
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDopamine levels determined in synthetic urine using an electrochemical tyrosinase biosensor based on ZnO@Au core–shell†
Thainá G. Beattoa,
Wyllerson E. Gomesa,
Augusto Etchegaraya,
Ruchi Gupta b and
Renata K. Mendes*a
b and
Renata K. Mendes*a
aPontíficia Universidade Católica de Campinas, Campinas, SP, Brazil. E-mail: renatavalente@puc-campinas.edu.br; Tel: +55 19 33437656
bSchool of Chemistry, University of Birmingham, Birmingham, B15 2TT, UK
First published on 14th November 2023
Abstract
This work presents a biosensor based on core–shell nanostructure formed by zinc oxide (ZnO) nanoparticles coated with gold (Au). The core–shell nanostructure served as a support for the immobilisation of tyrosinase on screen-printed carbon electrodes to measure dopamine using differential pulse voltammetry. While ZnO is a semiconductor with good electrical conductivity, Au offers high stability and biocompatibility, which is beneficial for maintaining enzyme activity. Atomic force microscopy (ATM), ultraviolet (UV) and infrared (IR) spectroscopy measurements confirmed that the core–shell was successfully formed. The biosensor comprising of ZnO@Au core–shell nanostructures with immobilised tyrosinase allowed the detection of dopamine in real samples with remarkable selectivity and accuracy with a relative error of 3.8%. The limit of detection and dynamic range of the biosensor for dopamine in real samples were 86 nmol L−1 and 0.1 to 500 μmol L−1, respectively. Thus, the results indicate that the proposed miniaturized biosensor device is promising for the monitoring of dopamine in real samples and can be used for disease diagnosis and prognosis. Furthermore, the reported electrochemical biosensor is of low-cost when compared to conventional techniques.
Introduction
Parkinson's disease (PD) is an incurable and progressive pathological disorder of the nervous system caused by the continued deficiency of the neurotransmitter dopamine (DA), leading to motor-related problems.1 These uncontrolled processes affect the central nervous system (CNS) resulting in tremors, balance and muscle disorders, and slowness of voluntary movements (akinesia and bradykinesia), heavily impacting the quality of life.2Urine is the most used fluid for DA analysis because the collection is non-invasive and stable under ambient conditions.3,4 The reference value of dopamine in urine ranges from 60 to 440 μg day−1, which corresponds to 0.2 to 1.4 μmol L−1 considering that approximately 2 L of urine is produced per day.5 Therefore, a measurement below the minimum value would be associated with a decrease in the performance of the dopaminergic system.6 The most widely used methods for the quantification of dopamine in body fluids and drug composition are spectrophotometry,7 chromatography,8 chemiluminescence9 and electrophoresis.10 However, the use of electrochemical biosensors for point-of-care measurements has increased in recent years because of low cost and reduced analysis time while still maintaining the usual figures of merit.11 Neurotransmitters such as DA are identifiable by electrochemical analysis because they are easily oxidized.12 However, several other species in real samples are also easily oxidized and may interfere with the selective detection of DA.
Selectivity can be improved using biomolecules such as enzymes (oxidases) that are used as the biological recognition element in DA-specific biosensors. For instance, laccase and tyrosinase can catalyse the oxidation of catecholamines including DA. Tyrosinase has the additional advantage of being relatively cheap due to its availability from plant extracts,13,14 thus further minimizing the device cost. The enzyme acts by catalysing the oxidation reaction of an o-diphenol to o-quinone, or to o-dopaquinone in the case of DA. These two processes can be measured by electrochemistry. Therefore, electrochemical biosensors can be a viable alternative to existing methods for the measurement of DA due to their high specificity, sensitivity, reduced operational costs, speed of analysis and the ability to be miniaturized, allowing real-time field analysis.15
Biosensors using enzymes as the biorecognition element require the use of support materials for enzyme immobilization. The support must offer a large surface area, be non-toxic, and preferably, contribute to electron transference; thus, improving the device's sensitivity.16,17 This latter property is an important parameter since low concentrations of DA in urine are indicative of Parkinson's disease.18
The state-of-the-art composites used as a support for the immobilisation of biomolecules are based on “core–shell” nanomaterials. These are composed of a nanoparticle-denominated “core (X)” that is covered with another material named “shell (Y)”. The core–shell may be represented as X@Y.19 ZnO stands out as a metallic oxide nanoparticle used as a core. These oxides are semiconductors with a gap energy of 3.37 eV at room temperature and good electrical conductivity, which are of great importance in electrochemical devices.20,21 In addition, they have low toxicity, are biodegradable and easily synthesized.22 Furthermore, when ZnO nanoparticles are coated with gold, and the material is used for biomolecule immobilization, the system gains increased stability while allowing easy functionalization of the biorecognition element. In addition, the biocompatibility of Au nanoparticles is beneficial for maintaining the activity of enzymes after immobilization.18 Thus, the properties of these nanomaterials are promising for constructing biosensors with high sensitivity. For example, Rasouli et al.23 obtained the linear range from 1 and 10 μmol L−1 and a limit of detection of 0.1 nmol L−1 for human papillomavirus type 16 (HPV-16) using a biosensor formed by Fe3O4@Au core–shell as DNA support. Similarly, Güler et al.24 immobilised glucose oxidase (GOx) on Au@Pd core–shell nanoparticles supported on carboxylated graphene oxide (cGO) to construct a glucose biosensor with a linear range from 0.2 to 4200 μmol L−1 and limit of detection of 10.4 μmol L−1.
To combine both the social aspect of public health and to obtain an early diagnosis of Parkinson's disease, the present work aimed to develop a novel tyrosinase biosensor. The core–shell ZnO@Au was used for tyrosinase immobilization, and the nanocomposite was successfully employed on screen-printed carbon electrodes to determine dopamine levels in samples of synthetic urine.
Experimental
All reagents used in this work were of analytical grade. Potassium chloride and ammonium chloride were bought from Exodus (Brazil). Sodium chloride, urea and calcium chloride dehydrate were bought from Dynamics (Brazil). Additional reagents were purchased from Sigma-Aldrich and were used as received. Phosphate buffer solutions were obtained by mixing solutions of 0.1 mol L−1 Na2HPO4 and 0.1 mol L−1 NaH2PO4 in ultrapure water. All solutions were prepared in deionized water (>18 MΩ cm, Milli-Q, Millipore). Synthetic urine was produced by following the recipe reported by Silva25 and contained 0.73 g NaCl; 0.40 g KCl; 0.27 g CaCl2·2H2O; 0.56 g Na2SO4; 0.35 g KH2PO4; 0.25 g NH4Cl and 6.25 g of urea in 250 mL deionized water.For the spectrometric characterisation of core–shell nanostructures, the Spectrophotometers UV-1800 (Shimadzu) and Infrared (FT-IR MB3000 – ABB) were used. Atomic force microscope measurements were carried out using the ThermoMicroscope AutoProbe CP and analysis was supported by the software TopoMetrix SPMLab 4.0 and Microcal Origin 6.0.
Core-shell production
For the synthesis of ZnO nanoparticles (ZnONPs), 1.0 mol L−1 NaOH solution was heated under constant stirring at 85 °C. Then, a 0.5 mol L−1 ZnCl2 solution was slowly added to the NaOH solution to precipitate ZnO. The solution was stirred for 1 hour keeping the temperature at 85 °C. Afterwards, the solution was filtered and washed at least 5 times with deionized water, until it reached pH 7, and then stored in deionized water.The method reported by Riahifar et al.26 was used for core–shell synthesis. Briefly, 20 mL of a suspension of ZnO nanoparticles was transferred to a 250 mL round-bottomed flask and heated until boiling. Then 10 mL of HAuCl4 (0.1%) and 5 mL of sodium citrate (1%) solutions prepared in deionized water were added to ZnO nanoparticles suspension. The system was closed and refluxed for 30 min. The original colour of the ZnO nanoparticles suspension changed instantly from white to purple. Subsequently, the heating was stopped, and the solution was cooled naturally to room temperature. Afterwards, the ZnO@Au nanoparticles were washed with deionized water, and the new suspension was centrifuged at 1800 rpm for 10 min. The supernatant was discarded, and this washing process was repeated until the suspension reached pH 7. The system was stored in deionized water.
Biosensor construction
The biorecognition element used in this work was the enzyme tyrosinase. The tyrosinase extract was obtained from yam according to the procedure reported by Signori.27 For tyrosinase immobilization, 40 μL of the water suspension of the core–shell (prepared as above) was transferred to an Eppendorf and 200 μL of the enzymatic extract (∼1 mU mL−1) was added to the suspension, and the mixture was left to settle for 45 min. Then the supernatant was removed, and the system was washed with 20 μL of 0.1 mol L−1 phosphate buffer solution pH 8.0, to remove weakly bound biomolecules and to keep the immobilized enzyme in active form. The supernatant was removed again and 5 μL of the obtained suspension of ZnO@Au/tyrosinase was dropped onto the screen-printed carbon electrode (Metrohm) and dried in air to produce the biosensor as shown in Scheme 1.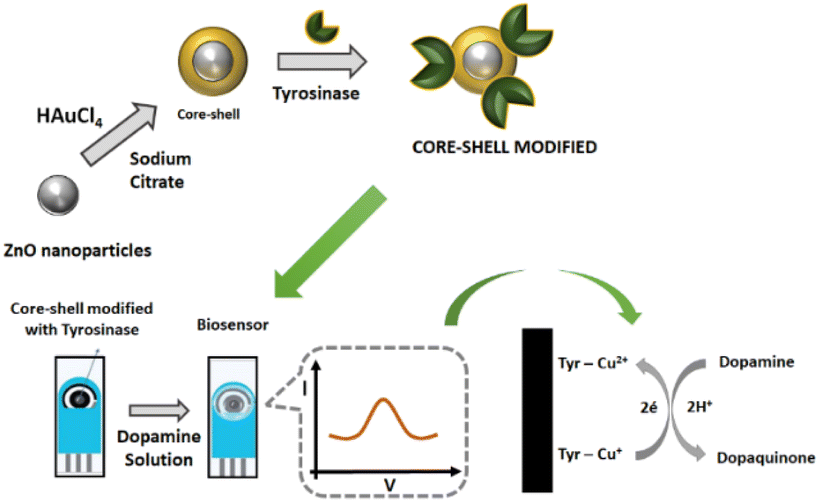 | ||
| Scheme 1 Schematic representation of the steps for biosensor construction. | ||
Electrochemical measurements
A sample of 50 μL of 2500 μmol L−1 dopamine solution was added to the obtained biosensor, followed by voltammetric analysis. The constructed biosensor should be disposable and exhibit fast response, so DA measurements were performed using the differential pulse voltammetry technique, in the range of −300 to 500 mV with 25 mV amplitude at 80 mV s−1. All experiments were carried out in an AUTOLAB micro potentiostat (Metrohm) PGSTAT 101.Atomic force microscopy (AFM) imaging
For AFM imaging, the suspensions of nanomaterials (ZnO and ZnO@Au) were deposited on a silicon surface (20 × 20 mm, 〈100〉), which, before use, was submerged in 50% m m−1 hydrofluoric acid and cleaned with deionized water. To obtain a tiny amount of sample deposit on the silicon surface, both the suspensions of ZnO and core–shell were diluted 10 times, agitated, and allowed to settle for approximately 10 min. Then an aliquot of 3 to 5 μL was collected from the top of each respectively settled suspension and deposited on the silicon surface. This was to obtain highly dispersed structures, as required for AFM measurements. Before analysis, each sample was air-dried for 2 hours.22 The small curvature tip (radius of 5 mm), located at the end of the silicon nitride cantilever, was driven in non-contact mode at a scanning rate of 1 to 4 Hz. The non-contact mode did not cause any damage to the structure of the nanoparticles while providing images with nanometric resolution. AFM measurements were performed using a Thermo Microscope AutoProbe CP-Research. Images were profiles of nanomaterials were obtained using the AFM software.Results and discussion
Core-shell characterisation
To confirm that the ZnO@Au core–shell was formed, the suspensions were analysed in the spectrophotometer using the ultraviolet-visible region. The spectra for pure ZnO nanoparticles and core–shell (ZnO@Au) are compared in Fig. 1.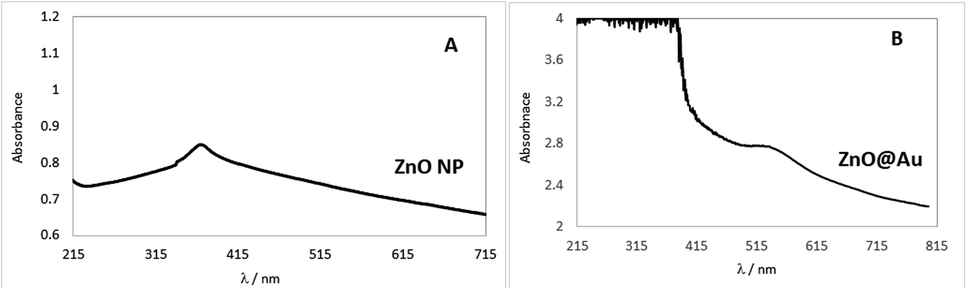 | ||
| Fig. 1 UV-vis spectra of ZnO nanoparticles (A) and ZnO@Au core–shell (B). | ||
An absorption band at 371 nm in Fig. 1A confirms the formation of ZnO and is in agreement with the findings of Bhushan et al.20 Thus, the synthesis of these nanoparticles was regarded as successful. On the other hand, the spectrum of ZnO@Au nanoparticles (Fig. 1B) showed a peak at 523 nm, which coincides with the peak absorption wavelength of gold as reported by Abdelhalim et al.28,29 Thus, there is evidence that the metal oxide nanoparticles were converted into the proposed core shell.
Atomic force microscopy (AFM) measurements were used to estimate the size and morphology of the Zno@Au core shell. Images of both the ZnO nanoparticles and ZnO@Au are provided in Fig. 2A and B. It is possible to verify that core–shell particles are larger than ZnO particles, confirming the production of ZnO@Au nanostructures. Based on Fig. 2, the diameter of the ZnO and ZnO@Au nanoparticles was respectively 13 ± 2 nm and 29 ± 2 nm.
 | ||
| Fig. 2 AFM 2D images of the ZnO nanoparticles (A) and ZnO@Au core–shell nanostructure (B) with 3 μm × 3 μm dimension with lighting angle change for highlighting. | ||
FTIR (Fourier Transform Infrared) was carried out to confirm the structural analysis of the ZnO@Au nanoparticles and core–shell. Fig. 3 shows the results.
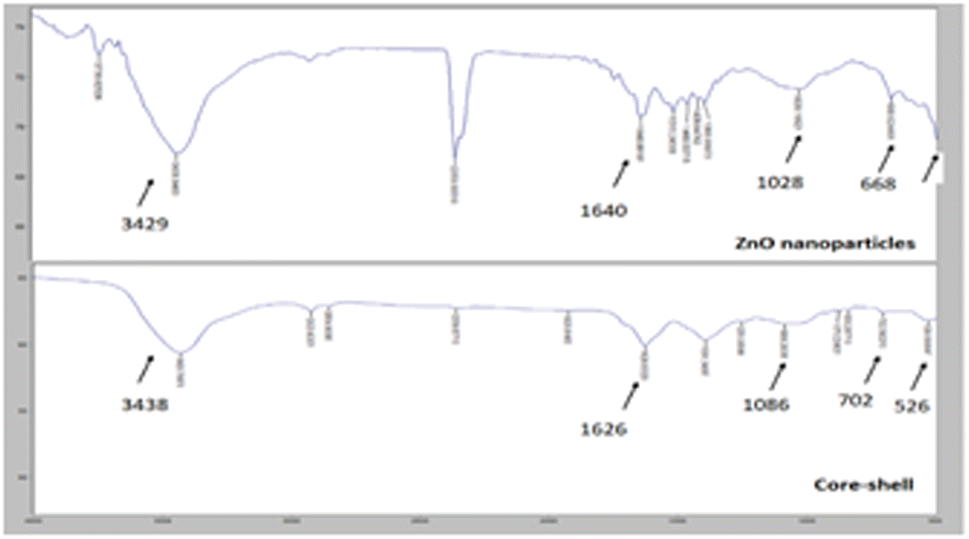 | ||
| Fig. 3 FT-IR spectra of ZnO nanoparticle and ZnO@Au core–shell. | ||
As shown, a change in transmittance at ≈3400 of ZnO-NPs and core–shell correlates to O–H peak.30 Core-shell presented a peak at 526 cm−1, which relates to Au-NPs31 and is not observed in the ZnO NP spectrum. It is not possible to observe the peak corresponding to the ZnO oxygen bond29 (≈425 cm−1) because the limit of detection of the spectrometer is 500 cm−1. However, a peak near 500 cm−1 can be detected, which possibly corresponds to the Zn–O bond.
Biosensor response
The voltammograms presented in Fig. 4 show that a biosensor with tyrosinase immobilized on ZnO@Au core–shell resulted in a higher response than that with ZnO nanoparticles.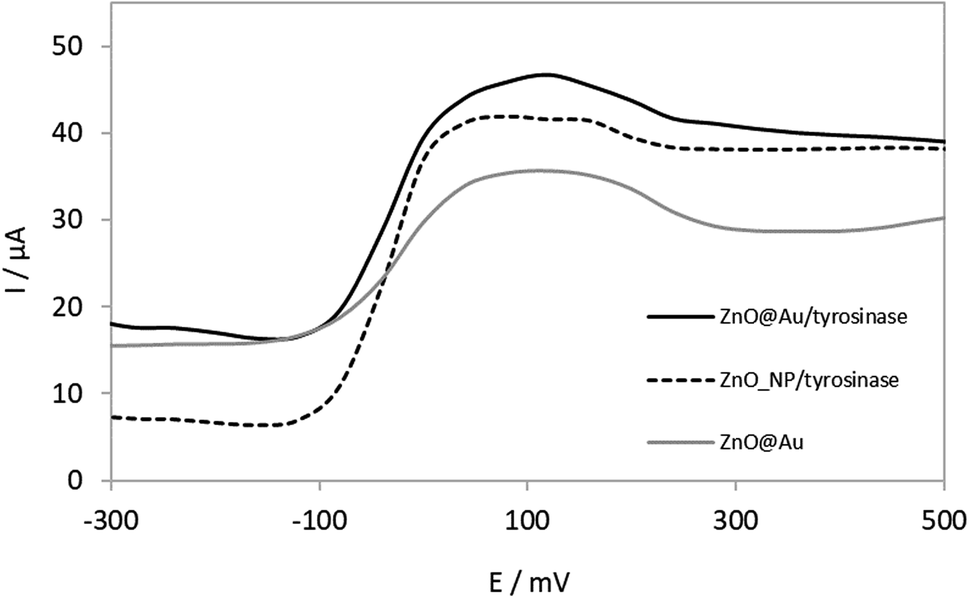 | ||
| Fig. 4 Differential pulse voltammograms obtained for ZnO nanoparticles with enzyme, core–shell ZnO@Au with and without enzyme in 2500 μmol L−1 dopamine solution, in 0.1 mol phosphate buffer L−1, pH 7.0, at 25 mV pulse amplitude and 80 mV s−1. The maximum signal for the core–shell indicates this platform as suitable for immobilization support for tyrosinase. | ||
According to Fig. 4, it is possible to observe that the biosensor constructed by ZnO@Au core–shell as a support for the enzymatic immobilization, presents an increase in the signal when compared to ZnONps. This is an indication that the coating by gold nanoparticles has promoted a synergic effect, favouring enzyme activity. Probably this is due to the biocompatibility of the inert material.32 Thus, ZnO@Au core–shell nanostructures had a positive impact on tyrosinase immobilization, which in turn was beneficial for biosensing of DA.
Optimization of experimental conditions
After the identification of the most suitable nanomaterials for enzyme immobilization, studies were carried out to optimize the immobilization conditions. Firstly, the influence of interaction time between the core–shell ZnO@Au and the tyrosinase was studied. For this, four different interaction times between core–shell and enzyme (1 mU mL−1) were tested: 20 min; 45 min; 2 hours and 24 hours. For each time, differential pulse voltammograms were obtained in 2500 μmol L−1 dopamine solution, in 0.1 mol L−1 phosphate buffer, pH 7.0.The maximum current value was obtained when the enzyme solution was incubated with ZnO@Au nanoparticles for 45 minutes (data in ESI†). When compared with the biosensor built using an incubation time of 20 minutes, the signal increase was only 3.5%, but it can be significant for analysis at low analyte concentration. At longer times, there was a decrease in the response of the biosensor. Increasing the incubation time to 2 and 24 hours would likely have resulted in a thicker biolayer27 which impairs the transfer of electrons on the electrode surface, with a consequent decrease in the current. Thus, the time of 45 minutes was selected for the subsequent experiments.
The effect of the pH of the buffer used for washing biosensors after incubating the enzyme solution with core–shell nanostructures was then studied. The pH of the wash buffer was increased in one unit, between 5 and 9, step by step. As the enzyme has amino acids in the structure, altering the pH will protonate or deprotonate the available amino and carboxyl groups, thus interfering with the immobilization process, which is based on electrostatic interactions. Differential pulse voltammograms were then obtained in 2500 μmol L−1 dopamine solution, in 0.1 mol L−1 phosphate buffer. The highest current was recorded at pH 8 (data in ESI†), which has been reported to be an optimal pH for the enzyme.33
Analytical parameters
After optimizing the immobilization conditions, the analytical performance of the developed sensor for the measurement of DA in real samples was determined. For this, novel devices were constructed using the optimized immobilization conditions and immersed in different concentrations (n = 6) of dopamine solutions. As we intend to apply the biosensor in physiological samples, the DA solutions were prepared at pH 7.4. Measurements were performed using differential pulse voltammetry (standard deviation 0.5 μA) and presented in Fig. 5A and the respective analytical curve in Fig. 5B.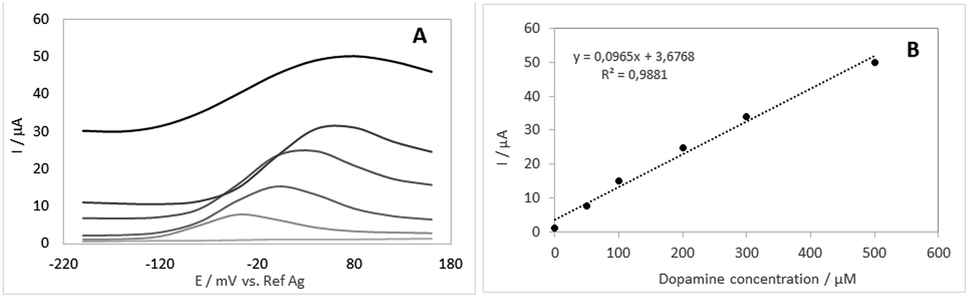 | ||
| Fig. 5 Differential pulse voltammograms were obtained with the proposed biosensor at different concentrations of dopamine (A) in 0.1 mol L−1 phosphate buffer at 25 mV pulse amplitude and 80 mV s−1 and the respective analytical curve (B). | ||
The calibration curve shows that the proposed biosensor has good linearity to DA concentration between 0.1 to 500 μmol L−1 with the equation of y(A) = 1 × 10−7x (μmol L−1) + 4 × 10−6 (R2 = 0.9881). It was also shown that after these concentration values of DA, the current remained constant, probably due to the saturation of the system. From the calibration curve, the limit of detection (LOD) for DA was determined to be 86 nmol L−1 based on the 3 SD/Slope of the analytical curve where SD is the standard deviation of ten measurements of blank.22
To verify if the method used in the biosensor construction was adequate, three devices were produced under the same experimental conditions, and they were applied in 250 μmol L−1 DA solution (the middle point within the linear range). The coefficient of variation was 1.05%, leading to the conclusion that the fabrication method resulted in biosensors with minimal device-to-device variability.
Table 1 presents a relationship between the limit of detection and the linear region of other biosensors found in the literature involving the determination of DA. The LOD achieved in the current work is lower for instance than that obtained by other authors using molecularly imprinted polymers (MIPs). However, the use of enzyme extracts for the preparation of our device is less expensive and uses water as extraction media, thus being more environmentally friendly and sustainable.
| Biosensor | Limit of detection/μmol L−1 | Linear range/μmol L−1 | Ref. |
|---|---|---|---|
| AuNPs@SiO2/molecularly imprinted polymer/glassy carbon | 0.02 | 0.048 to 50 | 34 |
| Ag@C/AuNPs/glassy carbon | 0.21 | 0.5 to 4278 | 35 |
| AuNPs@polyaniline/chitosan/glassy carbon | 5 | 10 to 1700 | 36 |
| AuNPs@ZnONPs/nanocone/graphene foam | 0.04 | 0 to 80 | 18 |
| AuNPs@Fe3O4/glassy carbon | 0.0027 | 0 to 0.8 | 32 |
| AuNPs@ molecularly imprinted polymer | 0.0078 | 0.02 to 0.54 | 37 |
| ZnO@Au/tyrosinase/screen-printed carbon electrode | 0.0855 | 0.1 to 500 | This work |
According to Table 1, the proposed biosensor has a wide linear region that covers the concentration range that is sought in clinical analysis. In contrast, previously reported biosensors did not provide a wide dynamic range to cover physiological levels of DA as presented here.
A brief survey in the literature was carried out in the Science Direct database, where it was possible to find other types of DA detection methods with core–shell nanoparticles made of other materials and with other types of structure, such as graphene foam nanocones18 and core–shell deposited on a glassy carbon electrode,38 which is not the case in this work, as we used imprinted carbon microelectrodes. However, in this database, no method to detect DA was found using core–shell ZnO@Au, on microelectrodes, highlighting the innovation of the proposed device.
Selectivity studies
To verify the biosensor selectivity to DA in the presence of substances commonly found in physiological urine samples, five biosensors were constructed, and all were applied to a sample that was spiked with 250 μmol L−1 of DA in 0.1 mol L−1 phosphate buffer, pH 7.4. The DA response was analysed with the concomitant presence of glucose, ascorbic acid, sodium oxalate (all at 250 μmol L−1) and a mixture of all these substances. The study was evaluated using differential pulse voltammograms. The currents obtained in each case were compared to the signal obtained for DA (250 μmol L−1) which was considered as 100%. The DA response in the presence of the studied interferents did not change the signal significantly (data in ESI†). The device showed a high selectivity presenting a difference of less than 7% in the presence of the studied interferents. This result is extremely relevant because ascorbic acid is a potential interferent in urine samples.12Biosensor stability
To verify how long the biosensor can be used without significant loss of response, a study was carried out to evaluate the stability of the proposed device. For this, a biosensor was constructed under optimized conditions. The biosensor was used daily to measure its response to 250 μmol L−1 DA in 0.1 mol L−1 phosphate buffer solution, pH 7.0. After each measurement, the DA drop was removed with clean paper, by capillarity, and the device washed with buffer. The biosensor was then stored at 4 °C until the next day. After 15 days, the biosensor performance decreased by approximately 20%, which is considered significant since the device is intended to determine low concentrations of DA in the samples (data in ESI†). During the first five days, the variation of biosensor response between measurements was approximately 11%. Therefore, this timing was assumed as the limit for the effective reusability of the biosensors. However, since we do not know if the cause was due to storage or continuous use, the miniaturized biosensor can be regarded as a disposable device.In addition, biosensors for the determination of catecholamines can adsorb the oxidized products on the electrode surface, generating unreliable responses. Thus, the same biosensor was tested at two different concentrations of DA: at high (250 μmol L−1) and low (100 μmol L−1) concentrations within the linear range. Four consecutive measurements were performed with the same device at the two above mentioned concentrations. The currents obtained at different concentrations varied around 4% (data in ESI†). Thus, it is confirmed that the measurement of high concentrations of DA does not influence the behaviour of the biosensor. The latter finding indicates that the biosensor does not adsorb oxidized products on the surface and that it has no memory effect.
Application of the proposed biosensor in a synthetic urine sample
To check the accuracy and selectivity of the biosensor in samples containing other substances along with the analyte, it was applied in synthetic urine spiked with 250 μmol L−1 of DA. This study was carried out in triplicate using differential pulse voltammetry.The mean value obtained was 29.943 μA which when substituted in the calibration curve provided an estimated concentration of 259.43 μmol L−1 of DA, with a relative error (%) of approximately 3.8%. The estimated and actual concentrations of DA as are in close agreement, confirming that the biosensor can measure DA levels in samples containing interferents such as urine.
Conclusions
According to the results obtained, it is possible to conclude that the biosensor, constructed from a core–shell nanostructure based on ZnO@Au, as a support for the tyrosinase immobilization, presented favourable characteristics in its use for the quantification of dopamine. The device showed efficient responses, with a low detection limit of 86 nmol L−1, presenting a wide linear region between 0.1 and 500 μmol L−1, being able in this range, to detect normal levels or the lack of dopamine excreted. When applied to synthetic urine samples, it showed a low relative error (3.8%), indicating that the other substances present did not interfere with the signal. Thus, the results indicate that the proposed miniaturized device is promising for monitoring dopamine in real samples and can contribute to the follow-up of patients and the diagnosis of neurological disease. It is noteworthy that it is a low-cost biosensor when compared to conventional techniques.Author contributions
Beatto: conceptualization; data curation; formal analysis; validation; writing – original draft. Gomes, Etchegaray and Gupta: writing – review & editing. Mendes: conceptualization; formal analysis; funding acquisition; investigation; methodology; writing – original draft; writing – review & editing; project administration; resources; supervision; visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001 – scholarship PNPD (Gomes, W.E.) and Prof. Dr David Mendez Soares for assistance with AFM measurements. Some of the equipment and material used in this work were acquired with a grant from FAPESP (2013/20570-6).References
- W. Poewe and P. Mahlknecht, Neurol. Clin., 2020, 38, 255–267 CrossRef PubMed.
- D. Galasko, Neurol. Clin., 2017, 35, 325–338 CrossRef PubMed.
- D. T. Marc, J. W. Ailts, D. C. A. Campeau, M. J. Bull and K. L. Olson, Neurosci. Biobehav. Rev., 2011, 35, 635–644 CrossRef CAS PubMed.
- S. Cohrs, Z. Guan, K. Pohlmann, W. Jordan, J. Pilz, E. Rüther and A. Rodenbeck, Neurosci. Lett., 2004, 360, 161–164 CrossRef CAS PubMed.
- C. Otte, T. C. Neylan, S. S. Pipkin, W. S. Browner and M. A. Whooley, Am. J. Psychiatry, 2005, 162, 2139–2145 CrossRef PubMed.
- Q. Hassan, S. Li, C. Ferrag and K. Kerman, Anal. Chim. Acta, 2019, 1089, 32–39 CrossRef CAS PubMed.
- H. Khajehsharifi, E. Pourbasheer, H. Tavallali, S. Sarvi and M. Sadeghi, Arabian J. Chem., 2017, 10, S3451–S3458 CrossRef CAS.
- M. Tsunoda, C. Aoyama, H. Nomura, T. Toyoda, N. Matsuki and T. Funatsu, J. Pharm. Biomed. Anal., 2010, 51, 712–715 CrossRef CAS PubMed.
- R. Li, D. Zhang, X. Li and H. Qi, Bioelectrochemistry, 2022, 146, 108148 CrossRef CAS PubMed.
- L. Zhang, S. Qv, Z. Wang and J. Cheng, J. Chromatogr. B, 2003, 792, 381–385 CrossRef CAS PubMed.
- N. O. Decarli, E. Zapp, B. S. Souza, E. R. Santana, J. P. Winiarski and I. V. Cruz, Biochem. Eng. J., 2022, 186, 108565 CrossRef CAS.
- P. Deepti, S. Mondal, C. Pal, A. Kumar and S. Majumder, Colloids Surf., A, 2023, 671, 131630 CrossRef CAS.
- C. Lete, B. Lakard, J. Y. Hihn, F. J. del Campo and S. Lupu, Sens. Actuators, B, 2017, 240, 801–809 CrossRef CAS.
- A. Roychoudhury, S. Basu and S. K. Jha, Biosens. Bioelectron., 2016, 84, 72–81 CrossRef CAS PubMed.
- C. Wardak, B. Paczosa-Bator and S. Malinowski, Mater. Sci. Eng., C, 2020, 116, 111199 CrossRef CAS PubMed.
- P. Liu, L. Yin and X. Qi, Int. J. Electrochem. Sci., 2020, 15, 5821–5832 CrossRef CAS.
- N. P. Shetti, S. D. Bukkitgar, K. R. Reddy, C. V. Reddy and T. M. Aminabhavi, Biosens. Bioelectron., 2019, 141, 111417 CrossRef CAS PubMed.
- H. Y. Yue, H. . J. Zhang, S. Huang, X. X. Lu, X. Gao, S. S. Song, Z. Wang and W. Q. Wang, Mater. Sci. Eng., C, 2020, 108, 110490 CrossRef CAS PubMed.
- W. Zhai, D. Wei, M. Cao, Z. Wang and M. Wang, Food Chem., 2023, 429, 136944 CrossRef CAS PubMed.
- M. Bhushan, R. Jha, R. Bhardwaj and R. Sharma, Mater. Today: Proc., 2022, 48, 629–632 CAS.
- V. Srivastava, D. Gusain and Y. C. Sharma, Ceram. Int., 2013, 39, 9803–9808 CrossRef CAS.
- R. Mendes, B. Arruda, E. de Souza, A. Nogueira, O. Teschke, L. Bonugli and A. Etchegaray, J. Braz. Chem. Soc., 2016, 28, 1212–1219 Search PubMed.
- E. Rasouli, W. J. Basirun, M. R. Johan, M. Rezayi, M. R. Mahmoudian and D. P. Poenar, Sensing Biosens. Res., 2023, 40, 100562 CrossRef.
- M. Güler, A. Zengin and M. Alay, Anal. Biochem., 2023, 667, 115091 CrossRef PubMed.
- L. P. Silva, B. C. Lourencao and O. Fatibello-Filho, Quim. Nova, 2015, 38, 801–806 CAS.
- V. Riahifar, N. Haghnazari, F. Keshavarzi and F. Nasri, Microchem. J., 2021, 166, 106217 CrossRef CAS.
- C. A. Signori and O. F. Filho, Quim. Nova, 1994, 17, 38–42 CAS.
- M. A. K. Abdelhalim and M. M. Mady, J. Nanomed. Nanotechnol., 2012, 3, 1–5 Search PubMed.
- D. V. Ponnuvelu, B. Pullithadathil, A. K. Prasad, S. Dhara, A. Ashok, K. Mohamed, A. K. Tyagi and B. Raj, Appl. Surf. Sci., 2015, 355, 726–735 CrossRef CAS.
- A. B. Meji, D. Usha, M. H. Sankar and B. M. Ashwin, Mater. Today, 2023 DOI:10.1016/j.matpr.2023.02.429.
- C. Poorshamohammad, L. Liu, X. Cheng, A. A. Momtazi-Borojeni and J. Shai, Arabian J. Chem., 2023, 16, 104386 CrossRef CAS.
- A. Thamilselvan, P. Manivel, V. Rajagopal, N. Nesakumar and V. Suryanarayanan, Colloids Surf., B, 2019, 180, 1–8 CrossRef CAS PubMed.
- E. Canbay and E. Akyilmaz, Anal. Biochem., 2014, 444, 8–15 CrossRef CAS PubMed.
- D. Yu, Y. Zeng, Y. Qi, T. Zhou and G. Shi, Biosens. Bioelectron., 2012, 38, 270–277 CrossRef CAS PubMed.
- X. Liu, Y. Fu, Q. Sheng and J. Zheng, Microchem. J., 2019, 146, 509–516 CrossRef CAS.
- L. Yang, S. Liu, Q. Zhang and F. Li, Talanta, 2012, 89, 136–141 CrossRef CAS PubMed.
- C. Xue, Q. Han, Y. Wang, J. Wu, T. Wen, R. Wang, J. Hong, X. Zhou and H. Jiang, Biosens. Bioelectron., 2013, 49, 199–203 CrossRef CAS PubMed.
- S. Xiang, Q. Meng, K. Zhang, Y. Gu, W. Liu and B. Yang, Chem. Res. Chin. Univ., 2019, 35, 924–928 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06277e |
| This journal is © The Royal Society of Chemistry 2023 |