
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNBS-mediated bromination and dehydrogenation of tetrahydro-quinoline in one pot: scope and mechanistic study†
Ruchun Yang ,
Yongge Xiong,
Si Deng,
Jiang Bai,
Xian-Rong Song* and
Qiang Xiao*
,
Yongge Xiong,
Si Deng,
Jiang Bai,
Xian-Rong Song* and
Qiang Xiao*
Institute of Organic Chemistry, Jiangxi Science & Technology Normal University, Key Laboratory of Organic Chemistry, Nanchang 330013, Jiangxi Province, China. E-mail: songxr2015@163.com; xiaoqiang@tsinghua.org.cn
First published on 15th November 2023
Abstract
A facile and general approach was developed for the efficient construction of functionalized bromoquinolines by the dehydrogenation of tetrahydroquinolines using NBS as the electrophile and as oxidant. The cascade transformation proceeded with good functional group tolerance under metal-free conditions with a short reaction duration. Various tetrahydroquinolines bearing either electron-rich or electron-deficient groups at different positions were successfully converted into the corresponding target products in moderate to high yields under mild conditions. It is worth noting that the obtained polybromoquinolines could further undergo classic metal-catalyzed cross-coupling reactions with good regioselectivity. The Sonagashira coupling reaction occurred regioselectively in the C-6 position of the obtained products followed by a Suzuki coupling reaction to give multifunctionalized quinolines. The mechanism indicated that electrophilic bromination/radical dehydrogenation sequences occurred in one pot.
Quinoline derivatives are very important structural skeletons that have long been known for their wide range of biological activities and chemotherapeutic activities.1–5 Of these compounds, bromo-quinolines are of interest to chemists as precursors for heterocyclic compounds with multi functionality, providing synthetic access to a wide variety of compounds.6–9 Generally, brominated quinolines can be synthesized from benzene derivatives substituted with nitrogen functional groups by electrophilic cyclization using NBS or Br2.10–14 However, these methods are mainly used to construct monobromo-quinolines; the synthesis of polybromoquinolines is rarely reported.
Polybromoquinolines are very important synthetic intermediates in organic synthesis because the aromatic ring contains several bromine atoms with different reactivities, which can provide a potential opportunity for the construction of other functionalized quinolines.15,16 In addition, multi-bromoquinolines also played an important role in pharmacological chemistry with effective antibacterial, anticancer activities and some metabolic enzyme inhibition activities (Fig. 1).17 Recently, our group reported the NBS-promoted cascade electrophilic cyclization of N-(3-phenylprop-2-ynyl) anilines for the construction of di-/tribromoquinolines (Scheme 1a).18 However, the regioselectivity was poor when the substituent was located in the meta-position of the propargyl amino group. Additionally, when there was a substituent in the ortho-position, the reaction could not occur due to steric hindrance. Therefore, it is necessary to develop a new strategy to solve the problem of regioselectivity for the synthesis of multi-brominated quinolines with diverse functional groups.
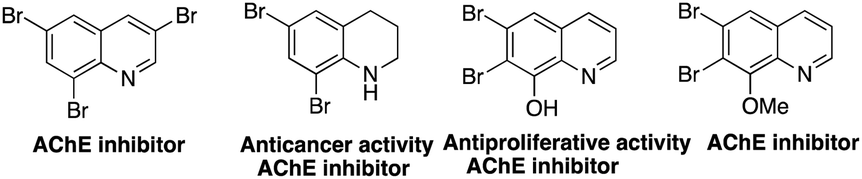 | ||
| Fig. 1 Several bioactive multi-bromoquinolines. | ||
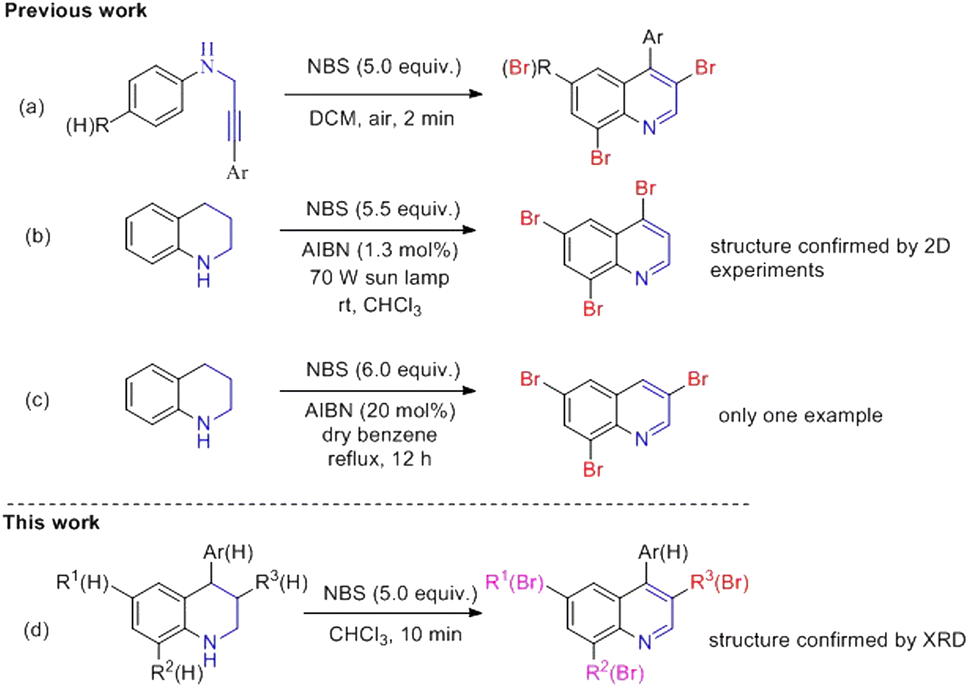 | ||
| Scheme 1 Synthesis of multi-bromo quinolines: (a) our group's previous work; (b) Sahin's work; (c) Langer's work; (d) this work. | ||
The dehydrogenation of tetrahydroquinolines, as an efficient and atom-economical approach towards quinolines, has been investigated extensively in recent years. Numerous examples include metal-complex catalysed acceptor less dehydrogenation,19–21 along with aerobic oxidation catalyzed by Ru(phd)3,22 Mn(Pc),23 Co3O4-NGr/C,24 and CuI/DEAD25 etc. In addition, metal free catalytic systems,26,27 photochemical methods,28–30 and electrochemical methods31 have also been reported. However, there are few reports on the simultaneous dehydrogenation and functionalization or halogenation of tetrahydroquinolines.
In 2008, Sahin15 reported an NBS-mediated bromination/dehydrogenation of 1,2,3,4-tetrahydroquinoline for the synthesis of 4,6,8-tribromoquinoline in the presence of AIBN in CHCl3 under sun lamp irradiation at 70 W (Scheme 1b). The product 4,6,8-tri-bromoquinoline was obtained in 75% yield with the structure confirmed by 2D experiments. Moreover, a very similar work was also reported by Langer et al. in which 1,2,3,4-tetrahydroquinoline reacts with 6.0 equivalents of NBS in the presence of AIBN in benzene at reflux (Scheme 1c).16 However, these reactions only occurred in the presence of radical initiator AIBN, and the substrate scope was not investigated in these two studies.
On basis of our continuing studies in the synthesis of quinolines,32,33 we herein reported an oxidative bromination/dehydrogenation of tetrahydroquinolines in the presence of NBS without a radical initiator under mild conditions. The mechanism indicated that NBS not only functioned as an electrophilic reagent to undergo electrophilic bromination, but also as an oxidant to achieve the oxidative dehydrogenation of tetrahydroquinoline via a radical pathway. Notably, the obtained multi-bromoquinolines could readily undergo the Sonagashira reaction and Suzuki coupling to afford the different substituted quinolines, which provide a method for constructing the library of quinolines (Scheme 1d).
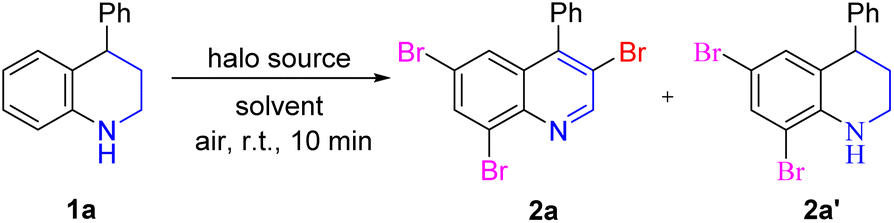
Initially, 4-phenyltetrahydroquinoline 1a was chosen as the model substrate with 5.0 equiv. NBS and DCM as the solvent (Table 1). Fortunately, the multi-bromination and dehydrogenation reactions occurred successfully, affording the desired product 3,6,8-tribromoquinoline (2a) in 50% yield. Next, some representative solvents were screened, and CHCl3 was selected as the best solvent for this transformation (entries 2–9). Notably, a great deal of heat was released from the reaction system when NBS was added at once, so NBS was added in batches, which was greatly increased the yield (entry 10). Next, the equivalent of NBS was also screened (entries 11–14). When the amount of NBS was 1.0 equiv. or 2.0 equiv., the desired product 2a was not observed. However, the side product 2a′, resulting from the 6,8-dibromination of tetrahydroquinoline, was isolated in 85% yield when 2.0 equiv. NBS was added. The reaction was incompleted by increasing the amount of NBS to 3.0 equiv. or 4.0 equiv., and both 2a and 2a′ were obtained simultaneously. Subsequently, when the reaction was carried out under an argon atmosphere or dark conditions, the reaction efficiency was slightly reduced (entries 15–16). Other halo sources N-bromoacetamide, tribromoisocyanuric acid, dibromoisocyanuric acid and Br2 were also conducted in this reaction and no better yield was obtained than NBS (entries 17–20). Interestingly, no product was observed when Br2 was used. However, product 2a was isolated in 65% yield after quenching for 12 h. When NBS was replaced by NCS or NIS, no corresponding product was not observed. When NCS was added, some raw materials decomposed, and only 22% of 6,8-dichlorotetrahydroquinoline was isolated. When NIS was used, most of the raw material decomposed quickly (entries 21–22). Therefore, the optimal reaction conditions were obtained, that is, 5.0 equiv. NBS, was added in batches, with CHCl3 as solvent under an air atmosphere.
|
|||
|---|---|---|---|
| Entry | Halo source | Solvent | Yieldb [%] |
| a Reaction conditions: 0.2 mmol 1a, 5.0 equiv. NBS, 2.0 mL CHCl3, r.t., air atmosphere.b Halo sources was added in batches.c 2a′ was isolated in 74% yield.d 2a′ was isolated in 15% yield.e Under argon atmosphere.f Under dark condition.g 65% product 2a was isolated after quenching for 12 h.h 22% 6,8-Dichlorotetrahydronquinoline was isolated, some raw material decomposed.i Mixture, the raw material decomposed. | |||
| 1 | NBS (5.0 eq.) | DCM | 50 |
| 2 | NBS (5.0 eq.) | Toluene | 67 |
| 3 | NBS (5.0 eq.) | EA | 45 |
| 4 | NBS (5.0 eq.) | CH3OH | Trace |
| 5c | NBS (5.0 eq.) | THF | 60 |
| 6 | NBS (5.0 eq.) | 1,4-Dioxane | 65 |
| 7 | NBS (5.0 eq.) | CH3CN | 50 |
| 8 | NBS (5.0 eq.) | DMSO | Trace |
| 9b | NBS (5.0 eq.) | CHCl3 | 70 |
| 10b | NBS (5.0 eq.) | CHCl3 | 80 |
| 11b | NBS (1.0 eq.) | CHCl3 | — |
| 12b, c | NBS (2.0 eq.) | CHCl3 | — |
| 13b, c | NBS (3.0 eq.) | CHCl3 | 10 |
| 14b, d | NBS (4.0 eq.) | CHCl3 | 68 |
| 15b, e | NBS (5.0 eq.) | CHCl3 | 79 |
| 16b, f | NBS (5.0 eq.) | CHCl3 | 78 |
| 17b | N-bromoacetamide (5.0 eq.) | CHCl3 | 35 |
| 18b | Tribromoisocyanuric acid (5.0 eq.) | CHCl3 | 20 |
| 19b | Dibromoisocyanuric acid (5.0 eq.) | CHCl3 | 38 |
| 20b, g | Br2 (5.0 eq.) | CHCl3 | — |
| 21b, h | NCS (5.0 eq.) | CHCl3 | — |
| 22b, i | NIS (5.0) | CHCl3 | — |
Based on these results, a one-pot construction of the multibromoquinolines from tetrahydroquinolines 1 was carried out. Various substitutions of tetrahydroquinolines 1, bearing electron-donating and electron-withdrawing groups, were investigated under the standard conditions, giving the target product quinolines 2a–2r in moderate to good yields, as shown in Scheme 2. First, a series of 4-phenyl-tetrahydroquinolines were tested under the standard conditions. Substrates with weak electron-withdrawing groups in the C6-position of 4-phenyl-tetrahydroquinoline, such as Br, Cl or F, afforded the corresponding 3,8-dibromo-4-phenyl-quinolines 2a–2c in good yields. When an alkyl group (tBu, nBu or CH3) or strong electron donating group (OCH3) was in the C6 position, the corresponding 3,8-dibromo-4-phenyl-quinolines 2d–2g were obtained in moderate yields. It was worth noting that substrate bearing trifluoromethoxy (OCF3) in the C6-position gave a good yield. When substrate lacked a substituent in the C6-position, the corresponding tribromide quinoline 2i was obtained in high yield. Next, the reaction was applied to the 4-phenyl tetrahydroquinoline with substituents at the C3 or C8 position. When an nPiv group was located in the C3 position, the resulting 6,8-dibromoquinoline derivative was obtained in 83% yield. The yield was relatively low when a methyl group was in the C3 position (2k–2m, 54–63%). When CH3 or OCH3 was at the C8 position of 4-phenyl-tetrahydroquinoline, the expected 3,6-dibromo-4-phenyl quinolines 2n–2p were successfully be obtained in moderate yields. Furthermore, unsubstituted tetrahydroquinoline 1q was also investigated under standard conditions, and the 3,6,8-tetrahydroquinoline 2q was obtained in 78% yield. Finally, when an ester group was at the C6 position of tetrahydroquinoline, the target product 2r was obtained in moderate yield.
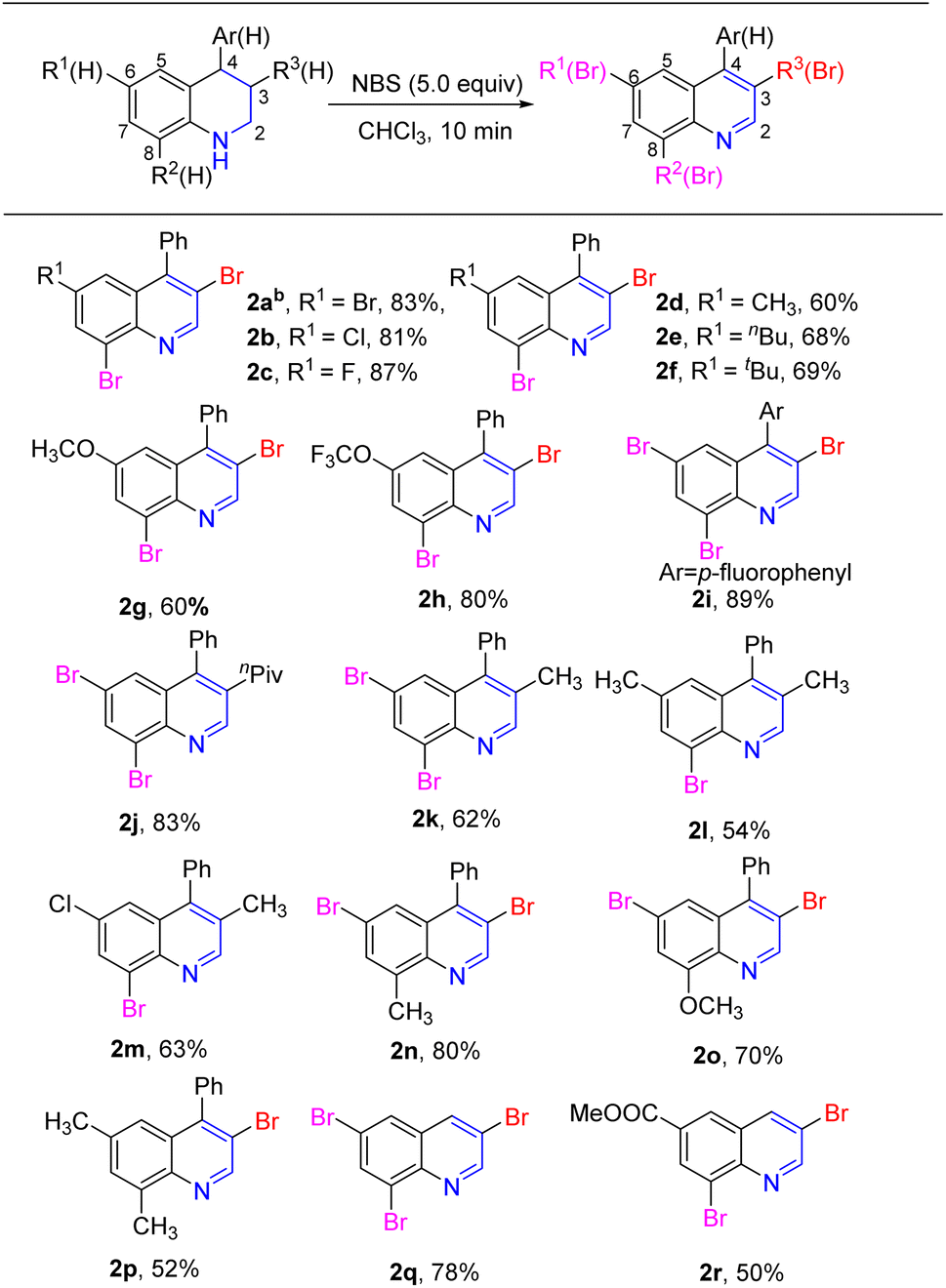 | ||
| Scheme 2 Substrate scope studies. a Reaction conditions: 0.2 mmol 1a–1r, 5.0 equiv. NBS (added one by one equivalent), in 2.0 mL CHCl3 at room temperature under air. b6-bromo-4-phenyl-1,2,3,4-tetrahydroquinoline was used as the substrate. | ||
To get insights into the mechanism of NBS-mediated bromination/dehydrogenation of tetrahydroquinolines, some control experiments were carried out (Scheme 3). First, substrate 1a was reacted with 5.0 equiv. 1,1-diphenylene under the standard conditions, and the side-product 2a′ was isolated in 81% yield (Scheme 3a). The reaction was also performed in the presence of 2,2,6,6-tetramethylpiperidine oxide (TEMPO) as a radical scavenger under the standard conditions, and a trace amount of the target product was detected. Two side products 2a1 and 2a2 were isolated in 9% and 12%, respectively. This may be due to the oxidative property of TEMPO (Scheme 3b). Based on these results, it was believed that the bromination of benzene may have occurred via an ionic manner. Because of the activation of the amino group, electrophilic substitution of the aromatic rings could readily occur. However, the dehydrogenation and 3-bromination of tetrahydroquinoline may have occurred via a radical pathway. Moreover, the 2a′ could easily be transformed to 2a in 87% yield in the presence of 3.0 equiv. NBS. This result indicated that compound 2a′ may be the intermediate in this reaction (Scheme 3c). Furthermore, when tetrahydroquinoline 1b was used as the substrate, two other by-products (2b1 and 2b2) were obtained in addition to main product 2b (Scheme 3d). These results all indicated that the dehydrogenation process of tetrahydroquinoline may have been via a radical pathway.
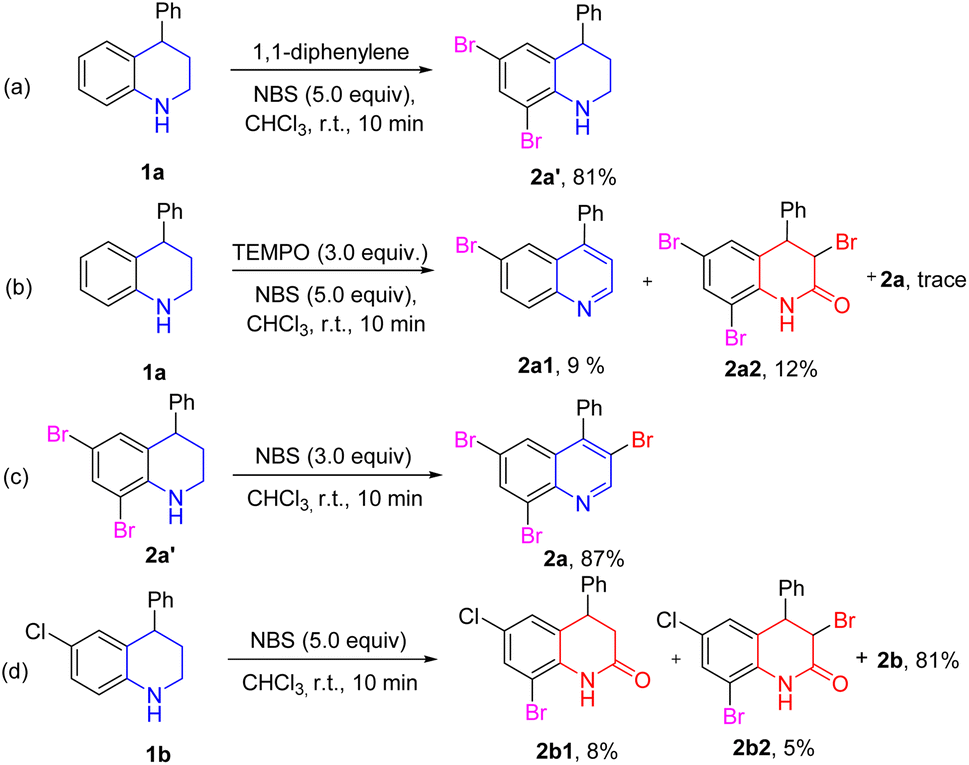 | ||
| Scheme 3 (a) 1,1-Diphenylene suppression; (b) TEMPO suppression; (c) intermediate reaction; (d) byproducts. | ||
According to the above results and previous literature,31,34–37 the possible reaction pathway from 4-phenyl-6-cholor-tetrahydroquinoline (1b) into 4-cholor-6,8-tribromo-quinolines (2b) was proposed as shown in Scheme 4. First, electrophilic bromination of the aryl ring occurred due to the activation of the amino group to give the 6-chloro-8-bromo-quinoline.34 After that, the electron-rich amino group was further attacked by NBS to form quaternary ammonium salt intermediate A. The nitrogen cation radical intermediate B was obtained by dehydrogenation under the action of the succinimide anion. Then, the key carbon radical intermediate C was generated after the rearrangement of the intermediate B.35 The intermediate C could be captured by Br˙, which was either followed by the removal of one HBr molecule to form 3,4-dihydroquinoline D (path a), or it could be captured by oxygen to form a by-product 2b1 (path b).35 In path a, the Wohl–Ziegler reaction was occurred in the ortho-position of 3,4-dihydroquinoline D leading to the formation of intermediate E.36 Then, 1,2-dihydroquinoline intermediate F was afforded through isomerization of intermediate E.31 The desired multi-bromination product 2b was formed through another dehydrogenation. In path b, the Wohl–Ziegler reaction occurred in the ortho-position of the carbonyl group to furnish the by-product 2b2.
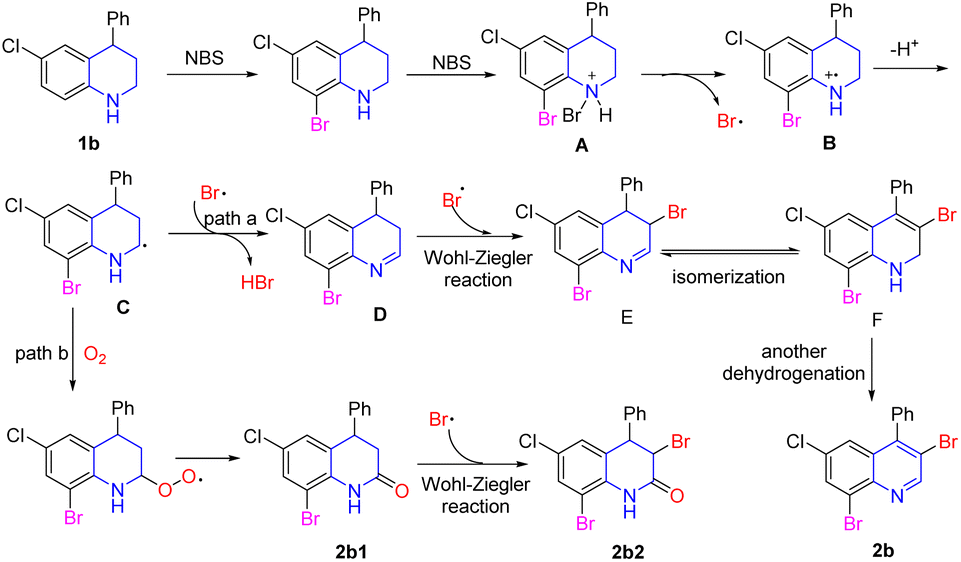 | ||
| Scheme 4 The proposed mechanism. | ||
It was worth noting that the reaction could be effectively scaled up with good efficiency. As shown in Scheme 5a, the desired product 2n was isolated in 78% yield by gram-scale reaction. Moreover, the obtained product 2n could be transformed into various quinoline derivatives via classic metal-catalyzed cross-coupling reactions (Scheme 5b). For example, the regioselective Sonogashira coupling reaction between the obtained product 2n and phenylacetylene occurred at the C6-position and the corresponding target product 3n was isolated in 65% yield. Notably, the structure of the product 3n was unambiguously verified by X-ray crystallography. Subsequently, the Suzuki–Miyaura coupling reaction of 3n with 4-methylphenylboronic acid in the presence of Pd (OAc)2/L1 gave the target product 4n in 45% yield.
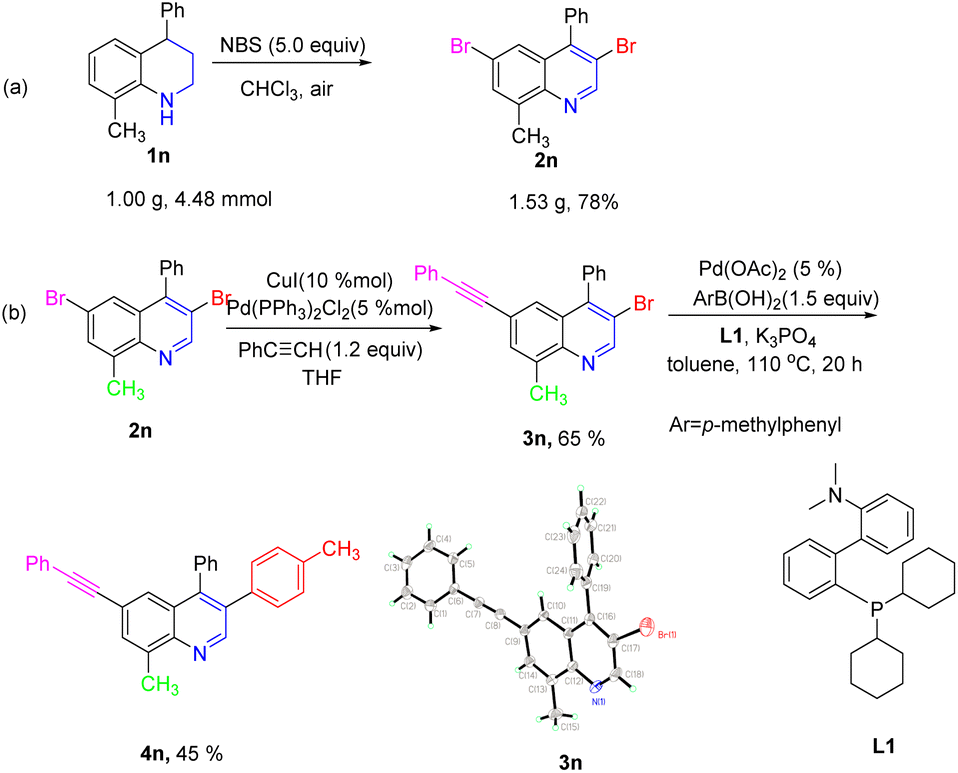 | ||
| Scheme 5 (a) Gram-scale reaction; (b) synthetic applications. | ||
Conclusions
In conclusion, an NBS-mediated bromination/dehydrogenation of tetrahydroquinolines under mild conditions was developed. Various quinolines bearing different functional groups were obtained in good yields. Furthermore, the Sonogashira reaction occurred regioselectively at the C6-position of dibromoquinoline. The mechanistic study revealed that the reaction may have been via electrophilic bromination and radical dehydrogenation to give the multibromoquinolines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (NSFC, No. 22261019 and 22001101) Jiangxi Provincial Natural Science Foundation (20232BAB203006 and 20212BAB213014), the Education Department of Jiangxi Province (GJJ2201340) and Jiangxi Science and Technology Normal University (2020BSQD015) for financial support.Notes and references
- J. P. Michael, Nat. Prod. Rep., 2008, 25, 166–187 RSC.
- A. Marella, O. P. Tanwar, R. Saha, M. R. Ali, S. Srivastava, M. Akhter and M. M. Alam, Saudi Pharm. J., 2013, 21, 1–12 CrossRef PubMed.
- R. S. Keri and S. A. Patil, Biomed. Pharmacother., 2014, 68, 1161–1175 CrossRef CAS PubMed.
- S. M. A. Hussaini, Expert Opin. Ther. Pat., 2016, 26, 1201–1221 CrossRef CAS PubMed.
- L. M. Nainwal, S. Tasneem, W. Akhtar, G. Verma, M. F. Khan, S. Parvez, M. Shaquiquzzaman, M. Akhter and M. M. Alam, Eur. J. Med. Chem., 2019, 164, 121–170 CrossRef CAS PubMed.
- Y. Benito, L. Canoira and J. G. Rodriguez, Appl. Organomet. Chem., 1987, 1, 535–540 CrossRef CAS.
- H. Brodnik, F. Pozgan and B. Stefane, Org. Biomol. Chem., 2016, 14, 1969–1981 RSC.
- L. N. R. Maddalin and R. J. Dhanorkar, Eur. J. Org Chem., 2014, 5214–5228 Search PubMed.
- T. Tagata and M. Nishida, J. Org. Chem., 2003, 68, 9412–9415 CrossRef CAS PubMed.
- V. A. Peshkov, O. P. Pereshivko, A. A. Nechaev, A. A. Peshkov and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3861–3896 RSC.
- S. V. Ryabukhin, V. S. Naumchik, A. S. Plaskon, O. O. Grygorenko and A. A. Tolmachev, J. Org. Chem., 2011, 76, 5774–5781 CrossRef CAS PubMed.
- Z. Huo, I. D. Gridnev and Y. A. Yamamoto, J. Org. Chem., 2010, 75, 1266–1270 CrossRef CAS PubMed.
- X. Zhang, M. A. Campo, T. Yao and R. C. Larock, Org. Lett., 2005, 7, 763–766 CrossRef CAS PubMed.
- X. Zhang, T. Yao, M. A. Campo and R. C. Larock, Tetrahedron, 2010, 66, 1177–1187 CrossRef CAS.
- A. Sahin, O. Cakmak, I. Demirtas, S. Okten and A. Tutar, Tetrahedron, 2008, 64, 10068–10074 CrossRef CAS.
- O. Akrawi, H. Mohammed and P. Langer, Synlett, 2013, 24, 1121–1124 CrossRef CAS.
- S. Ökten, A. Aydın, Ü. M. Koçyiğit, O. Çakmak, S. Erkan, C. A. Andac, P. Taslimi and İ. Gülçin, Arch. Pharm., 2020, 353, e2000086 CrossRef PubMed.
- S. Deng, W. Ouyang, J. Bai, X.-R. Song, R. Yang and Q. Xiao, Synthesis, 2021, 53, 2469–2476 CrossRef CAS.
- K.-H. He, F.-F. Tan, C.-Z. Zhou, G.-J. Zhou, X.-L. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 3080–3084 CrossRef CAS PubMed.
- Q. Wang, H. Chai and Z. Yu, Organometallics, 2018, 37, 584–591 CrossRef CAS.
- S. Wang, H. Huang, C. Bruneau and C. Fishermeister, ChemSusChem, 2019, 12, 2350–2354 CrossRef CAS.
- A. E. Wendlandt and S. S. Stahl, J. Am. Chem. Soc., 2014, 136, 11910–11913 CrossRef CAS PubMed.
- D. Jung, S. H. Jang, T. Yim and J. Kim, Org. Lett., 2018, 20, 6436–6439 CrossRef CAS PubMed.
- A. V. Iosub and S. S. Stahl, Org. Lett., 2015, 17, 4404–4407 CrossRef CAS.
- D. Jung, M. H. Kim and J. Kim, Org. Lett., 2016, 18, 6300–6303 CrossRef CAS PubMed.
- R. Yang, S. Yue, W. Tan, Y. Xie and H. Cai, J. Org. Chem., 2020, 85(11), 7501–7509 CrossRef CAS PubMed.
- S. Shang, Y. Li, Y. Lv and W. Dai, Carbon, 2022, 11, e202200126 CAS.
- S. Chen, Q. Wan and A. K. Badu-Tawiah, Angew. Chem., Int. Ed., 2016, 55, 9345–9349 CrossRef CAS PubMed.
- M. Zheng, J. Shi, T. Yuan and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 5487–5491 CrossRef CAS.
- H. Jin, S. Ju, H. Yu, L. Yang, W. Zheng and J. Wu, Green Chem., 2023, 25, 5296–5303 RSC.
- Y. Wu, H. Yi and A. Lei, ACS Catal., 2018, 8, 1192–1196 CrossRef CAS.
- X.-R. Song, R. Li, H. Ding, X. Chen, T. Yang, J. Bai, Q. Xiao and Y. M. Liang, Org. Chem. Front., 2018, 5, 1537–1541 RSC.
- F. Jin, T. Yang, X.-R. Song, J. Bai, R. Yang, H. Ding and Q. Xiao, Molecules, 2019, 24, 3999 CrossRef PubMed.
- M. C. Carreiio, J. L. G. Ruano, G. Sanz, M. A. Toledo and A. Urbano, J. Org. Chem., 1995, 60, 5328–5331 CrossRef.
- C. Huo, H. Xie, M. Wu, X. Jia, X. Wang, F. Chen and J. Tang, Chem.–Eur. J., 2015, 21, 5723–5726 CrossRef CAS PubMed.
- C. Djerassi, Chem. Rev., 1948, 43, 271–317 CrossRef CAS.
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837–7042 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2295384. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra06747e |
| This journal is © The Royal Society of Chemistry 2023 |