
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 3-hydroxypyridin-4-one derivatives bearing benzyl hydrazide substitutions towards anti-tyrosinase and free radical scavenging activities†
Bahareh Hassani‡
a,
Fateme Zare‡b,
Leila Emamib,
Mehdi Khoshneviszadeha,
Razieh Fazela,
Negin Kavea,
Razieh Sabet *a and
Hossein Sadeghpour*a
*a and
Hossein Sadeghpour*a
aDepartment of Medicinal Chemistry, Faculty of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran. E-mail: sabet_r@sums.ac.ir; Fax: +98-7132424126; Tel: +98-7132424127-8
bFaculty of Pharmacy and Pharmaceutical Sciences Research Center, Shiraz University of Medical Sciences, Shiraz, Iran. E-mail: sadeghpurh@sums.ac.ir; Fax: +98-7132424126; Tel: +98-7132424127-8
First published on 7th November 2023
Abstract
Tyrosinase is a vital enzyme in the biosynthesis of melanin, which has a significant role in skin protection. Due to the importance of the tyrosinase enzyme in the cosmetics and health industries, studies to design new tyrosinase inhibitors have been expanded. In this study, the design and synthesis of 3-dihydroxypyridine-4-one derivatives containing benzo hydrazide groups with different substitutions were carried out, and their antioxidant and anti-tyrosinase activities were also evaluated. The proposed compounds showed tyrosinase inhibitory effects (IC50) in the 25.29 to 64.13 μM range. Among all compounds, 6i showed potent anti-tyrosinase activity with an IC50 = 25.29 μM. Also, the antioxidant activity of derivatives by using DPPH radical scavenging indicates an EC50 value between 0.039 and 0.389 mM. Molecular docking studies were performed to reveal the position and interactions of 6i as the most potent inhibitor within the tyrosinase active site. The results showed that 6i binds well to the proposed binding site and forms a stable complex with the target protein. Furthermore, the physicochemical profiles of the tested compounds indicated drug-like and bioavailability properties. The kinetic assay revealed that 6i acts as a competitive inhibitor. Also, for the estimation of the reactivity of the best compound (6i), the density functional theory (DFT) was performed at the B3LYP/6-31+G**.
1. Introduction
Tyrosinase is a copper-containing enzyme that plays an important role in melanin formation via mono- and di-phenolase reactions.1 Melanin can prevent ultraviolet skin damage; on the other hand, excessive gathering of melanin leads to different skin discomforts, such as age spots, malignant melanoma, and melisma.2 Tyrosinase is responsible for the undesirable enzymatic browning of fruits and vegetables.3 Various tyrosinase inhibitors have been established today, but due to the different side effects of available tyrosine inhibitor agents, drug design and studies on this enzyme are necessary.4 So far, several natural and synthetic inhibitors for tyrosinase enzymes have been identified.5–8 The studies showed that the compounds can inhibit tyrosinase enzyme in several different ways, including reduction of dopaquinone to dopa (ascorbic acid),9 reduction of the amount of dopaquinone (thiol analogs),10 and much greater affinity to tyrosinase enzyme compared to the main substrate (phenolic compounds).11 Also, the alteration in the shape of the active site of the enzyme, such as compounds with acidic or basic properties,12 the inactivation of the enzyme through the covalent bond with the enzyme, and irreversible inhibition of the enzyme,13 and finally, the occupation of the active site and reversible inhibition of the enzyme14 are the other mechanisms of tyrosinase inhibition. In recent years, researchers have focused more on synthesizing compounds with a phenolic group,15 which showed more affinity to the tyrosinase enzyme than dopaquinone and inhibited melanin formation.15 One of the scaffolds that has received much attention from researchers is hydroxypyridine (HP). The HPs are N-heterocycles bearing a hydroxyl and a ketone group. These scaffolds included three subgroups: 1, 2-HP, 3, 2-HP, and 3, 4-HP.16 Due to their chelating properties, the 3,4-HP scaffolds were the more basic, and some of their derivatives are used in the clinic, such as diferiprone.17 The derivatives of HPs can be easily synthesized and functionalized in different positions. Based on this fact, the hydroxypyridinone-like scaffolds (HPOs) were extensively used for research with various biological activities such as antiproliferative,18 chelating agents,19 antimicrobial,20,21 monoamine oxidase,22 and cytotoxic agents.23 Due to their copper chelation ability, they were used as tyrosinase enzyme inhibitors.24–26 In a recent study to search the possible application of hydroxypyridine (HP) as a tyrosinase inhibitor, some novel hydroxy pyridinone (compound I and II) was reported as tyrosinase inhibitors with IC50 = 1.95 μM and 2.79 μM against tyrosinase in diphenolase activity, respectively.27 In 2016, hydroxypyridinone derivatives containing an oxime ether moiety (compound III and IV) were found to be another potent inhibitor with an IC50 value of 2.04 μM and 1.60 μM against monophenolase activity28 (Fig. 1). On the other hand, in previous studies, the benzyl hydrazide group derivatives were synthesized with proper tyrosinase inhibitory potential (compounds V and VI) (Fig. 1).29 Given the critical tyrosinase activities of 3-hydroxypyridine-4-one and benzyl hydrazide as tyrosinase inhibitors, we followed fragment-based hybridization of 3-hydroxypyridine-4-one and benzyl hydrazide groups to design a new series of tyrosinase inhibitors.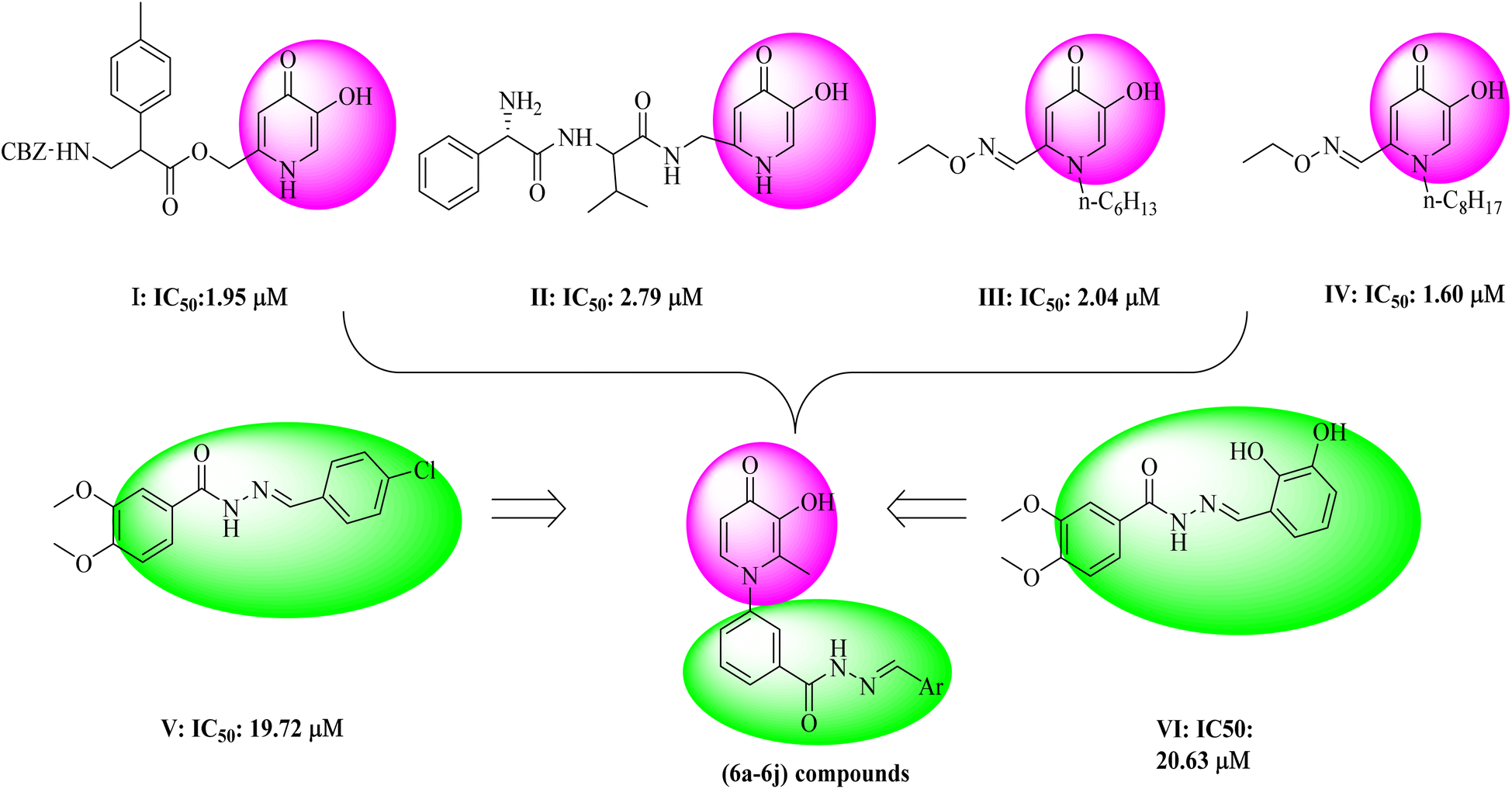 | ||
| Fig. 1 The design approach of synthesized compounds. | ||
In the present work, the synthesis of some 3-hydroxypyridine-4-one benzyl hydrazide derivatives was assigned. The activity of these compounds as tyrosinase inhibitors was evaluated, and also antioxidant activity was examined. In addition, molecular docking studies were applied to understand the binding poses and interactions of compounds in the active site of the enzyme. The physicochemical properties of these compounds were also determined. As well as DFT calculations were applied to investigate the electronic structure of molecules.
2. Results and discussion
2.1 Chemistry
Synthesis of 3-hydroxypyridine-4-one benzyl-hydrazide derivatives was executed in four steps according to our previously reported procedure.30 In this process, maltol (1) was used as the starting material that reacted with 3-aminobenzoic acid (2) to drive the compound (3). In the next step, the reaction of compound (3) with methanol in the presence of CDI as coupling agent and DMAP led to compound (4) in the form of an ester. Subsequently, the reaction proceeded to the desired intermediate with the acyl hydrazide motif (5) from the reaction of hydrazine hydrate and the compound (4). Eventually, the compound (5) was condensed with different aldehydes to yield the final products (6a–6j) through a Schiff base reaction (Fig. 2).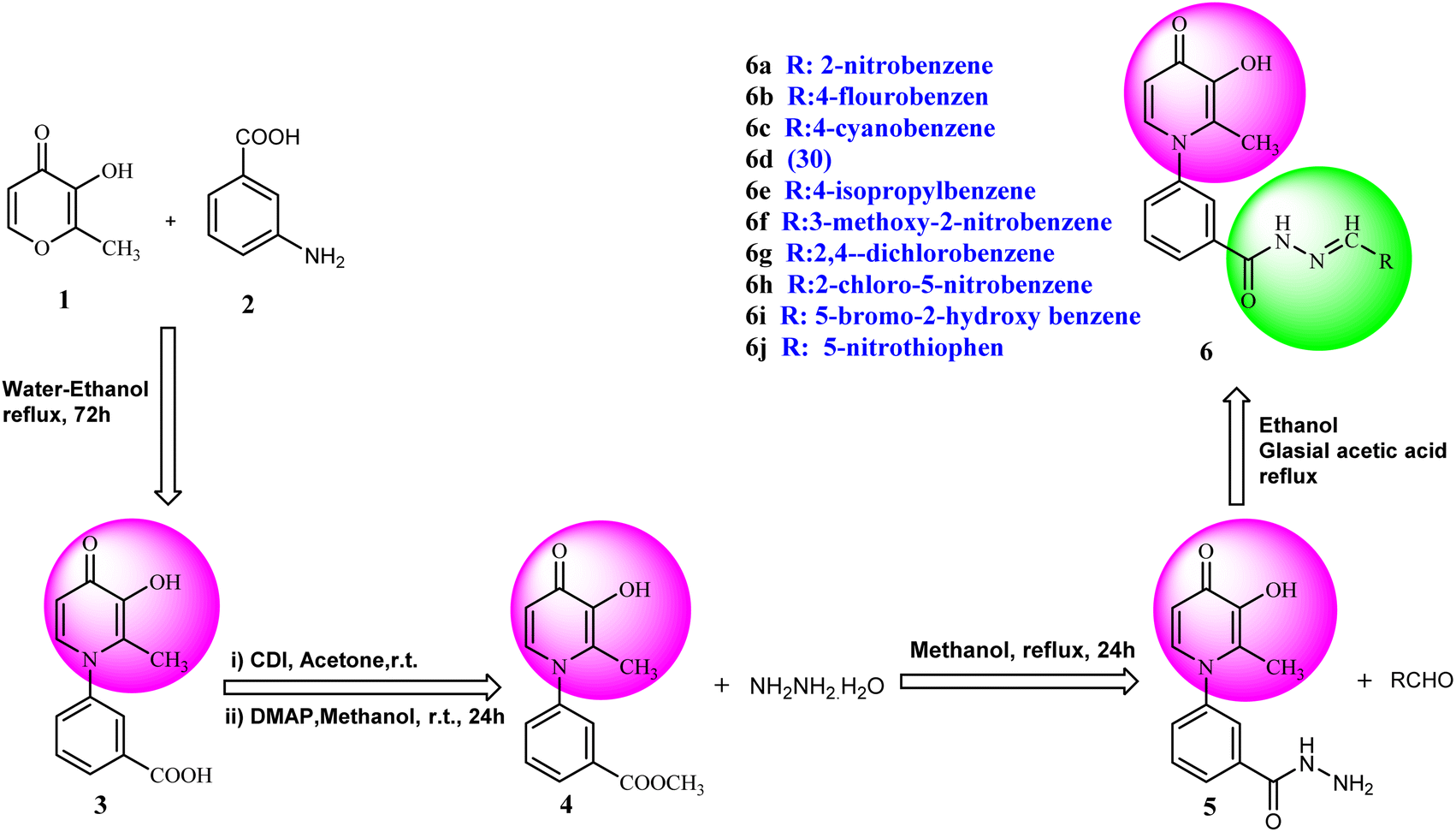 | ||
| Fig. 2 Outline for the synthesis of 3-hydroxypyridin-4-one derivatives (6a–6j). | ||
2.1.1.1 Synthesis of methyl 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)benzoate (4). Yield: 42.6%; m.p. 258 °C; IR (KBr, cm−1): 3424, 3080, 3050, 1720, 1626, 1574,1115, 1099; 1H-NMR (300 MHz, DMSO-d6) δH (ppm): 8.13 (s, 1H); 8.00 (s, 1H); 7.73–7.82 (m, 3H); 6.6 (d, 1H); 3.87 (s, 3H); 2 (s, 3H).
2.1.1.2 Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)benzohydrazide (5). Yield: 84%; m.p. 249 °C; IR (KBr, cm−1): 3329, 3219, 3050, 3020, 1666, 1632, 1576, 1524, 1206, 1096; 1H-NMR (300 MHz, DMSO-d6) δH (ppm): 9.9 (s, 1H); 7.95 (s, 1H); 7.8 (s, 1H); 7.62 (s, 2H); 7.575 (d, 1H); 6.215 (d, 1H); 4.6 (s, 2H); 1.96 (s, 3H).
2.1.1.3 Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N′-(2-nitrobenzylidene)benzohydrazide (6a). Yield: 75%; m.p. 235 °C; IR (KBr, cm−1): 3335, 3200, 3057, 3009, 1693, 1639, 1578, 1549; 1H-NMR (300 MHz, DMSO-d6) δH (ppm):12.35 (brs. 1H, NH), 8.90 (s, 1H), 8.20–8.14 (m, 3H), 8.06 (s, 1H), 7.89 (t, J = 6 Hz, 1H), 7.80–7.79 (m, 2H), 7.69–7.75 (m, 2H), 6.31 (d, J = 6 Hz, 1H), 3.83 (brs, 1H), 2.06 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δC (ppm): 173.25, 162.40, 148.74, 145.57, 144.02, 142.04, 138.41, 134.73, 134.29, 131.33, 131.10, 130.50, 129.07, 128.49, 126.56, 125.19, 111.51, 13.91; MS m/z (%): 392.2 (100).
2.1.1.4 Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N′-(4-fluorobenzylidene)benzohydrazide (6b). Yield: 67%; m.p. 265 °C; IR (KBr, cm−1): 3354, 3213, 3063, 3038, 1665, 1628, 1603, 1578, 1300; 1H-NMR (300 MHz, DMSO-d6) δH (ppm): 12.04 (s, 1H), 8.56 (s, 1H), 8.14–8.12 (m, 1H), 8.03 (s, 1H), 7.84–7.88 (m, 2H), 7.78 (d, J = 6 Hz, 2H), 7.69 (d, J = 9 Hz, 1H), 7.37 (t, J = 9 Hz, 2H), 6.32 (d, J = 6 Hz, 1H), 3.82 (brs, 1H), 2.06 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δC (ppm): 170.26, 164.92, 145.62, 142.05, 141.98, 138.53, 138.40, 135.15, 130.30, 130.12, 129.09, 128.11, 125.83, 123.89, 122.90, 116.65, 116.29, 111.56, 13.84; MS m/z (%): 199.1 (97), 365.3 (100).
2.1.1.5 Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N′-(4-cyanobenzylidene)benzohydrazide (6c). Yield: 82.6%; m.p. 205 °C; IR (KBr, cm−1): 3196, 2226, 1670; 1H-NMR (300 MHz, DMSO-d6) δH (ppm): 12.26 (brs, NH); 8.52 (s, 1H); 8.09 (s, 1H); 7.93 (m, 5H); 7.72 (s, 3H); 7.60 (s, 1H), 6.23 (s, 1H); 1.98 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δC (ppm): 170.25, 164.92, 162.42, 146.80, 145.63, 142.04, 139.05, 138.40, 135.18, 134.85, 133.27, 130.53, 130.20, 129.17, 128.19, 126.54, 119.11, 112.52, 111.53, 13.91; MS m/z (%): 372.2 (80); 244 (26); 226 (36); 199 (100); 154 (17); 128 (44) (100); 77.1 (44); 55.1 (64).
2.1.1.6 Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N′-(4-methylbenzylidene)benzohydrazide (6d). M.p. 330 °C; ref. 30.
2.1.1.7 Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N′-(4-isopropylbenzylidene)benzohydrazide (6e). Yield: 74.3%; m.p. 135 °C; 1H-NMR (300 MHz, DMSO-d6) δH (ppm): 1.26–1.28 (m, 6H); 2.06 (s, 3H); 2.98 (m, 1H); 6.32 (d, J = 9 Hz, 1H); 7.33–7.49 (m, 3H); 7.71–7.87 (m, 4H); 8.03–8.18 (m, 2H); 8.47 (s, 1H); 11.94 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δC (ppm): 170.24, 164.91, 162.07, 153.37, 148.84, 142.62, 142.01, 138.40, 135.23, 135.16, 130.29, 130.12, 129.12, 129.09, 128.12, 127.76, 127.34, 126.42, 125.83, 111.52, 33.87, 24.14, 13.85; MS m/z (%): 389.2 (49); 355 (18); 292 (13); 249 (28); 221 (33); 149 (100); 105 (62); 71 (43); 43 (68).
2.1.1.8 Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridine-1(4H)-yl)-N′-(3-methoxy-2-nitrobenzylidene)benzohydrazide (6f). Yield: 80%; m.p. 189 °C; IR (KBr, cm−1): 3210, 3094, 2990, 2930, 1682, 1630, 1574, 1555, 1533, 1377, 1302, 1202, 1076; 1H-NMR (300 MHz, DMSO-d6) δH (ppm): 12.14 (s, 1H); 8.36 (s, 1H); 8.07 (s, 1H); 7.97 (s, 1H); 7.59–7.78 (m, 5H); 7.43 (d, J = 9 Hz, 1H); 6.22 (d, J = 9 Hz, 1H); 3.93 (s, 3H); 2.00 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δC (ppm): 170.25, 162.18, 150.85, 145.62, 142.05, 141.45, 140.23, 138.39, 134.64, 132.36, 131.18, 130.53, 129.08, 126.52, 118.79, 115.34, 111.54, 57.31, 56.49, 19.02, 13.89; MS m/z (%): 422.2 (47.9); 244.1 (15); 228.1 (27.4); 200.1 (100); 76.1 (21.3).
2.1.1.9 Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N′-(2,4-dichlorobenzylidene)benzohydrazide (6g). Yield: 55%; m.p. 200 °C; IR (KBr, cm−1): 3300, 3208, 3067, 2924, 1668, 1626, 1433; 1H-NMR (300 MHz, DMSO-d6) δH (ppm): 12.21 (s, 1H), 8.80 (s, 1H), 8.1–8.09 (m, 1H), 8.06–8.03 (m, 1H), 8.00 (s, 1H), 7.75–7.74 (m, 2H), 7.64 (d, 1H, J = 9 Hz), 7.56–7.54 (m, 1H), 6.25 (s, 1H), 2.01 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δC (ppm): 170.27, 162.24, 145.71, 145.35, 142.55, 142.06, 138.39, 135.75, 134.81, 134.42, 131.11, 131.08, 131.01, 130.52, 129.90, 129.04, 128.60, 126.51, 111.52, 13.90; MS m/z (%): 415.1 (55), 228.1 (30), 199.1 (100), 154.1 (15).
2.1.1.10 Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N′-(2-chloro-5-nitrobenzylidene)benzohydrazide (6h). Yield: 70%; m.p. 316 °C; IR (KBr, cm−1): 3366, 3210, 3050, 1674, 1632, 1611, 1564, 1526, 1342, 1302, 1198, 1045; 1H-NMR (300 MHz, DMSO-d6) δH (ppm):12.38 (s, 1H); 8.83 (s, 1H); 8.695 (s, 1H); 8.23 (dd, J = 15 Hz, J = 3 Hz, 1H); 8.11 (s, 1H); 8.02 (s, 1H); 7.82 (d, 1H J = 15 Hz); 7.76 (s, 2H); 7.65 (d, 1H J = 6 Hz); 6.28 (d, 1H, J = 9 Hz); 2.02 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δC (ppm): 170.25, 162.04, 156.34, 146.01, 145.60, 142.04, 141.66, 138.42, 134.58, 134.24, 131.07, 130.43, 129.12, 126.49, 121.88, 119.14, 111.57, 111.00, 13.93; MS m/z (%): 331.1 (13.3); 244.1 (81.3); 226.1 (20); 199.1 (42.6); 163.1 (100); 133.1 (14.6); 90.1 (37.3).
2.1.1.11 Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N′-(5-bromo-2-hydroxybenzylidene)benzohydrazide (6i). Yield: 32%; m.p. 310 °C; IR (KBr, cm−1): 3356, 3213, 3073, 2932, 1684, 1630; 1H-NMR (300 MHz, DMSO-d6) δH (ppm): 12.22 (s, 1H), 8.62 (s, 1H), 8.1 (t, J = 3 Hz, 1H), 8.00 (s, 1H), 7.83 (d, J = 2.6 Hz, 1H), 7.74 (d, J = 3 Hz, 2H), 7.64 (d, J = 9 Hz, 1H), 7.44 (dd, J = 9 Hz, J = 3 Hz, 1H) 6.91 (d, J = 9 Hz, 1H), 6.26 (d, J = 9 Hz, 1H), 2.01 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δC (ppm): 170.23, 162.03, 156.86, 146.07, 145.62, 142.04, 138.41, 134.58, 134.22, 131.05, 130.52, 129.21, 129.07, 126.48, 121.87, 119.14, 111.58, 110.99, 109.30, 13.91; MS m/z (%): 441.1 (10), 227.1 (100), 201.1 (70), 170.1 (11), 76.1 (12.5).
2.1.1.12 Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N'-((5-nitrothiophen-2-yl)methylene)benzohydrazide (6j). Yield: 53%; m.p. 248 °C; 1H-NMR (300 MHz, DMSO-d6) δH (ppm): 12.4 (s, 1H), 8.69 (s, 1H), 8.26–7.98 (m, 4H), 7.62–7.45 (m, 3H), 6.26 (s, 1H), 3.60 (s, 1H), 2.00 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δC (ppm): 186.5, 162.5, 151.5, 146.9, 146.8, 145.6, 142.1, 138.4, 136.2, 134.6, 131.2, 130.9, 130.6, 130.5, 129.1, 126.6, 111.6, 13.9.
2.2 Inhibitory effect of analogs on the activity of mushroom tyrosinase
The tyrosinase inhibitory activity of all synthesized compounds is shown in Table 1. Seven derivatives present an appropriate inhibitory effect with an IC50 range of 25.29–64.13 μM compared to kojic acid as the positive control. The compounds 6i and 6d revealed the highest activity on tyrosinase inhibition with IC50 values of 25.29 and 26.36 μM. In the assessment of the mono-substituted group (6a–6e), it can be stated that the presence of an electron-donating group, such as methyl, at the para position of the phenyl moiety, can improve the tyrosinase inhibitor activity, just as the introduction of the electron-withdrawing group (NO2) in the ortho position showed good activity with an IC50 = 28.57 μM. Compounds 6b and 6e bearing fluoro and isopropyl groups at C4 of the phenyl ring also showed appropriate tyrosinase inhibitory activity. Finally, the presence of the cyanide group led to diminished activity (IC50 > 100 μM). In the case of di-substitution (6f–6i), compound 6i (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OH, Br) had the best anti-tyrosinase potency with an IC50 = 64.13 μM. The substitution of chloro at the meta and ortho positions led to significantly decreased activity in compound 6g. In general, it can be seen that most of the multi-substitution compounds such as 6f, 6h bearing R = 2-NO2, 3-OCH3 and R = 2-Cl, 5-NO2 had weak inhibitory activity, and compound 6j containing 2-nitro thiol ring also had low potential. Taken together, mono-substituted compounds had better diphenolase inhibitory properties than multi-substitution series (6f–6i). The SAR study indicated that positioning methyl at a phenyl ring, such as compound 6d, led to an increase in activity in series 1, and concerning series 2, placing the brome and hydroxyl groups at 2, 5 positions caused improved activity compared to other compounds.
OH, Br) had the best anti-tyrosinase potency with an IC50 = 64.13 μM. The substitution of chloro at the meta and ortho positions led to significantly decreased activity in compound 6g. In general, it can be seen that most of the multi-substitution compounds such as 6f, 6h bearing R = 2-NO2, 3-OCH3 and R = 2-Cl, 5-NO2 had weak inhibitory activity, and compound 6j containing 2-nitro thiol ring also had low potential. Taken together, mono-substituted compounds had better diphenolase inhibitory properties than multi-substitution series (6f–6i). The SAR study indicated that positioning methyl at a phenyl ring, such as compound 6d, led to an increase in activity in series 1, and concerning series 2, placing the brome and hydroxyl groups at 2, 5 positions caused improved activity compared to other compounds.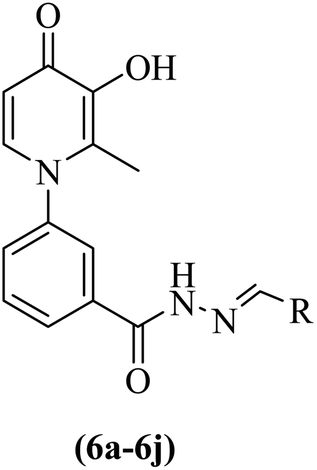
| ID | R | Inhibition%a ± SEMb | IC50c (μM) | ID | R | Inhibition%a ± SEMb | IC50c (μM) |
|---|---|---|---|---|---|---|---|
| a Values for tested compounds and kojic acid were measured at 50 μM.b Values for 3 repetitions of the experiment.c 50% inhibitory concentration (IC50). | |||||||
| 6a | 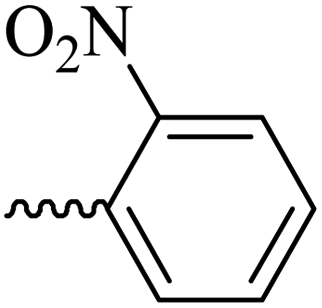 |
46.07 ± 8.21 | 28.57 | 6f | 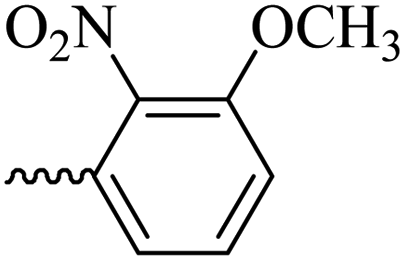 |
34.83 ± 3.83 | >100 |
| 6b | 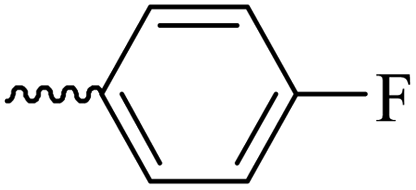 |
43.08 ± 3.41 | 33.41 | 6g | 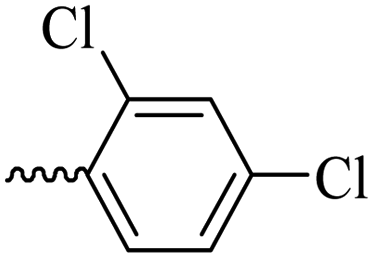 |
28.14 ± 3.97 | 43.65 |
| 6c | 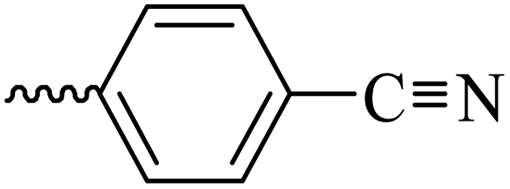 |
30.84 ± 2.85 | >100 | 6h | 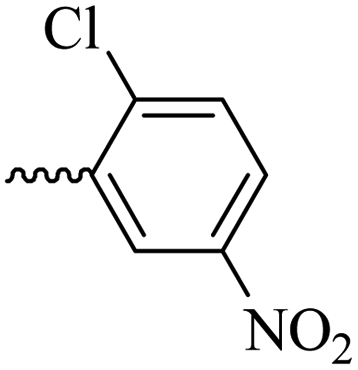 |
11.38 ± 2.59 | >100 |
| 6d | 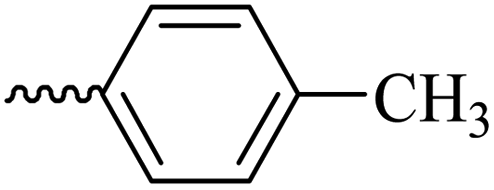 |
49.04 ± 7.51 | 26.36 | 6i | 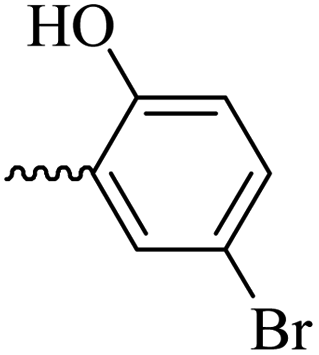 |
49.63 ± 12.63 | 25.29 |
| 6e | 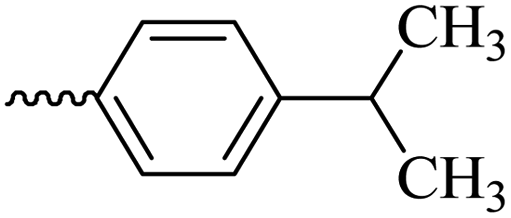 |
41.69 ± 4.07 | 34.27 | 6j | 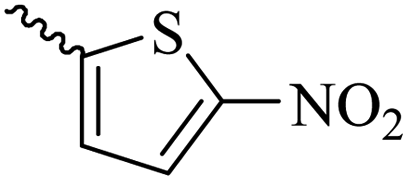 |
40.35 ± 5.00 | 64.13 |
| Kojic acid | — | 62.67 ± 7.88 | 16.68 | ||||
2.3 Determination of the inhibition type
The inhibition type on tyrosinase enzyme was determined for the compound 6i as the best antityrosinase. For consideration of the inhibition type, Lineweaver–Burk plots (plot of 1/V versus 1/[S]) were drawn with several concentrations of 6i (as the inhibitor) and L-Dopa (as the substrate).31 The results showed that the Km value was increased along with increasing the concentration of inhibitor while Vmax was not affected by the concentration. According to the obtained results, it can be concluded that compound 6i acts as the competitive inhibitor of tyrosinase enzyme. It can be assumed that the structural similarity of compound 6i and the substrate of the mushroom tyrosinase enzyme (L-Dopa), especially the presence of the phenolic group, has caused competition with the substrate to bind to the active site of the enzyme (Fig. 3).32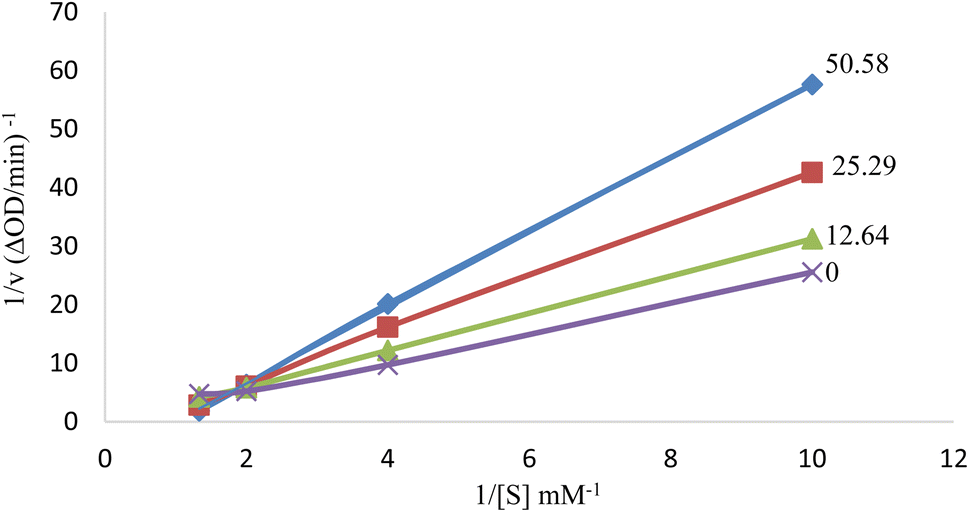 | ||
| Fig. 3 Lineweaver–Burk plot for tyrosinase enzyme inhibition by different concentrations of 6i in the presence of L-Dopa (double reciprocal plot, n = 3). | ||
2.4 Free radical scavenging activity
The radical scavenging activity toward DPPH was represented in Table 2 in EC50 values. The analogs (6a, 6b, and 6e) having 2-NO2, 4-F, and 4-iso-propyl moiety were found to have excellent radical scavenging potential with EC50 = 0.032, 0.012, and 0.014 mM, respectively. Further investigations demonstrated that the presence of cyanide, methyl, and multi-substitution groups also led to a decrease in activity. The structure–activity relationship revealed that the presence of an electron-donating group, such as isopropyl, and an electron-withdrawing group, such as a fluorine atom, on the phenyl ring caused an improvement in activity, and the existence of a nitro group at the ortho position of the phenyl ring increased the DPPH activity.| ID | DPPH (%) | EC50 (mM) | Compound | DPPH (%) | EC50 (mM) |
|---|---|---|---|---|---|
| 6a | 77.15 | 0.032 ± 0.01 | 6f | 43.67 | 0.168 ± 0.03 |
| 6b | 71.42 | 0.012 ± 0.06 | 6g | 39.11 | 0.191 ± 0.07 |
| 6c | 43.67 | 0.216 ± 0.08 | 6h | 31.91 | >400 |
| 6d | 39.13 | 0.369 ± 0.01 | 6i | 42.64 | 0.185 ± 0.13 |
| 6e | 61.44 | 0.014 ± 0.01 | 6j | 42.74 | 0.383 ± 0.12 |
| Quercetin | 74.33 | 0.0089 |
2.5 Molecular docking study
Molecular docking was used to predict the binding mode and interaction of potent compound (6i) in the active site of the tyrosinase enzyme (PDB ID: 2y9x). In the binding pocket of the tyrosinase enzyme, two copper atoms interacted with residues, including His85, His61, and His94 in one subunit and His259, His263, and His296 in another subunit.33 The copper atoms are essential to the enzyme's catalytic activity.34 If synthesized compounds interact with the residues of copper atoms, they can inhibit the tyrosinase enzyme. The molecular docking results on compound 6i showed that this compound involved His85 and His244 by hydrogen bonding, which were vital residues in the binding pocket.35 Also, the hydroxyl group in the 1,4-dihydroxypyridinone ring established a hydrogen bond with residues of Asn81 and Cys83. Another important interaction was the T-shaped interaction with Phe264. The 2D and 3D interactions for compound 6i are shown in Fig. 4.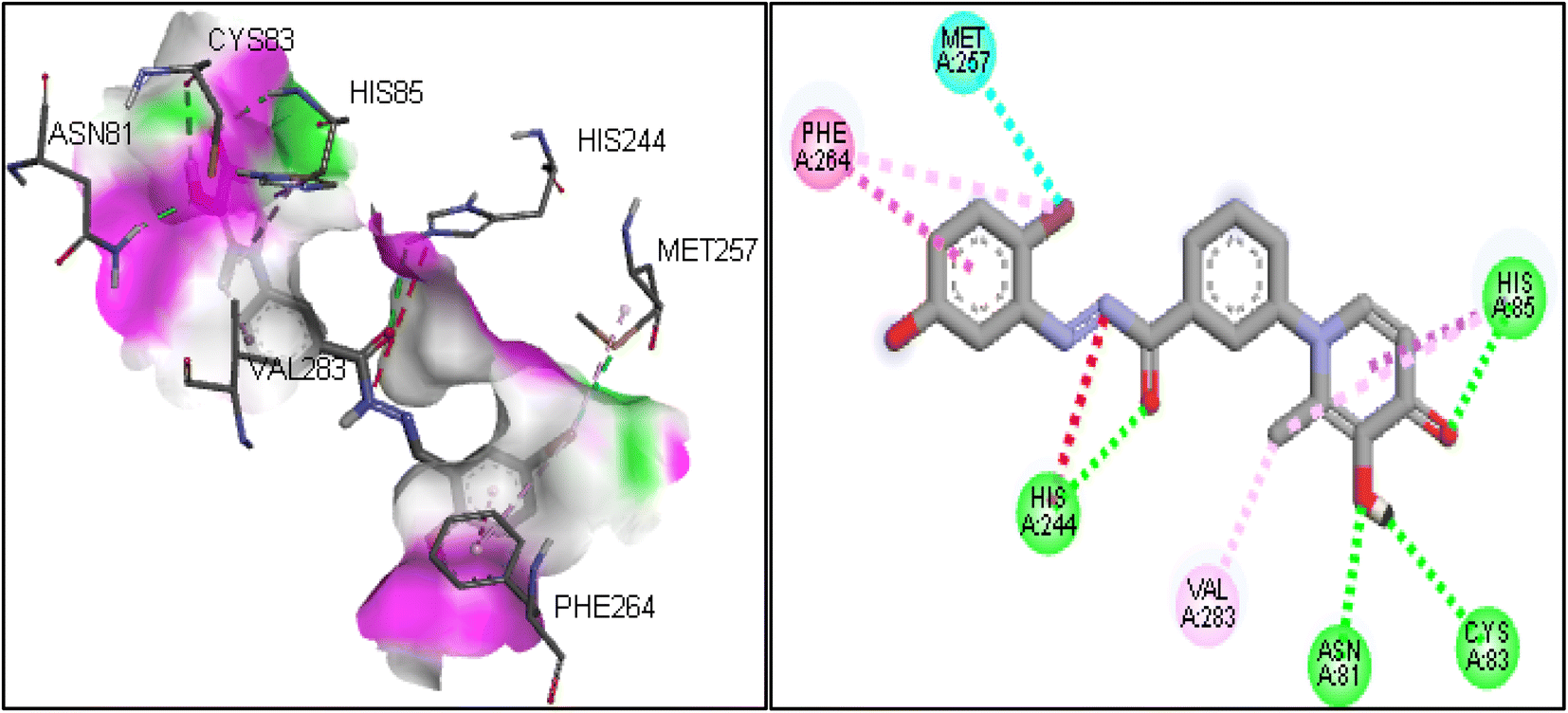 | ||
| Fig. 4 Interaction and binding mode of compound 6i in the active site of 2y9x. | ||
2.6 DFT analysis
The DFT analysis calculated the compound 6i as the best antityrosinase activity and the compound 6 h as one of the compounds with the lowest antityrosinase activity. This analysis is valuable because it predicts the reactivity of compounds in chemical reactions. One of the calculated parameters is the homo, LUMO energies, and the energy gap between them, which is shown for the compounds in Fig. 5. The HOMO–LUMO gap for 6h and 6i was obtained at 2.56 ev and 2.99 ev, respectively. As can be seen, the HOMO is on the 3-hydroxypyridine-4-one ring and the LUMO is located on the rest of the molecules for compounds of 6h and 6i. The electrostatic potential (ESP) surface or map of compounds is shown in Fig. 6. The negative and positive charge sites are indicated with red and blue colors, respectively. The thermochemical parameters were calculated and are presented in Table 3. The results for the total energy (E), enthalpy (H), and Gibbs free energy (G) showed that compound 6i is more thermodynamically stable than 6h. Also according to theoretical calculations, IR spectra for both compounds are depicted in Fig. 7. As it is clear from the spectra of the two compounds, the obtained results confirm the structure of both compounds, which is consistent with the laboratory results.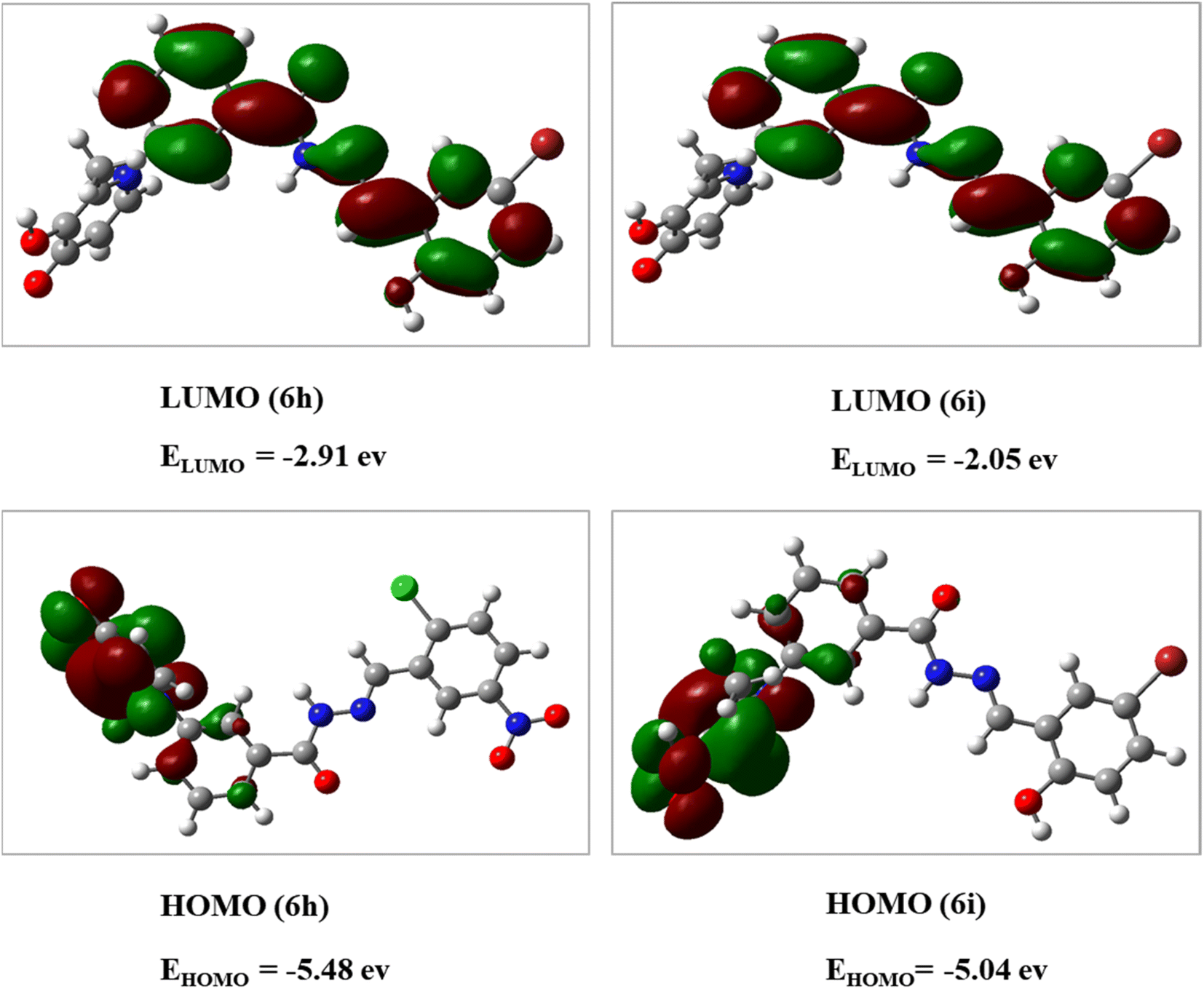 | ||
| Fig. 5 Calculated LUMO and HOMO for 6h and 6i at B3LYP/6-31+G** level of theory. | ||
 | ||
| Fig. 6 Electron density maps of 6h (left) and 6i (right) at B3LYP/6-31+G** level of theory. | ||
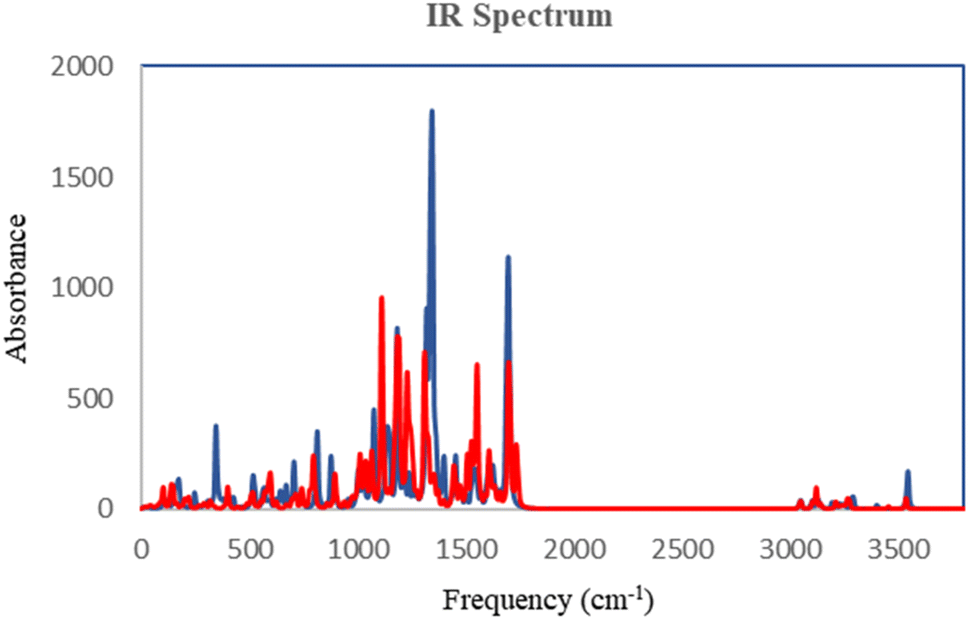 | ||
| Fig. 7 Calculated IR data for 6h (red) and 6i (blue) at B3LYP/6-31+G** level of theory. | ||
2.7 In silico ADME properties
The physicochemical properties of the studied compounds were obtained using the online software SwissADME. The pharmacokinetic properties of the tested compounds are given in Table 4. For a compound to pass the drug-likeness test, five characteristics of MW ≤ 500 Da, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) p < 5, nHBD ≤ 5, nHBA ≤ 10, and TPSA ≤ 140 Å must be stated in the range.36 As observed in Table 4, all compounds were within an acceptable range. In the logKp parameter (skin permeability), the range of −1 to −8 indicates penetration into the skin, and for our compounds, the values were obtained above −8. In the bioavailability radar shown in the table, the colored zone indicates the suitable physicochemical space for oral bioavailability. It proved that all the synthetic compounds were placed in a suitable space.37 The results showed that the oral bioavailability of derivatives was good and could be described as “drug-like”.
p < 5, nHBD ≤ 5, nHBA ≤ 10, and TPSA ≤ 140 Å must be stated in the range.36 As observed in Table 4, all compounds were within an acceptable range. In the logKp parameter (skin permeability), the range of −1 to −8 indicates penetration into the skin, and for our compounds, the values were obtained above −8. In the bioavailability radar shown in the table, the colored zone indicates the suitable physicochemical space for oral bioavailability. It proved that all the synthetic compounds were placed in a suitable space.37 The results showed that the oral bioavailability of derivatives was good and could be described as “drug-like”.
| ID | M.W. | nRB | nHBA | nHBD | TPSA | logp |
Lipinski rule/violation | logkp |
|---|---|---|---|---|---|---|---|---|
| 6a | 392.36 | 6 | 6 | 2 | 129.51 | 0.71 | 0 | −6.71 |
| 6b | 365.36 | 5 | 5 | 2 | 83.69 | 2.05 | 0 | −7.02 |
| 6c | 372.38 | 5 | 5 | 2 | 107.48 | 1.02 | 0 | −7.33 |
| 6d | 361.39 | 5 | 4 | 2 | 83.69 | 1.89 | 0 | −6.64 |
| 6e | 389.45 | 6 | 4 | 2 | 83.69 | 2.32 | 0 | −6.27 |
| 6f | 422.39 | 7 | 7 | 2 | 138.74 | 0.44 | 0 | −6.91 |
| 6g | 416.26 | 5 | 4 | 2 | 83.69 | 2.64 | 0 | −5.84 |
| 6h | 426.81 | 6 | 6 | 2 | 129.51 | 1.21 | 0 | −6.47 |
| 6i | 442.26 | 5 | 5 | 3 | 103.92 | 1.73 | 0 | −6.65 |
| 6j | 398.39 | 6 | 6 | 2 | 157.75 | 0.29 | 0 | −6.49 |
3. Material and methods
3.1 Apparatus
All reagents and solvents were purchased from commercial suppliers. Analytical thin layer chromatography (TLC) was performed on MERCK-precoated silica gel 60-F254 (0.5 mm) aluminum plates. Thin layer chromatography (TLC) was used to control the progress of the reaction on MERCK precoated silica gel 60-F254 (0.5 mm) aluminum plates. Melting points were determined on a Kofler hot stage apparatus. The IR spectra were obtained using Nicolet's FT-IR Magna 550 spectrometer (KBr disks). 1H and 13C NMR spectra were recorded on a Bruker 300 MHz NMR instrument using CDCl3 as solvent and TMS as an internal standard. The chemical shifts (δ) and coupling constants (J) are expressed in parts per million (ppm) and hertz (Hz), respectively.3.2 Synthesis
:1 chloroform/methanol).39 Consequently, the mixture was extracted with water and dichloromethane and concentrated to yield the white-colored intermediate4 (1 mmol) (20%, m.p. 160 °C). The compounds4 (0.5 mmol) and hydrazine hydrate (1 mmol) in the presence of methanol (5 mL) were reacted under reflux at 90 °C for 24 h. The obtained precipitate was filtered off and washed with cold methanol to furnish compound (5) (0.4 mmol) (80%, m.p.247 °C).:1). The crystalline products isolated by filtration were recrystallized in a desired ratio of ethyl acetate and petroleum ether to give the final products (6a–6j) (Fig. 2).3.3 Enzymatic assay for measurement of tyrosinase inhibition
The assessment of tyrosinase inhibition was measured using L-Dopa as the substrate according to previously reported methods.40 This method monitors the conversion of L-Dopa to dopachrome at 475 nm. Firstly, the serial dilution of derivatives was prepared and added to tyrosinase dissolved in phosphate buffer (pH = 6.8) in 96-well microplates. Then L-Dopa was added to the mixture after incubation for 20 min. The results were compared to kojic acid as a reference compound. The percentage of inhibition of the proposed compounds was calculated as follows:| % Inhibition = (Acontrol − Asample/Acontrol) × 100 |
3.4 Determination of the inhibition type
Kinetic analysis was performed on the best compound as antityrosinase activity, 6i. The concentrations of inhibitor were: 50.58, 25.29, 12.64, and 0 μM. Substrate (L-Dopa) concentrations were 0.5–10.0 mM in all kinetic studies. Determining the type of tyrosinase inhibition was done according to the protocol reported in previous studies. Maximum initial velocity was determined from the initial linear portion of absorbance up to 10 min after the addition of L-Dopa with 1 min interval. The Michaelis constant (Km) and the maximal velocity (Vmax) of the tyrosinase activity were determined by the Lineweavere–Burk plot at various concentrations of L-Dopa as a substrate. The inhibition type of the enzyme was evaluated by Lineweavere–Burk plots of the inverse of velocities (1/V) versus the inverse of substrate concentrations 1/[S] mM−1.3.5 Enzymatic assay for measurement of antioxidant
The antioxidant assay was evaluated using 1,1-diphenyl-2-picryl hydrazine (DPPH) radical scavenging according to the previously reported method.41 The decolorization of the DPPH solution after adding the tested compounds as antioxidant agents was measured at 517 nm. The percentage of DPPH radical scavenging activity was calculated as follows:DPPH radical scavenging, % = (A0 − As/A0) × 100 where A0 is the absorbance of the DPPH solution and as is the absorbance of the sample. The results were compared to ascorbic acid as a positive control.
3.6 Molecular docking study
Molecular docking was conducted using AutoDock 4.2 and AutoDock Tools 1.5.4 (ADT).42 Firstly, the 3D crystal structure of the tyrosinase enzyme complexed with tropolone was downloaded from the protein data bank (https://www.rcsb.org/) with PDB ID: 2y9x.43 Water, ions, and cocrystals were deleted to prepare the receptor file; hydrogens were added; and Gasteiger charges were computed. The designed structures were drawn, minimized using ChemBio 3D, and converted to pdbqt format. The grid box was set with dimensions of 40 × 40 × 40, and docking runs were set at 100. The interaction and binding modes were visualized by the Discovery Studio 2016 client.3.7 DFT analysis
The density functional theory (DFT) was performed by Gaussian 09 at the B3LYP/6-31+G** level of theory for the compounds of 6h and 6i. DFT analysis computed the HOMO and LUMO energies, electron surface potential (ESP), FT-IR vibrational frequencies, and the thermochemistry parameters with B3LYP/6-31+G**.3.8 In silico ADME profile measurement
The studied compounds' pharmacokinetic properties, including absorption, distribution, metabolism, and excretion, were obtained using SWISSADME online software.44 To determine drug-like and oral bioavailability, parameters of molecular weight, number of hydrogen bonds of acceptors, number of hydrogen bonds of donors, number of rotatable bonds, and TPSA were considered.4. Conclusions
A series of novel 3-hydroxypyridine-4-ones containing benzyl hydrazide with different substitutions were synthesized and assayed as antityrosinase and antioxidant agents. Compound 6i, containing 5-hydroxyl and 2-bromobenzyl hydrazide motives, had the best anti-tyrosinase activity among all compounds. Also, compound 6b showed potent activity against antioxidants. The SAR study showed that positioning methyl at the phenyl ring and the brome and hydroxyl groups at the 2, 5 position of phenyl enhanced the activity. In addition, molecular docking analysis revealed that the high tyrosinase inhibitory activity of 6i may be due to the formation of strong interactions with the critical residues in the active site of the tyrosinase enzyme. The result of the kinetic assay showed a competitive type of inhibition by 6i. The DFT analysis showed that the HOMO is located on a 3-hydroxypyridin-4-one ring for the studied compounds and also, the compound 6i as the best antityrosinase compound was thermodynamically stable. Finally, the ADME profile of the proposed compounds proved suitable for bioavailability and drug-likeness. The synthesized compounds can be considered promising leads in tyrosinase studies.Data availability
The data sets used and analyzed during the current study are available from the corresponding author upon reasonable request. We have presented all data in the form of Tables and Figures. The PDB code (2y9x) was retrieved from the protein data bank (https://www.rcsb.org/), https://www.rcsb.org/structure/2y9x.Author contributions
Mehdi Khoshneviszadeh supervised the biological tests. Fateme Zare prepared the manuscript and performed the docking study. Leila Emami wrote the biological part and contributed to the preparation of the manuscript. Bahareh Hassani synthesized the compounds. Razieh Fazel contributed to the synthesis of compounds. Negin Kave performed the biological assay. Razieh Sabet revised the manuscript and edited it. Hossein Sadeghpour supervised the study. All authors read and approved the final manuscript.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
Financial assistance from the Shiraz University of Medical Sciences through grant numbers 25721 and 24175 is gratefully acknowledged.References
- U. Ghani, Carbazole and hydrazone derivatives as new competitive inhibitors of tyrosinase: experimental clues to binuclear copper active site binding, Bioorg. Chem., 2019, 83, 235–241 CrossRef CAS PubMed.
- N. Fujieda, K. Umakoshi, Y. Ochi, Y. Nishikawa, S. Yanagisawa and M. Kubo, et al., Copper–oxygen dynamics in the tyrosinase mechanism, Angew. Chem., 2020, 132(32), 13487–13492 CrossRef.
- D. L. Abd Razak, N. H. M. Fadzil, A. Jamaluddin, N. Y. Abd Rashid, N. A. Sani and M. A. Manan, Effects of different extracting conditions on anti-tyrosinase and antioxidant activities of Schizophyllum commune fruit bodies, Biocatal. Agric. Biotechnol., 2019, 19, 101116 CrossRef.
- J. Li, L. Feng, L. Liu, F. Wang, L. Ouyang and L. Zhang, et al., Recent advances in the design and discovery of synthetic tyrosinase inhibitors, Eur. J. Med. Chem., 2021, 224, 113744 CrossRef CAS PubMed.
- S. Y. Lee, N. Baek and T.-g Nam, Natural, semisynthetic and synthetic tyrosinase inhibitors, J. Enzyme Inhib. Med. Chem., 2016, 31(1), 1–13 CrossRef CAS PubMed.
- S. Karimian, S. Ranjbar, M. Dadfar, M. Khoshneviszadeh, M. Gholampour and A. Sakhteman, et al., 4H-benzochromene derivatives as novel tyrosinase inhibitors and radical scavengers: synthesis, biological evaluation, and molecular docking analysis, Mol. Diversity, 2021, 25, 2339–2349 CrossRef CAS PubMed.
- S. Karimian, F. Kazemi, M. Attarroshan, M. Gholampour, S. Hemmati and A. Sakhteman, et al., Design, synthesis, and biological evaluation of symmetrical azine derivatives as novel tyrosinase inhibitors, BMC Chem., 2021, 15, 1–11 CrossRef PubMed.
- M. Hajimiri, M. Khosravikia, M. Khoshneviszadeh, K. Pedrood, S. Z. Hosseini and M. S. Asgari, et al., Rational design, synthesis, in vitro, and in silico studies of chlorophenylquinazolin-4 (3H)-one containing different aryl acetohydrazides as tyrosinase inhibitors, Chem. Biodiversity, 2022, 19(7), e202100964 CrossRef CAS PubMed.
- Y. T. Wen, Y. Q. Liang, W. M. Chai, Q. M. Wei, Z. Y. Yu and L. J. Wang, Effect of ascorbic acid on tyrosinase and its anti-browning activity in fresh-cut Fuji apple, J. Food Biochem., 2021, 45(12), e13995 CrossRef CAS PubMed.
- K. Hałdys and R. Latajka, Thiosemicarbazones with tyrosinase inhibitory activity, MedChemComm, 2019, 10(3), 378–389 RSC.
- H. K. Lee, J. W. Ha, Y. J. Hwang and Y. C. Boo, Identification of L-cysteinamide as a potent inhibitor of tyrosinase-mediated dopachrome formation and eumelanin synthesis, Antioxidants, 2021, 10(8), 1202 CrossRef CAS PubMed.
- Y. Matoba, K. Oda, Y. Muraki and T. Masuda, The basicity of an active-site water molecule discriminates between tyrosinase and catechol oxidase activity, Int. J. Biol. Macromol., 2021, 183, 1861–1870 CrossRef CAS PubMed.
- X. Zhai, R. A. Ward, P. Doig and A. Argyrou, Insight into the therapeutic selectivity of the irreversible EGFR tyrosine kinase inhibitor osimertinib through enzyme kinetic studies, Biochemistry, 2020, 59(14), 1428–1441 CrossRef CAS PubMed.
- D.-F. Li, P.-P. Hu, M.-S. Liu, X.-L. Kong, J.-C. Zhang and R. C. Hider, et al., Design and synthesis of hydroxypyridinone-L-phenylalanine conjugates as potential tyrosinase inhibitors, J. Agric. Food Chem., 2013, 61(27), 6597–6603 CrossRef CAS PubMed.
- P. García-Molina, F. García-Molina, J. A. Teruel-Puche, J. N. Rodríguez-López, F. García-Cánovas and J. L. Muñoz-Muñoz, Considerations about the kinetic mechanism of tyrosinase in its action on monophenols: a review, Mol. Catal., 2022, 518, 112072 CrossRef.
- J. Tang, H. T. Do, A. D. Huber, M. C. Casey, K. A. Kirby and D. J. Wilson, et al., Pharmacophore-based design of novel 3-hydroxypyrimidine-2,4-dione subtypes as inhibitors of HIV reverse transcriptase-associated RNase H: tolerance of a nonflexible linker, Eur. J. Med. Chem., 2019, 166, 390–399 CrossRef CAS PubMed.
- J. P. Holt-Martyn, R. Chowdhury, A. Tumber, T. L. Yeh, M. I. Abboud and K. Lippl, et al., Structure-activity relationship and crystallographic studies on 4-hydroxypyrimidine HIF prolyl hydroxylase domain inhibitors, ChemMedChem, 2020, 15(3), 270–273 CrossRef CAS PubMed.
- P. Shirvani, N. Fayyazi, S. Van Belle, Z. Debyser, F. Christ and L. Saghaie, et al., Design, synthesis, in silico studies, and antiproliferative evaluations of novel indolin-2-one derivatives containing 3-hydroxy-4-pyridinone fragment, Bioorg. Med. Chem. Lett., 2022, 128784 CrossRef CAS PubMed.
- S. Chaves, L. Piemontese, A. Hiremathad and M. A. Santos, Hydroxypyridinone derivatives: a fascinating class of chelators with therapeutic applications-an update, Curr. Med. Chem., 2018, 25(1), 97–112 CrossRef CAS PubMed.
- K. Chen, L.-L. Shao, Y.-F. Huo, J.-M. Zhou, Q. Zhu and R. C. Hider, et al., Antimicrobial and antioxidant effects of a hydroxypyridinone derivative containing an oxime ether moiety and its application in shrimp preservation, Food Control, 2019, 95, 157–164 CrossRef CAS.
- Z. Zarrabi, L. Saghaie, A. Fassihi, N. Pestechian and S. Saberi, Synthesis and comparison of anti-leishmania major activity of antimony and iron complexes of 3-hydroxypyran-4-one and 3-hydroxypyridine-4-one as bi-dentate ligands, J. Rep. Pharm. Sci., 2020, 9(2), 177 CrossRef CAS.
- C. Zhang, K. Yang, S. Yu, J. Su, S. Yuan and J. Han, et al., Design, synthesis and biological evaluation of hydroxypyridinone-coumarin hybrids as multimodal monoamine oxidase B inhibitors and iron chelates against Alzheimer's disease, Eur. J. Med. Chem., 2019, 180, 367–382 CrossRef CAS PubMed.
- H. Sadeghi-Aliabadi, M. A. Zanjanchi, L. Saghaie and M. Borzoei, Evaluation of the cytotoxic effect of hydroxypyridinone derivatives on HCT116 and SW480 colon cancer cell lines, Pharm. Chem. J., 2019, 53(5), 388–391 CrossRef CAS.
- L. R. Singh, Y.-L. Chen, Y.-Y. Xie, W. Xia, X.-W. Gong and R. C. Hider, et al., Functionality study of chalcone-hydroxypyridinone hybrids as tyrosinase inhibitors and influence on anti-tyrosinase activity, J. Enzyme Inhib. Med. Chem., 2020, 35(1), 1562–1567 CrossRef CAS PubMed.
- Y.-Z. Zhu, K. Chen, Y.-L. Chen, C. Zhang, Y.-Y. Xie and R. C. Hider, et al., Design and synthesis of novel stilbene-hydroxypyridinone hybrids as tyrosinase inhibitors and their application in the anti-browning of freshly-cut apples, Food Chem., 2022, 385, 132730 CrossRef CAS PubMed.
- W. Xia, K. Chen, Y. Z. Zhu, C. J. Zhang, Y. L. Chen and F. Wang, et al., Antioxidant and anti-tyrosinase activity of a novel stilbene analogue as an anti-browning agent, J. Sci. Food Agric., 2022, 102(9), 3817–3825 CrossRef CAS PubMed.
- D.-Y. Zhao, M.-X. Zhang, X.-W. Dong, Y.-Z. Hu, X.-Y. Dai and X. Wei, et al., Design and synthesis of novel hydroxypyridinone derivatives as potential tyrosinase inhibitors, Bioorg. Med. Chem. Lett., 2016, 26(13), 3103–3108 CrossRef CAS PubMed.
- L.-L. Shao, X.-L. Wang, K. Chen, X.-W. Dong, L.-M. Kong and D.-Y. Zhao, et al., Novel hydroxypyridinone derivatives containing an oxime ether moiety: synthesis, inhibition on mushroom tyrosinase and application in anti-browning of fresh-cut apples, Food Chem., 2018, 242, 174–181 CrossRef CAS PubMed.
- Z. Dehghani, M. Khoshneviszadeh, M. Khoshneviszadeh and S. Ranjbar, Veratric acid derivatives containing benzylidene-hydrazine moieties as promising tyrosinase inhibitors and free radical scavengers, Bioorg. Med. Chem., 2019, 27(12), 2644–2651 CrossRef CAS PubMed.
- R. Sabet, A. Fassihi, B. Hemmateenejad, L. Saghaei, R. Miri and M. Gholami, Computer-aided design of novel antibacterial 3-hydroxypyridine-4-ones: application of QSAR methods based on the MOLMAP approach, J. Comput. -Aided Mol. Des., 2012, 26, 349–361 CrossRef CAS PubMed.
- S. Nie, F. Wu, J. Wu, X. Li, C. Zhou and Y. Yao, et al., Structure-activity relationship and antitumor activity of 1,4-pyrazine-containing inhibitors of histone acetyltransferases P300/CBP, Eur. J. Med. Chem., 2022, 237, 114407 CrossRef CAS PubMed.
- I. Kubo and I. Kinst-Hori, Tyrosinase inhibitors from anise oil, J. Agric. Food Chem., 1998, 46(4), 1268–1271 CrossRef CAS.
- A. Iraji, T. Adelpour, N. Edraki, M. Khoshneviszadeh, R. Miri and M. Khoshneviszadeh, Synthesis, biological evaluation and molecular docking analysis of vaniline–benzylidenehydrazine hybrids as potent tyrosinase inhibitors, BMC Chem., 2020, 14(1), 1–11 CrossRef PubMed.
- S. Zolghadri, A. Bahrami, M. T. Hassan Khan, J. Munoz-Munoz, F. Garcia-Molina and F. Garcia-Canovas, et al., A comprehensive review on tyrosinase inhibitors, J. Enzyme Inhib. Med. Chem., 2019, 34(1), 279–309 CrossRef CAS PubMed.
- R. Fazel, B. Hassani, F. Zare, H. Jokar Darzi, M. Khoshneviszadeh and A. Poustforoosh, et al., Design, synthesis, in silico ADME, DFT, molecular dynamics simulation, anti-tyrosinase, and antioxidant activity of some of the 3-hydroxypyridin-4-one hybrids in combination with acylhydrazone derivatives, J. Biomol. Struct. Dynam., 2023, 1–11 CrossRef PubMed.
- C. A. Lipinski, Lead-and drug-like compounds: the rule-of-five revolution, Drug Discovery Today: Technol., 2004, 1(4), 337–341 CrossRef CAS PubMed.
- K. M. Al Azzam, E.-S. Negim and H. Y. Aboul-Enein, ADME studies of TUG-770 (a GPR-40 inhibitor agonist) for the treatment of type 2 diabetes using SwissADME predictor: in silico study, J. Appl. Pharm. Sci., 2022, 12(4), 159–169 CAS.
- C. Gasche, T. Ahmad, Z. Tulassay, D. C. Baumgart, B. Bokemeyer and C. Büning, et al., Ferric maltol is effective in correcting iron deficiency anemia in patients with inflammatory bowel disease: results from a phase-3 clinical trial program, Inflamm. Bowel Dis., 2015, 21(3), 579–588 CrossRef PubMed.
- G. Kontoghiorghes, Chelators affecting iron absorption in mice, Arzneim. Forsch., 1990, 40(12), 1332–1335 CAS.
- Ş. Akın, E. A. Demir, A. Colak, Y. Kolcuoglu, N. Yildirim and O. Bekircan, Synthesis, biological activities and molecular docking studies of some novel 2,4,5-trisubstituted-1,2,4-triazole-3-one derivatives as potent tyrosinase inhibitors, J. Mol. Struct., 2019, 1175, 280–286 CrossRef.
- Z. Peng, Y. Li, L. Tan, L. Chen, Q. Shi and Q.-H. Zeng, et al., Anti-tyrosinase, antioxidant and antibacterial activities of gallic acid-benzylidenehydrazine hybrids and their application in preservation of fresh-cut apples and shrimps, Food Chem., 2022, 378, 132127 CrossRef CAS PubMed.
- H. Syahputra, M. Masfria, P. Hasibuan and I. Iksen, In silico docking studies of phytosterol compounds selected from Ficus religiosa as potential chemopreventive agent, Rasayan J. Chem., 2022, 15(2), 1080–1084 CrossRef CAS.
- A. Moulishankar and K. Lakshmanan, Data on molecular docking of naturally occurring flavonoids with biologically important targets, Data Brief, 2020, 29, 105243 CrossRef PubMed.
- M. Abdullahi and S. E. Adeniji, In silico molecular docking and ADME/pharmacokinetic prediction studies of some novel carboxamide derivatives as anti-tubercular agents, Chem. Afr., 2020, 3(4), 989–1000 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06490e |
| ‡ These authors are equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |