–232 CrossRef CAS; (b) L. Sun, R. Wang, X. Wang, Y. Dang, W. Li and B. Yu, Org. Chem. Front., 2019, 6, 2385–2391 RSC; (c) L. Sun, D. Zhu, L. Beverborg, R. Wang, Y. Dang, M. Ma, W. Li and B. Yu, Chin. J. Chem., 2020, 38, 1091–1097 CrossRef CAS.
- (a) X. Ma, B. Yu, Y. Hui, Z. Miao and J. Ding, Carbohydr. Res., 2001, 334, 159–164 CrossRef CAS PubMed; (b) P. Tang, F. Mamdani, X. Hu, J. Liu and B. Yu, Bioorg. Med. Chem. Lett., 2007, 17, 1003–1007 CrossRef CAS PubMed.
- (a) X. Ma, B. Yu, Y. Hui, D. Xiao and J. Ding, Carbohydr. Res., 2000, 329, 495–505 CrossRef CAS PubMed; (b) L. S. Khasanova, F. A. Gimalova, S. A. Torosyan, A. A. Fatykhov and M. S. Miftakhov, Russ. J. Org. Chem., 2011, 47, 1125–1129 CrossRef CAS; (c) Z. Pakulski and P. Cmoch, Tetrahedron, 2015, 71, 4757–4769 CrossRef CAS; (d) K. Kuczynska, P. Cmoch, L. Rárová, J. Oklešt'ková, A. Korda, Z. Pakulski and M. Strnad, Carbohydr. Res., 2016, 423, 49–69 CrossRef CAS PubMed; (e) L. Sun, R. Wang, X. Wang, Y. Dang, W. Li and B. Yu, Org. Chem. Front., 2019, 6, 2385–2391 RSC.
- (a) R. Li, H. Tang, L. Wan, X. Zhang, Z. Fu, J. Liu, S. Yang, D. Jia and D. Niu, Chem, 2017, 3, 834–845 CrossRef CAS; (b) W. Shang, Z. Mou, H. Tang, X. Zhang, J. Liu, Z. Fu and D. Niu, Angew. Chem., Int. Ed., 2018, 57, 314–318 CrossRef CAS PubMed; (c) H. Tang, Y. Tian, H. Cui, R. Li, X. Zhang and D. Niu, Nat. Commun., 2020, 11, 5681 CrossRef CAS PubMed.
- (a) K. Sakurai, T. Takeshita, M. Hiraizumi and R. Yamada, Org. Lett., 2014, 16, 6318–6321 CrossRef CAS PubMed; (b) M. Hiraizumi, R. Komatsu, T. Shibata, Y. Ohta and K. Sakurai, Org. Biomol. Chem., 2017, 17, 3568–3570 RSC; (c) K. Sakurai, M. Hiraizumi, N. Isogai, R. Komatsu, T. Shibata and Y. Ohta, Chem. Commun., 2017, 53, 517–520 RSC.
- While compound 3a is reacted with ally chloroformate using triethylamine (Et3N) as a base, it produces the high yield product 4a, which provides more options for disaccharide modification
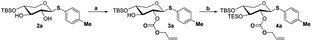 Scheme 7. Reaction condition: (a) allyl chloroformate, Et3N, DMF, room temperature, 2 h. (b) TESOTf, DMAP, Et3N, DMF, room temperature, 2 h..
Scheme 7. Reaction condition: (a) allyl chloroformate, Et3N, DMF, room temperature, 2 h. (b) TESOTf, DMAP, Et3N, DMF, room temperature, 2 h.. - J. Xue, P. Liu, Y. B. Pan and Z. W. Guo, J. Org. Chem., 2008, 73, 157–161 CrossRef CAS PubMed.
- Compound 13 as an accepter to glycosylate with donor 4 using N-iodosuccinimide (NIS) and BF3Et2O as promoters generated compound 15
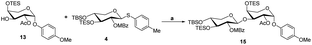 Scheme 5. Reaction condition: (a) BF3·Et2O (0.05 eq.), 4 Å MS, DCM, −40 °C..
Scheme 5. Reaction condition: (a) BF3·Et2O (0.05 eq.), 4 Å MS, DCM, −40 °C.. - (a) J. R. Williams, D. Chai and D. Wright, Steroids, 2002, 67, 1041–1044 CrossRef CAS PubMed; (b) D. Zheng, Y. Guan, X. Chen, Y. Xu, X. Chen and P. Lei, Bioorg. Med. Chem. Lett., 2011, 21, 3257–3260 CrossRef CAS PubMed; (c) C. Liu, A. Wang, L. Jin, Y. Guo, Y. Li, Z. Zhao and P. Lei, Tetrahedron, 2016, 72, 4091–4102 CrossRef CAS.
- (a) B. F. Shi, H. Wu, B. Yu and J. R. Wu, Angew. Chem., Int. Ed., 2004, 43, 4324–4327 CrossRef CAS PubMed; (b) J. W. Morzycki, A. Wojtkielewicz and S. Wołczyński, Bioorg. Med. Chem. Lett., 2004, 14, 3323–3326 CrossRef CAS PubMed; (c) A. Wojtkielewicz, M. Długosz, J. Maj, J. W. Morzycki, M. Nowakowshi, J. Renkiewicz, M. Strnad, J. Swaczynova, A. Z. Wilczewska and J. Wojcik, J. Med. Chem., 2007, 50, 3667–3673 CrossRef CAS PubMed; (d) T. Tschamber, S. Adam, Y. Matsuya, S. Masuda, N. Ohsawa, S. Maruyama, K. Kamoshita, H. Nemoto and J. Eustache, Bioorg. Med. Chem. Lett., 2007, 17, 5101–5106 CrossRef CAS PubMed; (e) J. Maj, J. W. Morzycki, L. Rarova, J. Oklest'kova, M. Strnad and A. Wojtkielewicz, J. Med. Chem., 2011, 54, 3298–3305 CrossRef CAS PubMed; (f) Y. Y. Guan, D. Zheng, L. Zhou, H. X. Wang, Z. Yan, N. Wang, H. Chang, P. P. She and P. S. Lei, Bioorg. Med. Chem. Lett., 2011, 21, 2921–2924 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05748h |
| ‡ Equal contribution as first-author. |
| This journal is © The Royal Society of Chemistry 2023 |