
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel 2-substituted-quinoxaline analogs with potential antiproliferative activity against breast cancer: insights into cell cycle arrest, topoisomerase II, and EGFR activity†
Manar G. Salem a,
Sara A. Abu El-atab,
Elsherbiny H. Elsayedb,
Suraj N. Malic,
Hussah Abdullah Alshwyehde,
Ghassan Almaimanif,
Riyad A. Almaimanig,
Hussain A. Almasmoumh,
Najla Altwaijryi,
Ebtesam Al-Olayanj,
Essa M. Saied*kl and
Mohamed F. Youssef*m
a,
Sara A. Abu El-atab,
Elsherbiny H. Elsayedb,
Suraj N. Malic,
Hussah Abdullah Alshwyehde,
Ghassan Almaimanif,
Riyad A. Almaimanig,
Hussain A. Almasmoumh,
Najla Altwaijryi,
Ebtesam Al-Olayanj,
Essa M. Saied*kl and
Mohamed F. Youssef*m
aDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt
bDepartment of Chemistry, Faculty of Science, Port Said University, Port Said, Egypt
cDepartment of Pharmaceutical Sciences and Technology, Birla Institute of Technology, Ranchi 835215, India
dDepartment of Biology, College of Science, Imam Abdulrahman Bin Faisal University, Dammam 31441, Saudi Arabia
eBasic & Applied Scientific Research Centre, Imam Abdulrahman Bin Faisal University, PO Box 1982, Dammam 31441, Saudi Arabia
fDepartment of Surgery, Faculty of Medicine, Umm Al-Qura University, Al Abdeyah, PO Box 7607, Makkah, Saudi Arabia
gDepartment of Biochemistry, Faculty of Medicine, Umm Al-Qura University, Al Abdeyah, PO Box 7607, Makkah, Saudi Arabia
hDepartment of Clinical Laboratory Sciences, Faculty of Applied Medical Sciences, Umm Al-Qura University, Al Abdeyah, PO Box 7607, Makkah, Saudi Arabia
iDepartment of Pharmaceutical Sciences, Princess Nourah Bint Abdulrahman University, PO Box 84428, Riyadh 11671, Saudi Arabia
jDepartment of Zoology, College of Science, King Saud University, Riyadh, Saudi Arabia
kDepartment of Chemistry (Biochemistry Division), Faculty of Science, Suez Canal University, Ismailia 41522, Egypt
lInstitute for Chemistry, Humboldt Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: saiedess@hu-berlin.de
mDepartment of Chemistry (Organic Chemistry Division), Faculty of Science, Suez Canal University, Ismailia 41522, Egypt
First published on 9th November 2023
Abstract
Breast cancer is a global health concern, with increasing disease burden and disparities in access to healthcare. Late diagnosis and limited treatment options in underserved areas contribute to poor outcomes. In response to this challenge, we developed a novel family of 2-substituted-quinoxaline analogues, combining coumarin and quinoxaline scaffolds known for their anticancer properties. Through a versatile synthetic approach, we designed, synthesized, and characterized a set of 2-substituted quinoxaline derivatives. The antiproliferative activity of the synthesized compounds was assessed toward the MCF-7 breast cancer cells. Our investigations showed that the synthesized compounds exhibit considerable antiproliferative activity toward MCF-7 cells. Notably, compound 3b, among examined compounds, displayed a superior inhibitory effect (IC50 = 1.85 ± 0.11 μM) toward the growth of MCF-7 cells compared to the conventional anticancer drug staurosporine (IC50 = 6.77 ± 0.41 μM) and showed minimal impact on normal cells (MCF-10A cell lines, IC50 = 33.7 ± 2.04 μM). Mechanistic studies revealed that compound 3b induced cell cycle arrest at the G1 transition and triggered apoptosis in MCF-7 cells, as evidenced by increasing the percentage of cells arrested in the G2/M and pre-G1 phases utilizing flow cytometric analysis and Annexin V-FITC/PI analysis. Moreover, compound 3b was found to substantially suppress topoisomerase enzyme activity in MCF-7 cells. Molecular modeling studies further supported the potential of compound 3b as a therapeutic candidate by demonstrating significant binding affinity to the active sites of both topoisomerase II and EGFR proteins. Taken together, the presented 2-substituted-quinoxaline analogues, especially compound 3b, show promise as potential candidates for the development of effective anti-breast cancer drugs.
Introduction
Breast cancer is considered the second leading cause of death among women.1 The significance of timely identification and intervention in breast cancer, before its metastasis, is of utmost importance, despite the ongoing discourse around the most effective approach to achieve this in recent times.2 The annual incidence of common breast cancer has reached a staggering two million cases worldwide, accounting for a significant proportion of all cancer types.3 It primarily originates in the lobules, which are responsible for producing milk, or the ducts that connect these lobules to the nipple.4 Hormones, alcohol, obesity, and nulliparity, which have garnered public attention, have a very modest relative risk (<2%) for breast cancer.5 Chemotherapy6 has been widely recognized as the foremost efficacious therapeutic modality for the management of localized breast and liver malignancies as it impedes their molecular action by the initiation of apoptosis,7 necrosis,8 and autophagy, resulting in the death of cancer cells.9 Besides chemotherapy, breast cancer patients have access to a wide range of treatment choices, including invasive and noninvasive biopsies, radiotherapy and molecular therapies.10 However, most available medications lack specificity, which brings up problems like the well-documented side effects of cancer treatment11 and the much more widespread phenomenon of multi-drug resistance.12DNA topoisomerases are enzymes that play a crucial role in regulating DNA twists and folds.13 Topoisomerases also play a vital role in preserving the structural integrity and stability of DNA,14 which is essential for many complex biological processes like transcription, replication, and recombination15 but can impede these activities and slow cell growth.16 DNA damage,17 slowed DNA replication,18 unrepaired DNA strand breakage,19 and cell death are all direct results of this blockade. Based on their ability to unwind both DNA strands at once, topoisomerases are categorized into two groups, topoisomerase I and topoisomerase II.20 Topoisomerases I and II (Top 1 and Top 2 as they are more often known) are largely considered to be significant molecular targets for the development of anticancer medicines.21 Irinotecan, topotecan, and camptothecin are well-known examples of topoisomerase I (Topo I) inhibitors.22 On the other hand, etoposide, doxorubicin, and epirubicin are all examples of topoisomerase II (Topo II) inhibitors.23 Due to its potential role as a therapeutic target and prognostic marker, Topo II has received a great deal of attention in the context of breast cancer.24 Topo II exists in two forms, called Topo IIα and Topo IIβ.25 DNA unwinding activity during replication is a hallmark of Topo IIα, and this protein has been considered to be a potential prognostic marker.26 Topo IIα is often overexpressed in HER2-positive breast cancer cells and thus has been recognized as a potential pharmacological target for the treatment of breast cancer.27,28
Heterocycles are recognized as a profoundly auspicious category of compounds in the quest for innovative anticancer agents.29 Quinoxaline has been identified as a prominent heterocyclic compound that has attracted considerable scientific attention.30–32 The wide substitution of quinoxaline and their derivatives, together with their many biological features such as antiviral, anticancer, and antileishmanial activity, contribute to their promising prospects in the field of medicinal chemistry. They are increasingly acknowledged as a distinctive category of chemotherapeutic agents exhibiting notable efficacy against various types of tumors.33 Quinoxaline-based compounds have shown significant anticancer efficacy by selectively inhibiting many cellular targets,34 including topoisomerase,35 VEGFR2,36 telomerase,37 farnesyltransferase,38 tyrosine kinase,39 and protein kinase CK-11 (Fig. 1). They hinder the proliferation and metastasis of breast cancer cells by targeting the PI3K/AKT/mTOR and MAPK/ERK pathways, as well as other proteins involved in angiogenesis. Additionally, quinoxaline derivatives impede the growth of ER-positive breast cancer cells by interfering with estrogen signaling at estrogen receptors.40,41 Currently, there are several quinoxaline analogues which are under clinical investigations for their applicability as anti-tumor drugs, including CQS, GK13, tirapazamine, and R-(+)-XK469 (ref. 31 and 42–44) (Fig. 1).
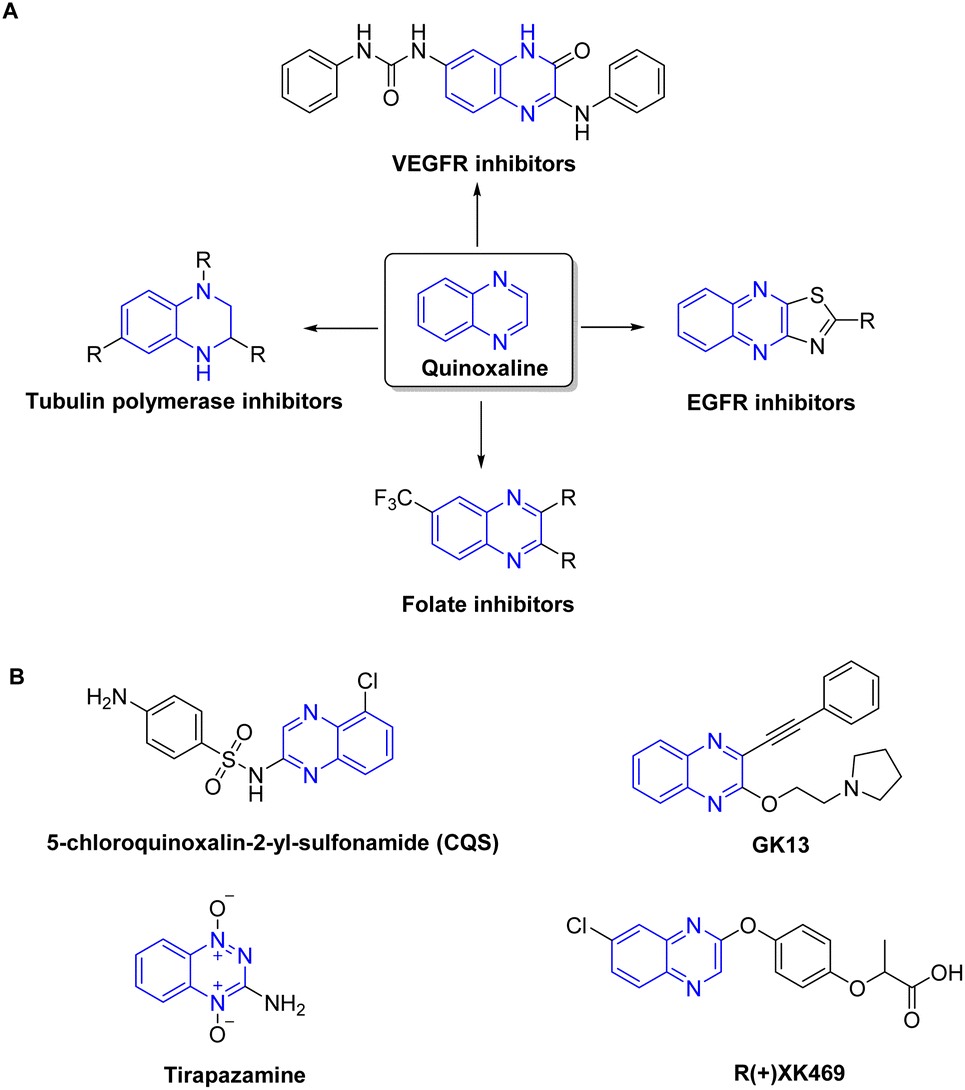 | ||
| Fig. 1 Representative quinoxaline-based analogs with potent anti-tumor properties (A) and being examined in clinical trials (B). | ||
The term “hybrid molecule” refers to the method used often in drug design to increase the potency, selectivity, and overall pharmacological characteristics of drugs by combining two distinct pharmacophores inside a single molecular scaffold.45,46 This strategy takes use of the pharmacophores' distinct characteristics to provide a synergistic impact, which may increase efficacy against a target or illness.47 Coumarins are heterocyclic polyphenolic chemicals found throughout plants.48,49 Systematic research found 1300 secondary metabolite chemotypes in plants, microbes, and fungi.50 The roots of Ferulago campestris,51 Murraya paniculata52 leaves, Aegle marmelos53 fruits, and tonka bean seeds contain derivatives of coumarins with several biological functions. Synthetic coumarin-based heterocyclic analogues such azoles, sulfonyl, furan, and pyrazole have promise anticancer, antitumor, and anti-proliferative properties.54,55 Natural coumarin-based compounds have demonstrated a potential anticancer activity by targeting several cellular targets including cycle arrest, angiogenesis, kinase activity, telomerase, and carbonic anhydrase.54,56
Considering the well-documented cytotoxic properties of coumarin- and quinoxaline-based compounds, our research hypothesis posits that the deliberate integration of these compounds into hybrid molecules may result in significant antitumor attributes worth exploring. Based on the abovementioned facts, and our persistent interest in synthesizing bioactive compounds,57–64 we aimed to design, synthesize, and characterized a new set of hybridized 2-substituted-quinoxaline derivatives bearing a phenyl/coumarin moiety (Fig. 2). The anti-proliferative activity of designed compound was assessed toward MCF-7 cells, and the mode of action was explored by investigating cell cycle arrest, and topoisomerase II activity. Finally, detailed computational analysis was performed utilizing molecular modelling and molecular simulation analysis to gain more insights to the mechanistic of this class of compounds.
 | ||
| Fig. 2 Illustration of the structural features of designed hybridized 2-substituted-quinoxaline analogues bearing phenyl or coumarin moiety. | ||
Results and discussion
Chemistry
In the current research, we have developed a synthetic method for synthesizing new 2-substituted quinoxaline derivatives containing either a phenyl or an 8-methoxy-coumarin-3-yl group, as illustrated in Schemes 1 and 2, respectively. The synthesis started by the bromination of 5-bromo-2-hydroxy-acetophenone (1a) or 3-acetyl-8-methoxy-coumarin (1b) in the presence of glacial acetic acid to afford aryl bromomethyl ketones (2a, b), which are crucial intermediates in the synthesis of quinoxaline derivatives.65,66 The reaction was conducted at a temperature of 60 °C with continuous stirring, following the established procedures described in the previous reports.31 Compounds 2a and 2b were subjected to a condensation procedure employing o-phenylene diamine and molten sodium acetate in an ethanol solvent to furnish 2-(5-bromo-2-hydroxyphenyl)-3,4-dihydroquinoxaline (3a) and 2-(8-methoxy-coumarin-3yl)-3,4-dihydroquinoxaline (3b), with satisfactory yields of 56% and 61%, respectively. The nuclear magnetic resonance (1H-NMR) spectra obtained for compounds 3a and 3b have revealed the presence of two distinct isomeric structures. The chemical confirmation of the structures of compounds 3a and 3b was achieved through a series of experimental procedures. Particularly, compounds 3a and 3b, were subjected to treatment with acetic anhydride under reflux conditions to afford 2-(2-acetoxy-5-bromo) phenyl quinoxaline (4a) and 2-(8-methoxy-coumarin-3-yl)-quinoxaline (4b) with yields of 73%, and 69%, respectively. The halogenation of compound 4a was carried out by using bromine in glacial acetic acid, resulting in the formation of the corresponding 2-(5-bromo-8-methoxy-coumarin-3-yl) quinoxaline (5) with a yield of 82%. | ||
| Scheme 1 Synthesis of 2-substituted-quinoxaline analogues bearing phenyl or coumarin moiety (3a, b-5). Regents and conditions: (a) Br2, glacial AcOH, 60 °C-r. t., 12 h; (b) o-phenylene diamine, sodium acetate, EtOH, reflux, 12 h; (c) acetic anhydride, reflux, 14 h; (d) bromine, acetic acid, reflux, 8 h. | ||
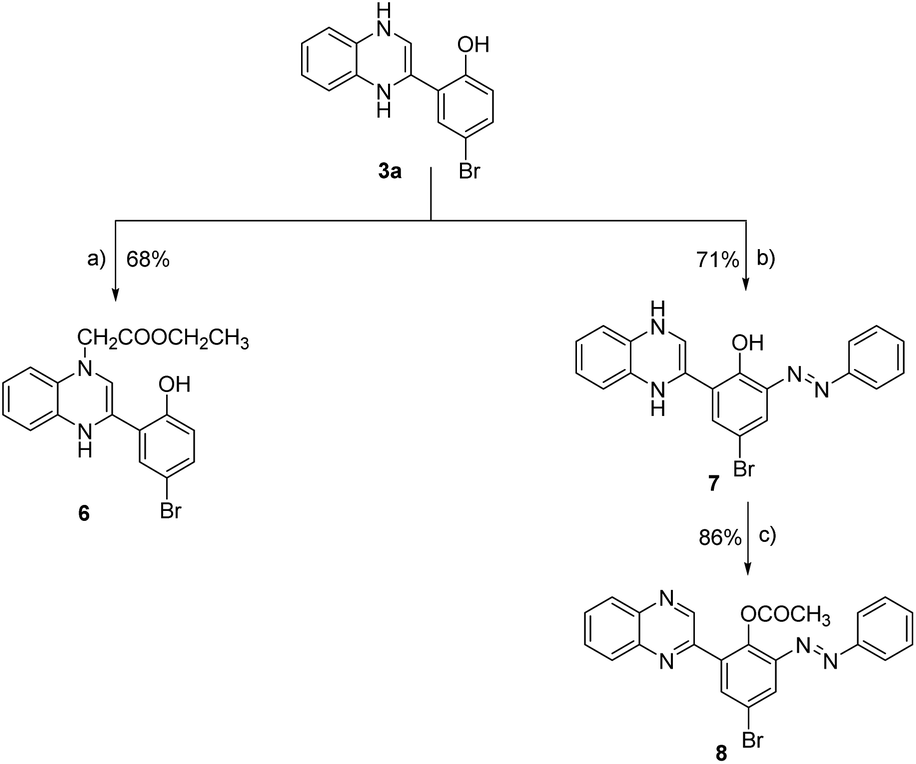 | ||
| Scheme 2 Synthesis of 2-phenyl-quinoxaline analogues (6–8). Reagent and conditions: (a) ethyl chloroacetate, triethyl amine, DMF, reflux, 12 h; (b) phenyl diazonium chloride, 10% NaOH, 0–5 °C, 24 h; (c) acetic anhydride, reflux, 16 h. | ||
We expanded our investigation to explore the structural features of the 2-phenyl-substituted quinoxaline scaffold in greater depth. As shown in Scheme 2, 2-(5-bromo-2-hydroxyphenyl)-3,4-dihydroquinoxaline (3a) was further reacted with ethyl chloroacetate in the presence of triethyl amine to provide ethyl [3-(5-bromo-2-hydroxyphenyl)-1,4-dihydroquinoxalin-1-yl] acetate (6) with a yield of 68%. Further, compound 3a was reacted with phenyl diazonium chloride in the presence of sodium hydroxide to afford 2-(5-bromo-3-phenylazo-2-hydroxyphenyl)-1,4-dihydroquinoxaline (7) with a yield of 71%. Finally, compound 7 was subjected to acetylation conditions using acetic anhydride under reflux to successfully furnish 2-(5-bromo-3-phenylazo-2-acetoxy) phenyl-quinoxaline-(8), with a yield of 86%.
Characterization of synthesized quinoxaline derivatives
The structural properties of all synthesized quinoxaline hybrids were thoroughly characterized using a variety of analytical techniques, such as nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry. The nuclear magnetic resonance (1H-NMR) spectra obtained for compounds 3a and 3b have provided valuable insights into the structural composition of these compounds, revealing the presence of two distinct isomers: 1,2-dihydroquinoxaline and 1,4-dihydroquinoxaline (Fig. S9†). This structural elucidation is visually represented in Scheme 1. The proton signal originating from the methylene (NCH2) group of the isomeric 1,2-dihydroquinoxaline compound was observed at chemical shifts of approximately 4.40 and 4.02 ppm. Similarly, the 13CNMR spectra displayed carbon signals corresponding to the methylene group at approximately 56.58 and 41.20 ppm. The proton signals originating from two NH groups in the isomeric form of 1,4-dihydroquinoxaline were observed within the chemical shift range of 9.60–9.72 ppm. The mass spectra analysis of dihydro quinoxaline derivatives (3a and 3b) exhibited highly prominent molecular ion peaks at m/z 302 and 306, which corresponded to the molecular formulas C14H11BrN2O and C18H14N2O3, respectively. The mass spectrum analysis of compound 3a revealed the presence of a molecular ion peak at m/z 302, which was identified as the base peak. In the instance of compound 3b, it is noteworthy that the base peak manifested at m/z 304, which aligns with the molecular ion of compound 3b following the elimination of hydrogen molecules (M+2). The 1H-NMR spectrum of compound 3a exhibited singlet signals at a chemical shift of 2.22 ppm, which can be attributed to the presence of a methyl group associated with the acetyl group (COCH3). Notably, there was an absence of any proton signal originating from the hydroxy group. The chemical compound known as azo methane (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) within the quinoxaline ring exhibited a proton signal at a chemical shift of approximately 9.26 ppm. Furthermore, it is worth noting that the proton signals originating from aromatic rings were detected within the spectral region spanning from δ 7.37 to 8.19 ppm, manifesting as multiplet signals. The 13C-NMR spectrum of compound 4a exhibited two distinct carbon signals at chemical shifts of 169.48 and 21.19 ppm, respectively. These chemical shifts can be attributed to the presence of the acetoxy group (OCOCH3) within the compound. The spectral analysis revealed the presence of carbon signals corresponding to quinoxaline and an aromatic ring, which manifested in the chemical shift regions of 149.51–119.31 ppm. It is noteworthy that the total number of carbon atoms in this system amounts to 14 carbons. The mass spectrum analysis of compound 4a revealed a highly pronounced molecular ion peak at m/z 342, which aligns precisely with the molecular formula C16H11BrN2O2. The molecular ion peak observed at m/z 342 exhibited fragmentation, resulting in the emergence of a base peak at m/z 300. This base peak corresponds to the molecular ion peak of 2-(2-hydroxy-5-bromo) phenyl-quinoxaline radical cation. The fragmentation process involved the detachment of a ketene molecule (CH2CO). The 1H-NMR analysis of compound 4b revealed the presence of three singlet signals at chemical shifts of 9.60, 8.89, and 3.97 ppm. These signals were attributed to the protons associated with the azomethine (CHN) group in the quinoxaline ring, the H-4 proton in the coumarin ring, and the methoxy group (OCH3), respectively. Furthermore, it is noteworthy that the protons associated with the quinoxaline and coumarin rings exhibited multiplet signals within the range of δ 6.56–8.16 ppm. Moreover, the structural verification of compound 4b was substantiated through the analysis of its 13C-NMR spectrum. Notably, the carbon signals observed at δ 56.67 and 56.16 ppm corresponded to the methoxy group (OCH3) present in both stereo isomers of the compound. In addition, it is worth noting that the carbon signals originating from the quinoxaline and coumarin rings have been detected within the spectral region spanning from 159.84 to 114.34 ppm. The mass spectrum analysis of compound 4b revealed the presence of a stable molecular ion peak at m/z 304. This peak corresponds to the molecular formula C18H12N2O3. The spectral analysis revealed the emergence of three distinct carbon signals at δ 168.79, 65.83, 61.38, and δ 14.47 ppm, which have been confidently attributed to the ethyl acetate fragment (NCH2COOCH2CH3). The 1H-NMR spectrum analysis of compound 7 revealed the presence of four singlet signals at chemical shift values of 14.88, 12.19, 9.68, and 4.52 ppm. These signals can be attributed to the protons associated with the hydroxyl (OH), amino (NH), and N-methylene (NCH2) functional groups within the compound. The multiplet signals observed in the region of approximately 6.47–8.19 ppm correspond to the proton signals (–CH) originating from the aromatic and quinoxaline ring structures. The 13C-NMR spectrum of compound 7 displayed carbon signals within the anticipated chemical shift range of 157.63–111.26 ppm. The 1H-NMR spectrum analysis of compound 8 revealed the absence of proton signals at chemical shifts of approximately 14.88, 12.19, 9.68, and 4.52 ppm in the presence of compound 7. Conversely, new proton signals emerged at chemical shifts of approximately 9.77, 9.24, 2.21, and 1.74 ppm, which can be attributed to the protons of the azomethine (CHN) group in quinoxaline and the acetoxy (OCOCH3) groups. These findings provide evidence for the existence of two stereoisomers in the compound under investigation. Moreover, the 13C NMR spectrum of compound 8 exhibited the emergence of two distinct carbon signals, corresponding to two isomers, at chemical shifts of 174.63 and 169.41 parts per million (ppm). These shifts are in reference to the carbon atom within the acetyl group (COCH3). Additionally, a separate carbon signal was observed at chemical shifts of 24.25 and 21.16 ppm, also referencing the carbon atom of the acetyl group. The mass spectrum analysis of compound 7 revealed the presence of a molecular ion peak at m/z 402, which exhibited a relatively low intensity. Conversely, the mass spectrum analysis of compound 8 indicated an unstable molecular ion peak. The fragmentation of compounds 7 and 8 resulted in the generation of a stable peak at m/z 221, which corresponds to the molecular ion of 2-(2-hydrophenyl) quinoxaline cation [C14H9NO2]+.
N) within the quinoxaline ring exhibited a proton signal at a chemical shift of approximately 9.26 ppm. Furthermore, it is worth noting that the proton signals originating from aromatic rings were detected within the spectral region spanning from δ 7.37 to 8.19 ppm, manifesting as multiplet signals. The 13C-NMR spectrum of compound 4a exhibited two distinct carbon signals at chemical shifts of 169.48 and 21.19 ppm, respectively. These chemical shifts can be attributed to the presence of the acetoxy group (OCOCH3) within the compound. The spectral analysis revealed the presence of carbon signals corresponding to quinoxaline and an aromatic ring, which manifested in the chemical shift regions of 149.51–119.31 ppm. It is noteworthy that the total number of carbon atoms in this system amounts to 14 carbons. The mass spectrum analysis of compound 4a revealed a highly pronounced molecular ion peak at m/z 342, which aligns precisely with the molecular formula C16H11BrN2O2. The molecular ion peak observed at m/z 342 exhibited fragmentation, resulting in the emergence of a base peak at m/z 300. This base peak corresponds to the molecular ion peak of 2-(2-hydroxy-5-bromo) phenyl-quinoxaline radical cation. The fragmentation process involved the detachment of a ketene molecule (CH2CO). The 1H-NMR analysis of compound 4b revealed the presence of three singlet signals at chemical shifts of 9.60, 8.89, and 3.97 ppm. These signals were attributed to the protons associated with the azomethine (CHN) group in the quinoxaline ring, the H-4 proton in the coumarin ring, and the methoxy group (OCH3), respectively. Furthermore, it is noteworthy that the protons associated with the quinoxaline and coumarin rings exhibited multiplet signals within the range of δ 6.56–8.16 ppm. Moreover, the structural verification of compound 4b was substantiated through the analysis of its 13C-NMR spectrum. Notably, the carbon signals observed at δ 56.67 and 56.16 ppm corresponded to the methoxy group (OCH3) present in both stereo isomers of the compound. In addition, it is worth noting that the carbon signals originating from the quinoxaline and coumarin rings have been detected within the spectral region spanning from 159.84 to 114.34 ppm. The mass spectrum analysis of compound 4b revealed the presence of a stable molecular ion peak at m/z 304. This peak corresponds to the molecular formula C18H12N2O3. The spectral analysis revealed the emergence of three distinct carbon signals at δ 168.79, 65.83, 61.38, and δ 14.47 ppm, which have been confidently attributed to the ethyl acetate fragment (NCH2COOCH2CH3). The 1H-NMR spectrum analysis of compound 7 revealed the presence of four singlet signals at chemical shift values of 14.88, 12.19, 9.68, and 4.52 ppm. These signals can be attributed to the protons associated with the hydroxyl (OH), amino (NH), and N-methylene (NCH2) functional groups within the compound. The multiplet signals observed in the region of approximately 6.47–8.19 ppm correspond to the proton signals (–CH) originating from the aromatic and quinoxaline ring structures. The 13C-NMR spectrum of compound 7 displayed carbon signals within the anticipated chemical shift range of 157.63–111.26 ppm. The 1H-NMR spectrum analysis of compound 8 revealed the absence of proton signals at chemical shifts of approximately 14.88, 12.19, 9.68, and 4.52 ppm in the presence of compound 7. Conversely, new proton signals emerged at chemical shifts of approximately 9.77, 9.24, 2.21, and 1.74 ppm, which can be attributed to the protons of the azomethine (CHN) group in quinoxaline and the acetoxy (OCOCH3) groups. These findings provide evidence for the existence of two stereoisomers in the compound under investigation. Moreover, the 13C NMR spectrum of compound 8 exhibited the emergence of two distinct carbon signals, corresponding to two isomers, at chemical shifts of 174.63 and 169.41 parts per million (ppm). These shifts are in reference to the carbon atom within the acetyl group (COCH3). Additionally, a separate carbon signal was observed at chemical shifts of 24.25 and 21.16 ppm, also referencing the carbon atom of the acetyl group. The mass spectrum analysis of compound 7 revealed the presence of a molecular ion peak at m/z 402, which exhibited a relatively low intensity. Conversely, the mass spectrum analysis of compound 8 indicated an unstable molecular ion peak. The fragmentation of compounds 7 and 8 resulted in the generation of a stable peak at m/z 221, which corresponds to the molecular ion of 2-(2-hydrophenyl) quinoxaline cation [C14H9NO2]+.
Assessment of cytotoxic activity against MCF-7 cells
We initially assessed the anti-proliferative activity of synthesized 2-substituted-quinoxaline derivatives (3b, 4a, 4b, 5, 6, and 7) on the viability of two different cell lines: MCF-7, which represents breast cancer cells, and MCF-10A, representing normal breast cells. To assess the anticancer potential of these compounds, we employed the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) colorimetric assay which is a widely utilized technique to evaluate cell viability and cytotoxicity. In our investigations, we incorporated staurosporine (STU) as a reference standard drug due to its well-established effectiveness as an anticancer agent. The MCF-7 and MCF-10A cell lines were subjected to different doses of synthesized quinoxaline derivatives, and the resulting cytotoxicity was evaluated in a dose-dependent manner (Fig. 3). Our results reveal that the developed 2-substituted quinoxaline analogues exhibit a broad range of anti-proliferative activity, spanning from moderate to highly potent. The evaluation of cytotoxic activity in 2-phenyl-substituted quinoxaline analogs (3a, 4a, 6, 7, and 8) highlighted essential structural characteristics that could be linked to the anti-proliferative properties of these compounds. As shown in Fig. 3, compound 3a with quinoxaline scaffold substituted at position 2 with 5-bromo-2-hydroxy-phenyl moiety exhibited a considerable cytotoxic activity with IC50 of 12.74 μM. Acylation of the hydroxyl-group at the 2-postion of the phenyl moiety of compound 3a afforded a compound with attenuated cytotoxic activity (4a, IC50 of 20.8 μM), suggesting the role of the hydroxyl-group on the anti-proliferative activity of the 2-phenyl substituted-quinoxaline scaffold. Similarly, alkylation of the quinoxaline scaffold at N4-position with ethylacetate moiety significantly reduced the cytotoxic activity of the quinoxaline analogue (compound 6, IC50 = 62.4 μM), indicating that the 1,4-quinoxaline ring play a substantial role in the observed anti-proliferative activity of this class. Interestingly, the addition of phenyl diazonium moiety at position-3 in the 2-phenyl-quinoxaline scaffold significantly improved the cytotoxic activity of the compound as indicated for compound 7 with IC50 of 8.6 μM. This finding indicates that the modification of 2-phenyl-quinoxaline can be extended for further investigations. Similar to compound 4a, acylation of the hydroxyl group at position 2 in the phenyl moiety of compound 7 substantially reduced the anti-proliferative activity of compound (compound 8, IC50 = 17.3 μM). These results further affirm the critical role of the OH-group in the activity of 2-phenyl-substituted quinoxaline scaffold. On the other hand, the evaluation of 2-(8-methoxy-coumarin-3-yl)-quinoxaline analogues (3b, 4b, and 5) revealed that these compounds possess a potential anti-proliferative activity. Our assessments indicated that compound 3b with 2-(8-methoxy-coumarin-3-yl)-moiety display a substantial anti-proliferative activity with IC50 of 1.85 μM. The observed cytotoxic activity of compound 3b was significantly attenuated upon conversion of quinoxaline moiety to 1,4-dihydroquinoxaline, as indicated in compound 4b with IC50 of 4.08 μM. These results support the crucial role of the quinoxaline ring and further indicate that the two-NH groups play a significant role in the binding of this class of compounds toward the target protein(s). Finally, bromination of the 8-methoxy-coumarin moiety significantly impaired the anti-proliferative activity of the 2-(8-methoxy-coumarin)-quinoxaline scaffold (compound 5, IC50 = 55.0 μM).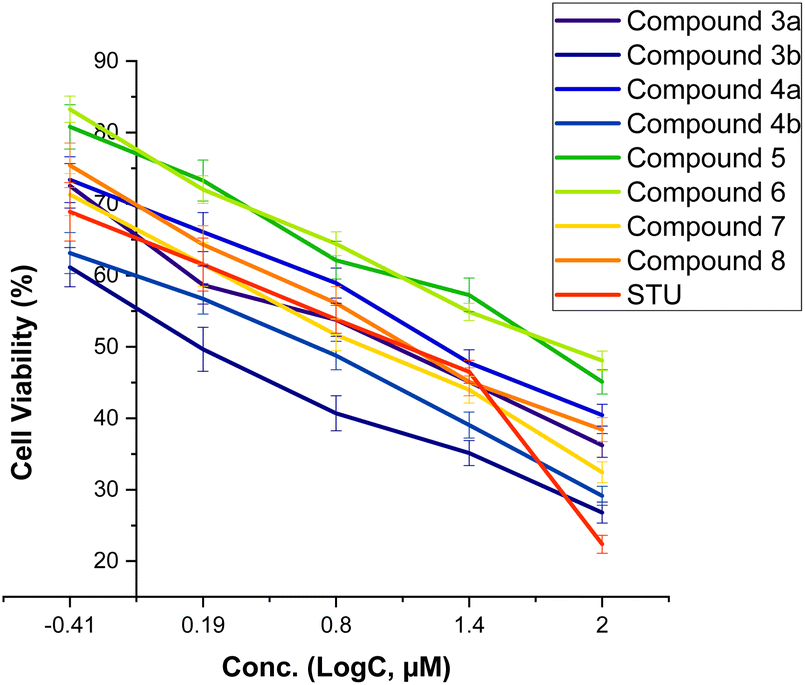 | ||
| Fig. 3 The dose-dependent effect of compounds (3–8) on the cell viability of MCF-7 cells. | ||
Among screened compounds, compound 3b exhibited the most potent inhibitory activity toward the growth of MCF7 cells with IC50 value of 1.85 ± 0.11 μM (Fig. 4). To provide a contextual framework, we conducted a comparative analysis between the activity of compound 3b and the anticancer drug staurosporine. STU demonstrated an IC50 value of 6.77 ± 0.41 μM on the proliferation MCF-7 cells. The observed difference in IC50 values suggests that compound 3b is more effective in inhibiting the proliferation of MCF-7 cells compared to the reference drug STU. Encouraged by these findings, we redirected our research efforts towards a more comprehensive investigation of the anti-proliferative effects of compound 3b on normal breast cells (MCF-10A). The evaluation of the selectivity of possible anticancer drugs is crucial to minimize potential harm to healthy cells. The results of our study suggest that compound 3b exerts minimal cytotoxicity on MCF-10A cells (IC50 = 33.7 ± 2.04 μM) compared to the widely recognized anticancer drug staurosporine (IC50 = 26.7 ± 1.62 μM). These results indicate that compound 3b exhibits a comparable level of selectivity towards cancer cells relative to staurosporine. Collectively, our research underscores the promise of the presented 2-substituted-quinoxaline analogues promising scaffold for the development of innovative anticancer agents for breast cancer. The potent inhibitory impact of compound 3b on MCF-7 cells, coupled with its relatively lower toxicity towards normal MCF-10A cells, underscores its promising potential as a foundational structure for the advancement of breast cancer therapeutics (Fig. 5).
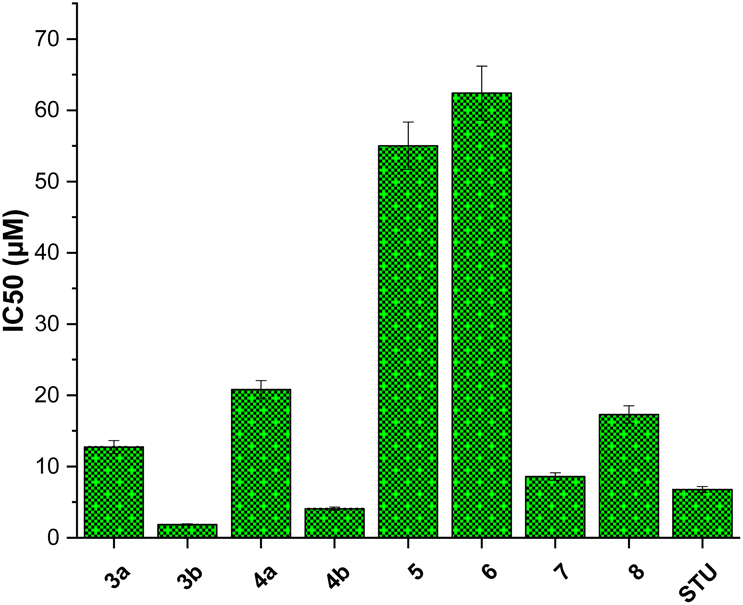 | ||
| Fig. 4 The inhibitory concentration values (IC50) of synthesized 2-substituted-quinoxaline analogues toward cellular viability of MCF-7 cells. | ||
 | ||
| Fig. 5 Impact of compound 3b (1.85 μM) on the cell cycle of MCF7 cells. Data in histogram format illustrating the distribution of cell cycles in both treated and untreated MCF-7 cells (A and B). (C) Presentation of DNA content percentages in distinct phases of the cell cycle for both untreated and compound 3b-treated MCF7 cells. | ||
Assessment of cell cycle arrest
Encouraged by the anti-proliferative activity of synthesized compounds, our research efforts were directed towards assessing the impact of compound 3b on the cellular progression through the cell cycle of MCF-7 cells, using flow cytometric techniques. The cells were subjected to compound 3b at a concentration equivalent to the IC50 value. Subsequently, an analysis of the cell cycle distribution was performed, and the obtained findings were compared to those of untreated control cells. As depicted in Fig. 6, our findings demonstrated a significant alteration in the cell cycle distribution following exposure to compound 3b. Treatment with compound 3b led to a noticeable shift in the cell population within the G0/G1 and S phases. Specifically, the percentage of cells in the G0/G1 phase decreased from the initial 55.46% to a final 46.36%. In contrast, the proportion of cells in the S phase increased from 32.89% to 47.12% after treatment. On the other hand, a substantial decrease in the cell population was observed at the G2/M phase, while a remarkable increase was seen in the pre-G1 phases. The data indicates a significant reduction in the percentage of cells in the G2/M phase, declining from 11.65% to 6.52%. Conversely, there was a substantial surge in the percentage of cells in the pre-G1 phase, rising dramatically from 2.02% to 42.39%. Our findings suggest that the administration of compound 3b leads to a substantial decrease in the proportion of cells in the G0/G1 and G1/S phases, implying that it slows down cellular proliferation, especially during these phases. The G0/G1 phase, also known as the first gap phase,67 is a crucial stage in the cell cycle68 during which cells undergo a period of rest69 and preparation for subsequent events such as DNA replication and cell division. It seems that the influence of compound 3b on the S phase, where DNA replication occurs, is relatively limited. Furthermore, it is worth noting that there has been a significant reduction in the number of cells in the G2/M phase, which indicates that compound 3b does not induce cell cycle arrest specifically at the G2/M checkpoint. The cell's meticulous oversight of DNA replication's completion and accuracy prior to entering mitosis represents a pivotal control point.70 However, compound 3b exhibits a distinct mechanism of action as an anticancer agent. The decline in cellular population within the G2/M phase, as observed, suggests that compound 3b exhibits inhibitory effects on cell cycle progression beyond this particular stage. This phenomenon may be attributed to the compound's potential influence on proteins associated with G2/M transition or the formation of the mitotic spindle.71 The most notable observation entailed a significant increase in the pre-G1 population subsequent to the administration of compound 3b. The pre-G1 phase, also known as the gap 0 phase, has been observed to be closely linked with the occurrence of apoptosis, a highly regulated and orchestrated process of programmed cell death.72 The observed substantial augmentation in the population of cells residing in the pre-G1 phase of the cell cycle strongly implies that the administration of compound 3b exerts a notable influence on the induction of programmed cell death in MCF-7 cells. These findings suggest that compound 3b induces the initiation of apoptotic signaling cascades, resulting in the fragmentation and degradation of cellular DNA, ultimately culminating in cellular demise. Overall, the presented flow cytometric analysis demonstrates that compound 3b exhibits a significant impact on cell growth, specifically by inducing cell cycle arrest, through its ability to alter the proportion of cells arrested in the G0-G1, S phase, G1/S and pre-G1 phases.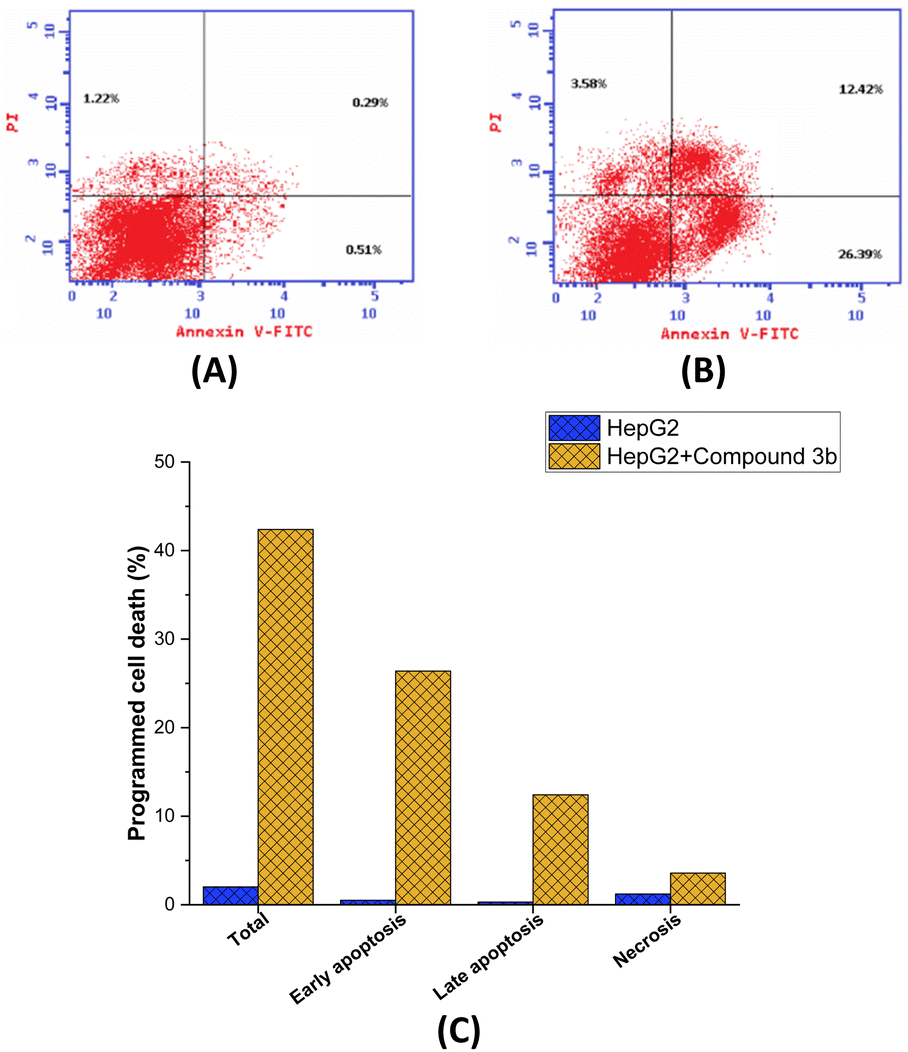 | ||
| Fig. 6 Induction of programmed cell death in MCF7 cells by compound 3b treatment. (A) Flow cytometric analysis of MCF7 cells (B) flow cytometric analysis of compound 3b-treated MCF7 cells (1.85 μM) (C) graphical analysis of the programmed cell death in MCF-7 cells (untreated and treated). | ||
Assessment of programmed cell death
Subsequently, an investigation was carried out utilizing Annexin V-FITC/PI screening methodology in order to delve deeper into the effects of compound 3b on the proliferation of MCF-7 cells and to comprehensively assess the occurrence of programmed cell death. The Annexin V-FITC/PI assay is a well-established and commonly employed technique in the field of cell biology.73 It serves as a valuable tool for distinguishing between apoptotic74 and necrotic75 in cells by assessing their respective membrane integrity and phospholipid exposure. As presented in Fig. 6, it is evident that the administration of compound 3b led to a significant augmentation in the overall proportion of programmed cell death observed in MCF-7 cells. After being exposed to compound 3b, cells saw a dramatic 21-fold increase in programmed cell death, from 2.02% in untreated cells to 42.39% in treated cell. The results also showed that compound 3b significantly increased the proportion of apoptosis in MCF-7 cells during both the early and late periods. Treatment with compound 3b resulted in a 51.7-fold increase in the proportion of cells experiencing early apoptosis, from 0.51% to 26.39% (control untreated to treated cells, respectively). The proportion of cells undergoing late-stage apoptosis also increased dramatically following treatment with compound 3b, elevating from 0.29% in the control cells to 12.42% in compound 3b-treated cells. However, compound 3b did not significantly affect the necrosis pathway in MCF-7 cells. After treatment with compound 3b, the proportion of cells suffering necrosis considerably increased from 1.2% in untreated cells to 3.58%. The mechanism by which compound 3b induces cell death in MCF-7 cells has been elucidated to a greater extent through the insights gained from the Annexin V FITC/PI screening. Compound 3b effectively promoted cell death in MCF-7 cells, as shown by a significant increase in the overall percentage of programmed cell death after treatment. The fact that compound 3b significantly upregulated early and late apoptosis shows that it activates the apoptotic pathway at various stages, ultimately resulting in cell death. Notably, compound 3b seems to promote apoptosis rather than necrosis in MCF-7 cells, since necrosis was not significantly affected. Overall, these findings suggest that the observed anti-proliferative activity of compound 3b in MCF7 cells may be ascribed to its significant induction of the apoptotic pathway within these cells.Assessment of DNA topoisomerase II activity
The enzyme family topoisomerase II, commonly known as DNA gyrase,76 plays a crucial role in preserving the three-dimensional structure of DNA.77 Their capacity to control DNA supercoiling,78 unlink DNA strands,79 and resolve DNA tangles80 is crucial to a number of cellular processes, including replication, transcription, and recombination. Two isoforms of topoisomerase II, topo II and topo II, have been identified.81 The N-terminal domain of topoisomerase is the DNA-binding region, which finds and binds to target sequences of DNA.82 The central catalytic domain is the enzyme's active site, where cleavage and re-ligation processes occur.14 The C-terminal domain contains areas important in dimerization (creating a functioning enzyme complex) and interactions with other proteins,83 while the N-terminal domain contains a conserved tyrosine residue that forms a covalent intermediate with the DNA during the cleavage process. The presence of this domain adds to the enzyme's interactions with other cellular components and helps stabilize the enzyme's overall structure.84 Toward this, we aimed to explore whether the anti-proliferative activity of compound 3b is associated to its ability to target topoisomerase II enzyme in MCF-7 cells. As depicted in Fig. 7, compound 3b exhibited substantial inhibitory activity against the topo II enzyme, demonstrating a 6.5-fold reduction in comparison to the untreated control cells. This suggests that compound 3b interferes with the enzyme's ability to alleviate DNA supercoiling or facilitate strand breakage and re-ligation. Consequently, this interference could disrupt DNA replication, transcription, and repair processes, potentially leading to cell cycle arrest and eventual cell death. These findings indicate that the anti-proliferative activity of compound 3b toward MCF-7 cells could be linked to its ability to target topoisomerase II enzyme.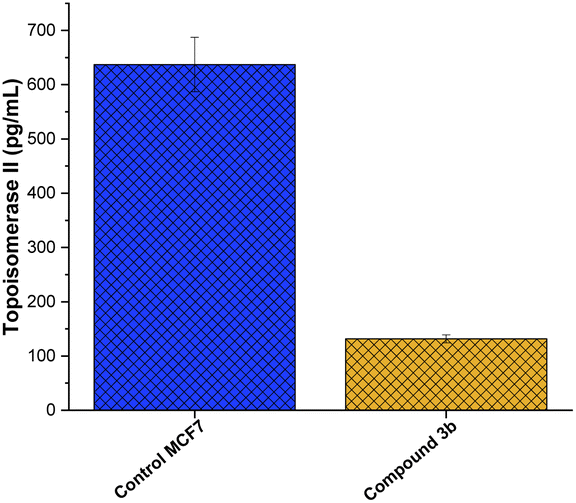 | ||
| Fig. 7 Inhibitory activity of compound 3b against topoisomerase II enzyme activity in MCF7 cells. | ||
Molecular docking analysis
Oncology research often aims to discover new molecular predictors for prognosis and treatment response.85 This shift will transition cancer therapy from non-targeted to targeted agents based on patients' tumor molecular characteristics. EGFR, also known as epidermal growth factor receptor, is a type of receptor protein categorized under the family of tyrosine.86 Tyrosine kinases are enzymes with the essential function of transmitting signals within cells by adding phosphate groups to specific tyrosine residues on target proteins. The activation of EGFR, triggered by binding with ligands, initiates a cascade of cellular reactions, including processes like cell growth, division, and differentiation.87 This receptor plays a critical role in various biological activities, both normal and pathological, such as cancer. In cancer therapy, a significant approach involves targeting the tyrosine kinase activity of EGFR to disrupt abnormal signaling pathways and hinder the proliferation of tumors. EGFR family members like EGFR, HER2, ErbB3, and ErbB4 are key cancer molecular targets.86 EGFR overexpression in breast cancer correlates with large tumor size, poor differentiation, and unfavorable outcomes, particularly in aggressive subtypes like triple-negative and inflammatory breast cancer. Considering the background literature,88 we carried out molecular docking simulations for the current set of compounds using the Glide module from Schrödinger, LLC, NY, 2023. The binding pocket for our synthesized compounds displayed analogous amino acid residues as seen from internal ligand. Fig. 8 depicts the overlay of the optimal docked candidate, 3b, with a docking score of −11.49 kcal mol−1 against the selected target (PDB ID: 1M17). The 3b (1,2-dihydro isomer) engaged with specific amino acid residues include Leu 694 (π-sigma), Leu 768 (alkyl), Leu 820 (alkyl), Ala 179, Lys 721 (alkyl or π-alkyl interactions), Met 769 and Thr 766 (conventional H-bonding), Pro 770 and Thr 830 (carbon hydrogen bond and Pi-donor H-bond, respectively). Met 742 showed π–sulfur interaction. These interactions encompassed diverse types such as hydrogen bonding, hydrophobic, and positively charged interactions, as detailed in Table 1. Conversely, the 3b (1,4-dihydro isomer) (docking score: −10.73 kcal mol−1), displayed considerable interactions (Fig. S10†). Additionally, Fig. 9 portrays the binding pocket for the selected standard, staurosporine (STU), which attained a docking score of −9.62 kcal mol−1. The native co-crystallized ligand, AQ4 exhibited only one H-bonding interaction with Met 769 amino acid. Other residues were Ala 719, Lys 764 and Lys 721 (alkyl typed), Leu 820 and Leu 964 (Pi-Sigma). Nevertheless, we achieved higher affinity of 3b towards the chosen target than the co-crystallized ligand, AQ4 as depicted in Fig. 8. | ||
| Fig. 8 Descriptive 2D and 3D binding modes* of AQ4, co-crystallized ligand (A and B) and compound 3b (C and D) inside the pocket of the kinase domain from the epidermal growth factor receptor (EGFRK) (PDB code: 1M17). *This figure was created by using Discovery Studio 4.0 Client (https://discover.3ds.com/discovery-studio-visualizer-download). | ||
| Residues | Interaction/residue type | Distance (Å) |
|---|---|---|
| Leu 694 | π-sigma | 3.98 |
| Leu 768, Leu 820 | Alkyl | 5.12, 4.76 |
| Ala 719, Lys 721 | π-alkyl | 4.85, 5.19 |
| Pro 770, Thr 830 | Hydrogen bond | 2.51, 3.44 |
| Met 742 | Pi-sulfur | 5.53 |
| Met 769 | Conventional H-bond | 2.98 |
| Thr 766 | Conventional H-bond | 2.84 |
 | ||
| Fig. 9 (A) Compound staurosporine (STU) (green) (standard) superimposed in the active site pocket (PDB ID: 1M17)*(3D); (B) 2D predicted binding mode of the compound STU in the active site (H-bond: green, hydrophobic interaction: pink). *This figure was created by using Discovery Studio 4.0 Client (https://discover.3ds.com/discovery-studio-visualizer-download). | ||
Furthermore, the evaluation of the compounds' impact on inhibiting human topoisomerase II (PDB ID: 5GWK) was conducted, with 3b (1,2 dihydro isomer) consistently emerging as the top docked candidate, achieving a docking score of −13.72 kcal mol−1. The alignment of the best docked 3b is depicted in Fig. 10. Remarkably, 3b maintained a notably higher docking score of −13.72 kcal mol−1 in contrast to the standard etoposide's score of −9.61 kcal mol−1. Noteworthy, interactions were observed, including conventional H-bonding interaction with Asp 543 amino acid residue. Amino acid residues Lys 489 and Glu 461 yielded alkyl interactions and carbon hydrogen bond interactions, respectively as detailed in Table 2. Further, the 3b (1,4-dihydro isomer) exhibited considerable interactions similar to that of 1,2-dihydro isomer (Fig. S11†) Fig. 10.
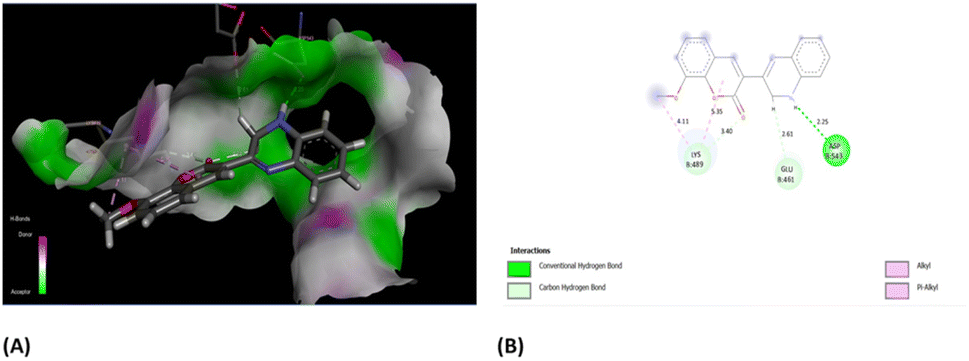 | ||
| Fig. 10 (A) Compound 3b (1,2-dihydro isomer) superimposed in the active site pocket (PDB ID: 5GWK)*(3D); (B) 2D predicted binding mode of the compound 3b in the active site. *This figure was created by using Discovery Studio 4.0 Client (https://discover.3ds.com/discovery-studio-visualizer-download). | ||
| Residues | Interaction/residue type | Distance (Å) |
|---|---|---|
| Glu 461 | Carbon hydrogen bond | 2.61 |
| Lys 489 | Alkyl | 4.11, 5.35 |
| Asp 463 | Conventional H-bond | 2.25 |
In order to have better insights from binding scores of native co-crystalized ligands, i.e., (EVP) and obtained best docked molecules, 3b, we carried out molecular docking of EVP on human topoisomerase II (PDB ID: 5GWK) (Fig. 11(A)). It was found that docking score of EVP was −12.58 kcal mol−1, which was lower than that of 3b. Furthermore, EVP interacted with similar amino acids as of 3b such as Asp 463, Gly 462, and Met 766. These findings strongly suggest the potential of compound 3b as an inhibitor for both EGFR and human topoisomerase II, underscoring the need for further exploration through in vivo experiments.
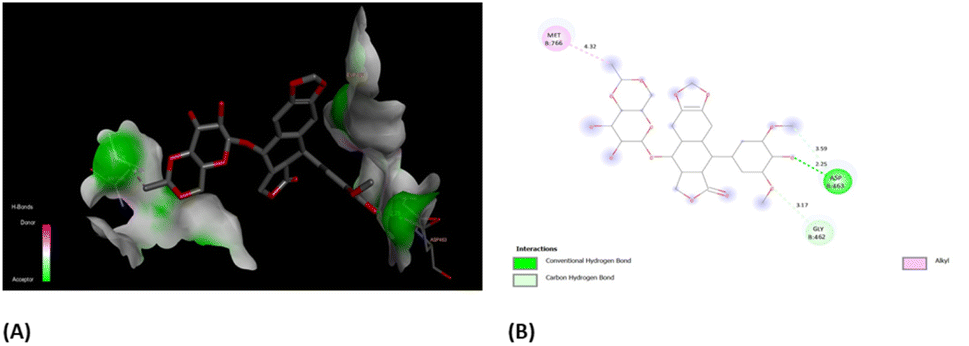 | ||
| Fig. 11 (A) 3D- and (B) 2D-binding pocket for the co-crystallized ligand, EVP within the target, human topoisomerase II (PDB ID: 5GWK). | ||
Molecular dynamics (MD) analysis
The molecular dynamics simulations (MD) were employed to evaluate the stability of the top docked ligand, 1M17_3b, in relation to the target. This analysis was conducted over a 150 ns simulation duration using the software ‘Desmond’ by Schrödinger, LLC, New York, 2023. The comprehensive MD system comprised 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 246 atoms, including 9896 water molecules, as illustrated in Fig. 12(a)–(f). In these simulations, we gauged the average displacements of atoms relative to a specific timeframe using the ‘RMSD’ (Root Mean Square Deviation) parameter. Our examination of the RMSD parameter disclosed a consistently stable conformation. When the ligand 3b (1,2-dihydro isomer) was bound to the target 1M17, the Cα-RMSD backbone RMSD values were consistently under 3.5 Å, and ‘Lig_fit_Prot’ values were maintained below 0.5 Å, as depicted in Fig. 12a. Throughout the 0–150 ns period, the entire complex exhibited enduring stability. Moreover, localized fluctuations within the protein chains were scrutinized through the ‘RMSF’ (Root Mean Square Fluctuation) plot, detailed in Fig. 12b. While there were minor spikes in fluctuation for specific amino acids, these variations didn't lead to significant changes, indicating their inherent flexibility. The analysis of the ‘protein–ligand’ interaction plot (Fig. 12c) revealed crucial interactions, including hydrophobic interactions with amino acids Leu 694, Phe 699, Lys 721, Met 742, Leu 764, Cys 773, and Leu 820. No ionic interactions were observed, but there were hydrogen bonds with Thr 830 and Asp 831, along with water bridges involving residues like Lys 721, Leu 694, Glu 738, Cys 751, Thr 766, Gln 767, Met 769, Thr 830, and Phe 832. Fig. 12f presented a timeline representation of these interactions with amino acid residues throughout the 150 ns simulation period. In Fig. 12e, the ‘ligand–protein’ contact plot highlighted interactions occurring more than 5.0% of the simulation time (Met 769: 31%, Lys 721: 12%, Glu 738: 52%, Asp 831: 8%, Thr 830: 28%, Thr 766: 23%, and Phe 699: 28%) within the chosen trajectory (0.00–150.00 ns). The ‘ligand torsions plot’ (Fig. 12d) summarized the conformational changes of each rotatable bond (RB) in the ligand during the simulation trajectory (0.00–150.00 ns). These results indicate that the ligand–protein conformation remained stable throughout the 150 ns simulation period. Additionally, we extended our analysis to explore the stability of the 1M17_3b (1,4-dihydro isomer), in relation to the target. This analysis was conducted over a 150 ns simulation duration. The comprehensive MD system comprised 34246 atoms, including 9896 water molecules, as illustrated in Fig. S12(a)–(f).†
246 atoms, including 9896 water molecules, as illustrated in Fig. 12(a)–(f). In these simulations, we gauged the average displacements of atoms relative to a specific timeframe using the ‘RMSD’ (Root Mean Square Deviation) parameter. Our examination of the RMSD parameter disclosed a consistently stable conformation. When the ligand 3b (1,2-dihydro isomer) was bound to the target 1M17, the Cα-RMSD backbone RMSD values were consistently under 3.5 Å, and ‘Lig_fit_Prot’ values were maintained below 0.5 Å, as depicted in Fig. 12a. Throughout the 0–150 ns period, the entire complex exhibited enduring stability. Moreover, localized fluctuations within the protein chains were scrutinized through the ‘RMSF’ (Root Mean Square Fluctuation) plot, detailed in Fig. 12b. While there were minor spikes in fluctuation for specific amino acids, these variations didn't lead to significant changes, indicating their inherent flexibility. The analysis of the ‘protein–ligand’ interaction plot (Fig. 12c) revealed crucial interactions, including hydrophobic interactions with amino acids Leu 694, Phe 699, Lys 721, Met 742, Leu 764, Cys 773, and Leu 820. No ionic interactions were observed, but there were hydrogen bonds with Thr 830 and Asp 831, along with water bridges involving residues like Lys 721, Leu 694, Glu 738, Cys 751, Thr 766, Gln 767, Met 769, Thr 830, and Phe 832. Fig. 12f presented a timeline representation of these interactions with amino acid residues throughout the 150 ns simulation period. In Fig. 12e, the ‘ligand–protein’ contact plot highlighted interactions occurring more than 5.0% of the simulation time (Met 769: 31%, Lys 721: 12%, Glu 738: 52%, Asp 831: 8%, Thr 830: 28%, Thr 766: 23%, and Phe 699: 28%) within the chosen trajectory (0.00–150.00 ns). The ‘ligand torsions plot’ (Fig. 12d) summarized the conformational changes of each rotatable bond (RB) in the ligand during the simulation trajectory (0.00–150.00 ns). These results indicate that the ligand–protein conformation remained stable throughout the 150 ns simulation period. Additionally, we extended our analysis to explore the stability of the 1M17_3b (1,4-dihydro isomer), in relation to the target. This analysis was conducted over a 150 ns simulation duration. The comprehensive MD system comprised 34246 atoms, including 9896 water molecules, as illustrated in Fig. S12(a)–(f).†
 | ||
| Fig. 12 MD simulation analysis for complex 1M17_3b (1,2-dihydro isomer) (a) RMSD plot; (b) RMSF analysis; (c) ligand–protein contact plot; (d) ligand torsion profile; (e) protein–ligand interaction plot; and (f) a timeline representation plot representing such interactions with amino acid residues over the simulation period of 150 ns. | ||
Taken together, these findings present compelling evidence for the therapeutic potential of the newly developed class of quinoxaline analogs. These analogs, including compound 3b, exhibited considerable anti-cancer properties against MCF-7 cells, surpassing established drugs like staurosporine with lower toxicity to normal cells. Compound 3b demonstrated its ability to significantly arrest the cell cycle, induce apoptosis, and inhibit topoisomerase II activity, disrupting critical processes in cancer cells. Molecular modeling and molecular simulation assessments showed its affinity for EGFR, strengthening its potential as a breast cancer treatment (Fig. 13). While promising, further research, including in vivo trials, is needed. Nonetheless, this study lays the groundwork for a novel and effective class of breast cancer treatments, offering insights into their mechanisms of action for future drug development.
 | ||
| Fig. 13 Summary of the anti-proliferative activity of compound 3b toward MCF7 cells. | ||
Experimental
Instruments and analytical techniques
The acquisition of the 1H-NMR and 13C-NMR spectra was performed utilizing a Bruker Avance Av-400 instrument. The chemical shifts observed in the spectra were meticulously recorded in parts per million (ppm) relative to the standard compound tetramethyl silane (TMS), a common reference point in the field of nuclear magnetic resonance (NMR) spectroscopy. The IR data was acquired utilizing the Shimadzu 470 spectrometer. The compounds underwent analysis utilizing an electron ionization (ET) mass spectrometer, specifically employing a probe Agilent MSD 5975 spectrometer. The instrument was configured to operate at an energy level of 70 electron volts (eV). The determination of the melting point was conducted utilizing an electrothermal 200 digital melting point apparatus, with the obtained results not being adjusted for potential temperature fluctuations. The elemental analysis was conducted utilizing an Aferkinelmer 2400 series CHN elemental analyzer. With the exception of any explicitly stated conditions, all chemicals were procured from a commercial supplier and utilized without undergoing subsequent purification procedures.89Synthetic procedure
2-(5-Bromo-2-hydroxyphenyl)-3,4-dihydroquinoxaline (3a). A orange crystals, yield 56%, m.p. 210 °C. IR (kBr) γmax = 3461 (br-OH), 3221 (NH), 1625 (C
N), 1605, 1588 (CC), 1046 (CO) cm−1. 1H-NMR (DMSO-d6, ppm, 400 MHz) δ: 4.40 (t, 2H, NCH2), 7.02–8.41 (m, 9H, Ar–H, NH, and H-3 of quinoxaline of two isomers), 9.68, 9.72 (s, 2H, 2*NH of 1,4-dihydroquinoxaline), 12.18, 12.45 (s, 1H, OH of two isomers). 13C-NMR (DMSO-d6, ppm, 100 MHz) δ: 159.04, 157.61, 152.26, 150.55, 145.82, 145.42, 141.04, 140.80, 140.19, 139.63, 136.77, 135.07, 140.19, 139.63, 136.77, 135.07, 133.49, 132.06, 131.92, 131.41, 131.37, 130.74, 130.42, 129.63, 129.33, 128.95, 128.59, 123.02, 120.08, 119.97, 118.02, 111.26, 56.50. MS analysis (m/z, %): 302 (100), 299 (43.17), 274 (6.68), 221 (31.52), 193 (29.27), 166 (5.73), 118 (2.87), 111 (2.80), 102 (4.56), 96 (4.02), 77 (6.35), 63 (2.98). Anal. calcd. for C14H11BrN2O (Mwt = 302): C, 55.63; H, 3.64; N, 9.27. Found: C, 55.48; H, 3.46; N, 9.09.
2-(8-Methoxy-coumarin-3yl)-3,4-dihydroquinoxaline (3b). A yellow crystals, yield 61%, m.p. 235 °C. IR (kBr) γmax = 3225 (NH), 1729 (C
O), 1631 (CN), 1605, 1589 (CC), 1172, 1083 (C–O) cm−1. 1H NMR (DMSO-d6, ppm, 400 MHz) δ: 3.95, 3.97 (s, 3H, OCH3 of two isomers), 4.02 (s, 2H, NCH2 of quinoxaline ring), 6.43–8.19 (m, 9H, Ar–H, H-3 of quinoxaline and NH), 8.89 (s, 1H, H-4 of coumarin ring), 9.60 (s, 1H, NH), 11.41 (s, 1H, NH). 13C-NMR (DMSO-d6, ppm, 100 MHz) δ: 159.84 (CO of coumarin ring), 148.22, 147.75, 147.31, 146.83, 146.70, 145.83, 145.27, 143.60, 141.89, 141.72, 141.13, 131.26, 131.15, 129.57, 129.35, 125.42, 125.32, 125.15, 124.16, 123.84, 122.12, 121.26, 120.10, 119.15, 118.89, 116.87, 115.82, 114.32, 112.59 (carbons of quinoxaline and coumarin rings of two isomers), 56.67, 56.57 (OCH3 of two isomers), 30.51 (NHCH2). MS analysis (m/z, %); 306 (16.30), 304 (100), 276 (16.95), 261 (7.32), 219 (9.08), 205 (12.88), 177 (10.56), 159 (3.55), 152 (8.82), 119 (3.83), 109 (3.04), 102 (17.32), 76 (8.40). Anal. calcd. for C18H14N2O3 (Mwt = 306): C, 70.59; H, 4.57; N, 9.15. Found: C, 70.33; H, 4.27; N, 9.01.
2-(2-Acetoxy-5-bromo) phenyl quinoxaline (4a). A pale orang crystal, yield 73%, m.p. 187 °C. IR (kBr) γmax = 1763 (C
O), 1632 (CN), 1608, 1584 (CC), 1113, 1066 (C–O) cm−1. 1H-NMR (DMSO-d6, ppm, 400 MHz) δ: 2.22 (s, 3H, COCH3), 7.37–7.39 (d, 1H, ArH), 7.83–7.96 (m, 3H, Ar–H), 8.15–8.19 (m, 3H, Ar–H), 9.26 (s, 1H, CHN of quinoxaline). 13C-NMR (DMSO-d6, ppm, 100 MHz) δ: 169.48 (CO), 149.51, 148.16, 145.74, 141.72, 141.28, 134.12, 133.84, 132.30, 131.40, 131.26, 129.74, 129.39, 126.71, 119.31 (C-aromatic and quinoxaline ring), 21.19 (CH3). MS analysis (m/z, %); 342 (2.50), 315 (3, 50), 302 (83.21), 300 (100), 272 (5.28), 221 (14,69), 193 (20.32), 164 (6.69), 102 (6.77). Anal. calcd. for C16H11BrN2O2 (Mwt = 342): C, 56.14; H, 3.21; N, 8.19. Found: C, 55.89; H, 3.01; N, 8.03.
2-(8-Methoxycoumarin-3-yl)-quinoxaline (4b). As pale-yellow crystal, yield 69%, m.p. 168 °C. IR (kBr) γmax = 1731 (C
O), 1633 (CN), 1609, 1592 (CC), 1213, 1073(C–O) cm−1. 1H-NMR (DMSO-d6, ppm, 400 MHz) δ: 3.92, 3.97 (s, 3H, OCH3 of two isomers), 6.56–6.18 (m, 7H, ArH), 8.89 (s, 1H, H-4 of coumarin), 9.60 (s, 1H, CHN of quinoxaline). 13C NMR (DMSO-d6, ppm, 100 MHz) δ: 159.84 (CO), 148.75, 148.23, 146.82, 145.83, 145.27, 143.60, 141.88, 141.72, 131.25, 131.15, 129.56, 129.35, 125.42, 124.17, 121.26, 120.11, 119.18, 118.02, 115.81, 114.34 (carbons of coumarin and quinoxaline rings), 56.67, 56.16, 21.19 (OCH3 of two stereo isomers). MS analysis (m/z, %); 304 (100), 276 (6.68), 240 (20.35), 205 (8.71), 191 (11.00), 153 (6.29), 144 (4.44), 132 (5.30), 102 (18.82), 76 (21.46), 50 (8.34). Anal. calcd. for C18H12N2O3 (Mwt = 304): C, 71.5; H, 3.95; N, 9.21. Found: C, 70.91; H, 3.66; N, 9.03.
Synthesis of 2-(5-bromo-8-methoxy-coumarin-3-yl)-quinoxaline (5). Compound 4b (0.01 mole) was dissolved in 20 mL of glacial acetic acid at ambient temperature. Subsequently, a solution containing 10 mL of bromine (equivalent to 0.01 moles) dissolved in glacial acetic acid was meticulously introduced in a dropwise manner to compound 4b, while maintaining a constant stirring rate within the temperature range of 40–60 °C. Following a time interval of 5–10 minutes, the characteristic hue of bromine was observed to dissipate, leaving behind a solution of a yellow nature. Subsequently, a volume of 1 mL of bromine solution was meticulously introduced into the experimental setup, while ensuring continuous agitation, and maintained at ambient temperature for a duration of 8 h. The resultant reaction mixture was carefully introduced into a ice-water, while being subjected to continuous agitation. Subsequently, the resultant solid precipitate was carefully collected through the process of filtration, followed by thorough rinsing with water and subsequent desiccation to achieve complete dryness. After a series of experimental procedures, the product underwent a crystallization process utilizing ethanol as the solvent, resulting in the formation of compound 5 in the form of vibrant orange crystals. The overall yield of this crystallization process was determined to be 82%. The melting point of compound 5 was 205 °C. IR (kBr) γmax = 1735 (C
O), 1632 (CN), 1608, 1587 (CC), 1121, 1064 (C–O) cm−1. 1H-NMR (DMSO-d6, ppm, 400 MHz) δ: 3.96, 4.12 (s, 3H, OCH3 of two isotopes of compound 5), 6.56–8.23 (m, 6H, ArH), 8.77, 8.87 (s, 1H, H-4 of coumarin), 9.61, 9.64 (s, 1H, CHN of quinoxaline of two isomers). 13C-NMR (DMSO-d6, ppm, 100 MHz) δ: 164.48 (CO), 163.30, 163.16, 155.18, 149.58, 147.37, 146.16, 145.90, 143.80, 141.90, 131.40, 129.35, 128.79, 116.35, 115.79 (carbons of coumarin and quinoxaline rings), 56.97, 56.70 (OCH3 of the two isomers). MS analysis (m/z, %); 384 (79.35), 383 (22.62), 382 (100), 354 (10.33), 311 (14.81), 303 (35.53), 285 (12.42), 275 (68.95), 247 (22.64), 232 (17.21), 219 (16.10), 204 (37.93), 191 (14.07), 177 (33.79), 153 (17.36), 144 (9.75), 129 (19.84), 116 (4.19), 102 (41.41), 96 (29.29), 82 (100), 80 (95.78), 79 (44.58), 76 (31.54). Anal. calcd. for C18H11BrN2O3 (Mwt = 382): C, 56.54; H, 2.88; N, 7.33. Found: C, 56.25; H, 2.59; N, 7.11.
Synthesis of ethyl [3-(5-bromo-2-hydroxy phenyl)-1,4-dihydroquinoxalin-1-yl] acetate (6). A solution of compound 3a (0.01 mole) and ethyl chloroacetate (0.01 mole) in dimethyl formamide (25 mL) was subjected to reflux for a duration of 12 hours in the presence of triethylamine (2 mL). The reaction mixture underwent a cooling process, followed by its transfer into a solution of ice-water. Subsequently, the mixture was subjected to neutralization using diluted aqueous hydrochloric acid (2%) under continuous stirring. The solid obtained from the experimental procedure was effectively isolated through the process of filtration and subsequently subjected to recrystallization using ethanol as the solvent. This recrystallization step yielded pale-yellow crystals (68%). The melting point of compound 6 was found to be 169 °C. IR (kBr) γmax = 3465 (br.OH), 3212 (NH), 1763 (C
O), 1605, 1587 (CC), 1121, 1093 (C–O) cm−1. 1H NMR (DMSO-d6, ppm, 400 MHz) δ: 1.22 (t, 3H, CH3), 4.20 (q, 2H, OCH2), 5.02 (s, 2H, NCH2CO), 7.03–8.31 (m, 8H, Ar–H and H-3 of quinoxaline of two stereo isomers), 9.53, 9.69 (s, 1H, NH of two isomers), 12.19 (s, 1H, OH). 13C-NMR (DMSO-d6, ppm, 100 MHz) δ: 168.79 (CO), 159.06, 157.63, 155.30, 150.84, 150.32, 147.30, 145.80, 142.23, 141.14, 141.05, 140.17, 135.06, 134.35, 133.87, 132.04, 131.36, 130.91, 130.78, 129.74, 129.30, 129.25, 128.04, 123.00, 120.09, 115.98, 113.84, 111.26 (carbons of aromatic and quinoxaline rings), 65.83 (NCH2CO), 61.38 (OCH2), 14.47 (CH3). Anal. calcd. for C18H17BrN2O3 (Mwt = 388): C, 55.67; H, 4.38; N, 7.22. Found: C, 55.44; H, 4.11; N, 7.02.
Synthesis of 2-(5-bromo-3-phenylazo-2-hydroxyphenyl)-1,4-dihydro quinoxaline (7). A solution of compound 3a (0.01 mole) was prepared by dissolving it in 10 mL of aqueous sodium hydroxide (10%). The resulting solution was then cooled using an ice bath to a temperature range of 0–5 °C. An aqueous solution (at −5 °C) containing the phenyl diazonium salt (0.01 mole) was meticulously introduced in a gradual manner with continuous agitation over a period of 15 minutes. Following the addition, the resulting reaction mixture was subjected to stirring for an additional duration of 30 minutes, after which it was subsequently allowed to stand at −5 °C for a period of 24 h. The resultant precipitate was meticulously collected through the process of filtration, followed by thorough washing with water. Subsequently, it was subjected to a meticulous drying procedure. In order to achieve optimal purity, the compound was further purified through a detailed crystallization process utilizing ethanol as the solvent. The purification process yielded compound 7 in the form of striking red crystals, with a yield of 71%. The compound exhibited a melting point of 242 °C. IR (kBr) γmax = 3467 (OH), 3227 (NH), 1605, 1588 (C
C), 1121, 1081 (C–O) cm−1. 1H NMR (DMSO-d6, ppm, 400 MHz) δ: 4.52 (s, 2H, NCH2), 6.47–8.30 (m, 12H, Ar–H and H-3 Of quinoxaline of two isomers), 9.68 (s, 1H, NH), 12.10 (S, 1H, OH), 14.88 (S, 1H, OH) PPM. 13C NMR (DMSO-d6, ppm, 100 MHz) δ: 157.63, 152.30, 151.21, 150.84, 145.80, 145.43, 141.05, 140.17, 135.05, 132.04, 131.35, 130.71, 129.90, 129.30, 128.93, 128.26, 122.99, 122.80, 120.09, 118.03, 114.35, 111.26 (carbon of aromatic and quinoxaline rings), 40.12 (NCH2). MS analysis (m/z, %); 406 (1.27), 404 (7.69), 377 (21.35), 326 (31.58), 313 (31.36), 300 (73.69), 271 (42.51), 237 (32.60), 221 (100), 193 (50.04), 178 (13.50), 169 (24.47), 152 (14.14), 139 (11.91), 102 (9.61), 93 (35.00), 77 (33.61). Anal. calcd. for C20H15BrN4O (Mwt = 406): C, 59.11; H, 3.69; N, 13.79. Found: C, 58.98; H, 3.33; N, 13.54.
Synthesis of 2-(2-acetoxy-3-phenylazo-5-bromo) phenyl quinoxaline (8). Compound 7 (0.01 mole) was dissolved in 25 mL of acetic anhydride. The resulting solution was subjected to reflux for a duration of 16 h, followed by a cooling process. The reaction mixture was carefully transferred into an ice-water bath under continuous agitation and allowed to incubate for a duration of 24 h at 0 °C. The resultant solid crude was separated from the solution through filtration, subsequently subjected to water rinsing, and then dried. To further refine its purity, the crude solid was further purified by re-crystallization technique using ethanol as a solvent. This process yielded compound 8 in the form of pale orange crystals, with a yield of 86%. The melting point assessed as 187 °C. IR (kBr) γmax = 1756 (C
O), 1638 (CN), 1610, 1588 (CC), 1131, 1063 (C–O) cm−1. 1H-NMR (DMSO-d6, ppm, 400 MHz) δ: 1.74, 2.21 (s, 3H, COCH3 of two isomers), 7.01–8.17 (m, 11H, Ar–H), 9.24 (s, 1H, CHN of quinoxaline), 9.77 (s, 1H, CHN of another isomer). 13C-NMR (DMSO-d6, ppm, 100 MHz) δ: 174.81, 169.41 (CO), 162.86, 159.81, 151.69, 149,49, 148.14, 146.70, 145.68, 141.72, 141.34, 141.26, 140.87, 134.51, 134.32, 133.80, 132.26, 131.42, 131.26, 130.84, 130.12, 129.72, 129.53, 129.37, 129.19, 128.51, 126.70, 124.33, 121.76, 120.85, 119.31 (carbon of aromatic and quinoxaline rings of two stereo isomers), 24.25, 21.16 (CH3 of two stereo isomers). MS analysis (m/z, %); 446 (M+, unstable), 404 (7.69%), 377 (15.60), 327 (17.78), 300 (49.93), 268 (10.03), 221 (100), 193 (40.95), 77 (14.18). Anal. calcd. for C22H15BrN4O2 (Mwt = 446); C, 59.19; H, 3.36; N, 12.56. Found: C, 58.98; H, 3.11; N, 12.22.
Assessment of anti-proliferative activity
The in vitro measurement of cytotoxic activity was conducted for the quinoxaline derivatives utilizing the MTT assay. The cells were seeded into a 96-well multiwall plate at a density of 105 cells per well and allowed to adhere for a period of 24 hours prior to the commencement of compound treatment. The test compounds underwent dissolution in dimethyl sulfoxide (DMSO). Various concentrations of the compound under investigation were introduced to the monolayer of cells. For each specific concentration, a total of three wells were meticulously prepared. The monolayer cells underwent incubation with the compound(s) for a duration of 48 hours at a temperature of 37 °C, within an atmosphere containing 5% carbon dioxide. Following a 48 hour incubation period, cellular specimens were subjected to fixation, thorough washing, and subsequent staining with a 40 μL volume of MTT solution (comprising 5 mg mL−1 of MTT in a 0.9% NaCl medium) per well. This staining process was carried out for an additional 4 hour duration under controlled incubation conditions. The MTT crystals were effectively dissolved through the addition of 180 μL of acidified isopropanol per well. The plate was then subjected to gentle agitation at ambient temperature, allowing for optimal solubilization. Subsequently, the absorbance at 570 nm was measured using an ELISA reader, enabling accurate quantification. The determination of the molar concentration necessary to achieve a 50% reduction in cell viability, commonly referred to as the IC50 value, was computed and subsequently juxtaposed with the reference pharmaceutical agent, staurosporine.90–92 The experimental procedures were conducted in triplicate to ensure robustness and reliability of the results.Assessment of cell cycle arrest
MCF-7 cells were seeded at a density of 3.0 × 105 cells per well and subsequently subjected to incubation at a temperature of 37 °C for a duration of 12 h. After cellular acquisition, the designated cellular entities were subjected to the administration of compound 3b at a concentration dose value equivalent to its IC50, followed by an incubation period of 48 h. Following the completion of the treatment, cellular specimens were gathered and subjected to fixation using a 75% ethanol solution at a temperature of 20 °C for the duration of 18 h. Subsequently, the cells underwent a thorough rinsing procedure utilizing PBS, followed by centrifugation. The cells were then exposed to a solution containing Rnase (Sigma, USA) at a concentration of 10 mg mL−1, along with propidium iodide (PI, Sigma) at a concentration of 5 mg mL−1. This preparation preceded the subsequent analysis of the cells using flow cytometry, specifically employing the FACS Calibur cytometer in conjunction with Cellquest software, both provided by BD Bioscience, USA.93,94Assessment of apoptosis and necrosis by Annexin-V assay
The MCF-7 cells were subjected to treatment with compound 3b at a concentration equivalent to its IC50 value for a duration of 48 h. Following the therapeutic intervention, cellular entities were procured and subjected to a dual washing procedure involving two cycles of centrifugation (180 g, 10 minutes, 4 °C) using phosphate-buffered saline (PBS). The suspension of each individual cell well was performed by resuspending it in 100 μL of binding buffer, followed by the addition of 5 μL of Annexin V-FITC. Following an incubation period of 10 minutes at ambient temperature, an additional 400 μL of binding buffer was introduced, resulting in a final volume of 500 μL. The cellular specimens were subjected to staining with propidium iodide (PI) in a prompt manner, just prior to the commencement of the measurement process. The cells were subjected to analysis utilizing the FACS Calibur flow cytometer, a cutting-edge instrument manufactured by Becton and Dickinson in Heidelberg, Germany. The acquired data were subjected to analysis using the Cell-Quest software, developed by Becton and Dickinson in Heidelberg, Germany.In vitro assessment of topoisomerase II activity
Compound 3b was chosen as the subject of evaluation in relation to topo II [MBS#942146], employing the utilization of human DNA topoisomerase II-β (TOPII-β) ELISA kit, in accordance with the guidelines provided by the manufacturer. All necessary reagents, working standards, and samples were appropriately prepared in accordance with established protocols and procedures. A total volume of 100 millilitres (mL) of both the standard and sample solutions were introduced into each well, followed by an incubation period of 2 h at a temperature of 37 °C. The aqueous solution was extracted from each well. Subsequently, a volume of 120 μL of biotin-antibody solution was introduced into each well, followed by an incubation period of 1 h at a temperature of 37 °C. The media was extracted utilizing an aspirator and subsequently subjected to a triple washing procedure employing a washing buffer. A volume of 100 pL of Horseradish Peroxidase (HRP-Avidin) was introduced into every well, followed by an incubation period of 1 h at a temperature of 37 °C. The repetition of the aspiration/wash process was performed a total of five cycles. A volume of 90 μL of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate was introduced into every well and subjected to an incubation period of 15–30 minutes at a temperature of 37 °C, ensuring protection from any light exposure. A volume of 50 μL of stop solution was introduced into every well, followed by the prompt measurement of the optical density of each well within a time frame of 5 minutes. This was accomplished using a ROBONEK P2000 enzyme-linked immunosorbent assay (ELISA) reader at a wavelength of 450 nm. The experimental procedures were conducted in triplicate to ensure robustness and reliability of the results.Molecular docking simulations
For molecular docking simulations, we downloaded a 3D-crystal structure of receptor EGFR tyrosine kinase (Epidermal Growth Factor Receptor tyrosine kinase) (PDB ID: 1M17; resolution: 2.60 Å) from the RCSB website.88,95 Similarly, a 3D-crystal structure of receptor human topoisomerase II was also downloaded from RCSB databank (PDB ID: 5GWK; resolution: 3.15 Å).45 We then carried out molecular docking simulations using the popular software module, Glide, Schrodinger, LLC, NY, 2023. The 3D structures of synthesized analogues (ligands) were drawn using the software ‘MarvinSketch®’ 5.8.3 (2012) and ‘ChemAxon’ tools. These structures were then imported into Schrödinger's Maestro workflow, and ligand preparation was conducted using the ‘LigPrep’ module from Schrödinger, LLC, New York (2023). During the docking process, the grid dimensions were maintained as documented in previous research. The docking calculations were performed using the ‘XP mode’ (Extra precision), with all default parameters. The most favorable docking positions were visualized using Discovery Studio 4.5 (Studio, 2008).96–101Molecular dynamics simulation (MDS) study
For the Molecular Dynamics Simulation (MDS) analysis, we conducted simulations on the complex labelled as ‘1M17_3b’ using the ‘Desmond’ module developed by Schrödinger, Inc., New York, USA (2023). In this simulation setup, we employed an explicit solvent model, which included TIP3P water molecules and the OPLS-2005 force field. The simulation was performed within a periodic boundary box with dimensions of 10 Å × 10 Å × 10 Å. To maintain a charge balance of 0.15 M, sodium ions (Na+) were introduced into the system. Additionally, NaCl solutions were incorporated to replicate physiological conditions. The simulation was carried out for a duration of 150 nanoseconds (ns). The specific details of the MD protocol were taken from a previous study conducted by Mishra et al., 2019.88Conclusions
The prevalence of breast cancer among women worldwide presents a significant global public health concern. This paper provided compelling evidence regarding the potential therapeutic effectiveness of a newly developed group of 2-substituted-quinoxaline analogs as potential anticancer agents toward breast cancer. These synthesized quinoxaline derivatives, incorporating phenyl and coumarin components, demonstrate notable antiproliferative properties against MCF-7 cells. Remarkably, compound 3b exhibited substantial inhibitory potential, with an IC50 value of 1.85 ± 0.11 μM, surpassing the inhibitory effects of the established anticancer drug staurosporine, while maintaining low toxicity towards normal cells. Furthermore, our study delved into the mechanisms responsible for the antiproliferative action of compound 3b, revealing its ability to induce cell cycle arrest at the G1/S phase and initiate apoptosis in MCF-7 cells, with minimal necrosis. Additionally, compound 3b significantly inhibited topoisomerase II activity, a crucial enzyme involved in DNA replication and repair, suggesting its capacity to disrupt essential biological processes linked to cancer cell development and survival. Moreover, molecular modeling and molecular simulation investigations affirmed the strong affinity of compound 3b for the active cavity of EGFR, a member of the tyrosine kinase receptor protein family, further supporting its potential as a promising therapeutic agent for breast cancer treatment. The data presented in this study underscores the therapeutic potential of the developed quinoxaline derivatives, particularly those incorporating phenyl and coumarin components, with a specific focus on compound 3b, in the context of breast cancer treatment. Although these findings are promising, further investigations, including in vivo trials, are imperative to validate the efficacy of compound 3b. Nevertheless, our study establishes a foundational framework for identifying a novel and highly effective class of compounds against breast cancer, while also shedding light on the underlying mechanisms of their action. These insights provide valuable guidance for future drug development endeavors.Author contributions
Conceptualization, M. G. S., S. A. A. E., S. N. M., R. A. A., E. M. S., and M. F. Y.; methodology, M. G. S., E. H. E, S. A. A. E., S. N. M., G. A., R. A. A., H. A. A., E. M. S., and M. F. Y.; software, S. N. M., E. H. E, H. A. A., H. A. A., N. A., E. A., E. M. S., and M. F. Y.; validation, S. N. M., H. A. A., G. A., R. A. A., N. A., E. A., M. F. Y.; formal analysis, M. G. S., E. H. E, S. A. A. E., S. N. M., H. A. A., N. A., E. A., and M. F. Y.; investigation, M. G. S., H. A. A., G. A., R. A. A., H. A. A., N. A., E. A., E. M. S., and M. F. Y.; resources, M. G. S., S. A. A. E., E. H. E, S. N. M., H. A. A., R. A. A., N. A., E. A., and M. F. Y.; data curation, M. G. S., S. A. A. E., E. H. E, S. N. M., G. A., R. A. A., E. M. S., and M. F. Y.; writing – original draft preparation, M. G. S., S. A. A. E., S. N. M., E. M. S., and M. F. Y.; writing – review and editing, M. G. S., S. A. A. E., E. H. E, S. N. M., H. A. A., G. A., R. A. A., H. A. A., N. A., E. A., E. M. S., and M. F. Y.; visualization, E. H. E, H. A. A., G. A., R. A. A., H. A. A., N. A., E. A., and M. F. Y.; supervision, M. G. S., S. N. M., E. M. S., and M. F. Y.; project administration, M. G. S., S. A. A. E., S. N. M., R. A. A., E. M. S., and M. F. Y.; funding acquisition, G. A., R. A. A., N. A., E. A., and E. M. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Princess Nourah Bint Abdulrahman University for funding this work through the Researchers Supporting Project number (PNURSP2023R89), Princess Nourah Bint Abdulrahman University, Riyadh, Saudi Arabia. This study was also supported by Researchers Supporting Project number (RSP2023R111), King Saud University, and Riyadh, Saudi Arabia.Notes and references
- G. N. Sharma, R. Dave, J. Sanadya, P. Sharma and K. K. Sharma, J. Adv. Pharm. Technol. Res., 2010, 1, 109–126 Search PubMed.
- R. Snijders, L. Brom, M. Theunissen and M. Van Den Beuken-van Everdingen, Cancers, 2023, 15, 591 CrossRef PubMed.
- F. Cardoso, S. Kyriakides, S. Ohno, F. Penault-Llorca, P. Poortmans, I. T. Rubio, S. Zackrisson and E. Senkus, Ann. Oncol., 2019, 30, 1194–1220 CrossRef CAS PubMed.
- K. P. McGuire, in Breast Disease, ed. A. Aydiner, A. İğci and A. Soran, Springer International Publishing, Cham, 2016, pp. 1–14 Search PubMed.
- M. E. Jones, M. J. Schoemaker, L. B. Wright, A. Ashworth and A. J. Swerdlow, Breast Cancer Res., 2017, 19, 118 CrossRef PubMed.
- M. J. Meegan and N. M. O'Boyle, Pharmaceuticals, 2019, 12, 134 CrossRef CAS PubMed.
- D. Acharya, S. Satapathy, P. Somu, U. K. Parida and G. Mishra, Biol. Trace Elem. Res., 2021, 199, 1812–1822 CrossRef CAS PubMed.
- P. Baindara and S. M. Mandal, Biochimie, 2020, 177, 164–189 CrossRef CAS PubMed.
- L. V. Spirina, A. V. Avgustinovich, S. G. Afanas'ev, O. V. Cheremisina, M. Yu. Volkov, E. L. Choynzonov, A. K. Gorbunov and E. A. Usynin, Curr. Drug Targets, 2020, 21, 713–721 CrossRef CAS PubMed.
- S. Łukasiewicz, M. Czeczelewski, A. Forma, J. Baj, R. Sitarz and A. Stanisławek, Cancers, 2021, 13, 4287 CrossRef PubMed.
- G. Radha and M. Lopus, Transl. Oncol., 2021, 14, 101166 CrossRef CAS PubMed.
- C. Duan, M. Yu, J. Xu, B.-Y. Li, Y. Zhao and R. K. Kankala, Biomed. Pharmacother., 2023, 162, 114643 CrossRef CAS PubMed.
- A. K. McClendon and N. Osheroff, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2007, 623, 83–97 CrossRef CAS PubMed.
- S. J. McKie, K. C. Neuman and A. Maxwell, BioEssays, 2021, 43, 2000286 CrossRef CAS PubMed.
- V. Brázda, R. C. Laister, E. B. Jagelská and C. Arrowsmith, BMC Mol. Biol., 2011, 12, 33 CrossRef PubMed.
- Y. Pommier, Curr. Med. Chem.: Anti-Cancer Agents, 2004, 4, 429–434 CrossRef CAS PubMed.
- Y. Pommier, J. M. Barcelo, V. A. Rao, O. Sordet, A. G. Jobson, L. Thibaut, Z. Miao, J. A. Seiler, H. Zhang, C. Marchand, K. Agama, J. L. Nitiss and C. Redon, in Progress in Nucleic Acid Research and Molecular Biology, Elsevier, 2006, vol. 81, pp. 179–229 Search PubMed.
- A. Ray Chaudhuri, Y. Hashimoto, R. Herrador, K. J. Neelsen, D. Fachinetti, R. Bermejo, A. Cocito, V. Costanzo and M. Lopes, Nat. Struct. Mol. Biol., 2012, 19, 417–423 CrossRef CAS PubMed.
- J. J. Reynolds and G. S. Stewart, DNA Repair, 2013, 12, 588–599 CrossRef CAS PubMed.
- S. Lamba and A. Roy, Biochem. Pharmacol., 2022, 203, 115158 CrossRef CAS PubMed.
- A. Dhiman, R. Sharma and R. K. Singh, Acta Pharm. Sin. B, 2022, 12, 3006–3027 CrossRef CAS PubMed.
- E. Martín-Encinas, A. Selas, F. Palacios and C. Alonso, Expert Opin. Drug Discovery, 2022, 17, 581–601 CrossRef PubMed.
- C. Bailly, Cytokine, 2023, 168, 156234 CrossRef CAS PubMed.
- F. Ye, S. Dewanjee, Y. Li, N. K. Jha, Z.-S. Chen, A. Kumar, Vishakha, T. Behl, S. K. Jha and H. Tang, Mol. Cancer, 2023, 22, 105 CrossRef CAS PubMed.
- T. T. Le, M. Wu, J. H. Lee, N. Bhatt, J. T. Inman, J. M. Berger and M. D. Wang, Nat. Chem. Biol., 2023, 19, 641–650 CrossRef CAS PubMed.
- L. Oelschläger, P. Stahl, F. Kaschani, R. H. Stauber, S. K. Knauer and A. Hensel, Cells, 2023, 12, 363 CrossRef PubMed.
- M. Tufail, J. Cui and C. Wu, Am. J. Cancer Res., 2022, 12, 2920–2949 CAS.
- Z. Pang, X. Dong, H. Deng, C. Wang, X. Liao, C. Liao, Y. Liao, W. Tian, J. Cheng, G. Chen, H. Yi and L. Huang, Oncogene, 2022, 41, 3064–3078 CrossRef CAS PubMed.
- M. Aatif, M. A. Raza, K. Javed, S. M. Nashre-ul-Islam, M. Farhan and M. W. Alam, Antibiotics, 2022, 11, 1750 CrossRef CAS PubMed.
- J. Baranwal, S. Kushwaha, S. Singh and A. Jyoti, Curr. Phys. Chem., 2023, 13, 2–19 CrossRef CAS.
- S. K. Suthar, N. S. Chundawat, G. P. Singh, J. M. Padrón and Y. K. Jhala, Eur. J. Med. Chem. Rep., 2022, 5, 100040 CAS.
- R. S. Keri, S. S. Pandule, S. Budagumpi and B. M. Nagaraja, Arch. Pharm., 2018, 351, 1700325 CrossRef PubMed.
- R. L. Lucaciu, A. C. Hangan, B. Sevastre and L. S. Oprean, Molecules, 2022, 27, 6485 CrossRef CAS PubMed.
- D. Deng, Y. Yang, Y. Zou, K. Liu, C. Zhang, M. Tang, T. Yang, Y. Chen, X. Yuan, Y. Guo, S. Zhang, W. Si, B. Peng, Q. Xu, W. He, D. Xu, M. Xiang and L. Chen, J. Med. Chem., 2023, 66, 9495–9518 CrossRef CAS PubMed.
- Y. Bonakolluru, S. K. Nukala, G. Dasari, V. Badithapuram, R. Manchal and S. Bandari, Polycyclic Aromat. Compd., 2023, 43, 6319–6335 CrossRef CAS.
- X. Mu, W. Shi, L. Sun, H. Li, Z. Jiang and L. Zhang, Molecules, 2012, 17, 6854–6868 CrossRef CAS PubMed.
- M.-H. Hu and J.-H. Lin, Int. J. Biol. Macromol., 2023, 241, 124548 CrossRef CAS PubMed.
- Y. Zhang, G. Li and Y. Zhao, Bioorg. Med. Chem., 2023, 90, 117337 CrossRef CAS PubMed.
- G. Dasari, S. Bandari, S. Kumar Nukala, N. Swamy Thirukovela, N. Sirassu, V. Badithapuram and R. Manchal, ChemistrySelect, 2022, 7, e202200681 CrossRef CAS.
- I. Balderas-Renteria, P. Gonzalez-Barranco, A. Garcia, B. K. Banik and G. Rivera, Curr. Med. Chem., 2012, 19, 4377–4398 CrossRef CAS PubMed.
- M. A. Moga, O. G. Dimienescu, A. Bălan, L. Dima, S. I. Toma, N. F. Bîgiu and A. Blidaru, Molecules, 2021, 26, 1054 CrossRef CAS PubMed.
- S. D. Undevia, F. Innocenti, J. Ramirez, L. House, A. A. Desai, L. A. Skoog, D. A. Singh, T. Karrison, H. L. Kindler and M. J. Ratain, Eur. J. Cancer, 2008, 44, 1684–1692 CrossRef CAS PubMed.
- S.-H. Lee, N. Kim, S.-J. Kim, J. Song, Y.-D. Gong and S.-Y. Kim, J. Cancer Res. Clin. Oncol., 2013, 139, 1279–1294 CrossRef CAS PubMed.
- L. Marcu and I. Olver, Curr. Clin. Pharmacol., 2006, 1, 71–79 CrossRef CAS PubMed.
- Z. Wang and B. Yang, in Polypharmacology, Springer International Publishing, Cham, 2022, pp. 43–72 Search PubMed.
- I. H. El Azab, E. M. Saied, A. A. Osman, A. E. Mehana, H. A. Saad and N. A. A. Elkanzi, Future Med. Chem., 2021, 13, 1743–1766 CrossRef CAS PubMed.
- A. Kabir and A. Muth, Pharmacol. Res., 2022, 176, 106055 CrossRef CAS PubMed.
- J. Sharifi-Rad, N. Cruz-Martins, P. López-Jornet, E. P.-F. Lopez, N. Harun, B. Yeskaliyeva, A. Beyatli, O. Sytar, S. Shaheen, F. Sharopov, Y. Taheri, A. O. Docea, D. Calina and W. C. Cho, Oxid. Med. Cell. Longevity, 2021, 2021, 6492346 Search PubMed.
- M. J. Matos, L. Santana, E. Uriarte, O. A. Abreu, E. Molina, E. G. Yordi, M. J. Matos, L. Santana, E. Uriarte, O. A. Abreu, E. Molina and E. G. Yordi, in Phytochemicals – Isolation, Characterisation and Role in Human Health, IntechOpen, 2015 Search PubMed.
- S. D. Sarker and L. Nahar, in Progress in the Chemistry of Organic Natural Products 106, ed. A. D. Kinghorn, H. Falk, S. Gibbons and J. Kobayashi, Springer International Publishing, Cham, 2017, pp. 241–304 Search PubMed.
- S. Fiorito, L. Palumbo, F. Epifano, D. Fraternale, C. Collevecchio and S. Genovese, Biomass Convers. Biorefin., 2022 DOI:10.1007/s13399-022-03309-z.
- A. Khalid, W. Khan, K. Zia, Azizuddin, W. Ahsan, H. A. Alhazmi, A. N. Abdalla, A. Najmi, A. Khan, A. Bouyahya, Z. Ul-Haq and A. Khan, Front. Pharmacol, 2023, 14, 1133809 CrossRef CAS PubMed.
- Herbs, Shrubs and Trees of Potential Medicinal Benefits, ed. A. Husen, CRC Press, Boca Raton, 1st edn, 2022 Search PubMed.
- A. Rawat and A. Vijaya Bhaskar Reddy, Eur. J. Med. Chem. Rep., 2022, 5, 100038 CAS.
- T. Al-Warhi, A. Sabt, E. B. Elkaeed and W. M. Eldehna, Bioorg. Chem., 2020, 103, 104163 CrossRef CAS PubMed.
- J. C. Menezes and M. Diederich, Future Med. Chem., 2019, 11, 1057–1082 CrossRef CAS PubMed.
- E. M. Saied and C. Arenz, Int. J. Mol. Sci., 2021, 22, 8171 CrossRef CAS PubMed.
- E. M. Saied, S. Diederich and C. Arenz, Chem.–Asian J., 2014, 9, 2092–2094 CrossRef CAS PubMed.
- E. M. Saied, T. L.-S. Le, T. Hornemann and C. Arenz, Bioorg. Med. Chem., 2018, 26, 4047–4057 CrossRef CAS PubMed.
- E. M. Saied, S. Banhart, S. E. Bürkle, D. Heuer and C. Arenz, Future Med. Chem., 2015, 7, 1971–1980 CrossRef CAS PubMed.
- R. D. Healey, E. M. Saied, X. Cong, G. Karsai, L. Gabellier, J. Saint-Paul, E. Del Nero, S. Jeannot, M. Drapeau, S. Fontanel, D. Maurel, S. Basu, C. Leyrat, J. Golebiowski, G. Bossis, C. Bechara, T. Hornemann, C. Arenz and S. Granier, Angew. Chem., Int. Ed., 2022, 61, e202109967 CrossRef CAS PubMed.
- S. M. Khirallah, H. M. M. Ramadan, H. A. A. Aladl, N. O. Ayaz, L. A. F. Kurdi, M. Jaremko, S. Z. Alshawwa and E. M. Saied, Pharmaceuticals, 2022, 15, 1576 CrossRef CAS PubMed.
- E. M. Radwan, E. Abo-Elabass, A. E. Abd El-Baky, H. A. Alshwyeh, R. A. Almaimani, G. Almaimani, I. A. A. Ibrahim, A. Albogami, M. Jaremko, S. Z. Alshawwa and E. M. Saied, Front. Chem., 2023, 11, 1231030 CrossRef CAS PubMed.
- S. M. Khirallah, H. M. M. Ramadan, A. Shawky, S. H. Qahl, R. S. Baty, N. Alqadri, A. M. Alsuhaibani, M. Jaremko, A.-H. Emwas and E. M. Saied, Molecules, 2022, 27, 6271 CrossRef CAS PubMed.
- A. Olyaei, E. Feizy and A. Aghajanzadeh, J. Heterocycl. Chem., 2021, 58, 757–765 CrossRef CAS.
- G. A. Hampannavar, R. Karpoormath, M. B. Palkar, M. S. Shaikh and B. Chandrasekaran, ACS Med. Chem. Lett., 2016, 7, 686–691 CrossRef CAS PubMed.
- C. Davies, C. M. Brown, D. Westphal, J. M. Ward and V. K. Ward, J. Virol., 2015, 89, 6057–6066 CrossRef CAS PubMed.
- O. Hayes, B. Ramos, L. L. Rodríguez, A. Aguilar, T. Badía and F. O. Castro, Anim. Reprod. Sci., 2005, 87, 181–192 CrossRef CAS PubMed.
- F. Heitz, P. Harter, N. Ewald-Riegler, M. Papsdorf, S. Kommoss and A. Du Bois, Expert Rev. Anticancer Ther., 2010, 10, 1125–1136 CrossRef CAS PubMed.
- E. V. Mirkin and S. M. Mirkin, Microbiol. Mol. Biol. Rev., 2007, 71, 13–35 CrossRef CAS PubMed.
- J. Y. Kim, K.-W. Lee, S.-H. Kim, J. J. Wee, Y.-S. Kim and H. J. Lee, Planta Med., 2002, 68, 119–122 CrossRef CAS PubMed.
- H. El Hajj, B. Khalil, B. Ghandour, R. Nasr, S. Shahine, A. Ghantous, R. Abdel-Samad, A. Sinjab, H. Hasegawa, M. Jabbour, W. W. Hall, G. Zaatari, G. Dbaibo, C. Pisano, A. Bazarbachi and N. Darwiche, Blood, 2014, 124, 2072–2080 CrossRef CAS PubMed.
- C. M. Henry, E. Hollville and S. J. Martin, Methods San Diego Calif, 2013, 61, 90–97 CrossRef CAS PubMed.
- B. Schutte, R. Nuydens, H. Geerts and F. Ramaekers, J. Neurosci. Methods, 1998, 86, 63–69 CrossRef CAS PubMed.
- Z. Wu, L. Wu, L. Zou, M. Wang and X. Liu, J. Chemother., 2023, 35, 131–141 CrossRef CAS PubMed.
- A. Plaper, M. Golob, I. Hafner, M. Oblak, T. Šolmajer and R. Jerala, Biochem. Biophys. Res. Commun., 2003, 306, 530–536 CrossRef CAS PubMed.
- D. A. Burden and N. Osheroff, Biochim. Biophys. Acta, Gene Struct. Expression, 1998, 1400, 139–154 CrossRef CAS PubMed.
- I. Laponogov, D. A. Veselkov, I. M.-T. Crevel, X.-S. Pan, L. M. Fisher and M. R. Sanderson, Nucleic Acids Res., 2013, 41, 9911–9923 CrossRef CAS PubMed.
- J. V. Walker and J. L. Nitiss, Cancer Invest., 2002, 20, 570–589 CrossRef CAS PubMed.
- J. E. Deweese and N. Osheroff, Nucleic Acids Res., 2009, 37, 738–748 CrossRef CAS PubMed.
- C. O. Okoro and T. H. Fatoki, Int. J. Mol. Sci., 2023, 24, 2532 CrossRef CAS PubMed.
- A. C. Dougherty, M. G. Hawaz, K. G. Hoang, J. Trac, J. M. Keck, C. Ayes and J. E. Deweese, ACS Omega, 2021, 6, 25892–25903 CrossRef CAS PubMed.
- M. Antoniou-Kourounioti, M. Mimmack, A. Porter and C. Farr, Int. J. Mol. Sci., 2019, 20, 1238 CrossRef CAS PubMed.
- V. Singh, T. Afshan, P. Tyagi, P. K. Varadwaj and A. K. Sahoo, Int. J. Biol. Macromol., 2023, 226, 473–484 CrossRef CAS PubMed.
- H. Masuda, D. Zhang, C. Bartholomeusz, H. Doihara, G. N. Hortobagyi and N. T. Ueno, Breast Cancer Res. Treat., 2012, 136, 331–345 CrossRef CAS PubMed.
- P. Wee and Z. Wang, Cancers, 2017, 9, 52 CrossRef PubMed.
- R. Kshatriya, P. Shelke, S. Mali, G. Yashwantrao, A. Pratap and S. Saha, ChemistrySelect, 2021, 6, 6230–6239 CrossRef CAS.
- V. R. Mishra, C. W. Ghanavatkar, S. N. Mali, H. K. Chaudhari and N. Sekar, J. Biomol. Struct. Dyn., 2019, 1–14 CrossRef PubMed.
- C. Kirschbaum, E. M. Saied, K. Greis, E. Mucha, S. Gewinner, W. Schöllkopf, G. Meijer, G. von Helden, B. L. J. Poad, S. J. Blanksby, C. Arenz and K. Pagel, Angew. Chem., Int. Ed., 2020, 59, 13638–13642 CrossRef CAS PubMed.
- B. A. Abdel-Wahab, H. F. Abd El-Kareem, A. Alzamami, C. A. Fahmy, B. H. Elesawy, M. Mostafa Mahmoud, A. Ghareeb, A. El Askary, H. H. Abo Nahas, N. G. M. Attallah, N. Altwaijry and E. M. Saied, Metabolites, 2022, 12, 715 CrossRef CAS PubMed.
- S. Banhart, E. M. Saied, A. Martini, S. Koch, L. Aeberhard, K. Madela, C. Arenz and D. Heuer, Antimicrob. Agents Chemother., 2014, 58, 5537–5546 CrossRef PubMed.
- S. Koch-Edelmann, S. Banhart, E. M. Saied, L. Rose, L. Aeberhard, M. Laue, J. Doellinger, C. Arenz and D. Heuer, Cell. Microbiol., 2017, 19, e12752 CrossRef PubMed.
- L. S. Kalinichenko, C. Mühle, T. Jia, F. Anderheiden, M. Datz, A.-L. Eberle, V. Eulenburg, J. Granzow, M. Hofer, J. Hohenschild, S. E. Huber, S. Kämpf, G. Kogias, L. Lacatusu, C. Lugmair, S. M. Taku, D. Meixner, N. Tesch, M. Praetner, C. Rhein, C. Sauer, J. Scholz, F. Ulrich, F. Valenta, E. Weigand, M. Werner, N. Tay, C. J. Mc Veigh, J. Haase, A.-L. Wang, L. Abdel-Hafiz, J. P. Huston, I. Smaga, M. Frankowska, M. Filip, A. Lourdusamy, P. Kirchner, A. B. Ekici, L. M. Marx, N. P. Suresh, R. Frischknecht, A. Fejtova, E. M. Saied, C. Arenz, A. Bozec, I. Wank, S. Kreitz, A. Hess, T. Bäuerle, M. D. Ledesma, D. N. Mitroi, A. M. Miranda, T. G. Oliveira, E. Gulbins, B. Lenz, G. Schumann, J. Kornhuber and C. P. Müller, Mol. Psychiatry, 2021, 26, 7403–7416 CrossRef CAS PubMed.
- T. D. de Oliveira, G. H. Ribeiro, J. Honorato, C. M. Leite, A. C. da S. Santos, E. D. Silva, V. R. A. Pereira, A. M. Plutín, M. R. Cominetti, E. E. Castellano and A. A. Batista, J. Inorg. Biochem., 2022, 234, 111906 CrossRef CAS PubMed.
- L. Huang and L. Fu, Acta Pharm. Sin. B, 2015, 5, 390–401 CrossRef PubMed.
- D. Samaha, H. H. Hamdo, X. Cong, F. Schumacher, S. Banhart, Ö. Aglar, H. M. Möller, D. Heuer, B. Kleuser, E. M. Saied and C. Arenz, Chem.–Eur. J., 2020, 26, 16616–16621 CrossRef CAS PubMed.
- D. I. Mohamed, D. A. Abou-Bakr, S. F. Ezzat, H. F. A. El-Kareem, H. H. A. Nahas, H. A. Saad, A. E. Mehana and E. M. Saied, Pharmaceuticals, 2021, 14, 1222 CrossRef CAS PubMed.
- D. I. Mohamed, S. F. Ezzat, W. M. Elayat, O. A. El-Kharashi, H. F. A. El-Kareem, H. H. A. Nahas, B. A. Abdel-Wahab, S. Z. Alshawwa, A. Saleh, Y. A. Helmy, E. Khairy and E. M. Saied, Pharmaceuticals, 2022, 15, 832 CrossRef CAS PubMed.
- S. Fathy Elhabal, M. A. El-Nabarawi, N. Abdelaal, M. F. M. Elrefai, S. A. Ghaffar, M. M. Khalifa, P. M. Mohie, D. S. Waggas, A. M. E. Hamdan, S. Z. Alshawwa, E. M. Saied, N. A. Elzohairy, T. Elnawawy, R. A. Gad, N. Elfar, H. Mohammed and M. A. Khasawneh, Drug Deliv., 2023, 30, 2241665 CrossRef PubMed.
- N. Coant, J. D. Bickel, R. Rahaim, Y. Otsuka, Y.-M. Choi, R. Xu, M. Simoes, C. Cariello, C. Mao, E. M. Saied, C. Arenz, T. P. Spicer, T. D. Bannister, P. J. Tonge, M. V. Airola, L. Scampavia, Y. A. Hannun, R. C. Rizzo and J. D. Haley, Bioorg. Chem., 2023, 139, 106747 CrossRef CAS PubMed.
- D. I. Mohamed, D. Alaa El-Din Aly El-Waseef, E. S. Nabih, O. A. El-Kharashi, H. F. Abd El-Kareem, H. H. Abo Nahas, B. A. Abdel-Wahab, Y. A. Helmy, S. Z. Alshawwa and E. M. Saied, Pharmaceutics, 2022, 14, 529 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06189b |
| This journal is © The Royal Society of Chemistry 2023 |