
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual anticancer and antibacterial activity of fluorescent naphthoimidazolium salts†
Dung Ngoc Tran‡
a,
Thi Thuy Hang Hoang‡b,
Sondavid Nandanwarc,
Vuong Thi Thanh Xuan Hod,
Van Thong Phame,
Huy Duc Vuf,
Xuan Ha Nguyeng,
Huy Trung Nguyenh,
Trang Van Nguyenh,
Thuy Kieu Van Nguyeni,
Dai Lam Tranh,
Myeongkee Parkj,
Songyi Lee *ij and
Thanh Chung Pham*h
*ij and
Thanh Chung Pham*h
aFaculty of Chemistry, Hanoi National University of Education, Hanoi, Vietnam
bDepartment of Chemistry, Soongsil University, Seoul 06978, South Korea
cEco-friendly New Materials Research Center, Korea Research Institute of Chemical Technology, 141 Gajeong-ro, Yuseong-gu, Daejeon City, Republic of Korea
dFaculty of Pharmacy, Dong A University, Danang, Vietnam
eR&D Center, Vietnam Education and Technology Transfer JSC, Cau Giay, Hanoi, Vietnam
fDepartment of Radiology, School of Medicine, Daegu Catholic University, Daegu 42472, Korea
gInstitute of Natural Products Chemistry, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam
hInstitute for Tropical Technology, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam. E-mail: ptchung@itt.vast.vn
iIndustry 4.0 Convergence Bionics Engineering, Pukyong National University, Busan 48513, Korea. E-mail: slee@pknu.ac.kr
jDepartment of Chemistry, Pukyong National University, Busan 48513, Korea
First published on 14th December 2023
Abstract
Cancer has emerged as a significant global health challenge, ranking as the second leading cause of death worldwide. Moreover, cancer patients frequently experience compromised immune systems, rendering them susceptible to bacterial infections. Combining anticancer and antibacterial properties in a single drug could lead to improved overall treatment outcomes and patient well-being. In this context, the present study focused on a series of hydrophilic naphthoimidazolium salts with donor groups (NI-R), aiming to create dual-functional agents with antibacterial and anticancer activities. Among these compounds, NI-TPA demonstrated notable antibacterial activity, particularly against drug-resistant bacteria, with MIC value of 7.8 μg mL−1. Furthermore, NI-TPA exhibited the most potent cytotoxicity against four different cancer cell lines, with an IC50 range of 0.67–2.01 μg mL−1. The observed high cytotoxicity of NI-TPA agreed with molecular docking and dynamic simulation studies targeting c-Met kinase protein. Additionally, NI-TPA stood out as the most promising candidate for two-photo excitation, fluorescence bioimaging, and localization in lysosomes. The study findings open new avenues for the design and development of imidazolium salts that could be employed in phototheranostic applications for cancer treatment and bacterial infections.
Introduction
Cancer is a complex and devastating disease characterized by the aberrant proliferation of cells, resulting in the formation of malignant tumors that can infiltrate and disrupt normal body tissues. It arises from genetic mutations in any cell type and can occur in various anatomical locations, leading to its pervasive nature throughout the body.1–4 As the second-leading cause of mortality worldwide, cancer poses formidable challenges to medical research such as drug resistance and side effects, necessitating continuous efforts in the development of effective drugs and therapies.5–11 Compounding the burden of cancer and its treatment is the impact on the patient's immune system, rendering patients more susceptible to opportunistic infections, particularly bacterial pathogens. The concomitant presence of multiple diseases further exacerbates the severity of each condition, leading to a challenging clinical scenario.12–16 Thus, incorporating therapeutic agents with dual functionality, possessing both anti-cancer and antibacterial activities, holds promise in enhancing patient quality of health and life.The emergence of infectious bacteria poses a growing threat to human health.17–19 Of particular concern is the prevalence of multi-drug resistance in bacterial strains, exemplified by methicillin-resistant Staphylococcus aureus (MRSA), which presents a significant challenge in the management of infectious diseases.20–22 The development of untreatable bacterial infections is a pressing issue that demands attention in the near future. The molecular design of antibacterial agents encompasses various mechanisms, which can be broadly classified into four categories: inhibition of cell wall synthesis, inhibition of nucleic acid synthesis, and inhibition of protein synthesis, disruption of the cell membrane.23,24 Among antibacterial agents, lipophilic complexes have shown promising interactions with the lipophilic layer of bacteria, thereby increasing the membrane permeability of bacteria and subsequent antibacterial activity.25 Enhancing the lipophilicity of compounds can improve the effectiveness of existing antibacterial drugs currently in clinical use and exploring drug combinations represent potential strategies.
Moreover, recent advances have led to the development of non-invasive diagnostic and therapeutic techniques.26,27 Notably, the concept of optical biopsy has garnered substantial attention as it fulfills the medical demand for minimally invasive alternatives.28,29 In this context, optical spectroscopic instruments have been harnessed to transform traditional pathology into real-time spectroscopy in conjunction with biomedical materials. Of particular significance is the emergence of certain chemotherapeutic agents with inherent biocompatible fluorophore properties, such as doxorubicin (DOX), camptothecin, and SN-38.30,31 Their dual functionality as both therapeutic agents and biocompatible fluorophores makes them valuable tools in the field of medical imaging and targeted therapies.32–35
Among therapeutic agents, imidazolium salts have been recognized for antibacterial and/or anticancer activity and their potential biomedicine applications owing to their hydrophilic nature.36–40 These observations align with our recent research endeavor, which involved the synthesis of a naphthoimidazolium-based fluorescent chemosensor specifically designed for hypochlorite-activated antibacterial activity.41 Our investigations have revealed that the imidazolium family, encompassing imidazolium, benzimidazolium, and halogenated imidazolium derivatives, has been extensively explored for its antibacterial and anticancer properties.36–42 However, the exploration of naphthoimidazolium derivatives remains relatively limited.43,44 Moreover, the past and current design strategies primarily revolve around the introduction of functional groups into the two nitrogen atoms, with little attention given to the examination of the substituent's impact at the carbon position situated between these two nitrogen atoms (Fig. 1).36–45 Furthermore, it is noteworthy that π-system-expanded imidazolium salts exhibit a redshift in absorption and emission wavelength.41,43,44,46,47 Our recent inquiry has revealed that NI-H and NI-Cbz naphthoimidazolium, manifest heightened fluorescence emission in both organic and highly aqueous environments.47 The photophysical attributes exhibited by naphthoimidazolium render it a promising candidate for applications in fluorescent imaging and phototheranostics.
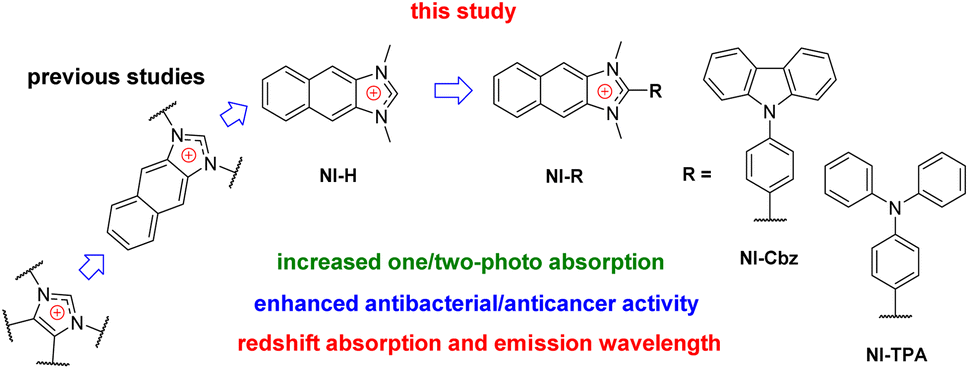 | ||
| Fig. 1 Molecular design toward NI-R. | ||
Hence, our investigative efforts continue to involve the synthesis, photophysical examination, theoretical calculation and biological activities of NI-H and its derivatives by incorporating donor groups, specifically carbazolyl phenyl and triphenylamine, resulting in NI-Cbz and NI-TPA, respectively. Among these compounds, NI-TPA was evaluated for its potential application in fluorescence bioimaging and lysosome localization using HeLa cells. Moreover, we assessed the antibacterial activity of these compounds against ten bacterial strains, consisting of five Gram-positive and five Gram-negative strains. Furthermore, we explored their anticancer efficacy by studying their cytotoxicity against four different cancer cell lines. Our results revealed that NI-TPA exhibited remarkable cytotoxicity against both cancerous cells and bacteria, including MRSA, indicating its potential as a dual-function agent with both anticancer and antibacterial activities. To gain insights into the underlying mechanisms of its anticancer activity, molecular docking, and dynamic simulation were employed to study the interaction between NI-TPA and specific proteins in cancer cells.
Results and discussion
Synthetic process and structural characterization
NI-R was synthesized using three key transformations (Scheme 1). The synthesis commenced with the cyclization reaction of 2,3-diaminonaphthalene to form the first intermediate compound (1-R). Subsequently, 1-R underwent methylation by reacting with iodomethane in the presence of sodium hydride in DMF, yielding the second intermediate compound (2-R). Finally, the target compound NI-R salts were obtained through the refluxing of 2-R with iodomethane in acetonitrile. The detailed experimental procedures for each step of the synthesis, along with the structural spectra data are documented in the Experimental section and ESI.†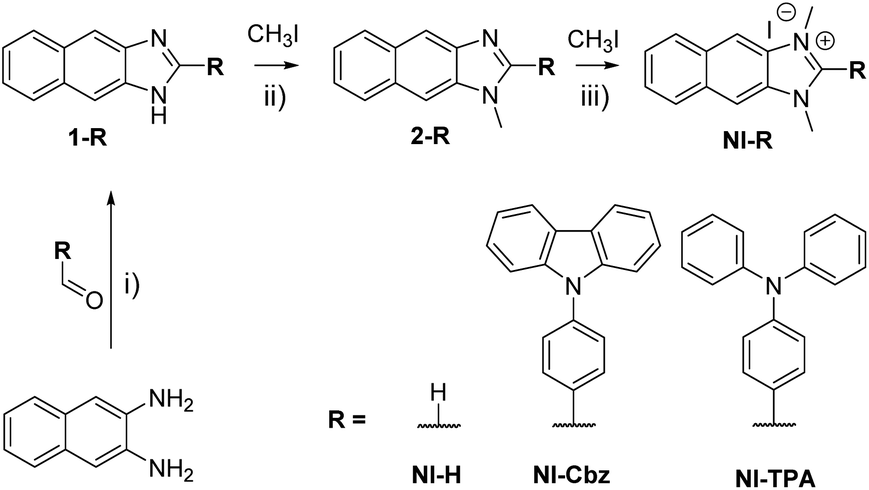 | ||
| Scheme 1 Synthetic process for NI-R structures. (i) DMF, acetic acid, 80 °C, 9 h; (ii) NaH, DMF, argon, RT, 18 h; (iii) CH3CN, reflux, 12–24 h. | ||
The 1H NMR spectra of NI-H displayed a single peak, characterized by the most prominent chemical shift recorded at 9.85 ppm. This resonance was ascribed to a hydrogen moiety directly bound to a carbon atom positioned intermediary between two nitrogen atoms. Upon the introduction of electron-donating groups at this specific carbon site, the 1H NMR signal associated with this proton manifestation ceased to manifest. This phenomenon serves as compelling substantiation for the manifestation of conjugative interactions amidst the substituent groups within both the NI-Cbz and NI-TPA molecular configurations. Furthermore, discernible alterations were discerned in the resonance patterns of the methyl group protons associated with NI-R, in contrast to their precursors in the 2-R configuration. This disparity not only underscores the establishment of said salts but also implies that the proton group equivalency within the NI-R construct signifies the liberation of positive charge within the naphthoimidazole framework.
Photophysical properties
In our antecedent report, we undertook an examination of the photophysical properties and theoretical calculations pertaining to NI-H and NI-Cbz.47 The investigation into the corresponding attributes of NI-Cz is herein expounded. The findings of both analyses have been succinctly compiled and are delineated in Table 1, Fig. 2 and 3.47 The absorption and fluorescence emission peaks of NI-H were observed at 324 nm and 433 nm (ΦF = 0.33), respectively, in ethanol. The introduction of donor groups into the naphthoimidazolium core resulted in a redshift of the absorption and fluorescence emission peaks. Specifically, NI-Cbz exhibited absorption peaks at 335 nm and fluorescence emission peaks at 535 nm, while NI-TPA showed absorption peaks at 370 nm and fluorescence emission peaks at 554 nm. Furthermore, the molar absorption coefficients of NI-H, NI-Cbz, and NI-TPA were found to increase significantly from 8.8 × 103 to 23.6 × 103 and further to 39.2 × 103 M−1 cm−1, respectively. In addition, the augmentation in the TP absorption cross-section (σ2) denoted by values of 11 GM, 36 GM, and 75 GM at 780 nm for NI-H, NI-Cbz, and NI-TPA, respectively. The redshift in the absorption and fluorescence emission peaks as well as enhanced TP absorbance can be attributed to the enhancement of push–pull effects and intramolecular charge transfer (ICT) resulting from the incorporation of strong donor groups. This is consistent with the observed decrease in the energy gap (Eg) between LUMO and HOMO from 3.99 to 3.43 and then 3.32 eV. Moreover, molecular orbital (MO) analysis revealed that the HOMO and LUMO of NI-H were in the naphthoimidazolium core (Fig. 3). Conversely, for NI-Cbz and NI-TPA, the HOMO was mainly localized in the carbazolyl phenyl and triphenylamine groups, respectively, while the LUMO remained primarily situated in the naphthoimidazolium core. This confirms the presence of a donor–acceptor framework in the structures of NI-Cbz and NI-TPA. Additionally, the fluorescence emission quantum yields (ΦF) of NI-Cbz and NI-TPA were calculated to be 0.04 and 0.06, respectively. Overall, based on the observed photophysical properties including absorption and emission wavelength, molar absorption coefficients, and fluorescence, NI-TPA demonstrated the most promising potential for fluorescence bioimaging among the three NI-R salts studied.| λabs (nm) | ε (M−1 cm−1) (×103) | λems (nm) | ΦF | σ2α (GM) | Eg (eV) | |
|---|---|---|---|---|---|---|
| a ε (molar absorption coefficient); ΦF (fluorescence emission quantum yield) was determined utilizing 9,10-diphenylanthracene (ΦF = 0.90 in cyclohexane) as reference;46 Eg (energy gap between HOMO and LUMO) was obtained by density functional theory (DFT) calculation using B3LYP/6-31+G(d,p) level of theory in ethanol/PCM solvent model. | ||||||
| NI-H | 324 | 8.8 | 433 | 0.33 | 11 | 3.99 |
| NI-Cbz | 335 | 23.6 | 535 | 0.04 | 36 | 3.43 |
| NI-TPA | 370 | 39.2 | 554 | 0.06 | 75 | 3.32 |
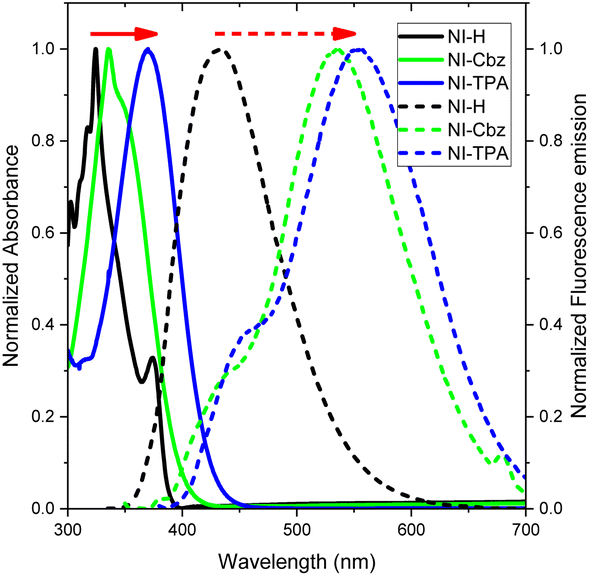 | ||
| Fig. 2 UV-Vis absorption (solid line) and fluorescence emission spectra (dotted line) of NI-R in ethanol. | ||
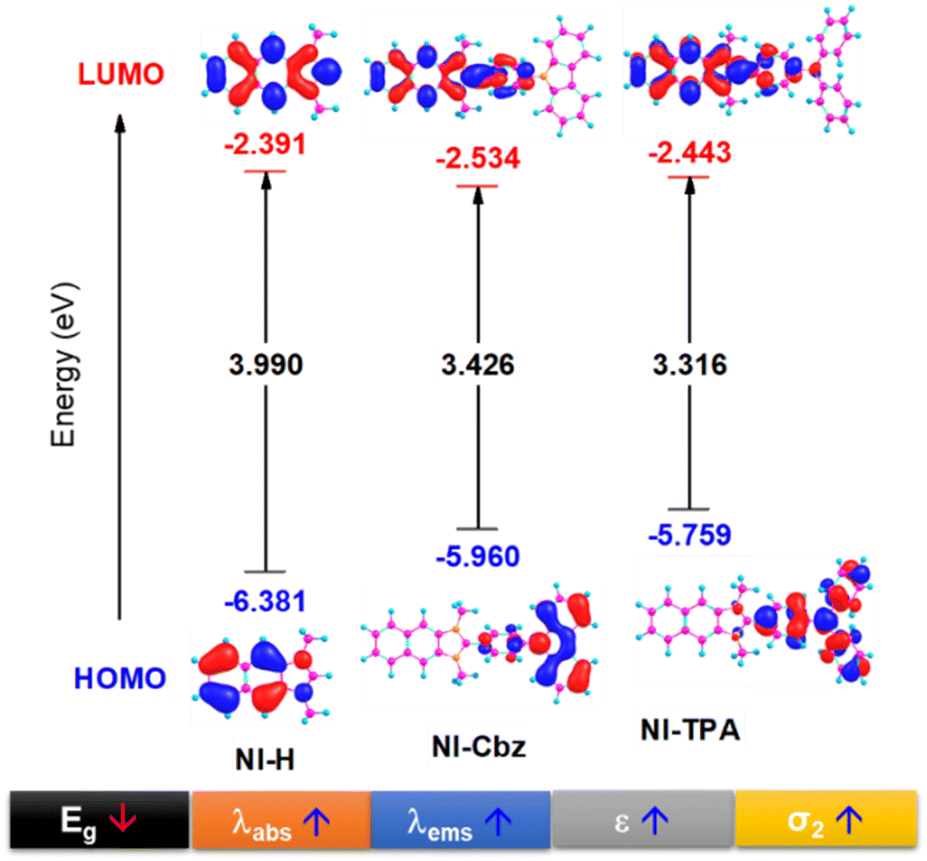 | ||
| Fig. 3 HOMO and LUMO image along with the energy gap (Eg) between HOMO and LUMO of NI-R in ethanol solvent. | ||
Fluorescence imaging in living cell
Motivated by the fluorescence emission of NI-TPA in both organic and aqueous solvents, the present research investigates its potential as a fluorescence agent for live-cell imaging, employing confocal laser scanning microscopy (CLSM). This study involves the examination of the cellular uptake efficiency of NI-TPA in HeLa cells after a 30 minute incubation period. Fig. 4a displays a remarkable fluorescent emission within the HeLa cells, indicating a rapid internalization of the NI-TPA. This notable fluorescence capability in living cells can be attributed to NI-TPA's ability to emit fluorescence across a wide range of solvent environments. In addition, NI-TPA shows a high TP absorption cross-section (σ2 = 75 GM) that holds significant advantageous implications for the field of near infrared (NIR) light-excited fluorescence bioimaging through two-photon excitation technique.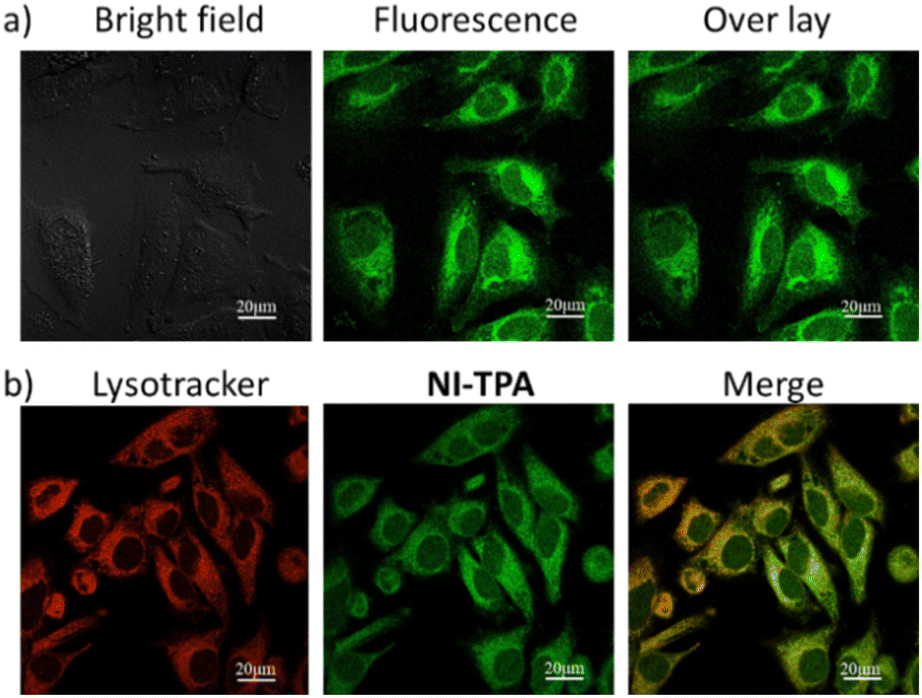 | ||
| Fig. 4 CLSM image of HeLa cells (a) after incubation with NI-TPA (10 μM), (b) after co-incubation of NI-TPA (10 μM) with Lysotracker Red (50 nM). | ||
To gain further insights into the selective localization of NI-TPA within specific cellular organelles, colocalization experiments were conducted. Specifically, HeLa cells were co-stained with NI-TPA and Lysotracker Red. Fig. 4b demonstrates the tendency of NI-TPA to localize in lysosomes, showing a high Pearson correlation coefficient of 0.78. Lysosomes are membrane-bound organelles responsible for vital cellular processes such as degradation and recycling of cellular waste, cellular signalling, and energy metabolism.48 Thus, NI-TPA shows promise as a potential lysosome-targeted fluorescent drug for cancer chemotherapy.
Antibacterial activity test
Table 2 provides a comprehensive summary of MICs observed for the NI-R series, elucidating their antibacterial activity. Notably, naphthoimidazolium salt (NI-H) did not exhibit growth inhibition against the tested bacteria. However, the introduction of carbazolyl phenyl and triphenylamine groups significantly enhanced the antibacterial activity of naphthoimidazolium salt against Gram-positive bacteria, such as MRSA, S. aureus, and L. monocytogenes, with MIC values ranging from 2 to 16 μg mL−1 (Table 2). In particular, NI-TPA demonstrated higher antibacterial activity against MRSA and S. aureus compared to NI-Cbz. Conversely, NI-Cbz exhibited stronger antibacterial activity against L. monocytogenes in comparison to NI-TPA. The augmented growth inhibition of NI-Cbz and NI-TPA against Gram-positive bacteria can be attributed to the electrostatic interaction between the positively charged salt and the negatively charged bacterial cell membrane. This electrostatic interaction mechanism may explain the non-antibacterial activity of both compounds against nearly all Gram-negative bacteria, as evidenced in Table 2. However, they did exhibit growth inhibition against S. typhimurium. It is worth noting that the cell walls of Gram-positive and Gram-negative bacteria exhibit distinct structural characteristics. Gram-positive bacteria possess a multilayered composition of peptidoglycan and teichoic acids within their cell walls. In contrast, Gram-negative bacteria possess a relatively thinner cell wall comprising a limited number of peptidoglycan layers, which is further enveloped by an outer lipid membrane consisting of lipopolysaccharides and lipoproteins. Consequently, the susceptibility of different compounds toward Gram-positive and Gram-negative bacteria may vary due to these structural disparities.49,50| Bacteria | NI-H | NI-Cbz | NI-TPA | |
|---|---|---|---|---|
| Gram-positive | 1 | — | 16 ± 0.004 | 7.8 ± 0.003 |
| 2 | — | 7.8 ± 0.002 | 3.9 ± 0.001 | |
| 3 | — | — | — | |
| 4 | — | — | — | |
| 5 | — | 2 ± 0.001 | 3.9 ± 0.001 | |
| Gram-negative | 6 | — | — | — |
| 7 | — | — | — | |
| 8 | — | 7.8 ± 0.003 | 7.8 ± 0.002 | |
| 9 | — | — | — | |
| 10 | — | — | — |
In summary, NI-TPA demonstrated the strongest growth inhibition against S. aureus and MRSA, with MIC values of approximately 3.9 μg mL−1 and 7.8 μg mL−1, respectively. In addition, NI-Cbz exhibited the highest antibacterial activity against L. monocytogenes, with a MIC value of approximately 2 μg mL−1. Both NI-Cbz and NI-TPA exhibited similar antibacterial activity against S. typhimurium, with a MIC value of approximately 7.8 μg mL−1. These MIC values show a low range in the comparison with those reported in the existing literature for imidazolium salts.12,37,38,45 Hence, NI-Cbz and NI-TPA possess the potential to serve as promising therapeutic agents for the treatment of various infections, including drug-resistant bacterial strains.
Anticancer activity test
The antibacterial properties of NI-Cbz and NI-TPA compounds have demonstrated efficacy against both Gram-positive and Gram-negative bacterial strains, which has inspired us to investigate their cytotoxic effects on four distinct cancer cell lines: MCF7, A549, HepG2, and HeLa. Our initial analysis revealed that the NI-H control structure displayed anticancer activity, with an IC50 range of 9.03–18.40 μg mL−1. Subsequently, the introduction of a carbazolyl phenyl group in NI-Cbz resulted in enhanced cytotoxicity towards MCF7 and HepG2 cells, with IC50 values of 1.76 and 1.45 μg L−1, respectively. However, NI-Cbz exhibited low cytotoxicity towards HeLa cells (IC50 = 36.11 μg mL−1) and negligible cytotoxicity towards A549 cells (IC50 > 100 μg mL−1).Significantly, the incorporation of the triphenylamine moiety into NI-H results in the generation of the NI-TPA product, manifesting pronounced improvements in cytotoxic efficacy against a spectrum of four distinct cancer cell lines, with IC50 values ranging from 0.31 to 2.01 μg mL−1. Notably, the IC50 value of NI-TPA was the lowest among the three products tested. The IC50 of NI-TPA against HepG2 cells was lowest among four cancer cell lines (Table 3). These findings underscore the potential application of NI-TPA as an effective anticancer drug. Given the promising cytotoxicity of NI-TPA, we further investigated the interaction mechanism between NI-TPA and HepG2 cells using MD simulations. This computational approach will provide valuable insights into the molecular-level interactions between NI-TPA and cancer cells, elucidating the underlying mechanisms of its potent anticancer activity.
| NI-H | NI-Cbz | NI-TPA | Ellipticine | |
|---|---|---|---|---|
| MCF7 | 18.40 ± 0.77 | 1.76 ± 0.07 | 0.67 ± 0.03 | 0.39 ± 0.02 |
| A549 | 6.92 ± 0.42 | >100 | 1.07 ± 0.08 | 0.46 ± 0.03 |
| HepG2 | 9.38 ± 0.43 | 1.45 ± 0.12 | 0.31 ± 0.03 | 0.35 ± 0.01 |
| HeLa | 9.03 ± 0.37 | 36.11 ± 0.73 | 2.01 ± 0.15 | 0.44 ± 0.03 |
Molecular docking and dynamics analysis
The geometric structure of NI-R was optimized utilizing Density Functional Theory (DFT) calculations at the B3LYP/6-31+G(d,p) level of theory within the Gaussian 16 program. The architectural arrangements of NI-R structures exhibited a consistent configuration within the naphthoimidazolium core. Notably, both NI-Cbz and NI-TPA displayed highly identical rotation angles between donor groups and the naphthoimidazolium core, as depicted in Fig. S3.† Consequently, the introduction of electron-donating groups was observed to enhance the antibacterial and anticancer activities of the compounds. Furthermore, it was observed that the diphenyl amino group exhibited a greater affinity towards cancer cells in comparison to the carbazole group. To gain a more profound insight into the mechanisms underlying the action of NI-R structures, molecular docking simulations were conducted. These simulations aimed to elucidate the potential anticancer properties of NI-R salts the protein c-Met kinase,51,52 which is one of the pathways in human cancer that is dysregulated most commonly. Tumor angiogenesis, metastasis, and tumor cell proliferation, survival, migration, and invasion are all facilitated by an active c-Met signaling system.53 Inhibiting c-Met kinase can help control the growth of many human malignancies, making c-Met kinase inhibitors a valuable approach in cancer therapy.54 Consequently, c-Met has drawn a lot of interest as a possible target for therapeutic intervention in oncology.Initially, the co-crystallized ARQ 197 ligand was re-docked into the catalytic domain of c-Met kinase using AutoDock Vina v1.2.0 (ref. 55) to validate the docking protocol. The results of the ARQ 197 ligand redocking revealed a RMSD value of 0.362 Å, indicating a precise co-crystallized ligand's binding conformation prediction. After that, NI-H, NI-Cbz, and NI-TPA were docked into the binding pocket of the c-Met kinase protein. Interestingly, the docking results of these compounds exhibited favorable affinities towards the c-Met kinase protein, as demonstrated by their high binding energies of −8.646, −10.490, and −11.03 kcal mol−1 for NI-H, NI-Cbz, and NI-TPA, respectively. The evaluation of NI-H compounds' interaction capabilities with the active site of c-Met kinase revealed the formation of six pi–alkyl and alkyl interactions with Arg1227, Lys1110, Met1211, Ala1226, Val1092, and Ala1108, along with two pi–sigma interactions with Phe1089 and Phe1223 (Fig. 5 and Table 4). Meanwhile, NI-Cbz forms pi–alkyl, pi–pi T-shaped, and pi–sigma interactions focused on a carbazole group with amino acid residues, namely Lys1110, Ala1226, Leu1157, Met1211, Ala1108 (pi–alkyl interactions), Phe1089 (pi–pi T-shaped interaction) and Val1092 (pi–sigma interaction). Regarding NI-TPA, this compound formed pi–alkyl, pi–sulfur, pi–pi T-shaped, and pi–ion interactions. Namely, it engages in pi–alkyl interaction with residues Ala1108, Ile1084, Arg1208, pi–sulfur interaction with residue Met1211 and pi–ion interaction with Arg1227 and Asp1164 (Fig. 5). The results on binding energies and interactions between ligands and c-Met kinase protein show that NI-TPA ligand binds better with c-Met kinase protein. These findings are consistent with the in vitro experimental results, where NI-TPA exhibits the lowest IC50 compared to NI-H and NI-Cbz (Table 3).
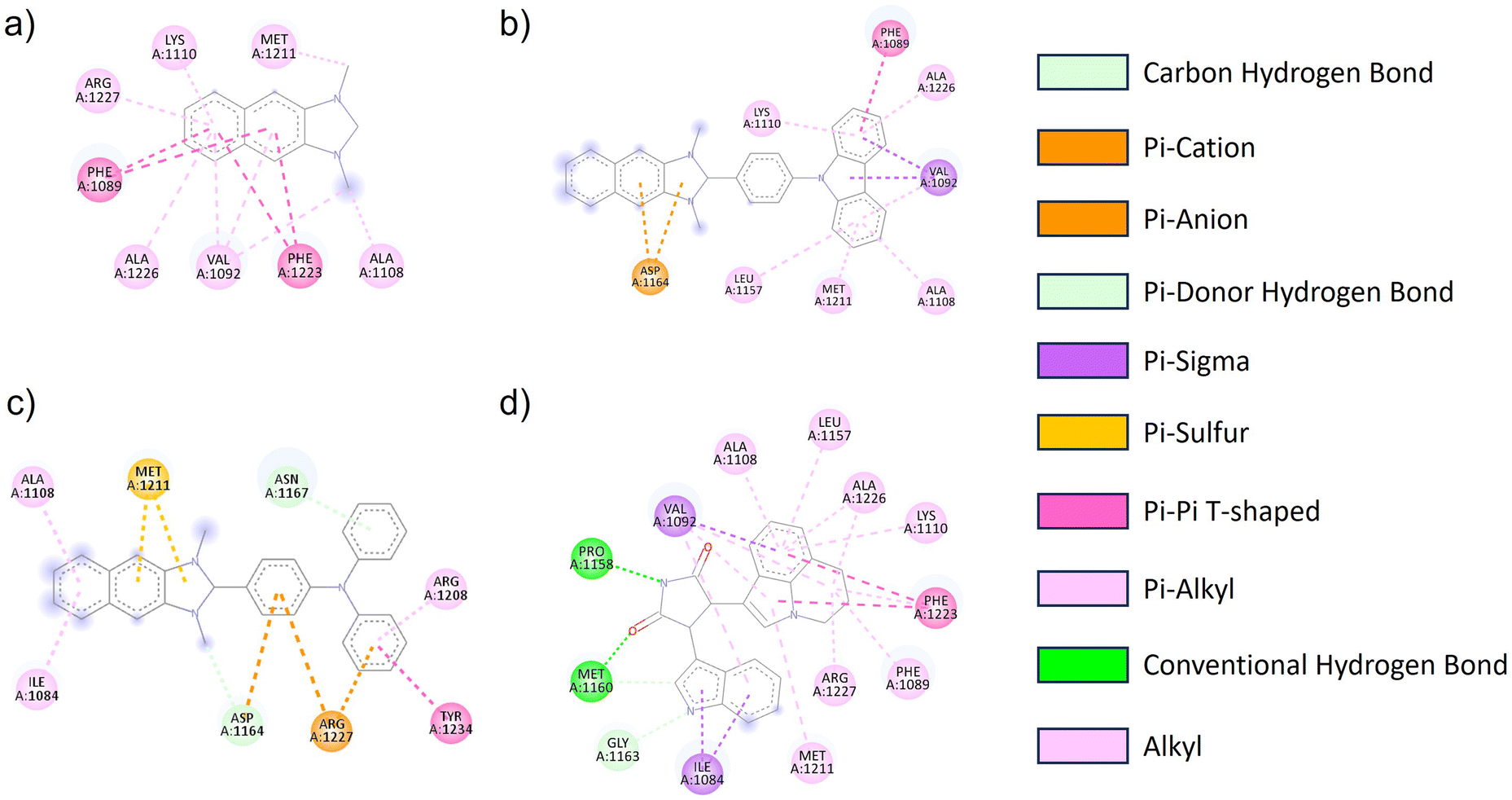 | ||
| Fig. 5 2D diagrams of the interactions between the (a) NI-H, (b) NI-Cbz, (c) NI-TPA, (d) erlotinib and residues in the c-Met kinase receptor. | ||
| Binding energy (kcal mol−1) | Type of interaction | Amino acid residues | |
|---|---|---|---|
| NI-H | −8.646 | Pi–alkyl and alkyl | Arg1227, Lys1110, Met1211, Ala1226, Val1092, Ala1108 |
| Pi–pi stacked | Phe1089, Phe1223 | ||
| NI-Cbz | −10.49 | Pi–alkyl | Lys1110, Ala1226, Leu1157, Met1211, Ala1108 |
| Pi–pi stacked | Phe1089 | ||
| Pi–sigma | Val1092 | ||
| Pi–anion | Asp1164 | ||
| NI-TPA | −11.03 | Pi–alkyl | Ala1108, Ile1084, Arg1208 |
| Pi–pi T-shaped | Tyr1234 | ||
| Pi–ion | Arg1227, Asp1164 | ||
| Pi–sulfur | Met1211 | ||
| ARQ 197 | −13.24 | Pi–alkyl and alkyl | Ala1108, Leu1157, Ala1226, Lys1110, Arg1227, Phe1089, Met1211 |
| Pi–sigma | Val1092, Ile1084 | ||
| Pi–pi T-shaped | Phe1223 | ||
| Hydrogen bond | Pro1158, Met1160 |
To further evaluate the stability of the complexes, the RMSD and Rg were calculated for all the saved structures during the MD simulation, and the changes in these factors were measured and are illustrated in Fig. 6. The RMSD of the c-Met kinase protein remains below 3 Å throughout the simulation, indicating its stable structure. The average RMSD values of the c-Met kinase protein in complex with NI-H, NI-Cbz, NI-TPA, and ARQ 197 are 0.280, 0.271, 0.258, and 0.261 nm, respectively while the average RMSD values of NI-H, NI-Cbz, NI-TPA, and ARQ 197 in complex with c-Met kinase protein is 0.029, 0.076, 0.074, and 0.037 nm, respectively. All the results are below 0.3 nm56 indicate the stability of NI-H, NI-Cbz, NI-TPA, and ARQ 197 in the active site of the c-Met kinase protein during the simulation. The Rg was used to assess the compactness of the protein during the simulation. The Rg values of the c-Met kinase protein in all complexes fall within a narrow range of 1.90–2.02 nm and do not show any abnormal changes during the simulation. The average Rg values of c-Met kinase protein is 2.044, 2.053, 2.030, and 2.041 nm in the complexes with the c-Met kinase protein bound to NI-H, NI-Cbz, NI-TPA, and ARQ 197. The RMSD and Rg results indicate that throughout the simulation, ligands stably bound and didn't leave the active site of the target protein and the whole system was in a relaxed conformation.57
 | ||
| Fig. 6 (a) RMSD of the backbone protein, (b) RMSD of the ligand, and (c) Rg analysis for the MD simulations. | ||
Conclusion
We synthesized a hydrophilic naphthoimidazolium salt (NI-H) and its derivatives, namely NI-Cbz and NI-TPA, by incorporating donor groups. The introduction of these donor groups resulted in redshifts of both the UV-Vis absorption and fluorescence emission peaks along with TP absorption cross-section from NI-H to NI-Cbz and further to NI-TPA. Importantly, NI-TPA demonstrated fluorescence emission and lysosome localization after incubation with HeLa cells, indicating its potential as a promising agent for fluorescence bioimaging applications. Furthermore, NI-Cbz and NI-TPA exhibited the antibacterial activity against three Gram-positive bacteria (L. monocytogenes, S. aureus, and MRSA) and one Gram-negative bacterium (S. typhimurium). Notably, NI-Cbz showed MIC of 2 μg mL−1 against L. monocytogenes, while NI-TPA displayed the lowest MIC (3.9 μg mL−1) against both L. monocytogenes and S. aureus. These findings underscore the potential of these naphthoimidazolium derivatives as antibacterial agents, particularly NI-TPA. Additionally, we investigated the cytotoxicity of NI-H, NI-Cbz, and NI-TPA against four different cancer cell lines (MCF7, A549, HepG2, and HeLa). Among these structures, NI-TPA demonstrated the highest cytotoxicity, with IC50 values ranging from 0.67–2.01 μg mL−1, further highlighting its potent anticancer activity. Molecular interactions between NI-R salts and c-Met kinase protein in cancer cells were also explored by docking and MD simulation, shedding light on the underlying mechanisms of their anticancer properties. In conclusion, the incorporation of donor groups, particularly the triphenylamine group, into the naphthoimidazolium salt led to enhanced one-photon absorption, emission wavelengths and TP absorption cross section, as well as improved antibacterial and anticancer activities. This study presents a novel design of imidazolium salts with promising potential for phototheranostic applications in cancer treatment and bacterial infections.Experimental
Synthetic process
NI-R was synthesized using a three-step process starting from 2,3-diaminonaphthalene as the initial material. Previously reported procedures were employed to prepare NI-H and NI-Cbz.47 The synthesis of NI-TPA is detailed below.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 as the eluent, resulting in the isolation of a white powder of the desired product (yield 45%). 1H NMR (400 MHz, chloroform-d) δ 8.81 (s, 2H), 8.07 (ddd, J = 8.4, 6.8, 1.5 Hz, 4H), 7.70 (ddd, J = 8.4, 6.6, 1.6 Hz, 2H), 7.67–7.58 (m, 8H), 7.21–7.14 (m, 4H).:2) as the eluent, leading to the isolation of a white powder of the desired product (yield 50%). 1H NMR (400 MHz, chloroform-d) δ 8.23 (d, J = 1.1 Hz, 1H), 8.03–7.96 (m, 1H), 7.96–7.90 (m, 1H), 7.73–7.66 (m, 3H), 7.45–7.35 (m, 2H), 7.35–7.26 (m, 4H), 7.21–7.14 (m, 6H), 7.14–7.05 (m, 2H), 3.93 (s, 3H).
:2 as the eluent, resulting in the isolation of a white powder of the desired product (yield 45%). 1H NMR (400 MHz, chloroform-d) δ 8.81 (s, 2H), 8.07 (ddd, J = 8.4, 6.8, 1.5 Hz, 4H), 7.70 (ddd, J = 8.4, 6.6, 1.6 Hz, 2H), 7.67–7.58 (m, 8H), 7.21–7.14 (m, 4H).:2) as the eluent, leading to the isolation of a white powder of the desired product (yield 50%). 1H NMR (400 MHz, chloroform-d) δ 8.23 (d, J = 1.1 Hz, 1H), 8.03–7.96 (m, 1H), 7.96–7.90 (m, 1H), 7.73–7.66 (m, 3H), 7.45–7.35 (m, 2H), 7.35–7.26 (m, 4H), 7.21–7.14 (m, 6H), 7.14–7.05 (m, 2H), 3.93 (s, 3H).Antibacterial activity tests
We aimed to assess the antibacterial properties of NP-R against a panel of diverse bacterial strains, encompassing both Gram-positive and Gram-negative bacteria. The Gram-positive strains that were investigated included Methicillin Resistant Staphylococcus aureus (MRSA), Staphylococcus aureus (S. aureus), Bacillus subtilis (B. subtilis), Enterococcus faecalis (E. faecalis), and Listeria monocytogenes (L. monocytogenes). The Gram-negative strains tested in this study comprised Extended Spectrum Beta-Lactamase (ESBL), Escherichia coli (E. coli), Salmonella typhimurium (S. typhimurium), Klebsiella pneumoniae (K. pneumoniae), and Vibrio parahaemolyticus (V. parahaemolyticus). To assess the effectiveness of NP-R against these bacterial strains, we determined the Minimal Inhibitory Concentration (MIC) following protocol outlined in our previous report.23Anticancer activity tests
The investigation into anticancer activity was conducted at the Institute of Biological Technology, Vietnam Academy of Science and Technology. This study aimed to evaluate the in vitro cytotoxicity of the NP-R series on four different cancer cell lines: MCF7 (human breast carcinoma), A549 (hepatocellular carcinoma), HepG2 (human liver carcinoma) and HeLa (human cervical carcinoma). The determination of the half maximal inhibitory concentration (IC50) and the subsequent detailed cell experiments were performed following our previous publication.58Molecular dynamic simulation
The anticancer potential of NI-R salts was subjected to further screening for molecular docking analysis to examine their binding modes and suppression against the human mesenchymal–epithelial transition factor (c-Met) protein (PDB ID 3RHK).59 The crystal structure of the c-Met kinase protein co-crystallized with ARQ 197 (ref. 60) ligand was downloaded from the RCSB Protein Data Bank (https://www.rcsb.org/structure/3RHK). Using PyMOL v2.5 software,61 water molecules, and ARQ 197 ligand were eliminated from the c-Met macromolecule. Hydrogen atoms in the protein structure were added, and Kollman charges were calculated using AutoDockTools (ADT). To prepare the input for docking simulations, the PDB structure file was converted to PDBQT format. A grid box of dimensions 24 × 24 × 24 Å3 was generated around the active site of the c-Met kinase protein with a spacing of 1 Å, assisted by ADT. The center of the grid box was set at the center of the c-Met kinase–Erlotinib complex. During the docking process, the c-Met kinase protein was kept rigid, while NI-H, NI-Cbz, and NI-TPA were set to be flexible. ADT and the AutoDock Vina v1.2.0 package were utilized for the molecular docking process.62 To evaluate the binding energies of the inhibitory complexes produced by NI-H, NI-Cbz, and NI-TPA with the c-Met kinase and to study their interaction potential, the output findings were examined using Discovery Studio Visualizer software. These complexes were then further characterized by molecular dynamics (MD). GROMACS v2022.5 package was used to simulate the molecular dynamics of the protein–ligand complexes.63 The force field for proteins was chosen to be the AMBER-f99SB-ILDN forcefield, and the Swissparam server created the ligand topologies.64 TIP3P water molecules were used in a rectangular box to dissolve all of the compounds.65 The necessary quantity of Na+ and Cl− ions was introduced, together with salt concentrations of 0.15 mol L−1 in each system, to make the simulation system electrically neutral. All the solvated systems underwent 50000 steps of energy minimization using the steepest descent method. The production run, NVT, and NPT equilibrations were then performed in the MD simulation. These equilibrations were carried out for a total of 300 ps at a temperature of 300 K and a pressure of 1 atm. In the simulation, the V-rescale thermostat and the C-rescale barostat were chosen. Finally, an MD simulation run of 100 ns was carried out. After that, a comparison analysis was done to evaluate the stability of the complexes by measuring the root mean square deviation (RMSD) and the radius of gyration (Rg).
Author contributions
N. D. T. and T. H. N. synthesized structures, T. T. H. H. and H. D. V. performed cell experiments, S. N. and V. T. T X. H. conducted bacterial experiments, V. T. P. and X. H. N. prepared molecular docking and dynamic simulation, V. T. N. and T. K. V. N. performed DFT calculation, M. P. measured TP absorption, D. L. T. and S. L. analysed NMR, T. C. P. prepared manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanh Chung Pham was funded by the Postdoctoral Scholarship Programme of Vingroup Innovation Foundation (VINIF), code VINIF.2022.STS.10. This work was supported by the Vietnam Academic of Science and Technology (VAST) under grant number THTEXS.03/23-25. We thank Centre for Informatics and Computing, Vietnam Academy of Science and Technology (CIC-VAST) for helping us use their High-Performance Computing (HPC) system.Notes and references
- M. Labrie, J. S. Brugge, G. B. Mills and I. K. Zervantonakis, Nat. Rev. Cancer, 2022, 22, 323–339 CrossRef CAS PubMed
.
- Y. Cheng, L. Zhao, Y. Li and T. Xu, Chem. Soc. Rev., 2011, 40, 2673–2703 RSC
- L. Wang, L. Lankhorst and R. Bernards, Nat. Rev. Cancer, 2022, 22, 340–355 CrossRef CAS PubMed
- K. Engle and G. Kumar, Eur. J. Med. Chem., 2022, 239, 114542 CrossRef CAS PubMed
- Y. Lu, X. Ma, X. Chang, Z. Liang, L. Lv, M. Shan, Q. Lu, Z. Wen, R. Gust and W. Liu, Chem. Soc. Rev., 2022, 51, 5518–5556 RSC
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC
- T. Zou, C. T. Lum, C.-N. Lok, J.-J. Zhang and C.-M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC
- M. Tilaoui, H. Ait Mouse and A. Zyad, Front. Pharmacol, 2021, 12, 719694 CrossRef CAS PubMed
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed
- S. H. Hassanpour and M. Dehghani, J. Cancer Res. Pract., 2017, 4, 127–129 CrossRef
- D. Osmaniye, B. N. Sağlık, N. Khalilova, S. Levent, G. Bayazıt, Ü. D. Gül, Y. Özkay and Z. A. Kaplancıklı, ACS Omega, 2023, 8, 6669–6678 CrossRef CAS PubMed
- M. R. Felício, O. N. Silva, S. Gonçalves, N. C. Santos and O. L. Franco, Front. Chem., 2017, 5, 5 Search PubMed
- A. F. Mohamed, M. Nasr, M. E. Amer, T. M. M. Abuamara, W. M. Abd-Elhay, H. F. Kaabo, E. E. R. Matar, L. E. El Moselhy, T. A. Gomah, M. A. E. L. F. Deban and R. I. Shebl, Infect. Agents Cancer, 2022, 17, 4 CrossRef CAS PubMed
- Y. Li, P. S. Kumar, S. Tan, C. Huang, Z. Xiang, J. Qiu, X. Tan, J. Luo and M. He, RSC Adv., 2022, 12, 24130–24138 RSC
- B. Pucelik, A. Sułek, M. Borkowski, A. Barzowska, M. Kobielusz and J. M. Dąbrowski, ACS Appl. Mater. Interfaces, 2022, 14, 14981–14996 CrossRef CAS PubMed
- C. Barton Behravesh, T. F. Jones, D. J. Vugia, C. Long, R. Marcus, K. Smith, S. Thomas, S. Zansky, K. E. Fullerton, O. L. Henao, E. Scallan and G. FoodNet Working, J. Infect. Dis., 2011, 204, 263–267 CrossRef PubMed
- D. Han, X. Liu and S. Wu, Chem. Soc. Rev., 2022, 51, 7138–7169 RSC
- W. Li, F. Separovic, N. M. O'Brien-Simpson and J. D. Wade, Chem. Soc. Rev., 2021, 50, 4932–4973 RSC
- S. Sethupathy, L. Vigneshwari, A. Valliammai, K. Balamurugan and S. K. Pandian, RSC Adv., 2017, 7, 23392–23406 RSC
- S. Gowrishankar, A. Kamaladevi, K. S. Ayyanar, K. Balamurugan and S. K. Pandian, RSC Adv., 2015, 5, 95788–95804 RSC
- J. Xi, L. An, G. Wei, Y. Huang, D. Li, L. Fan and L. Gao, Biomater. Sci., 2020, 8, 6225–6234 RSC
- S. K. Nandanwar, S. B. Borkar, B. N. Wijaya, J. H. Cho, N. H. Tarte and H. Jun Kim, ChemistrySelect, 2020, 5, 3471–3476 CrossRef CAS
- S. K. Nandanwar and H. J. Kim, ChemistrySelect, 2019, 4, 1706–1721 CrossRef CAS
- A. M. Abu-Dief and I. M. A. Mohamed, Beni-Suef Univ. J. Basic Appl. Sci., 2015, 4, 119–133 Search PubMed
- Z. Xie, T. Fan, J. An, W. Choi, Y. Duo, Y. Ge, B. Zhang, G. Nie, N. Xie, T. Zheng, Y. Chen, H. Zhang and J. S. Kim, Chem. Soc. Rev., 2020, 49, 8065–8087 RSC
- V.-N. Nguyen, Z. Zhao, B. Z. Tang and J. Yoon, Chem. Soc. Rev., 2022, 51, 3324–3340 RSC
- N. S. H. Motlagh, P. Parvin, F. Ghasemi and F. Atyabi, Biomed. Opt. Express, 2016, 7, 2400–2406 CrossRef CAS PubMed
- I. J. Bigio and S. G. Bown, Cancer Biol. Ther., 2004, 3, 259–267 CrossRef CAS PubMed
- C. Hu, S. Xu, Z. Song, H. Li and H. Liu, Chemosensors, 2023, 11, 125 CrossRef CAS
- M. L. Kabir, F. Wang and A. H. A. Clayton, Biology, 2022, 11, 1135 CrossRef CAS PubMed
- J. Han, M. Won, J. H. Kim, E. Jung, K. Min, P. Jangili and J. S. Kim, Chem. Soc. Rev., 2020, 49, 7856–7878 RSC
- J. H. Kim, P. Verwilst, M. Won, J. Lee, J. L. Sessler, J. Han and J. S. Kim, J. Am. Chem. Soc., 2021, 143, 14115–14124 CrossRef CAS PubMed
- A. Sharma, M.-G. Lee, M. Won, S. Koo, J. F. Arambula, J. L. Sessler, S.-G. Chi and J. S. Kim, J. Am. Chem. Soc., 2019, 141, 15611–15618 CrossRef CAS PubMed
- C.-H. Whang, E. Yoo, S. K. Hur, K. S. Kim, D. Kim and S. Jo, Chem. Commun., 2018, 54, 9031–9034 RSC
- J.-L. Yang, Y.-H. Ma, Y.-H. Li, Y.-P. Zhang, H.-C. Tian, Y.-C. Huang, Y. Li, W. Chen and L.-J. Yang, ACS Omega, 2019, 4, 20381–20393 CrossRef CAS PubMed
- P. Ganapathi, K. Ganesan, M. Dharmasivam, M. M. Alam and A. Mohammed, ACS Omega, 2022, 7, 44458–44469 CrossRef CAS PubMed
- L. A. Ismail, R. Zakaria, E. M. Hassan, M. Y. Alfaifi, A. A. Shati, S. E. I. Elbehairi, A. A. El-Bindary and R. F. M. Elshaarawy, RSC Adv., 2022, 12, 28364–28375 RSC
- G. Deng, B. Zhou, J. Wang, Z. Chen, L. Gong, Y. Gong, D. Wu, Y. Li, H. Zhang and X. Yang, Eur. J. Med. Chem., 2019, 168, 232–252 CrossRef CAS PubMed
- B. D. Wright, M. C. Deblock, P. O. Wagers, E. Duah, N. K. Robishaw, K. L. Shelton, M. R. Southerland, M. A. DeBord, K. M. Kersten, L. J. McDonald, J. A. Stiel, M. J. Panzner, C. A. Tessier, S. Paruchuri and W. J. Youngs, Med. Chem. Res., 2015, 24, 2838–2861 CrossRef CAS PubMed
- T. C. Pham, V.-N. Nguyen, Y. Choi, D. Kim, O.-S. Jung, D. J. Lee, H. J. Kim, M. W. Lee, J. Yoon, H. M. Kim and S. Lee, Front. Chem., 2021, 9, 713078 CrossRef CAS PubMed
- L.-X. Liu, X.-Q. Wang, B. Zhou, L.-J. Yang, Y. Li, H.-B. Zhang and X.-D. Yang, Sci. Rep., 2015, 5, 13101 CrossRef PubMed
- N. Kwon, G. Baek, K. M. K. Swamy, M. Lee, Q. Xu, Y. Kim, S.-J. Kim and J. Yoon, Dyes Pigm., 2019, 171, 107679 CrossRef CAS
- X. Zhang, G. Ko, J. F. Joung, M. Li, Y. Jeong, K. M. K. Swamy, D. Lee, Y. Liu, S. Lee, S. Park, T. D. James and J. Yoon, Chem. Commun., 2018, 54, 13264–13267 RSC
- S. N. Riduan and Y. Zhang, Chem. Soc. Rev., 2013, 42, 9055–9070 RSC
- T. C. Pham, D. J. Lee, D. H. Kim, J. Yoon, T. D. Lam, H. M. Kim and S. Lee, Chem. Commun., 2023, 59, 4503–4506 RSC
- T. C. Pham, T. T. H. Hoang, D. N. Tran, G. Kim, T. V. Nguyen, T. V. Pham, S. Nandanwar, D. L. Tran, M. Park and S. Lee, ACS Appl. Mater. Interfaces, 2023, 15, 47969–47977 CrossRef CAS PubMed
- S. R. Bonam, F. Wang and S. Muller, Nat. Rev. Drug Discovery, 2019, 18, 923–948 CrossRef CAS PubMed
- P. O. Asekunowo, R. A. Haque, M. R. Razali and S. Budagumpi, Appl. Organomet. Chem., 2015, 29, 126–137 CrossRef CAS
- A. A. El-Sherif, J. Solution Chem., 2010, 39, 1562–1581 CrossRef CAS
- J. G. Christensen, J. Burrows and R. Salgia, Cancer Lett., 2005, 225, 1–26 CrossRef CAS PubMed
- A. Danilkovitch-Miagkova and B. Zbar, J. Clin. Invest., 2002, 109, 863–867 CrossRef CAS PubMed
- X. Liu, R. C. Newton and P. A. Scherle, Trends Mol. Med., 2010, 16, 37–45 CrossRef CAS PubMed
- J. P. Eder, G. F. Vande Woude, S. A. Boerner and P. M. LoRusso, Clin. Cancer Res., 2009, 15, 2207–2214 CrossRef CAS PubMed
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed
- S. Rampogu, B. Shaik, J. H. Kim, T. S. Jung, M. W. Ha and K. W. Lee, Heliyon, 2023, 9, e13324 CrossRef CAS PubMed
- S. Rampogu, T. S. Jung, M. W. Ha and K. W. Lee, Sci. Rep., 2023, 13, 10583 CrossRef CAS PubMed
- N. T. T. Chi, V. T. Pham and H. V. Huynh, Organometallics, 2020, 39, 3505–3513 CrossRef CAS
- J. H. Park, Y. Liu, M. A. Lemmon and R. Radhakrishnan, Biochem. J., 2012, 448, 417–423 CrossRef CAS PubMed
- S. Eathiraj, R. Palma, E. Volckova, M. Hirschi, D. S. France, M. A. Ashwell and T. C. K. Chan, J. Biol. Chem., 2011, 286, 20666–20676 CrossRef CAS PubMed
- W. L. DeLano, CCP4 Newsletter on Protein Crystallography, 2002, vol. 40, pp. 82–92 Search PubMed
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed
- V. Zoete, M. A. Cuendet, A. Grosdidier and O. Michielin, J. Comput. Chem., 2011, 32, 2359–2368 CrossRef CAS PubMed
- D. J. Price and C. L. Brooks III, J. Chem. Phys., 2004, 121, 10096–10103 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06555c |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2023 |