DOI:
10.1039/D3RA07140E
(Review Article)
RSC Adv., 2023,
13, 36346-36363
α-Lipoic acid chemistry: the past 70 years
Received
20th October 2023
, Accepted 23rd November 2023
First published on 13th December 2023
Abstract
α-Lipoic acid (ALA) is a naturally occurring sulfur-containing fatty acid with high antioxidant activity. It is also used to treat diabetes, nerve pain, weight loss, heart disease, and primary mitochondrial disorders. Moreover, numerous therapeutic agents have been studied for managing other clinical conditions, including for anticancer, anti-HIV, anti-inflammatory, and anti-AD treatments. The medicinal importance of ALA, especially its biologically active form (R-ALA), has attracted considerable attention from synthetic chemists in industrial and academic fields. In this review, we discuss synthetic approaches to ALA and R-ALA over the past 70 years (1952 to the present), which will help medicinal chemists further develop novel routes for their synthesis.
 Jia-Qi Wang | Jia-Qi Wang studied applied chemistry at Northeast Forestry University and obtained her BS degree in 2011. In 2015, she got her MS degree from the School of Donghua University, focusing on the synthesis of waterborne fluorinated polyurethane. In 2020, she commenced her doctoral studies in the research group of Prof. Fen-Er Chen at Fudan University, working on developing novel and efficient strategies for the synthesis of pharmaceutical compounds. |
 Xu Ling | Xu Ling studied pharmaceutical science from the School of Pharmacy at Chongqing Medical University and obtained his BS degree in 2007. In 2010, he got his MS degree from the School of Pharmacy at Chongqing Medical University, focusing on the study of targeted drug delivery. In 2022, he obtained his PhD degree from the West China School of Pharmacy, Sichuan University under the supervision of Prof. Fen-Er Chen, engaging on the design and synthesis of antiviral drugs. In 2023, he joined Prof. Shen-You Nie's group at Chongqing Medical University as a Postdoctoral Fellow, focusing on the design and synthesis of antiviral drugs and relative methodology development. |
 Hui-Jing Wang | Hui-Jing Wang obtained her bachelor's degree in pharmaceutical science from Sichuan University in 2012. In 2017, she obtained her PhD degree in Chongqing University under the supervision of Prof. Yong Qin, working on total synthesis of natural product and relative methodology development. From 2017 to 2019, she worked with Prof. Dionicio Siegel in University of California, San Diego as a postdoctoral researcher. In 2020, she joined the group of Prof. Fen-Er Chen at Sichuan University as an associate professor. Her current research interests center on development of novel enzymatic reactions and chemoenzymatic strategies, total synthesis of bioactive natural products and pharmaceuticals. |
 Fen-Er Chen | Professor Fer-Er Chen received his MS in Pharmacy and PhD in organic chemistry from Sichuan University. He joined Wuhan Institute of Technology in 1988 and was promoted to professor in 1996. In 1998, he moved to Fudan University as full professor. His current research focuses on the development of new asymmetric catalysis, asymmetric total synthesis of natural products, process chemistry and computer assisted mechanism-based drug design (CADD). Prof. Fen-Er Chen has been as a visiting professor at numerous prestigious universities, including Washington University, King's College London, etc. He is currently a member of Academician of Chinese Academy of Engineering. |
1. Introduction
Nature is extremely generous in offering life-saving therapies using diverse natural products. α-Lipoic acid, also known as thioctic acid or ALA, is a naturally occurring, sulfur-containing fatty acid of great pharmacological importance. ALA was first isolated as an amphipathic molecule from liver tissue in 1951 by Reed et al.1 and was originally identified as an enzymatic cofactor of dihydrolipoate acyltransferase in the mitochondrial tricarboxylic acid cycle that manages gene transcription. Since then, various biological activities of ALA have been demonstrated. It is commonly known to function as a powerful antioxidant and is manufactured as a medicine or supplement to prevent aging.2 In addition, ALA is used in antidiabetic treatments because of its excellent glycemic control. It has been approved for the treatment of diabetic polyneuropathy in Germany.3 Moreover, it has great therapeutic potential in managing numerous clinical conditions, including anti-cancer, anti-HIV, anti-inflammatory, and anti-AD treatments. Some reviews have summarized the biological activities and therapeutic potentials of ALA.4
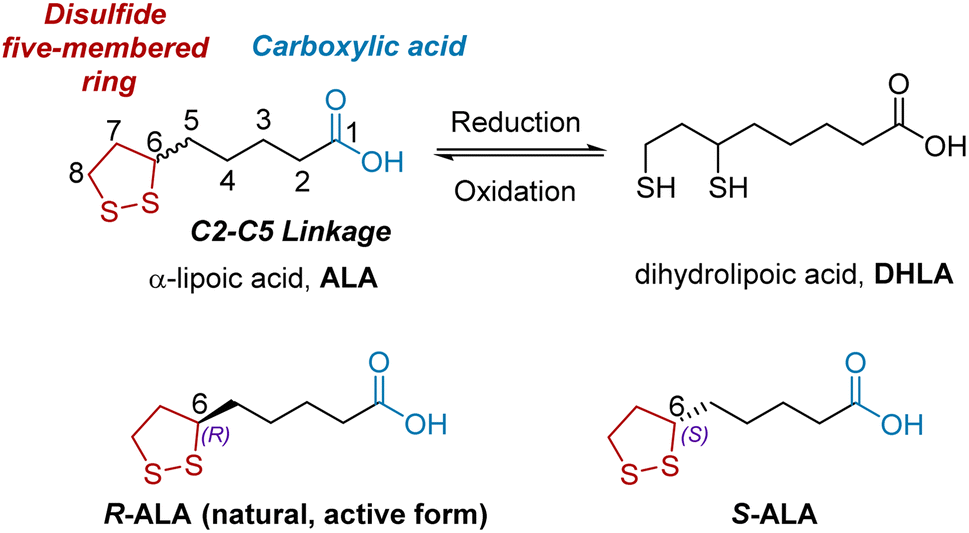
ALA is composed of three main structural units (Fig. 1): a disulfide five-membered ring (red), a C2–C5 linkage (black), and carboxylic acid (blue). ALA has a single chiral center at C6, resulting in two enantiomeric forms: R- and S-ALA.5 Notably, ALA exists in nature as the R enantiomer, which is the biologically active form. However, ALA is often marketed in its racemic form because S-ALA shows no significant biological side effects. The reduced form of ALA is an active metabolite, called dihydrolipoic acid (DHLA), which can be converted to ALA under various oxidative conditions such as FeCl3/O2 and I2/KI.
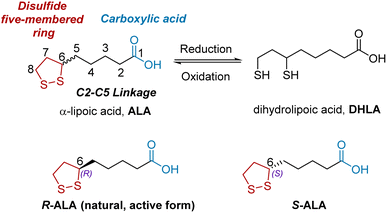 |
| | Fig. 1 The chemical structure of racemic ALA, optical isomers of ALA, and its active metabolite DHLA. | |
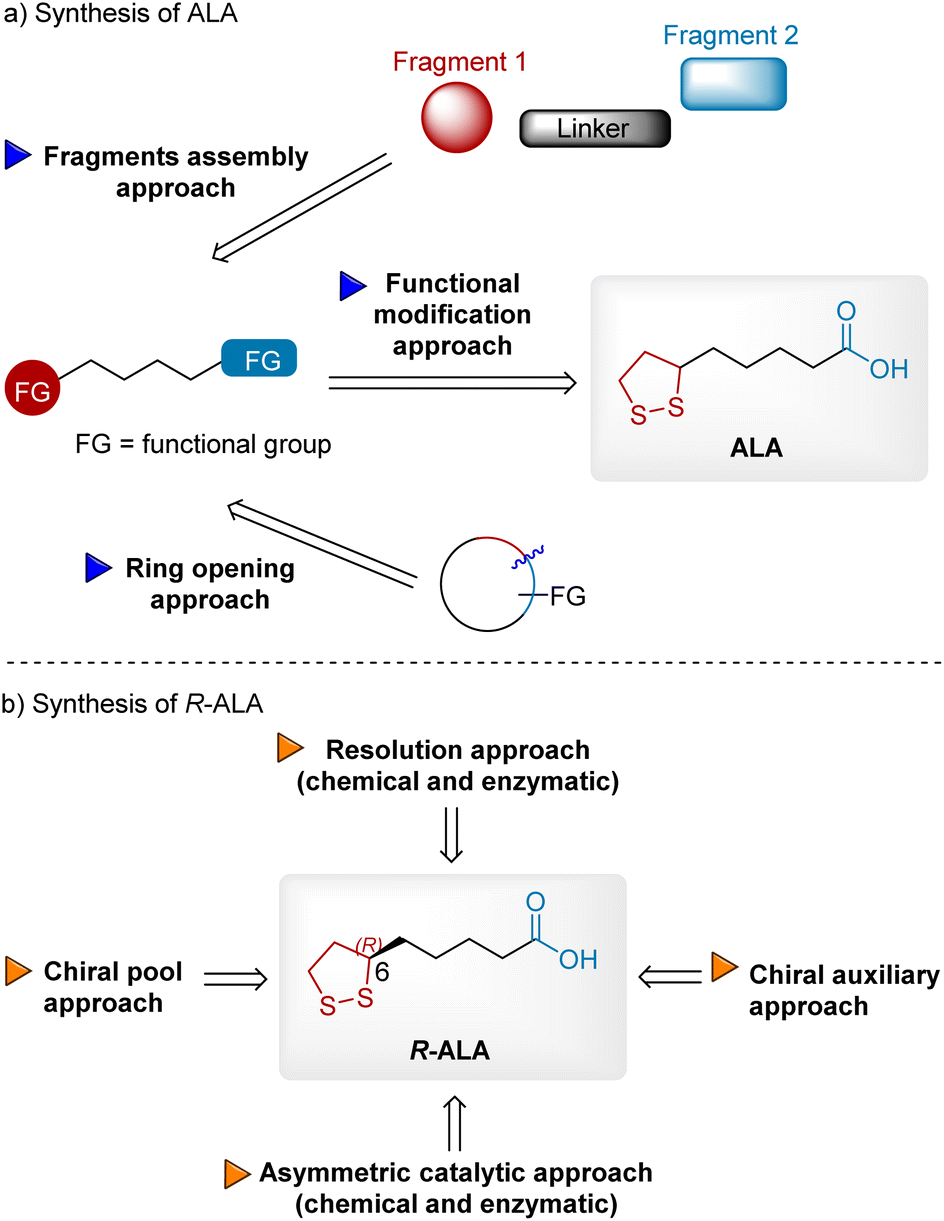
The high medicinal importance of ALA, especially R-ALA,6 has attracted great attention from synthetic chemists in industrial and academic fields. The global ALA market size was valued at USD 106.89 million in 2022 and is expected to expand at a CAGR of 3.86 percent during the forecast period, reaching USD 134.15 million by 2028. The extensive demand for ALA in global markets 3000–3500 tons per year necessitates the development of efficient synthetic approaches. Over the past 70 years, numerous synthetic approaches have been patented and reported. To the best of our knowledge, only a few reviews have summarized ALA's synthesis approaches. In 1990, Yadav et al. published an excellent review of synthetic studies on ALA.7 Schaefer et al. published another review in 2019 but it was relatively brief.8 In this review, we provide comprehensive advances in the fields of ALA and R-ALA synthesis to inspire the scientific community to develop innovations (Fig. 2). First, the synthesis of ALA is summarized and discussed according to three different approaches: functional modification, fragment assembly, and ring opening (Fig. 2a). In the functional modification approach, ALA can be readily synthesized from precursors bearing inherent C2–C5 linkage and two terminal functional groups. Some of these precursors are commercially available, but some must be synthesized from simple building blocks or cyclic compounds, resulting in fragment assembly and ring-opening approaches. In the second part, stereoselective approaches to R-ALA are summarized and classified into four sections: chiral resolution (chemical and enzymatic), chiral pool, chiral auxiliary, and asymmetric catalysts (chemical and enzymatic) to introduce chirality into the molecule (Fig. 2b).
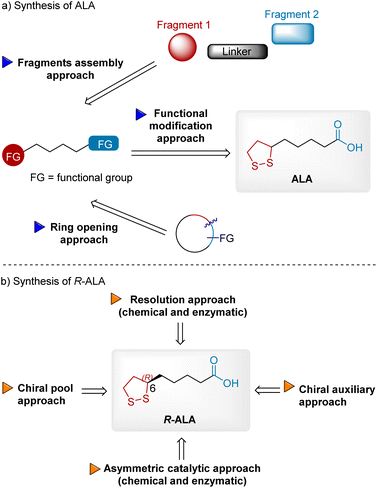 |
| | Fig. 2 Synthetic strategies for synthesizing ALA and R-ALA. (a) Synthesis of ALA; (b) synthesis of R-ALA. | |
2. Synthesis of racemic ALA
2.1 Functional modification
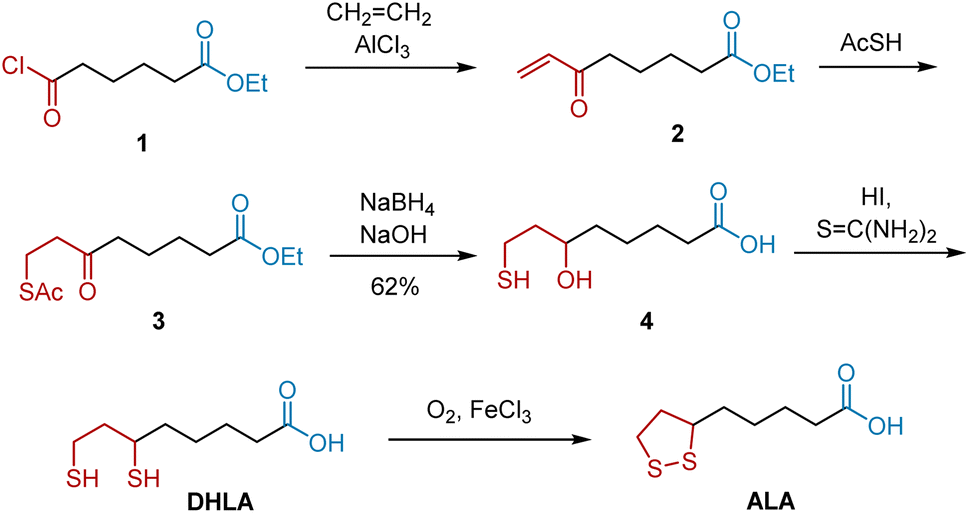
In 1952, Bullock et al. briefly reported the first total synthesis of racemic ALA.9 Later, they published the full article describing their synthetic approach which confirmed the structure of ALA.,10 Dicarbonyl compounds have great potential to be transformed into five-membered disulfide rings and carboxylic acids, which are key moieties in ALA. As shown in Scheme 1, compound 1, ethyl 6-chloro-6-oxohexanoate was selected as the starting material, because its inherent structure could be transformed into that of ALA through functional modifications. Initially, the Lewis acid-catalyzed insertion of ethene, followed by the spontaneous elimination of hydrogen chloride, afforded unsaturated ketone 2. Treatment of 2 with thiolacetic acid afforded compound 3, whose ketone group was reduced to a tertiary alcohol group. Subsequent hydrolysis delivered compound 4, which reacted with excess thiourea and hydriodic acid to yield DHLA. Finally, DHLA was oxidized smoothly in the presence of oxygen and ferric chloride to deliver ALA. The yields of most of these steps have not been previously reported.
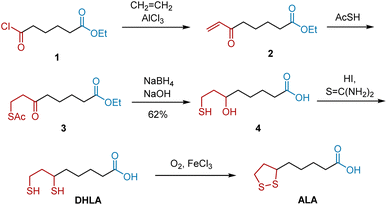 |
| | Scheme 1 Bullock's synthesis of ALA. | |
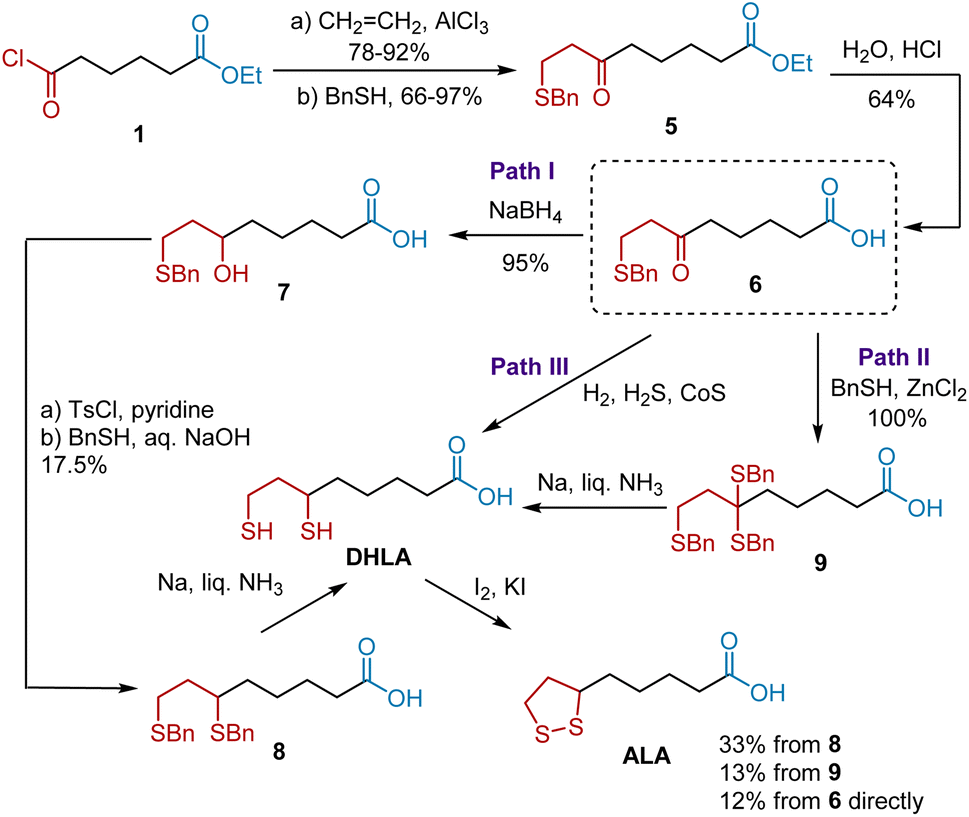
Inspired by this pioneering work, Soper et al. expanded the first generation of synthetic approaches, especially enriching the paths from keto acids, such as compound 6, to ALA (Scheme 2).11 Keto acid 6 was synthesized from 1 according to Bullock's protocol. Having synthesized keto acid 6, three divided paths were developed, providing fundamental benefits for the subsequent synthesis. Path I began with the NaBH4-mediated reduction of 6 to yield hydroxy acid 7 with a 95% yield. The esterification of 7 with tosyl, followed by treatment with benzyl mercaptan and a sodium hydroxide solution gave 6,8-dibenzylmercaptoöctanoic acid 8 in 17.5% yield over two steps. After deprotection, the DHLA was obtained. The subsequent oxidation of DHLA directly delivered ALA. Path II was developed using tribenzyl mercaptocaprylic acid 9 as the key intermediate, obtained from 6 in the presence of benzyl mercaptan and anhydrous zinc chloride. The treatment of 9 with liquid ammonia and solid sodium in anhydrous ether afforded DHLA, which was then oxidized to ALA. Alternatively, they developed a more efficient two-step synthetic route to ALA from 6 (path III). Hydrogenation of 6 using a sulfur/cobalt sulfide catalyst in the presence of H2 followed by the oxidation of DHLA afforded ALA in 12% yield over two steps.
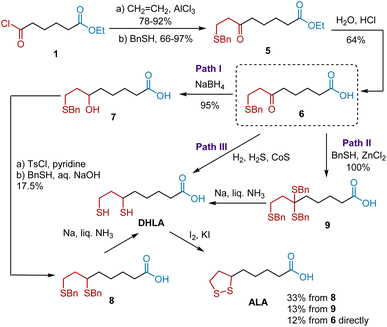 |
| | Scheme 2 Soper's synthesis of ALA. | |
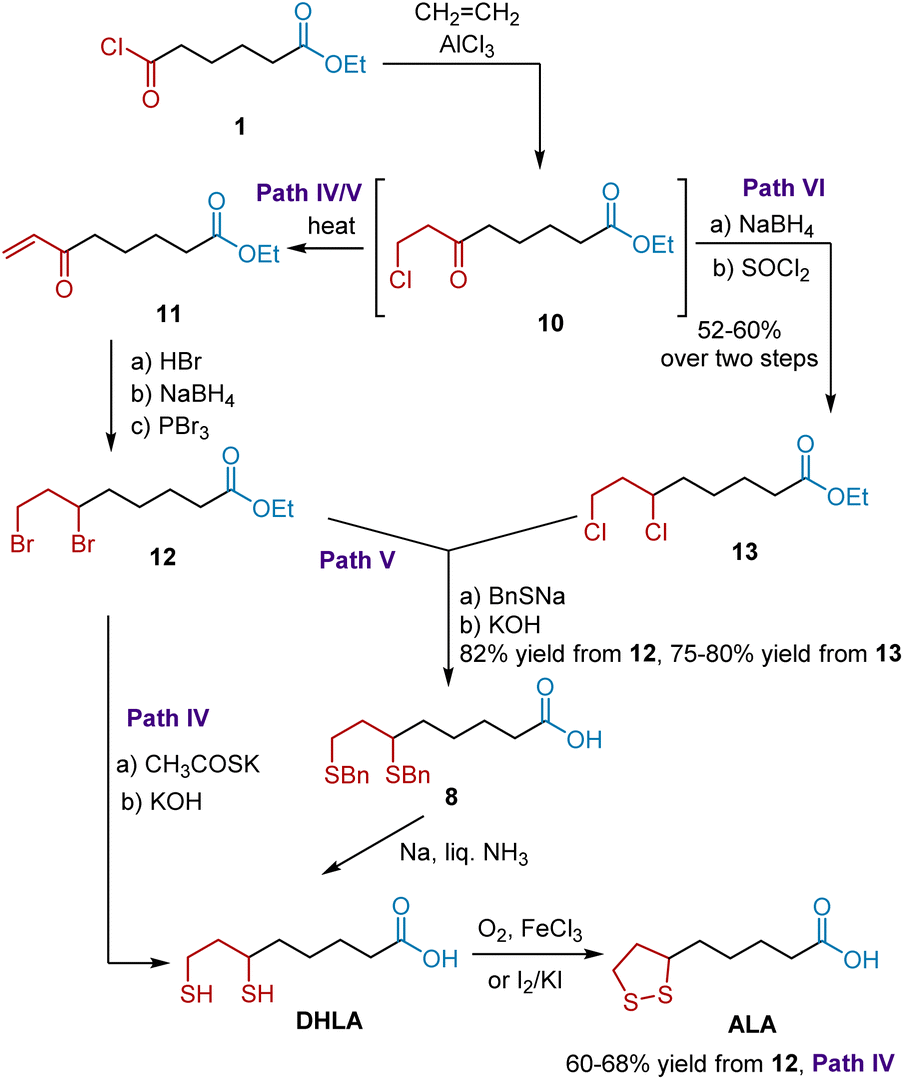
Since 6,8-dibenzylmercaptoöctanoic acid 8 and DHLA are useful intermediates for synthesizing ALA, an improved synthesis of 8 and DHLA was further developed by Reed et al. in 1955.12 As shown in Scheme 3, the synthesis also commenced with acid chloride 1, which was transformed into 8-chloro-6-oxoctanoöate 10. Without purification, compound 10 was transformed into dibromo esters 12 and dichloro esters 13 as key intermediates. The former paths IV and V started from heating 10 to give α,β-unsaturated ketone 11. The treatment of ketone 11 with anhydrous hydrogen bromide, followed by reduction and bromination, generated dibromo ester 12, which was readily transformed into DHLA and 8. In path IV, ALA was synthesized in 60–68% yields based on DHLA via thiolation and oxidation. In path V, dibromo ester 12 was converted to 8 in 82% yield by treatment with sodium benzyl mercaptide, followed by alkaline hydrolysis. ALA was synthesized in high yield according to Soper's protocol. In addition, path VI readily converted dichloro ester 13 to ALA. Later, these paths were widely used to synthesize ALA and were published in many patents13 and in the literature.14
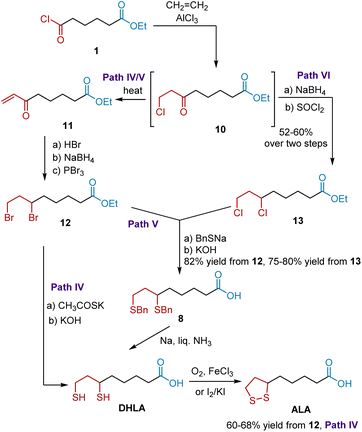 |
| | Scheme 3 Reed's synthesis of ALA. | |
Walton et al. reported the synthesis of ALA starting from 7-carbethoxy-2-heptenoic acid 14 by introducing a thiol group at C6, as shown in Scheme 4.15 The addition of thiolacetic acid to 14 yielded carboxylic acid 15 in 93% yield, which was then converted into its acid chloride 16. The following two-step reactions involving NaBH4-mediated reduction and alkaline hydrolysis proceeded smoothly to generate 8-hydroxy-6-thioloctanoic acid 17. The replacement of 17 followed by oxidation gave rise to ALA. R-ALA and S-ALA were synthesized using the corresponding chiral isomers of 15.
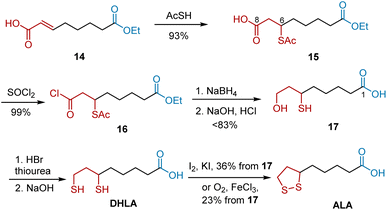 |
| | Scheme 4 Walton's synthesis of ALA. | |
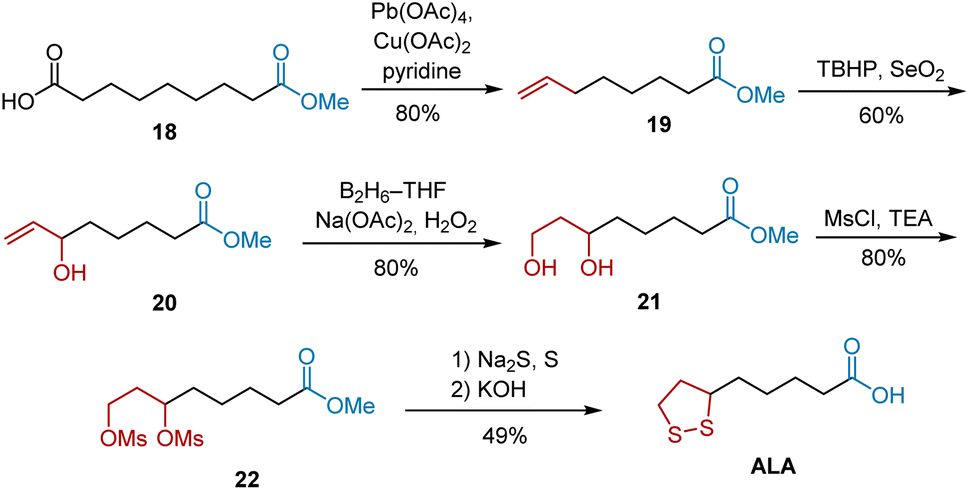
In 1987, Rao et al. developed a highly regioselective allylic oxidation of olefins using tert-butyl hydroperoxide (TBHP) and selenium dioxide (SeO2) to construct a secondary allylic alcohol, followed by the hydroboration–oxidation of the terminal double bond to construct a 1,3-diol system. By using this method, as illustrated in Scheme 5.16 Olefin methyl 7-octenoate 19 was synthesized from azelaic acid 18 via decarboxylative elimination using Pb(OAc)4/Cu(OAc)2/pyridine. In the presence of selenium dioxide and tert-butyl hydroperoxide, the regioselective allylic oxidation of 19 afforded secondary allylic alcohol 20 in 60% yield, which was treated with a borane–tetrahydrofuran complex to yield 21. Finally, the conversion of 21 to dimesylate 22, reaction with Na2S/S, and hydrolysis of the ester completed the synthesis of ALA.
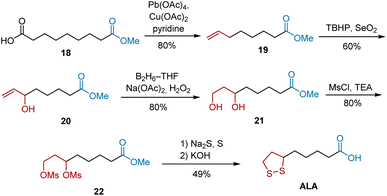 |
| | Scheme 5 Rao's synthesis of ALA. | |
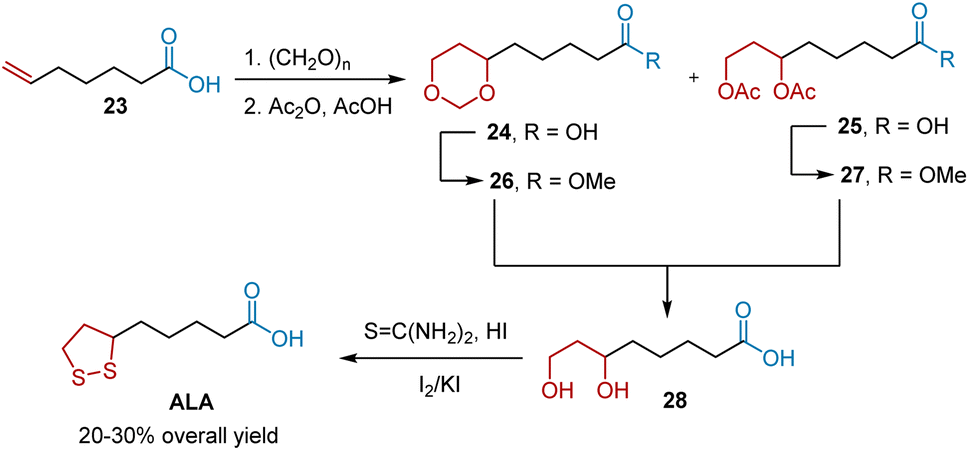
The Prins reaction is an acid-catalyzed condensation of olefins with aldehydes to produce 1,3-diols, 1,3-dioxanes, or unsaturated alcohols. Among them, 1,3-diols and 1,3-dioxanes can be readily transformed into five-membered disulfide rings. In 1956, Braude et al. reported a route to ALA from hept-6-enoic acid 23 using the Prins reaction as the key step (Scheme 6).17 The Prins reaction of 23 generated a mixture of 1,3-dioxane 24 and acyl-protected 1,3-diol 25 in a ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The conversions of Prins reaction products 24 and 25 into methyl esters 26 and 27 were performed to avoid lactonization during the synthesis of diol 28. Diol 28 was transformed into ALA as previously described. The authors reported that the overall ALA yield was 20–30%.
:1. The conversions of Prins reaction products 24 and 25 into methyl esters 26 and 27 were performed to avoid lactonization during the synthesis of diol 28. Diol 28 was transformed into ALA as previously described. The authors reported that the overall ALA yield was 20–30%.
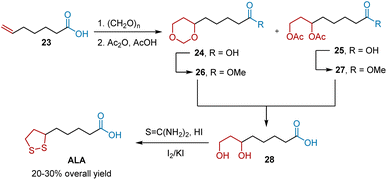 |
| | Scheme 6 Braude's synthesis of ALA. | |
2.2 Fragments assembly
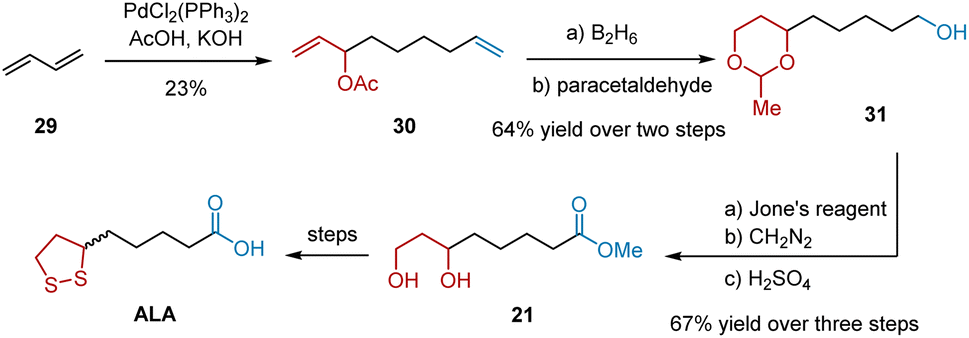
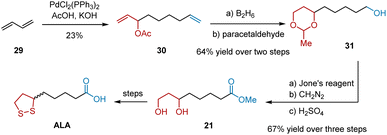
In 1978, Tsuji et al. reported a synthetic route to ALA using butadiene telomer 1 as the starting material (Scheme 7).18 Telomer 30, bearing the eight-carbon chain necessary for ALA synthesis, was synthesized from 29 using PdCl2(PPh3)2/AcOH/KOH. Hydroboration of the two terminal double bonds and subsequent protection yielded 31 in 64% yield over two steps. Oxidation of the unprotected terminal alcohol was carried out using the Jones reagent, giving carboxylic acid. Next, it was methylated with diazomethane to prevent lactone formation. After deprotection, 1,3-diol 21 was obtained in 67% yield in three steps. 1,3-Diol 21 can be transformed into ALA, as described in a previously published protocol.
 |
| | Scheme 7 Tsuji's synthesis of ALA. | |
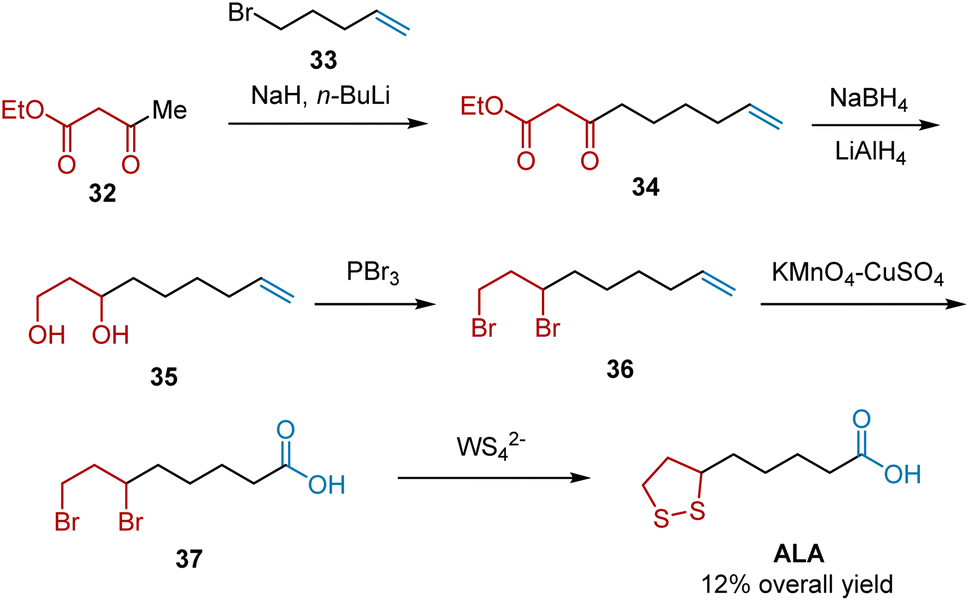
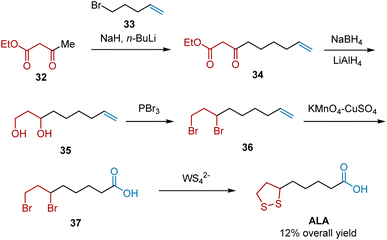
In 1992, Chandrasekaran et al. developed a methodology for the construction of cyclic disulfides from alkyl halides using tetrathiometalates (MS42) as sulfur transfer reagents. Subsequently, by employing this reaction as the key step, ALA was synthesized (Scheme 8).19 The synthesis began with the alkylation of ethylacetoacetate 32 and 5-bromopent-1-ene 33. The obtained intermediate 34 was reduced stepwise to 1,3-diol 35, whose alcohol groups were brominated to yield 6,8-dibromooctanoic acid 36. Oxidative cleavage of the terminal double bond in the presence of KMnO4 afforded compound 37 as a precursor to cyclic disulfides. Subsequently, the MS42−-mediated sulfur transfer reaction of 37 occurred, generating ALA in 12% overall yield.
 |
| | Scheme 8 Chandrasekaran's synthesis of ALA. | |
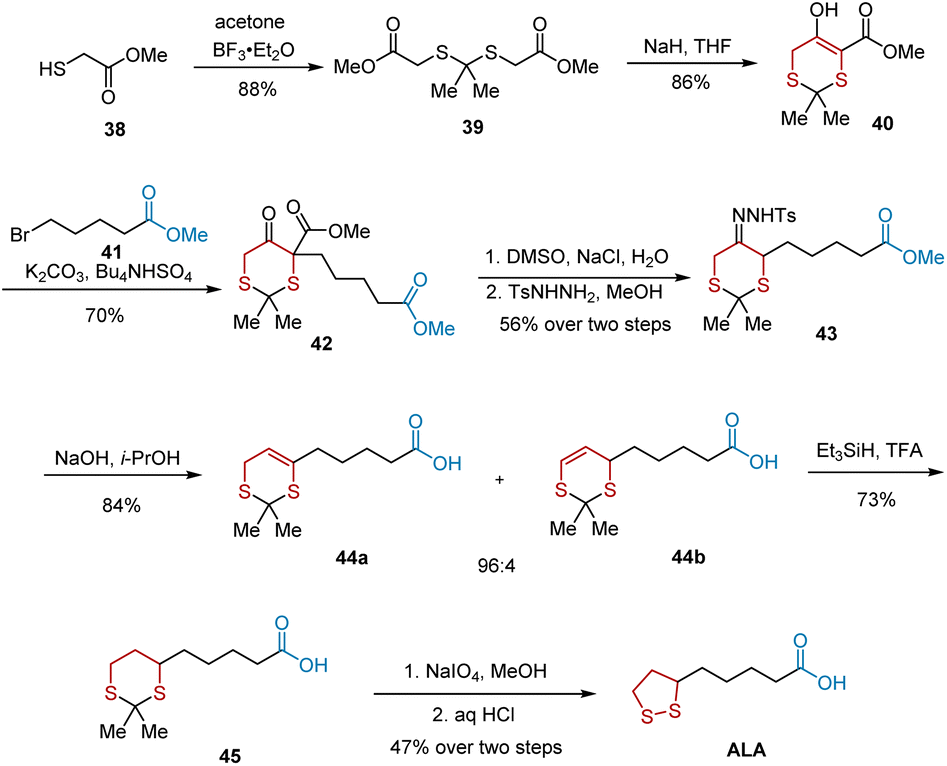
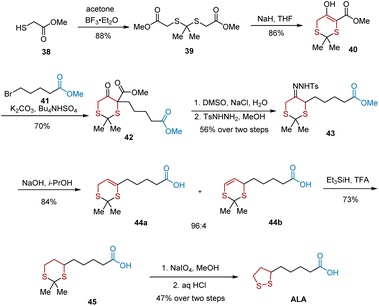
In 2005, Chavan et al. developed the regioselective decomposition of tosylhydrazones to form olefins and utilized this method to accomplish the total synthesis of ALA.20 As illustrated in Scheme 9, the synthesis began with acetonide protection of methyl thioglycolate 38 to give 88% yield of diester 39, which was subjected to Dieckmann condensation to afford cyclic compound 40 in 86% yield. The C-alkylation of 40 was carried out using K2CO3 in the presence of Bu4NHSO4 as a phase transfer catalyst, leading to the formation of C-alkylated product 42 in 70% yield. A two-step sequence involving decarboxylation and tosylhydrazone formation resulted in tosylhydrazone 43. Using NaOH in isopropyl alcohol, tosylhydrazone 43 was decomposed to olefins 44a and 44b with a regioselectivity of 96:4 in 84% yield. After the reduction of the mixture of olefins 44a and 44b, dithiane acid 45 was obtained. Subsequent oxidation and treatment with aqueous HCl in benzene culminated in the production of ALA in 47% yield.
 |
| | Scheme 9 Chavan's synthesis of ALA. | |
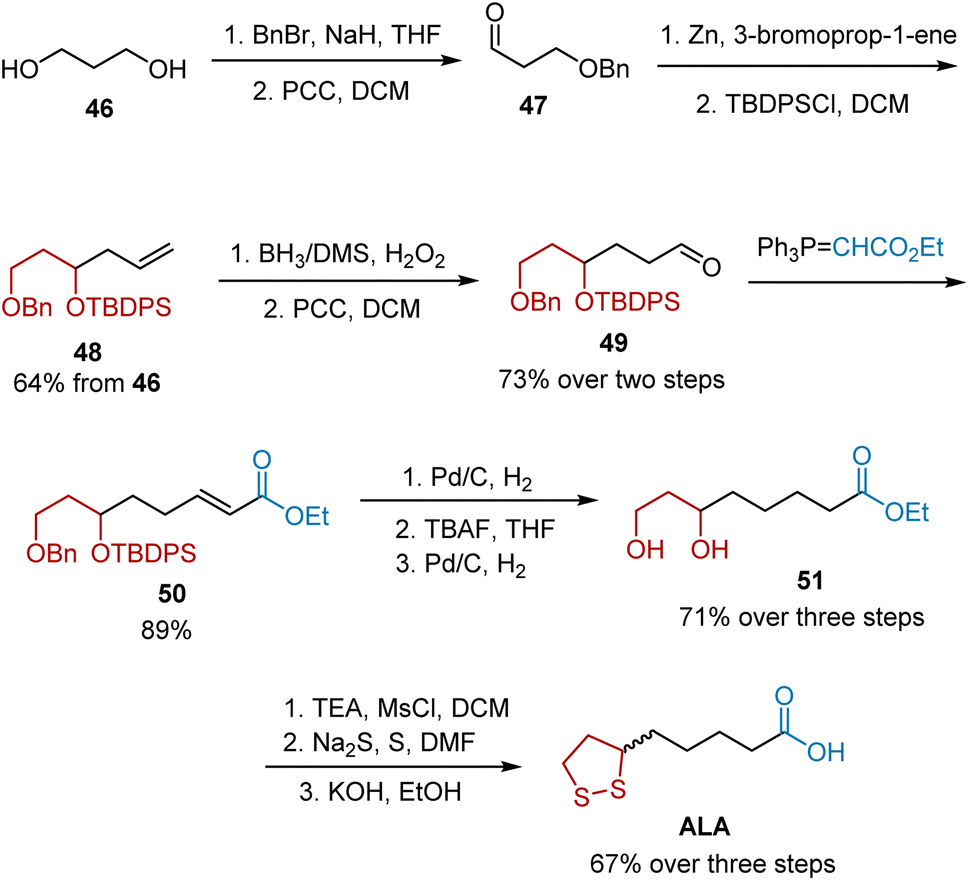
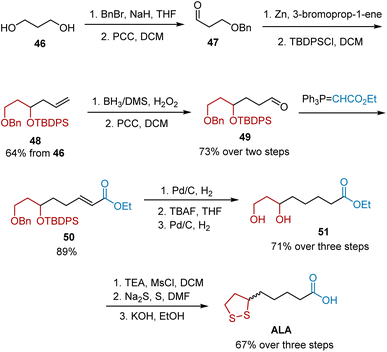
In 2015, Purude et al. reported their efforts to synthesize either racemic ALA or its chiral isomers, R- and S-ALA (Scheme 10).21 Selective benzyl protection of commercially available diol 46 and subsequent oxidation afforded aldehyde 47, which upon treatment with allyl bromide in the presence of Zn, followed by TBDPS protection, yielded terminal alkene 48. Oxidation and the Wittig reaction gave unsaturated ester 50, which possesses fundamental functional groups to afford ALA. Ester 50 was converted into diol 51 through a three-step sequence, including selective hydrogenation of the double bond and deprotection. Racemic ALA was obtained under the known protocol from diol 51. Using this strategy, the synthesis of R-ALA and S-ALA was also accomplished using this strategy.
 |
| | Scheme 10 Purude's synthesis of ALA. | |
2.3 Ring opening
Some cyclic compounds are readily transformed into 1,3-diol, which is a precursor for the formation of five-membered disulfide rings. Thus, efforts were made to synthesize ALA from cyclic compounds, followed by introducing the C2–C5 linkage and the carboxylic acid group at any stage of the synthesis.
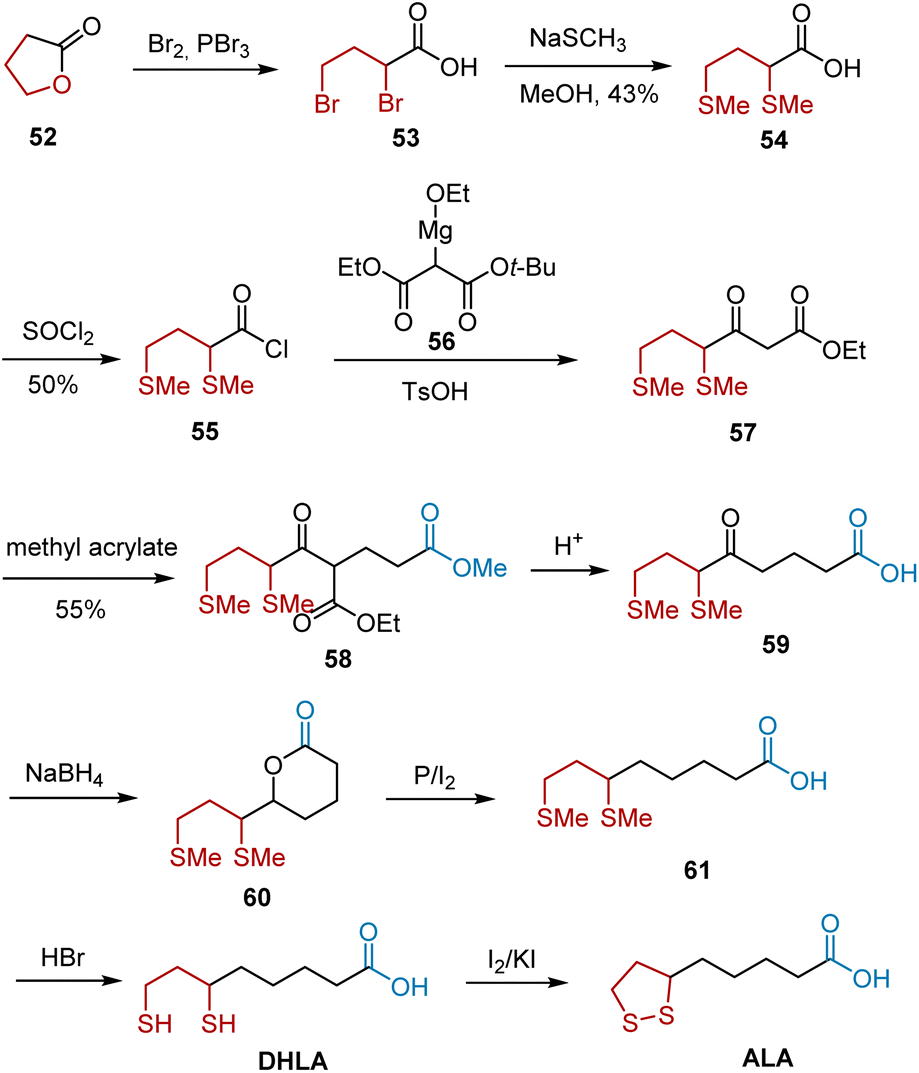
In 1955, Wagner et al. reported a synthesis of ALA by adopting γ-butyrolactone 52 as the starting material (Scheme 11).22 The first sulfur-containing intermediate 54 was synthesized from lactone 52, which was then converted into its acid chloride 55. Acylation, elimination of the t-butyl group, and decarboxylation in the presence of a catalytic amount of TsOH transformed 55 to 57, followed by condensation with methyl acrylate. Hydrolysis and decarboxylation of 57 under acidic conditions generated keto acid 59. The reduction of 59 to 61 was accomplished in two steps. Finally, demethylation and subsequent oxidation occurred, leading to ALA. The yields of most of these steps have not been reported in the literature.
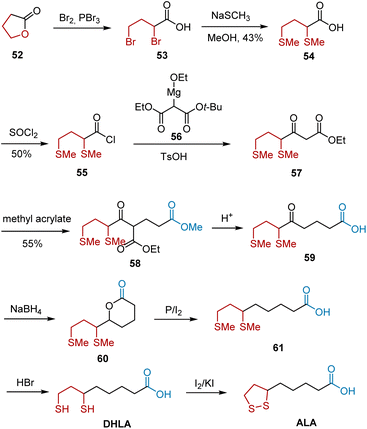 |
| | Scheme 11 Wagner's synthesis of ALA. | |
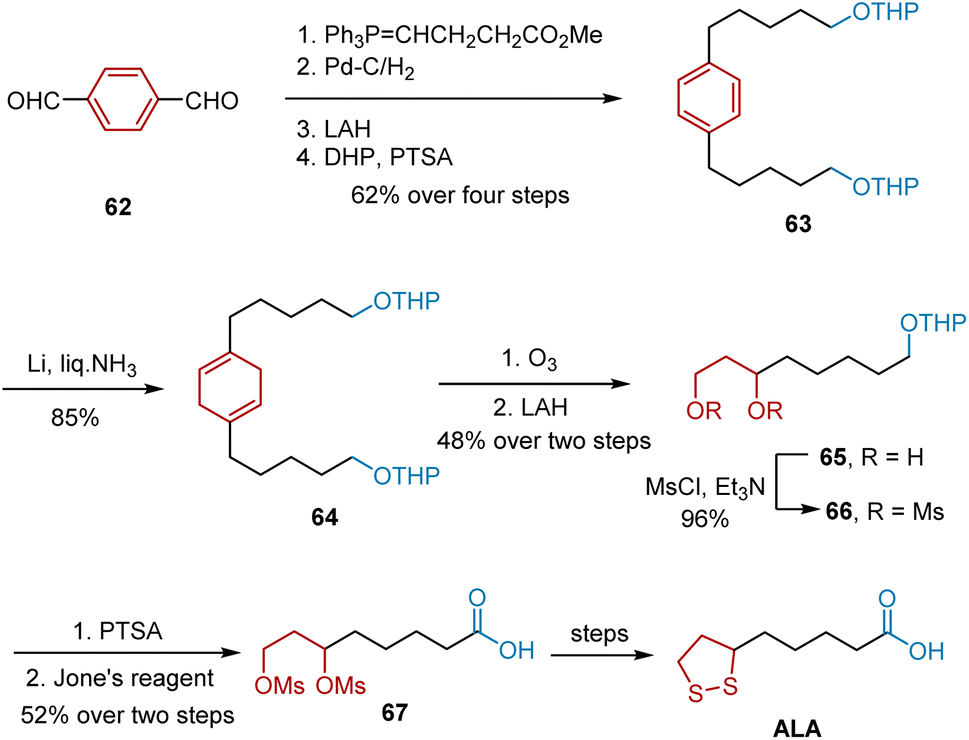
Another synthetic work was reported by Rao et al. in 1995 using Birch reduction as the key step (Scheme 12).23 The synthesis commenced with a Wittig reaction of terephthalaldehyde 62 and the ylide obtained from (3-carbomethoxy)propyl triphenylphosphonium bromide to yield an olefinic intermediate. Subsequent Pd–C-mediated hydrogenation, LAH-mediated reduction, and protection of the alcohols with tetrahydropyran gave diTHP ether 63 in 62% yield. The birch reduction of 63 afforded 1,4-diene 64 in 85% yield. Ozonolysis of 64 and quenching of the ozonide with LAH afforded 65 in 48% yield over two steps. The conversion of 65 to its dimesylate 66 was accomplished using triethylamine and mesyl chloride. Deprotection of the THP group and oxidation by Jones resulted in the formation of a carboxylic acid group in ALA, yielding 67 in 52% yield. In the last stage, ALA was synthesized according to a known procedure.
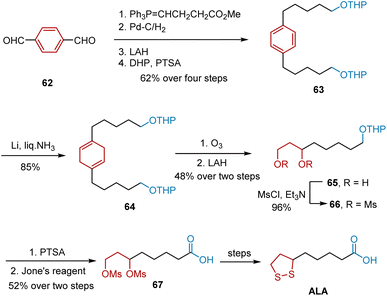 |
| | Scheme 12 Rao's synthesis of ALA. | |
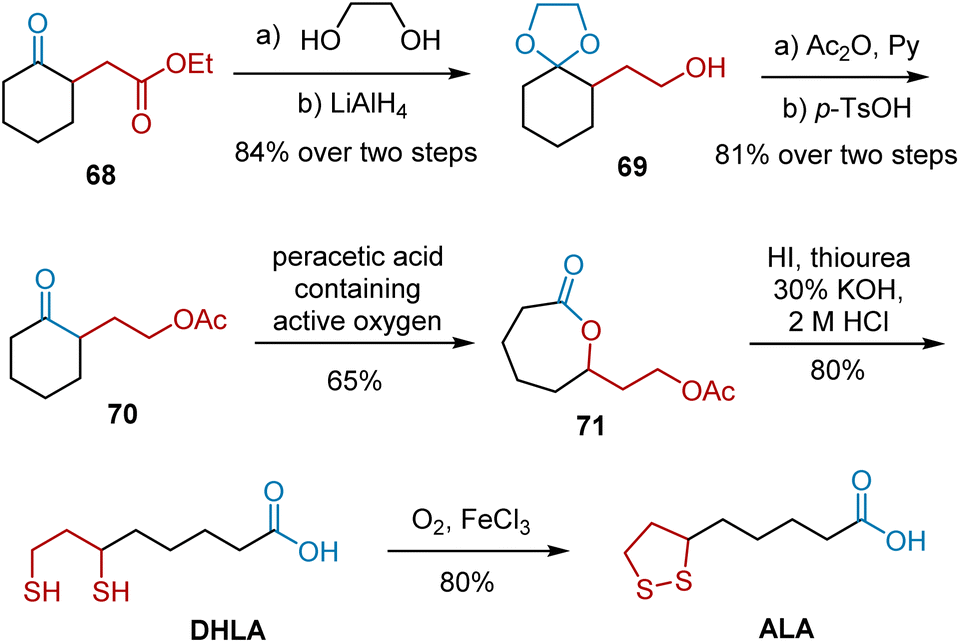
In 1957, Segre et al. selected ethyl cyclohexanone-2-acetate 68 as a precursor for ALA synthesis (Scheme 13).24 The synthesis featured the Baeyer–Villiger oxidation as the key step. A series of protection, reduction, and deprotection transformed 68 into ketone 70, which was then treated with a solution of peracetic acid (containing active oxygen) to produce seven-membered cyclolactone 71 in 65% yield. The treatment of lactone 71 with thiourea in a strong mineral acid via alkaline hydrolysis of the isothiuronium salt easily yielded DHLA, which afforded the desired ALA after oxidation.
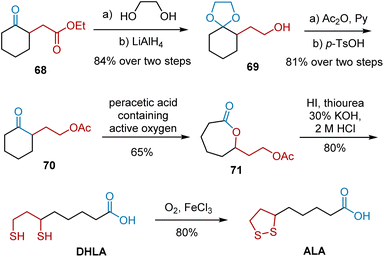 |
| | Scheme 13 Segre's synthesis of ALA. | |
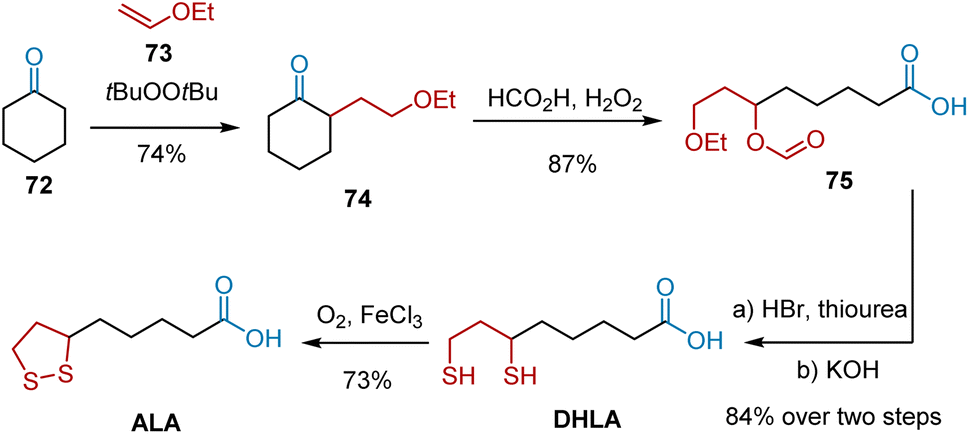
In 1999, Paust et al. followed the Baeyer–Villiger strategy to develop an optimized synthesis.25 As shown in Scheme 14, initially, the key intermediate 74 was synthesized in one stage from the base chemicals, cyclohexanone 72 and vinyl ethyl ether 73, catalyzed by di-tert-butyl peroxide. The key Baeyer–Villiger oxidation was performed using performic acid generated in situ from formic acid and 30% aqueous hydrogen peroxide. 8-Ethoxy-6-formyloxyoctanoic acid 75 was obtained as the main product in 87% yield. To introduce sulfur at C6 and C8, 75 was heated with 62% hydrohalic acid in the presence of thiourea and hydrolyzed to form DHLA, which was oxidized to ALA according to an established protocol. This route delivered ALA from cyclohexanone 72 at an overall yield of 40%.
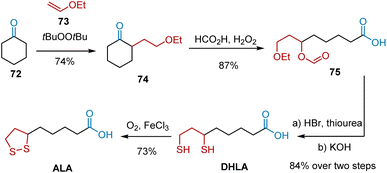 |
| | Scheme 14 Paust's synthesis of ALA. | |
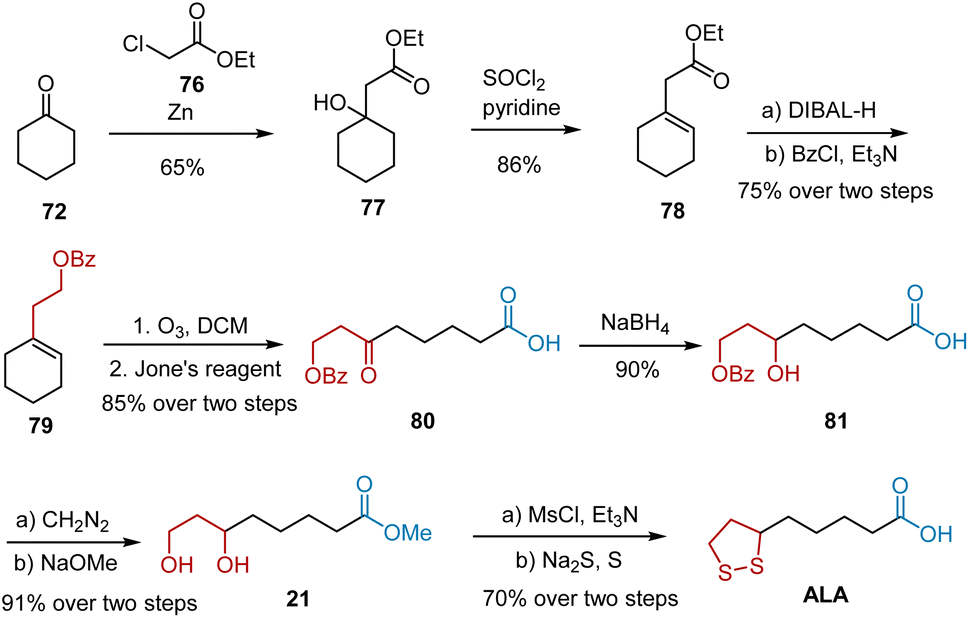
Chavan et al. have reported the synthesis of ALA via a modified Reformatsky reaction (Scheme 15).26 The synthesis commenced with the Reformatsky reaction of cyclohexanone 72 with ethyl chloroacetate 76 in the presence of activated Zn powder, followed by the elimination of the hydroxyl group to give alkene 78. DIBAL-H-mediated reduction and subsequent protection yielded compound 79, which was bubbled through O3 gas, followed by Jones oxidation to generate carboxylic acid 80. Under NaBH4-mediated reduction conditions, the tertiary alcohol 81 was obtained, which was esterified and treated with sodium methoxide to produce diol 21. Racemic ALA was obtained from diol 21 based on commonly used procedures.
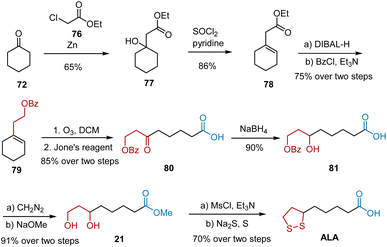 |
| | Scheme 15 Chavan's synthesis of ALA. | |
3. Synthesis of R-ALA
3.1 Chemical resolution
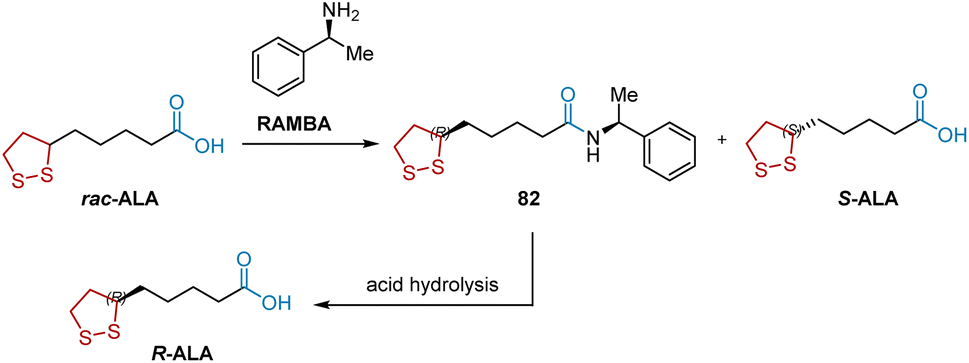
Currently, the synthesis of R-ALA on an industrial scale depends on the direct chemical resolution of its racemic mixture, ALA, using R-(+)-methylbenzylamine (RAMBA) as the chiral resolution reagent (Scheme 16).27 In the initial resolution step, RAMBA was added to the racemic ALA solution to form 82, which was separated by crystallization. After acid hydrolysis, R-ALA was obtained.
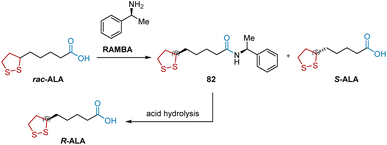 |
| | Scheme 16 RAMBA-mediated resolution of ALA to synthesize R-ALA. | |
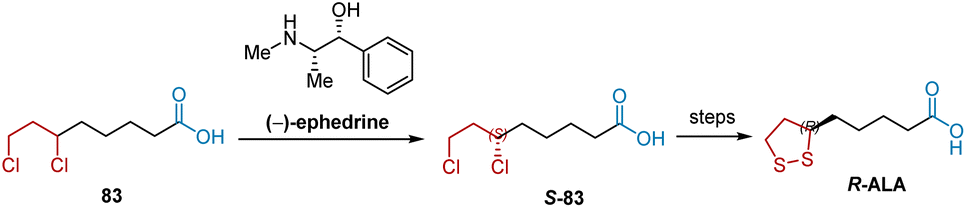
Alternatively, the resolution of racemic intermediates via a synthetic route is another method for synthesizing R-ALA. As shown in Scheme 17, S-83, a key intermediate for synthesizing R-ALA can be obtained by the optical resolution of racemic 6,8-dichlorooctanoic acid (DCA) 83 with (−)-ephedrine.14,28
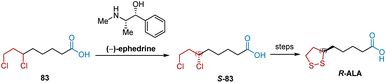 |
| | Scheme 17 (−)-Ephedrine-mediated resolution of DCA to synthesize R-ALA. | |
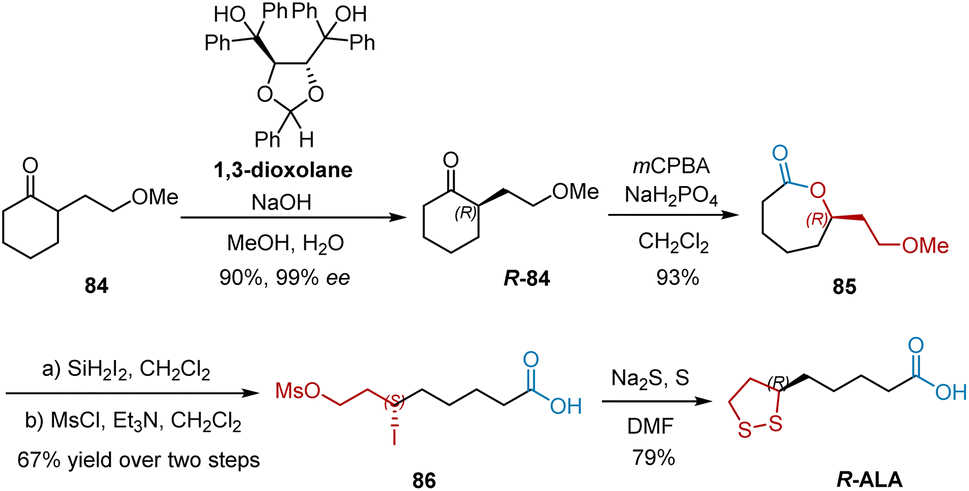
The aforementioned studies mainly involved esterification of the carboxylic group located four carbon atoms away from the stereogenic center, which remains a difficult task. In 2010, Kaku et al. developed the thermodynamically controlled deracemization of racemic monosubstituted cycloalkanones and utilized this methodology to accomplish the synthesis of R-ALA (Scheme 18).29 Using chiral 1,3-dioxolane as the chiral host molecule with sodium hydroxide in aqueous methanol, cycloalkanone 84 was converted into its R-isomers R-84 with 99% ee and 90% yield. R-84 was subjected to Baeyer–Villiger oxidation using 3-chloroperoxybenzoic acid and anhydrous sodium dihydrogen phosphate to afford lactone 85 in 93% yield. Ring-opening iodination of 85 and subsequent mesylation afforded 86, which was transformed into R-ALA in the presence of sodium sulfide and sulfur in DMF.
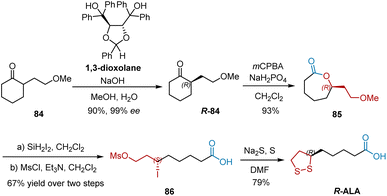 |
| | Scheme 18 Kaku's synthesis of R-ALA. | |
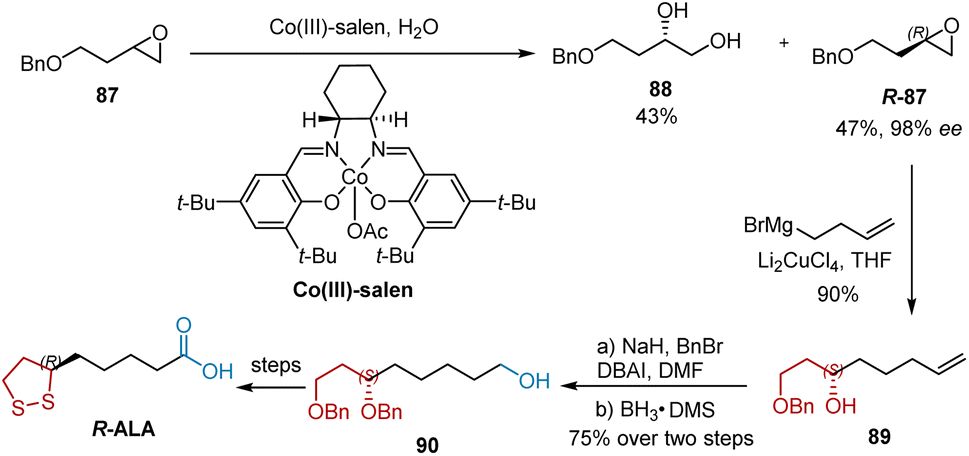
Kinetic resolution is a widely used method that provides easy access to catalysts, separation and recovery of catalysts, high enantiomeric excess, and high practical feasibility. Jacobsen's hydrolytic kinetic resolution (HKR) method uses a readily accessible Co-based chiral salen complex as the catalyst and is an efficient approach for the preparation of highly enantioselective chiral epoxides and diols with high optical purity in excellent yields. In 2006, Bose utilized the HKR of 1 in the synthesis of R-ALA.30 As shown in Scheme 19, racemic epoxide 87 was treated with (R,R)-salen–Co(III)–OAc complex and H2O at ambient temperature to afford a mixture of 1,2-diol 88 in 43% yield and epoxide R-87 in 47% yield and 98% ee. The regiospecific opening of epoxide R-87 with a Grignard reagent delivered chiral alcohol 89 in 90% yield. Subsequent benzyl protection and borohydride oxidation transformed 89 to 90, which was used to synthesize R-ALA by conventionally reported transformations including oxidation, deprotection, and mesylation.
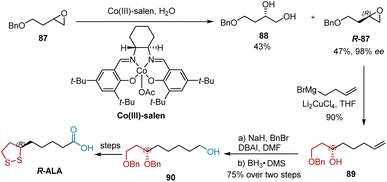 |
| | Scheme 19 Bose's synthesis of R-ALA. | |
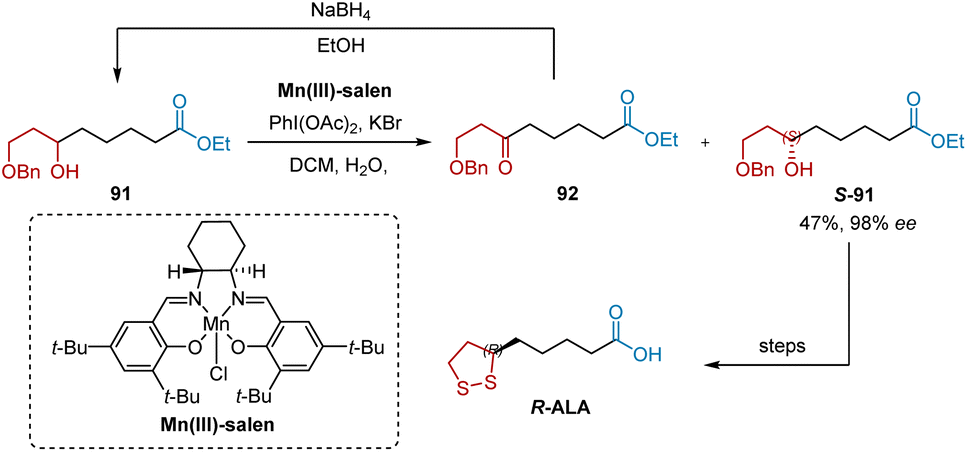
Mn(III)-salen is the most widely used chiral catalyst in the oxidative resolution of secondary alcohols. In 2015, Kalkote and Chavan reported the use of Mn(III)-salen-catalyzed oxidative kinetic resolution as the key step in obtaining enantiomerically pure R- and S-ALA (Scheme 20).21 The oxidative kinetic resolution of racemic 91 was performed on Mn(III)-salen to produce S-91 in 98% ee and 47% yield. The construction of R-ALA was achieved under the aforementioned conditions.
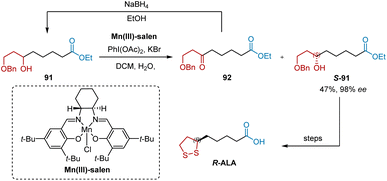 |
| | Scheme 20 Kalkote and Chavan's synthesis of R-ALA. | |
3.2 Enzymatic resolution
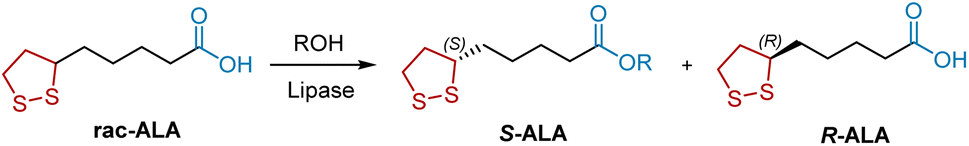
Enzyme-catalyzed organic reactions have provided great impetus for organic synthesis owing to several advantages, including mild and environmentally friendly reaction conditions and remarkable chemo-, regio-, and stereoselectivity. Lipases are routinely used for the treatment of racemic alcohols and carboxylic acids. R-ALA was synthesized through the resolution of racemic ALA by enzymatic esterification of the carboxylic group, which consists of four carbon atoms away from the stereogenic center (Scheme 21). In 1997, Fadnavis et al. used the lipase from Candida rugosa (E.C.3.1.1.3) to catalyze the enantioselective esterification of racemic ALA with aliphatic alcohols in hexane.31 The enzyme shows enantioselectivity towards the S-ALA (72% ee). In 2009, Yan et al. reported that the lipase from Aspergillus oryzae WZ007 is another potential candidate for enzymatic resolution of ALA.32 In 2015, Liu et al. combined an ionic liquid co-lyophilized lipase with microwave irradiation to improve enzyme performance in the enantioselective esterification of ALA.33 Under optimal reaction conditions, the ionic liquid co-lyophilized lipase exhibited satisfactory enzyme performance (E value, 41.2; enzyme activity, 178.1 μmol h−1 mg−1).
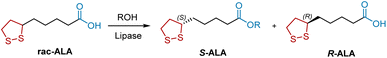 |
| | Scheme 21 Lipase-mediated resolution of ALA. | |
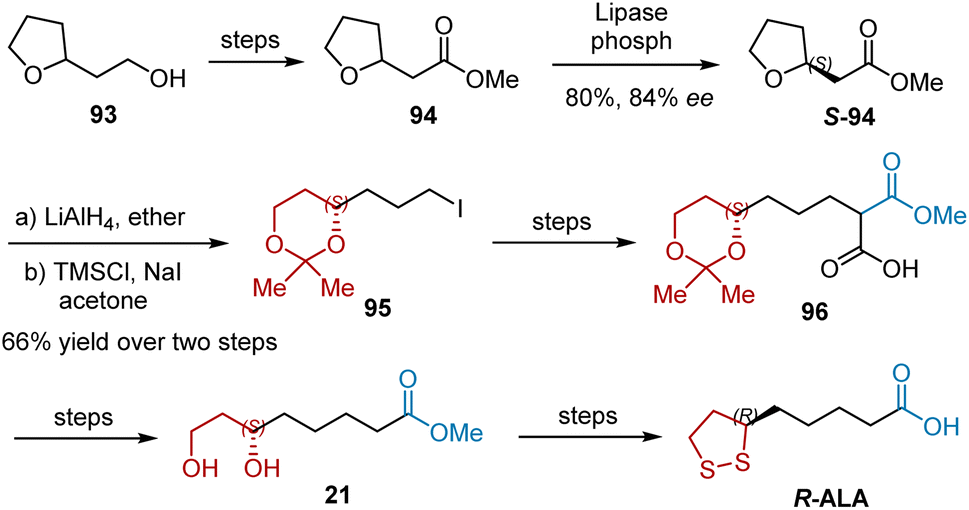
Enzymatic resolution involving a remote chiral center is a difficult task. The enzymatic resolution of racemic precursors for R-ALA is an alternative route. In 1996, Iyengar et al. reported a synthetic sequence for the preparation of 21, the precursor of R-ALA, using the kinetic resolution of 94 as the key step (Scheme 22).34 Kinetic resolution of racemic compound 94 with lipase from Candida cylindracea afforded S-94 with good enantiomeric purity and good yield (84% ee, 80% yield). Lithium aluminum hydride-mediated reduction and regioselective opening with iodotrimethylsilane gave rise to 95, the desired six-carbon synthon with a protected diol system. The alkylation of benzylmethyl malonate with 95 and subsequent debenzylation generated 96, which was converted into a diol 21 precursor via decarboxylation, and deprotection.
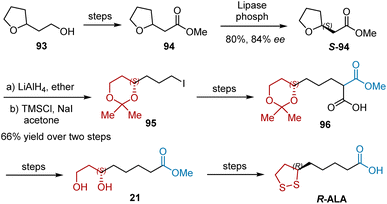 |
| | Scheme 22 Iyengar's synthesis of R-ALA. | |
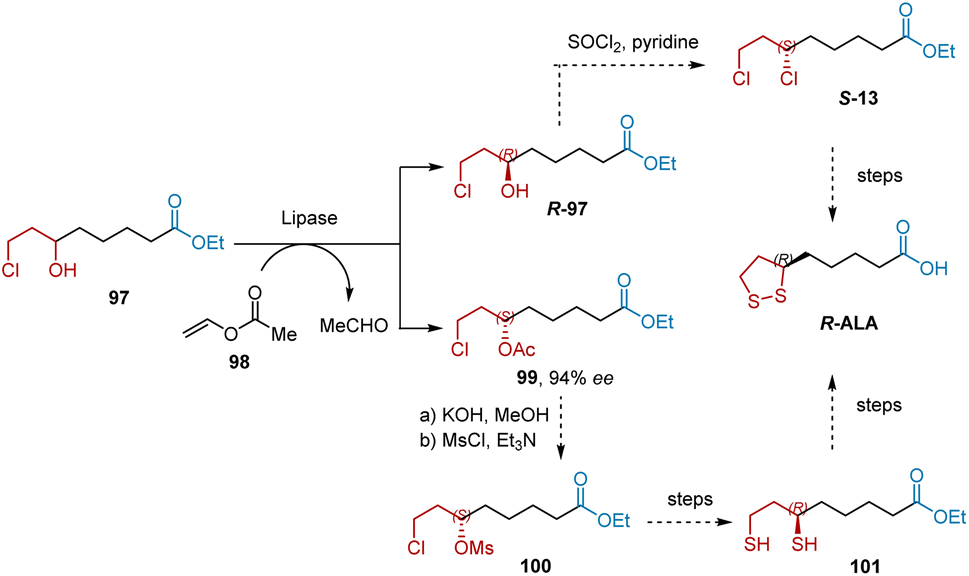
In 2014, Zhou et al. developed a lipase-catalyzed transacylation process to resolve ethyl 8-chloro-6-hydroxy octanoate 97 and produced two important chiral precursors, R-97 and 99, for the synthesis of R-ALA (Scheme 23).35 After systematically screening, the optimized condition of optical resolution was determined, resulting in Novozym 435 as catalyst, vinyl acetate as acyl donor, DIPE as solvent. The space-time yield of 99 was 38 g L−1 d−1 with 94% ee. According to a known procedure, R-ALA can be synthesized from R-97 or 99.
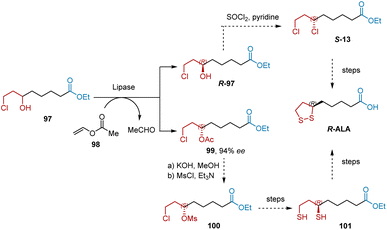 |
| | Scheme 23 Zhou's synthesis of R-ALA. | |
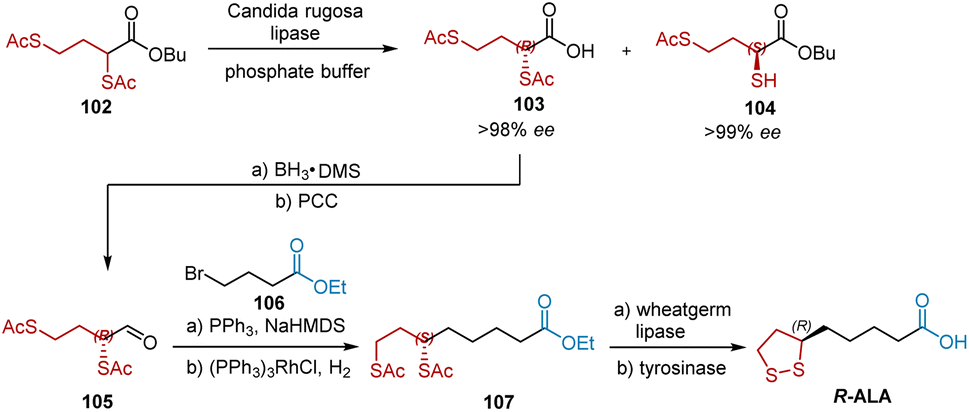
In 1998, Fadnavis et al. developed the enantioselective synthesis of pure R-2,4-dithioacetyl butyric acid 103 (ee > 98%) and S-butyl 2-thio-4-thioacetyl butyrate 104 (ee > 99%) by hydrolysis of the n-butyl ester of 2,4-dithioacetyl butanoic acid 102 using the native lipase of Candida rugosa (Scheme 24).36 R-ALA was synthesized using 103 as the chiral building block. The reduction of 103 with BH3·DMS provided an alcohol intermediate, which was oxidized by PCC to aldehyde 105. The subsequent Wittig reaction and hydrogenation afforded ethyl ester 107, whose ethyl and thioacetyl groups were removed by hydrolysis with the non-enantiospecific enzyme wheat germ lipase in phosphate buffer. R-ALA was obtained by treating the hydrolyzed product with the oxidative enzyme, mushroom tyrosinase, in the same reaction vessel. The yield of each step is not shown in this study.
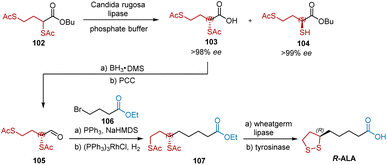 |
| | Scheme 24 Fadnavi's synthesis of R-ALA. | |
3.3 Chiral pool
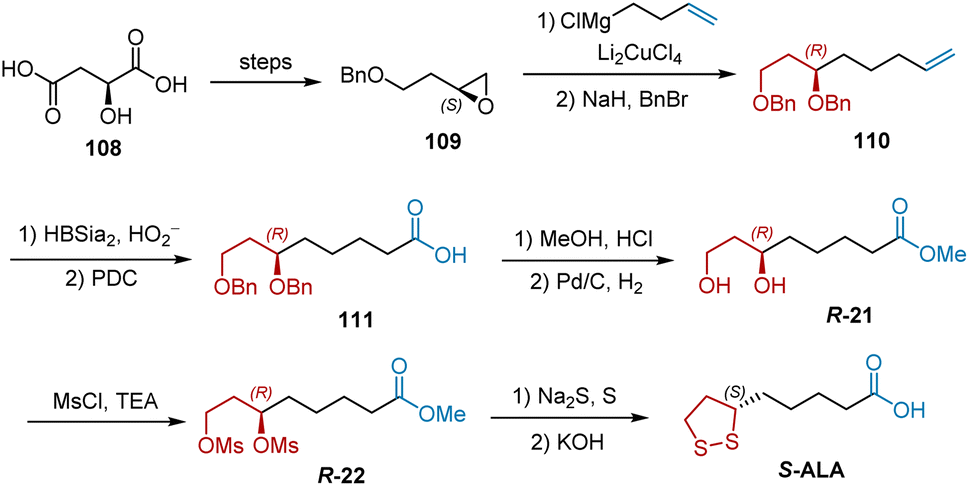
In 1983, Golding et al. selected S-2-hydroxysuccinic acid 108 as the starting material to synthesize S-ALA (Scheme 25).5 Epoxide 109 was obtained according to a previously published procedure. Treatment of epoxide 109 with 3-butenylmagnesium bromide and Li2CuCl4 gave the terminal alkene 110 after protection. Carboxylic acid 111 was generated by double-bond cleavage and PDC-mediated oxidation and was then converted into methyl ester R-21 by esterification and deprotection. The treatment of R-21 with MsCl and Et3N afforded dimethanesulfonate R-22. The final two-step transformation of R-22 completed the synthesis of S-ALA. The yield of each step is not shown in this study.
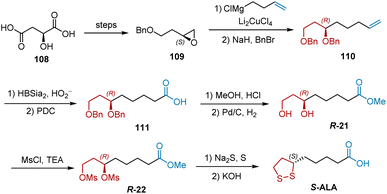 |
| | Scheme 25 Golding's synthesis of S-ALA. | |
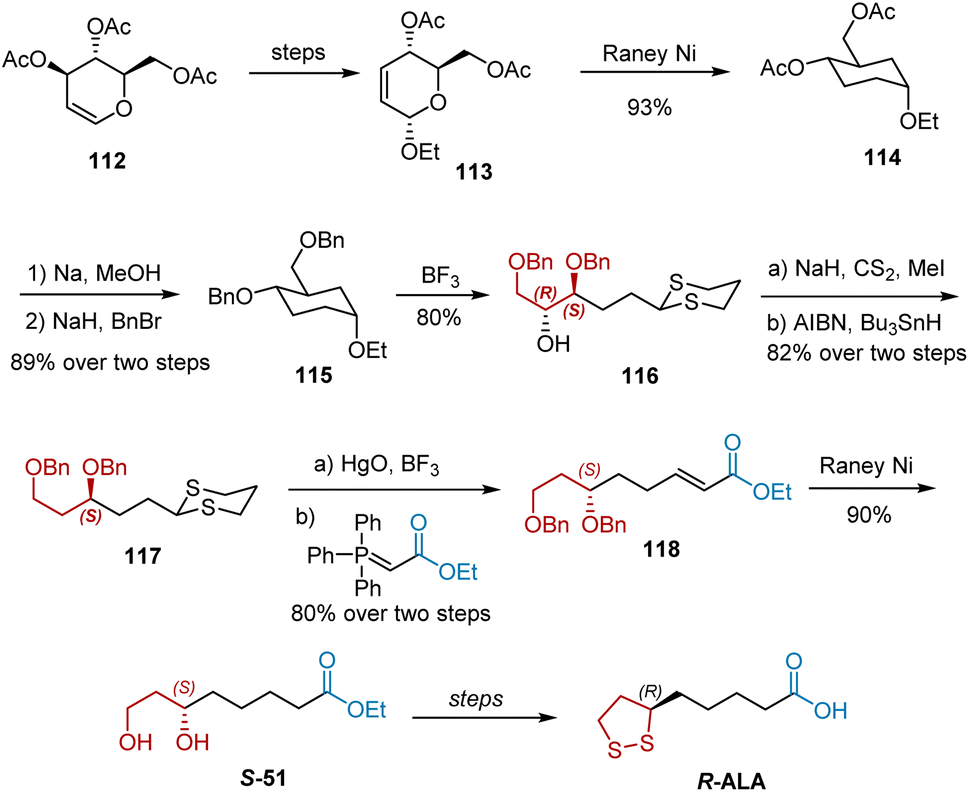
Rao et al. reported an enantiospecific synthesis of natural R-ALA in 13 steps (Scheme 26).37 3,4,6-Tri-O-acetyl-D-glucal 112, a chiral building block, was selected as the starting material and converted into compound 113, which was smoothly hydrogenated by treatment with RANEY® to give compound 114. The two acyl groups of 114 were replaced with Bn groups, yielding compound 115. The treatment of 115 with propanedithiol–boron trifluoride etherate–dichloromethane at room temperature afforded 80% of the dithiane compound 116. Barton–McCombie deoxygenation of 116 was performed to remove the hydroxyl group. With the compound 117 in hand, hydrolysis of 117 with mercuric oxide–boron trifluoride etherate in aqueous acetone yielded the desired aldehyde, which was converted into unsaturated ester 118 via the Wittig reaction. Hydrogenation of 118 over excess RANEY® reduced the double bond and debenzylation gave 90% diol S-51, which was transformed into R-ALA following a known strategy.
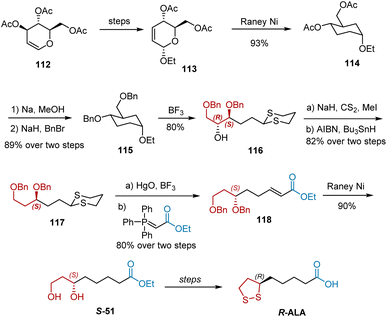 |
| | Scheme 26 Rao's asymmetric synthesis of R-ALA. | |
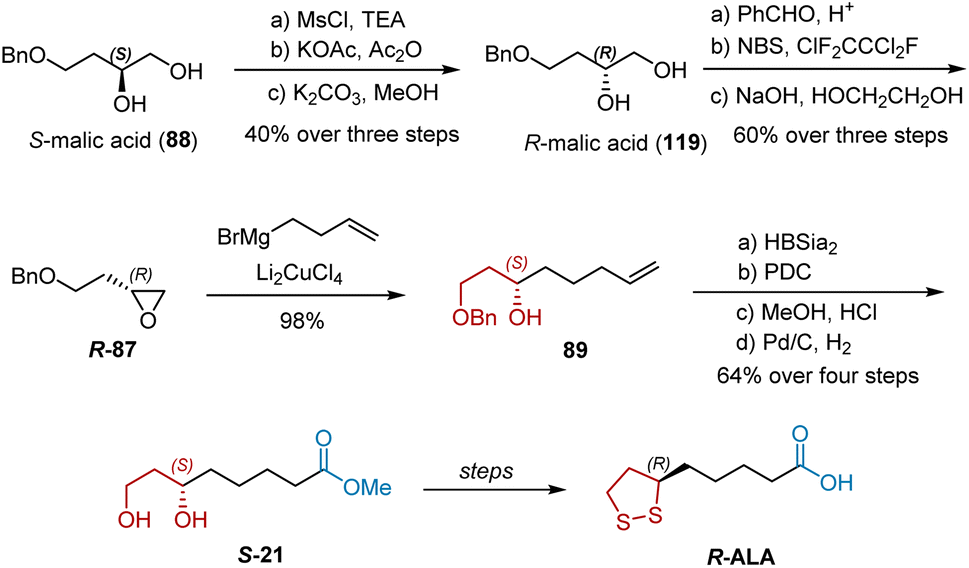
In 1988, Golding et al. revealed their synthesis of R-ALA and S-ALA from S-malic acid 88, a member of the “chiral pool” (Scheme 27).38 Notably, R-ALA could be directly synthesized from R-malic acid 119. They chose 88 as the starting material because 119 is relatively expensive. The total synthesis commenced with the stereospecific conversion of 88 to 119 using Takano's methodology. Compound 119 was converted into epoxide R-87, which was allowed to react with the Grignard reagent to give secondary alcohol 89. The following reaction sequences, including the benzylation of the secondary hydroxy group, hydroboration of the vinyl group, oxidation of the primary hydroxy group, esterification, and hydrogenolysis of the benzyl groups, transformed 89 into S-21 successively. R-ALA was obtained via the known transformations of S-21. S-ALA was synthesized using a similar route.
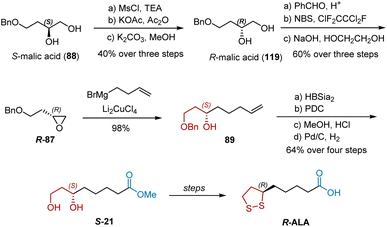 |
| | Scheme 27 Golding’ synthesis of R-ALA. | |
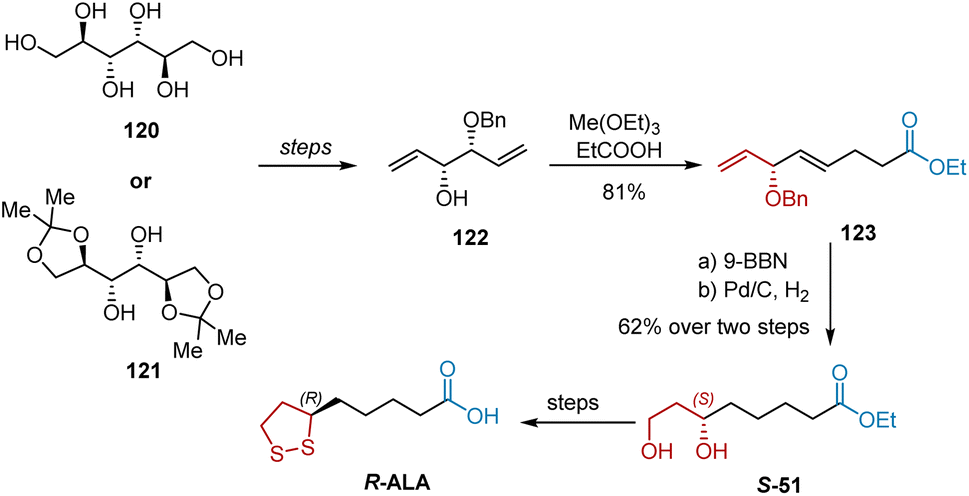
In 1989, Yadav et al. reported the synthesis of R-ALA from D-mannitol 120 and its derivatives 121, with diene 122 serving as a common intermediate (Scheme 28).39 With diene 122 in hand, the Claisen ester rearrangement with excess triethyl orthoacetate and a catalytic amount of propionic acid afforded diene ester 123 in 81% yield. Selective hydroboration–oxidation of the terminal double bond in 123 was accomplished with 9-BBN to afford an intermediate, whose hydrogenation proceeded smoothly to give diol S-51. Finally, the transformation of S-51 into R-ALA was performed using a previously described procedure.
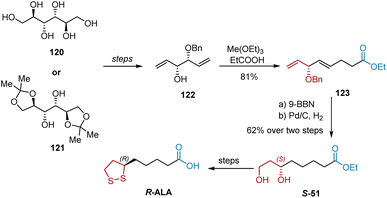 |
| | Scheme 28 Yadav's synthesis of R-ALA. | |
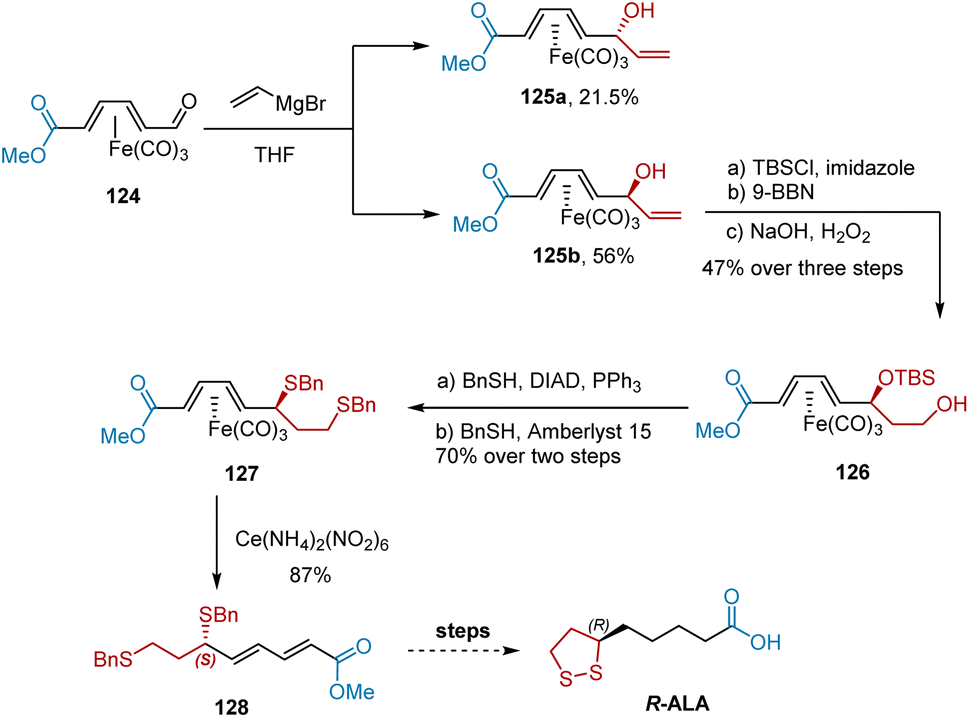
Tricarbonyl(diene)iron complexes are chiral and have various applications in the asymmetric synthesis of organic molecules. In 1998, Grée et al. utilized tricarbonyl(diene)iron complex 125b for the formal synthesis of R-ALA, as shown in Scheme 29.40 Complex 125b was obtained in an optically active form after Grignard addition-mediated resolution of 124. Complex 125b was used for the subsequent transformation. Subsequent protection of the hydroxyl group and hydroboration–oxidation afforded alcohol 126. A two-step transformation was performed to obtain compound 127 in high yield. Finally, oxidative decomplexation with ceric ammonium nitrate produced diene 128 in 87% yield, which could be used to deliver R-ALA.
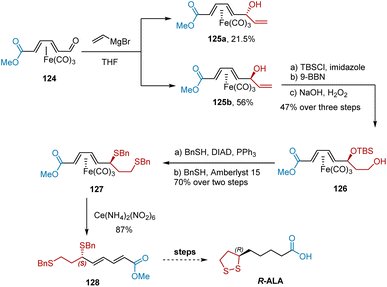 |
| | Scheme 29 Grée's synthesis of R-ALA. | |
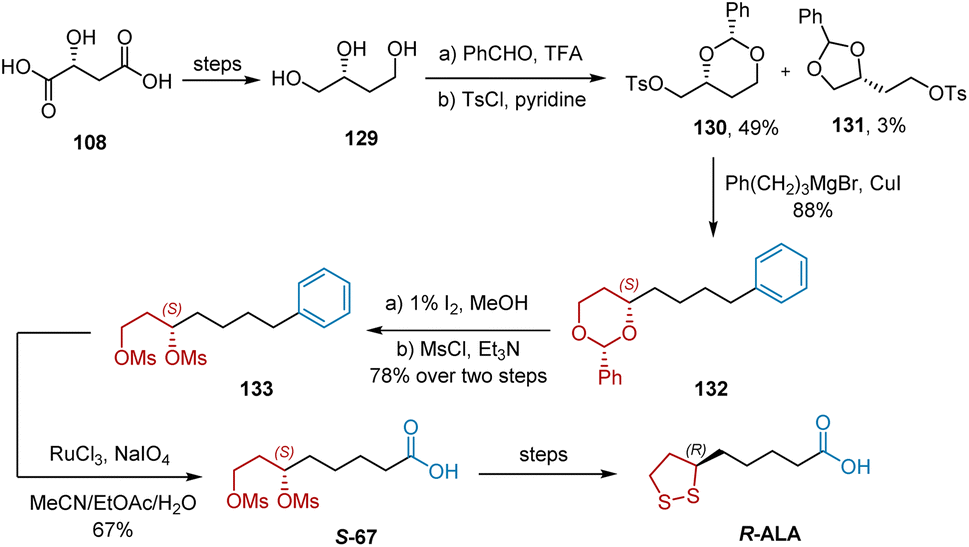
In 2009, Huang et al. developed a synthetic approach to R-ALA starting from 108 (Scheme 30).41 The synthesis began with the known compound 129, which is easily available from 108. In the presence of trifluoroacetic acid, treatment of 129 with benzaldehyde, followed by Ts-protection, afforded compounds 130 and 131, which could be readily separated by recrystallization. The subsequent coupling of tosylate 130 with phenylpropyl magnesium bromide in the presence of a catalytic amount of CuI afforded chain elongation product 132. The acetal groups of dioxane 132 were removed using I2/MeOH. Bismesylation yielded bismesylate 133. The oxidative cleavage of the phenyl group into a carboxylic acid group using the developed Sharpless conditions (RuCl3/NaIO4/EtOAc/MeCN/H2O) furnished S-67 in 67% yield. Finally, R-ALA was synthesized using the known transformations.
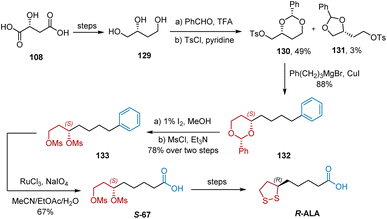 |
| | Scheme 30 Huang' synthesis of R-ALA. | |
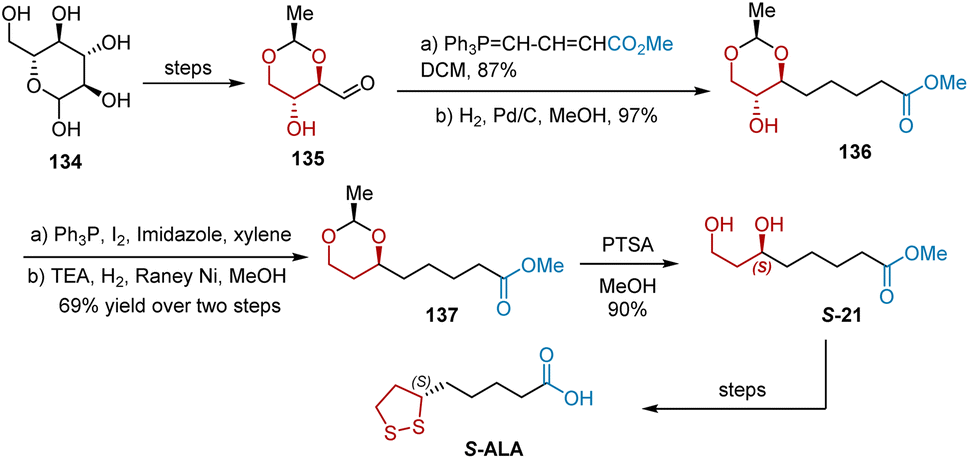
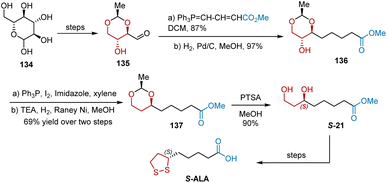
In 2015, Chavan et al. reported the synthesis of S-ALA from D-glucose (Scheme 31).42 The key reactions involved in this synthesis were splitting, iodination, and hydrogenation The synthesis commenced with the production of hydroxy aldehyde 135 from D-glucose 134, according to an established protocol. The following 4-carbon Wittig reaction of hydroxy aldehyde 135 took place to yield an inseparable mixture of cis and trans isomers, whose double bonds were reduced under the conditions of Pd/C/H2/MeOH to give hydroxyl ester 136 in 84% yield over two steps. The chiral hydroxyl group in 136 was removed to provide 137 via a two-step transformation involving iodination and reductive deiodination. Subsequently, the acetal group in ester 137 was deprotected using PTSA in methanol to obtain 1,3-diol S-21, which was then transformed into S-ALA according to previously reported steps.
 |
| | Scheme 31 Chavan' synthesis of S-ALA. | |
3.4 Chiral auxiliary
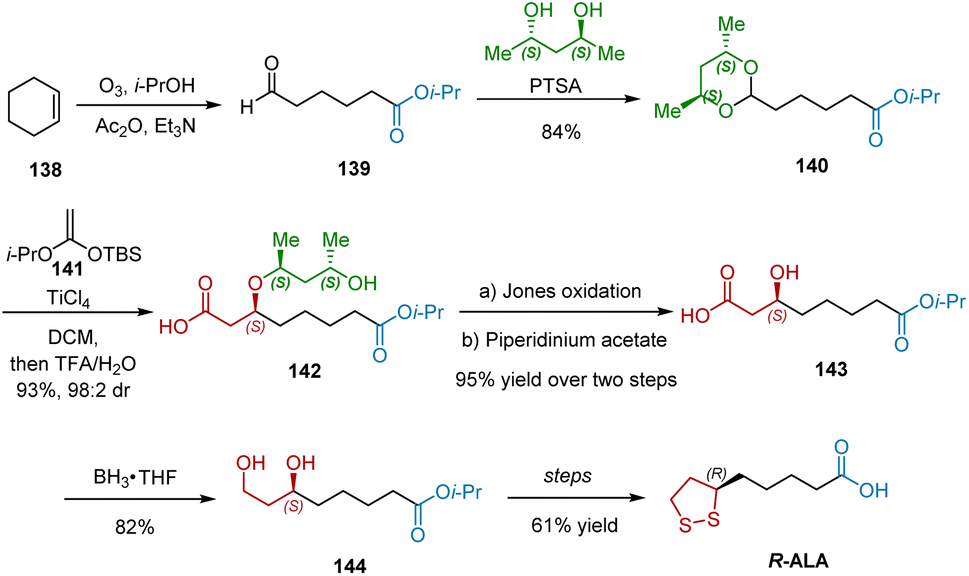
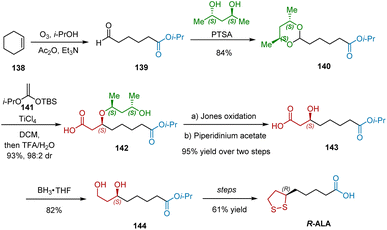
In 1984, Johnson and Elliott reported the use of R,R-2,4-pentanediol (PTSA) as a chiral auxiliary in a TiCl4-catalyzed aldol-type reaction to synthesize aldol esters in excellent yields and high diastereoselectivities.43 One year later, they extended this work by employing ketene acetal 141 as the nucleophile, and utilized the developed methodology as the key step in the synthesis R-ALA (Scheme 32).44 The initial transformation began with the synthesis of aldehyde 139 from cyclohexene 138. Acetalization of aldehyde 139 with PTSA as the chiral auxiliary proceeded smoothly to give 140 in 84% yield. TiCl4 mediated coupling of 140 with ketene acetal 141 afforded 142 in excellent yield and diastereoselectivity (93% yield, 98:2 dr). The following Jone's oxidation and β-elimination afforded chiral alcohol 143 in 95% yield over two steps, which was reduced to diol 144 by BH3·THF complex. Following the known procedure, R-ALA was harvested.
 |
| | Scheme 32 Johnson and Elliott’ synthesis of R-ALA. | |
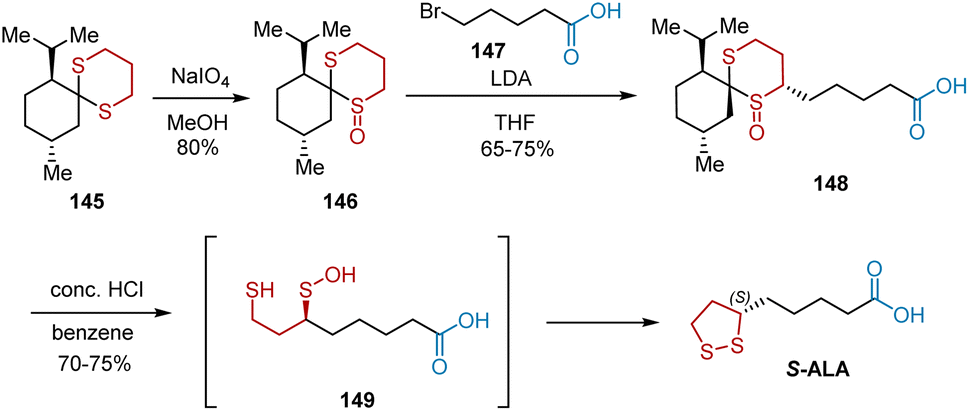
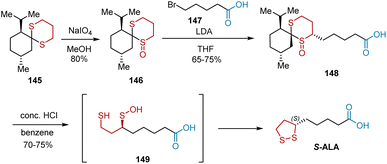
In 1987, a short and highly stereospecific synthesis of S-ALA using menthone as a recyclable chiral auxiliary was presented by Menon et al. (Scheme 33).45 Dithiane 145, the starting material, served as the chiral template that controlled the regiochemistry of the subsequent oxidation to provide 146 as a single regioisomer in 80% yield. The stereoselective alkylation of 146 with 5-bromovaleric acid 147 in the presence of lithium diisopropylamide as a base in THF resulted in the carboxylic acid derivative 148, which was hydrolyzed in aqueous HCl to yield S-ALA.
 |
| | Scheme 33 Menon's synthesis of S-ALA. | |
3.5 Chemical asymmetric catalysis
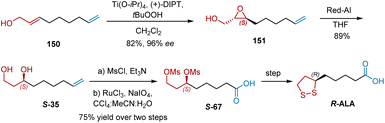
In 1986, Sutherland et al. reported the enantioselective synthesis of R-ALA from achiral precursor 150 using Sharpless asymmetric epoxidation as the key step (Scheme 34).46 Allylic alcohol 150 was synthesized by alkylation of the lithio-dianion of propargyl alcohol by dissolving the metal reduction of the resultant disubstituted acetylene in situ. The sharpless asymmetric epoxidation of 150 was carried out smoothly using Ti(Oi-Pr)4/(+)-DIPT/tBuOOH, giving epoxy alcohol 151 in 82% yield and 96% ee. The reduction of 151 with Red-Al resulted in the selective formation of diol S-35 in 89% yield. Under conditions using MsCl/Et3N, S-35 was sulfonated. Ruthenium tetroxide oxidation of the terminal double bond formed carboxylic acid S-67 in 75% yield over two steps. Finally, the disulfide displacement of the methanesulfonate groups of the potassium salt of S-67 afforded R-ALA in 52% yield.
 |
| | Scheme 34 Sutherland's asymmetric synthesis of R-ALA. | |
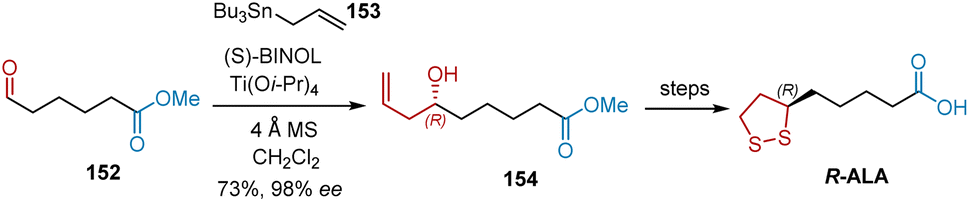
Chiral binaphthol-derived (BINOL) metal species have been widely used as catalysts for enantioselective allylation reactions. In 2000, Zimmer et al. used enantioselective allylation as the key step in the formal synthesis of R-ALA (Scheme 35).47 Starting with the readily accessible methoxycarbonyl aldehyde 152, (S)-BINOL/Ti(OiPr)4 catalyzed enantioselective allylation was performed with commercially available allyltributylstannane 153, giving 154 in 73% yield and 98% ee. According to a previously reported protocol, R-ALA was targeted from 154.
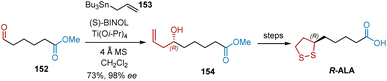 |
| | Scheme 35 Zimmer's asymmetric synthesis of R-ALA. | |
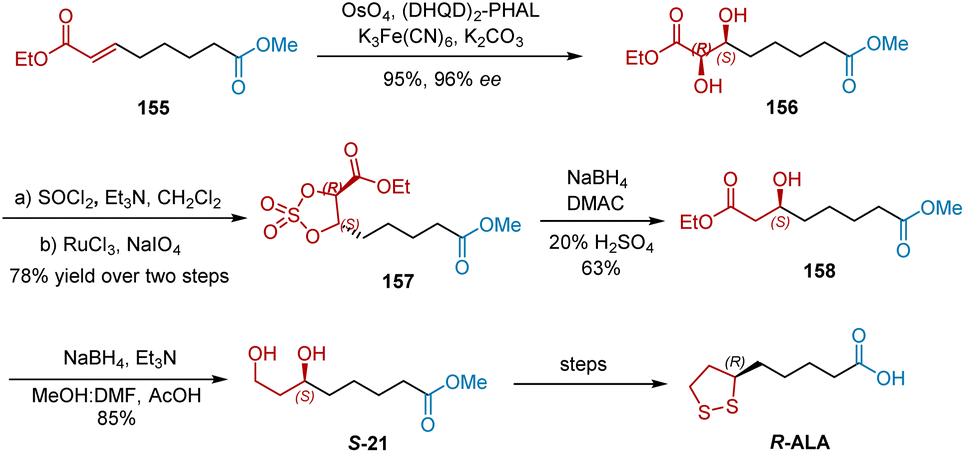
In 2001, Sudalai et al. developed two asymmetric catalytic methods to synthesize R-ALA, as shown in (Schemes 36 and 37).48 First, asymmetric dihydroxylation was adopted as the key step in the construction of the chiral center of R-ALA. Olefinic diester 1 was subjected to OsO4-catalyzed asymmetric dihydroxylation using hydroquinidine 1,4-phthalazinediyl diether [(DHQD)2–PHAL] as the chiral ligand, affording chiral diol 156 in 95% yield and 96% ee. Diol 156 was then converted into cyclic sulfate 157, which was reduced to give chiral alcohol 158 in 63% yield. Further selective reduction of one of the ester groups in 158 afforded S-21 in 85% yield, which was converted to R-ALA, according to a previously reported protocol.
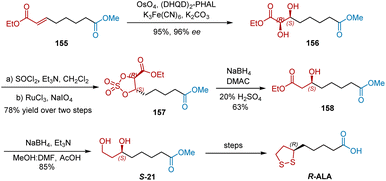 |
| | Scheme 36 Sudalai's first asymmetric synthesis of R-ALA. | |
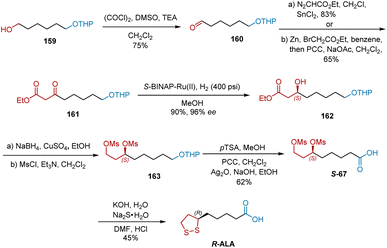 |
| | Scheme 37 Sudalai's second asymmetric synthesis of R-ALA. | |
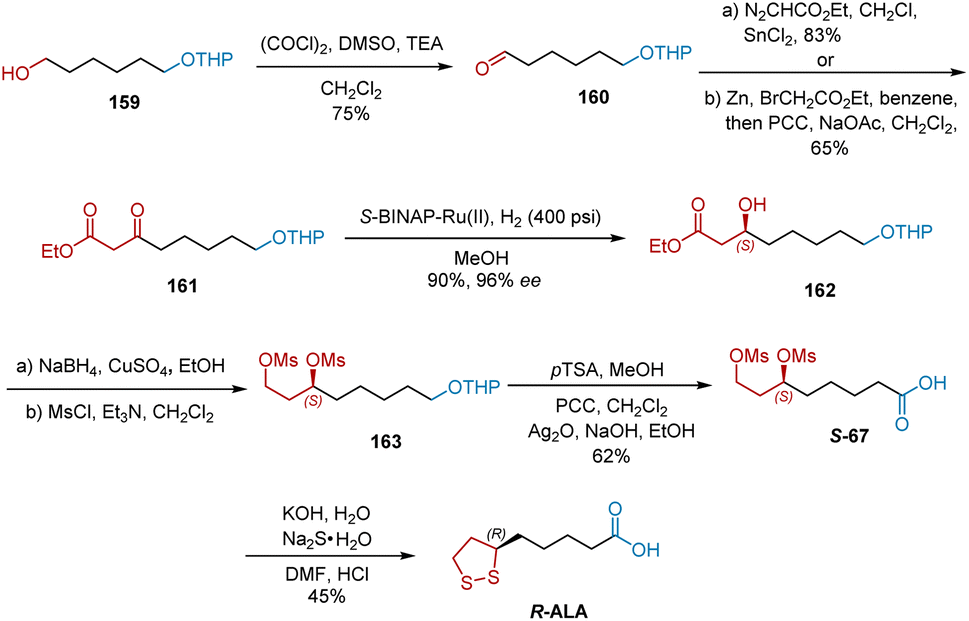
Sudalai developed another synthesis commencing with the Swern oxidation of alcohol 159, affording aldehyde 160 in 75% yield (Scheme 37).48 The C–H insertion of ethyl diazoacetate with 160 in the presence of a catalytic amount of anhydrous SnCl2 or the Reformatsky reaction of 160 followed by PCC-mediated oxidation gave keto ester 161 in 65% yield. Under the atmosphere of H2 (400 psi), S-BINAP–Ru-catalyzed asymmetric hydrogenation of 161 proceeded smoothly to generate 162 in 90% yield and 96% ee. The reduction of the ester functional group in 162 using NaBH4–CuSO4 in EtOH yielded the diol intermediate, which was subsequently mesylated under standard conditions to yield 163. Carboxylic acid S-67 was obtained from 163 in three steps of deprotection and oxidation. Accordingly, the disulfide displacement of S-67 delivered R-ALA.
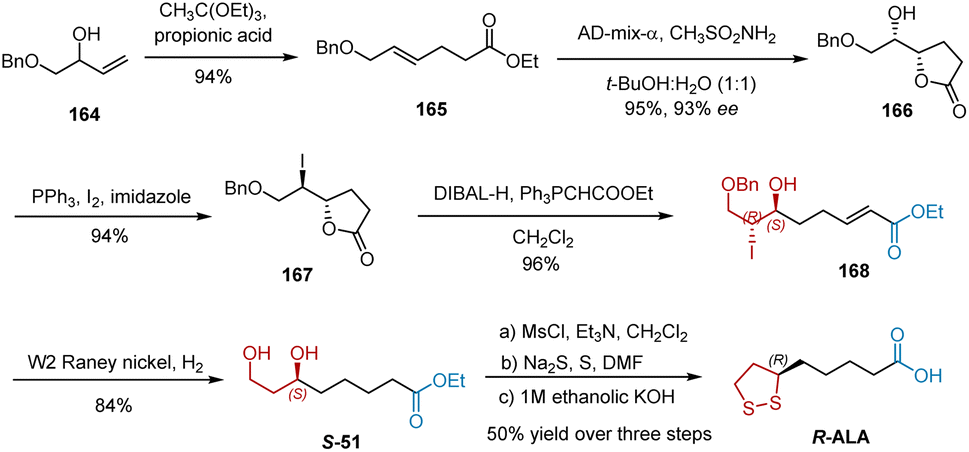
In 2004, Chavan et al. reported the synthesis of R-ALA by employing Claisen orthoester rearrangement and Sharpless asymmetric dihydroxylation as key steps (Scheme 38).49 Initially, Claisen rearrangement of monoprotected allylic alcohol 159 with triethyl orthoacetate in the presence of catalytic propionic acid afforded unsaturated ester 164 in 94% yield. Sharpless asymmetric dihydroxylation using AD-mix-α and the in situ cyclization of 165 furnished hydroxy lactone 166 in 95% yield and 93% ee. Lactone 166 was treated with triphenylphosphine, iodine, and imidazole to give iodolactone 167, whose reduction was followed by an in situ two-carbon Wittig reaction to give unsaturated ester 168. Under the conditions of W2 RANEY® and hydrogen, the benzyl protection and iodine were removed, and the double bond was reduced in one pot, giving diol S-51 as a known intermediate for the synthesis of R-ALA. R-ALA was synthesized according to the established protocol.
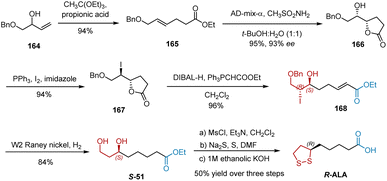 |
| | Scheme 38 Chavan's asymmetric synthesis of R-ALA. | |
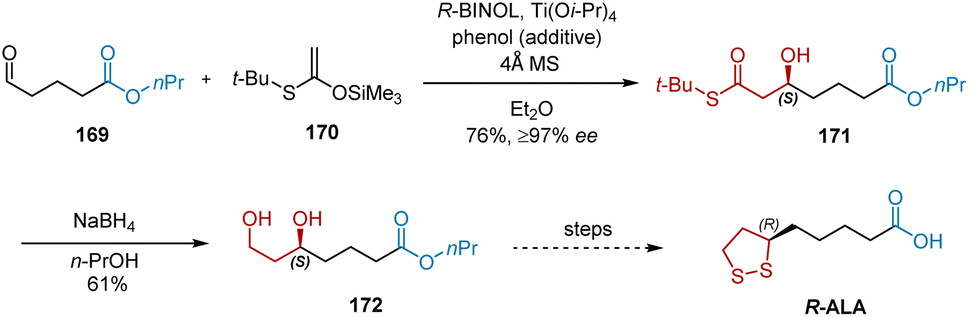
In 2002, Zimmer et al. developed the asymmetric synthesis of R-ALA using an enantioselective aldol reaction as the key transformation.50 As shown in Scheme 39, starting with the easily accessible substrate 169, enantioselective aldol reactions were performed with S-ketene silyl acetal 170 in the presence of R-BINOL, Ti(Oi-Pr)4 and phenol, affording chiral alcohol 171 in 76% yield and ≥97% ee. The subsequent NaBH4-mediated reduction of 171 generated the key intermediate 172 in 61% yield, which was converted into enantiopure R-ALA according to a previously reported protocol.
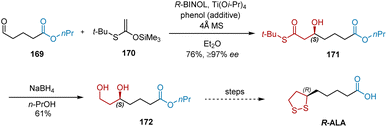 |
| | Scheme 39 Zimmer's asymmetric synthesis of R-ALA. | |
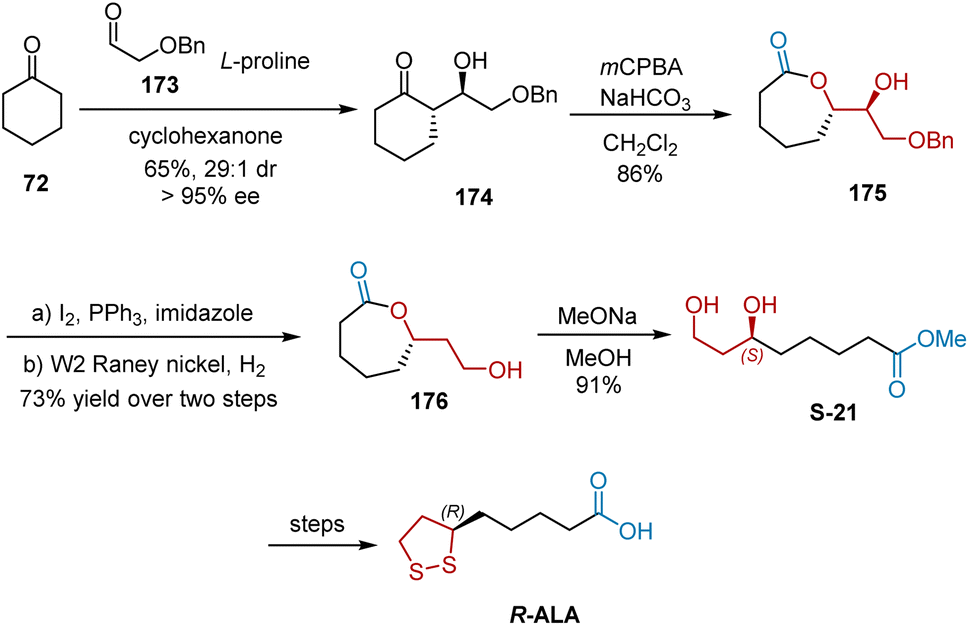
In 2008, Wang et al. unveiled an efficient, highly stereocontrolled formal synthesis of R-ALA utilizing L-proline-catalyzed, highly enantio- and diastereoselective cross-aldol reaction as the key step (Scheme 40).51 The L-proline-promoted aldol reaction between ketone 72 and aldehyde 173 afforded chiral aldol adduct 174 in 65% yield with 95% ee and dr of 29:1. The regioselective Baeyer–Villiger oxidation of ketone 174 gave lactone intermediate 175, which could be reduced to chiral compound 176. Finally, treatment of 176 with sodium methoxide in methanol afforded chiral diol S-21 in 91% yield. The target R-ALA was obtained using known synthetic sequences.
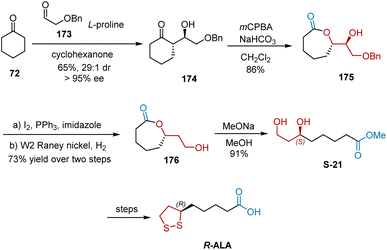 |
| | Scheme 40 Wang and Duan's asymmetric synthesis of R-ALA. | |
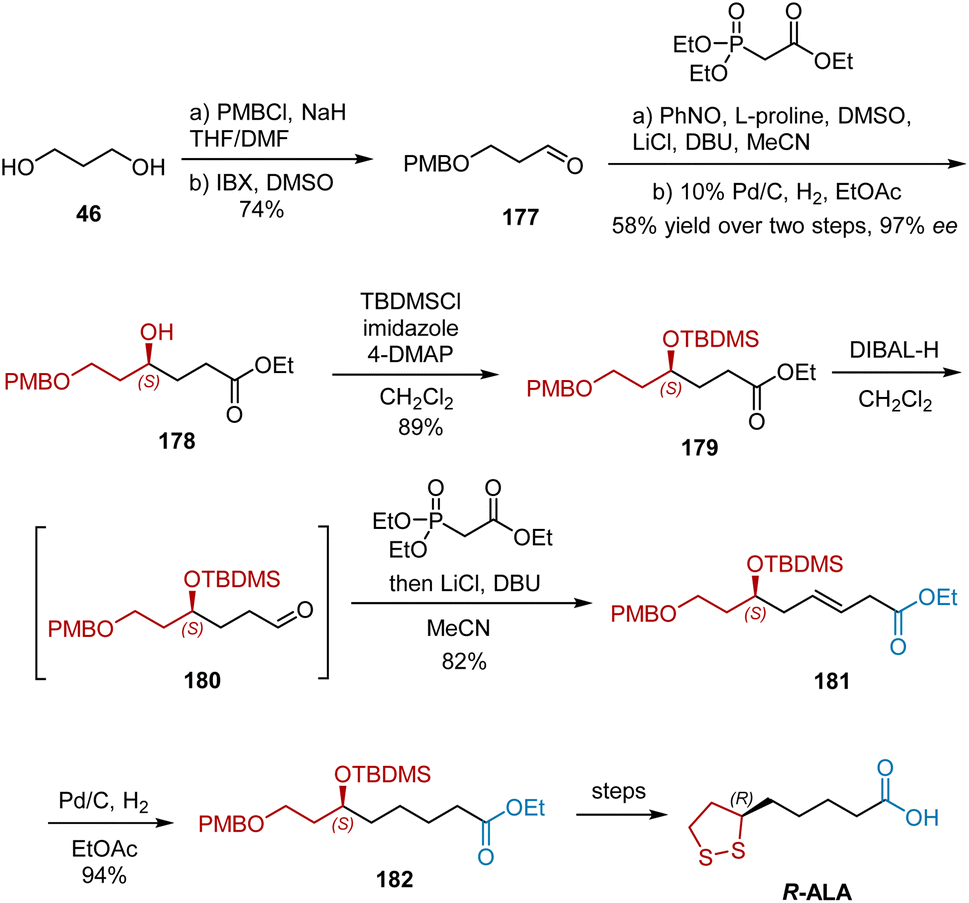
In 2010, Kalkote et al. reported an organocatalytic enantioselective approach for the synthesis of R-ALA using an L-proline-catalyzed sequential aminoxylation–HWE olefination reaction of aldehydes as the key step.52 The synthesis began with commercially available diol 46 as shown in Scheme 41. The mono-hydroxyl protection of diol 46 and subsequent oxidation provided aldehyde 177 in 74% yield in two steps. Aldehyde 177 was subjected to the following L-proline-catalyzed aminoxylation/HWE olefination/hydrogenation sequence to yield chiral hydroxyester 178 in 58% yield and 97% ee. After the protection of 178, ester 179 was then reduced to aldehyde 180 with DIBAL-H and subjected to HWE olefination to obtain α,β-unsaturated ester 181, which was hydrogenated to afford saturated ester 182. Finally, R-ALA was synthesized from 182 according to a previously reported protocol.
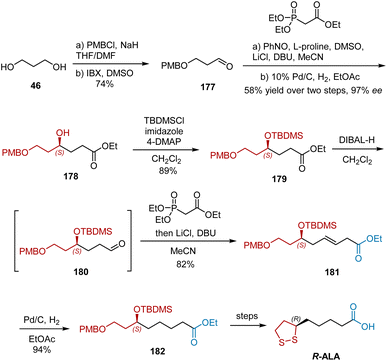 |
| | Scheme 41 Kalkote's asymmetric synthesis of R-ALA. | |
3.6 Enzymatic asymmetric catalysis
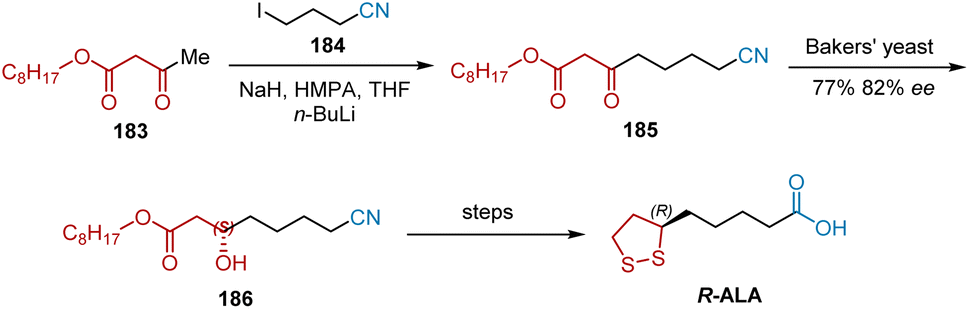
The reduction of functionalized β-keto esters by baker's yeast may be a valuable procedure for the preparation of a variety of chiral trifunctional synthons. In 1989, Gopalan et al. utilized baker's yeast reduction to obtain a valuable synthetic intermediate that could be converted to R-ALA in a short sequence of steps.53 As shown in Scheme 42, substrate 185 for baker's yeast was prepared by the alkylation of the dianion from alkyl acetoacetate 183 with 4-iodobutyronitrile 184 The Baker's yeast-catalyzed asymmetric reduction of 185 was performed to yield chiral alcohol 186 in 77% yield and 82% ee, which could be transformed into R-ALA via a published route.
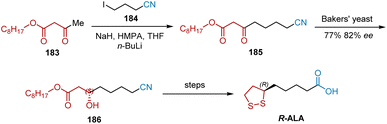 |
| | Scheme 42 Gopalan's asymmetric synthesis of R-ALA. | |
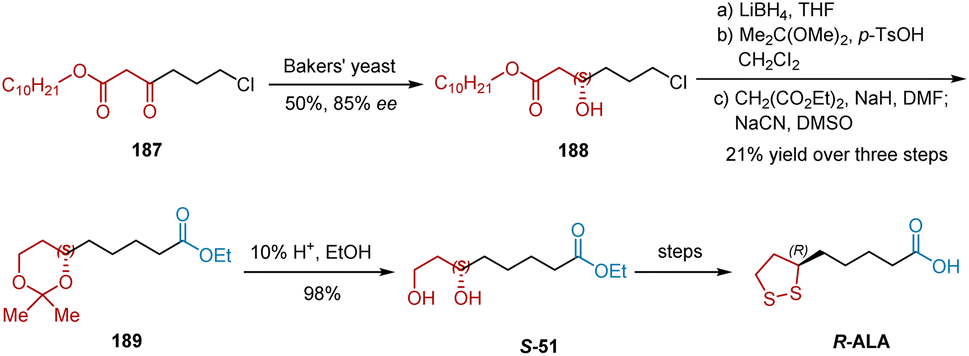
In 1990, Gopalan et al. also reported their studies on the baker's yeast reduction of readily prepared β-keto esters 187 and applied it to accomplish the total synthesis of R-ALA (Scheme 43).54 The keto ester 187 was reduced to the chiral alcohol 188 in 55% yield 85% ee using baker's yeast. Subsequent reduction, acetal protection, alkylation and deprotection generated the diol S-51, which was in turn converted into R-ALA following published procedures.
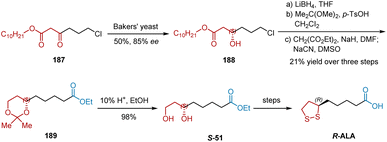 |
| | Scheme 43 Gopalan's asymmetric synthesis of R-ALA. | |
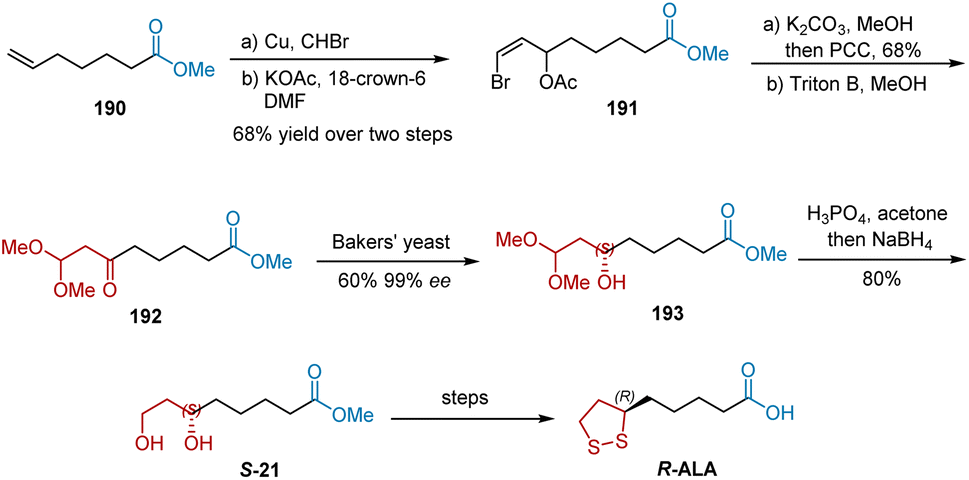
In 1990, Dasaradhi et al. reported the stereo-controlled reduction of keto acetals by baker's yeast immobilized on calcium alginate beads to synthesize R-ALA (Scheme 44).55 The Cu-catalyzed bromoform addition of 190 followed by treatment with potassium acetate and 18-crown-6 initiated the synthesis of R-ALA, yielding compound 191 in 68% yield over two steps. The subsequent hydrolysis of 191 in the presence of K2CO3 in methanol and oxidation with pyridinium chlorochromate afforded ketovinyl bromide, which was then transformed into ester 192 using Triton B in methanol. Ester 192 was enantiospecifically reduced using a glucose solution containing Baker's yeast (Saccharomyces cerevisiae NCIM 3044) immobilized on calcium alginate beads to generate crude product 193 in 60% yield and 99% ee. R-ALA was obtained after deprotection, NaBH4 reduction, mesylation, and hydrolysis R-ALA was achieved.
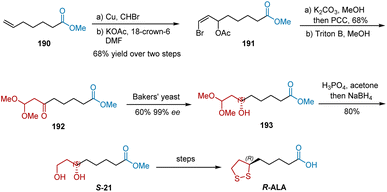 |
| | Scheme 44 Dasaradhi's asymmetric synthesis of R-ALA. | |
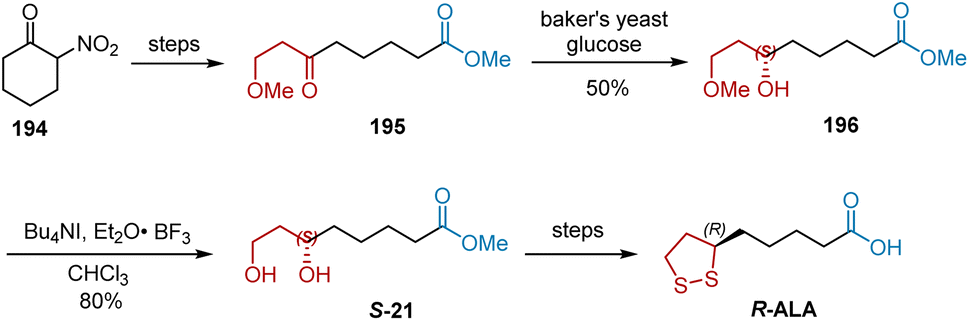
In 1996, Bezbarua et al. reported the synthesis of R-ALA by employing the baker's yeast reduction of ketones as a key step.56 As shown in Scheme 45, the substrate of baker's yeast reduction, ketone 195, was obtained from 2-nitrocyclohexanone 194. Ketone 195 was enantiospecifically reduced using Baker's yeast to yield chiral alcohol 196. The selective hydrolysis of 196 with tetrabutylammonium iodide in CHCl3 afforded chiral diol S-21 in 80% yield, which was converted to R-ALA according to a previously reported approach.
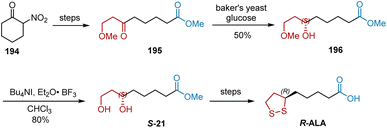 |
| | Scheme 45 Bezbarua's asymmetric synthesis of R-ALA. | |
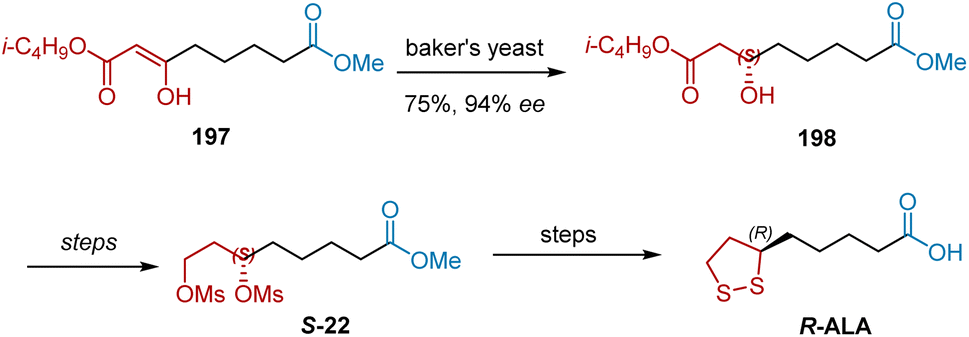
In 1999, Bringmann et al. reported an approach to gain R-ALA by exploiting 3-oxodiesters 197 as a starting material (Scheme 46).57 The reduction of 197 took place using baker's yeast, giving chiral alcohol 198 in 75% yield and 94% ee. R-ALA was achieved from 198 by using previously reported methods.
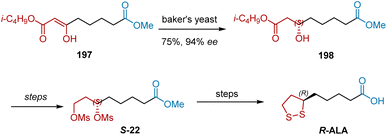 |
| | Scheme 46 Bringmann's asymmetric synthesis of R-ALA. | |
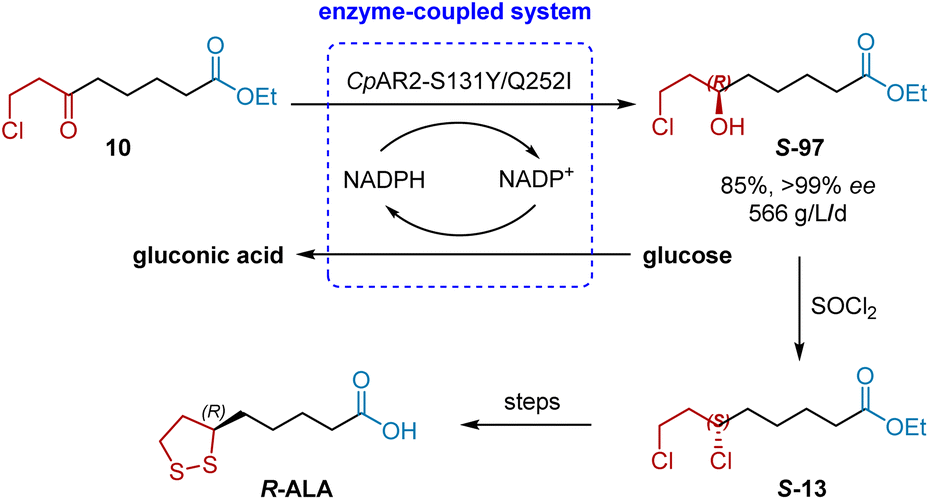
In 2015, Xu and Zheng et al. discovered a ketoreductase CpAR2 from Candida parapsilosis that was able to highly stereoselective reduce ethyl 8-chloro-6-oxooctanoate 1 to (R)-8-chloro-6-hydroxyoctanoate 2 (in 85% yield with >99% ee), an advanced chiral intermediate to R-ALA, with a space-time yield of 530 g L−1 d−1 (Scheme 47).27a However, high biocatalyst loading (10 g L−1 of dry E. coli cells coexpressing CpAR2 and a glucose dehydrogenase BmGDH) is required. Generally, the biocatalyst loading should be less than 5 g L−1 for a green and economical chemical process. Thus, a directed evolution approach was adopted to increase the enzyme activity and thermal stability simultaneously.58 By using error-prone PCR, the combined mutant S131Y/Q252I was obtained, resulting improved activity (214 U mg−1 versus 120 U mg−1 for wild-type enzyme) and the half-deactivating temperature (T50, for 15 min incubation, increased by 2.3 °C). Gratifyingly, as low as 2 g L−1 of the lyophilized E. coli cells coexpressing CpAR2S131Y/Q252I and GDH was sufficient to completely reduce 110 g L−1 of 1 and achieve the higher space-time yield (566 g L−1 d−1), affording the desired 2 in 85% yield with >99% ee.
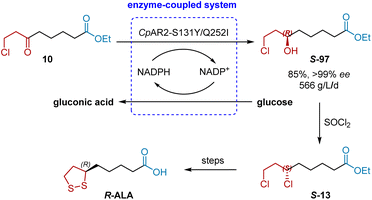 |
| | Scheme 47 Xu and Zheng's asymmetric synthesis of R-ALA. | |
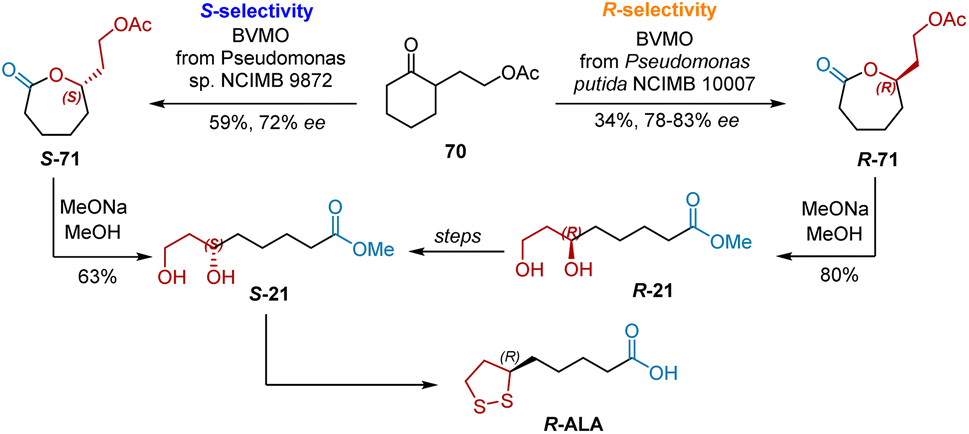
Enzymatic Baeyer–Villiger Oxidations of ketone provide chiral lactones which are precursors to readily construct chiral alcohol and carbonyl groups via ring-opening. Two Baeyer–Villiger monooxygenases were used in the synthesis of R-ALA, resulting in different stereoselectivities. As shown in Scheme 48, Adger et al. used Baeyer–Villiger monooxygenase from Pseudomonas putida NCIMB 10007 to catalyze the oxidation of 2-substituted cycloalkanone 70, yielding R-71 with good enantioselectivities (78–83% ee).59 Reductive ring opening was performed to yield R-21. Inversion by the Mitsunobu reaction afforded S-21, which was transformed into R-ALA according to a published procedure. Willetts et al. found that the Baeyer–Villiger oxidation of 70 was preferentially carried out to yield S-21 by monooxygenases from Pseudomonas sp. NCIMB 9872.60 Likewise, the reductive ring opening of S-71 yielded S-21 directly, which was the precursor for the synthesis R-ALA.
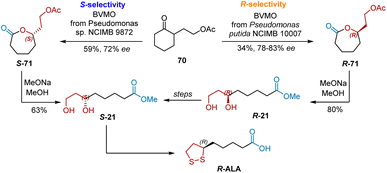 |
| | Scheme 48 Adger and Willetts's asymmetric synthesis of R-ALA. | |
4. Conclusions
This study demonstrates significant progress made over the past 70 years in the field of total ALA synthesis. Diverse routes and methods for synthesizing ALA have been described in the literature, including racemic synthesis, chemical and enzymatic resolution synthesis, asymmetric chiral pool synthesis, asymmetric chiral auxiliary synthesis, enzyme-catalyzed asymmetric synthesis, chemical-catalyzed asymmetric synthesis. While these methods generally focus on basic scientific research, few of them have proposed synthetic pathways for industrial production.
Accordingly, with the ever-growing demand for ALA, practical, concise, and general synthetic strategies and advanced technologies will continue to be explored and exploited. For example, continuous-flow chemistry, enzyme engineering, and other new technologies could have broadly applications in the total synthesis of ALA to achieve efficient, economical, and energy-saving industrial production. The ability to combine new technologies advances numerous possibilities for creating cheaper and higher quality products to fulfill the needs of the population. The development of these new technologies will undoubtedly benefit future applications. Furthermore, new technologies and strategies will be implemented in the synthesis of ALA to provide ideas and insights for scientific researchers and peers, associated with these realms and aspects in the long run.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for the financial support from Sichuan Science and Technology Program (2022NSFSC0615), National Natural Science Foundation of China (22201192).
Notes and references
- L. J. Reed, B. G. DeBusk, I. C. Guusalus and C. S. Horuberger Jr, Crystalline α-Lipoic Acid: A Catalytic Agent Associated with Pyruvate Dehydrogenase, Science, 1951, 114, 93 CrossRef CAS PubMed.
-
(a) D. Han, G. Handelman, L. Marcocci, C. K. Sen, S. Roy, H. Kobuchi, H. J. Tritschler, L. Flohé and L. Packer, Lipoic acid increases de novo synthesis of cellular glutathione by improving cystine utilization, Biofactors, 1997, 6, 321–338 CrossRef CAS PubMed;
(b) A. Gorąca, H. Huk-Kolega, A. Piechota, P. Kleniewska, E. Ciejka and B. Skibska, Lipoic acid–biological activity and therapeutic potential, Pharmacol. Rep., 2011, 63, 849–858 CrossRef PubMed.
- D. Ziegle, M. Reljanovic, H. Mehnert and F. A. Gries, Alpha-lipoic acid in the treatment of diabetic polyneuropathy in Germany: current evidence from clinical trials, Exp. Clin. Endocrinol. Diabetes, 1999, 107, 421–430 CrossRef PubMed.
- Selected reviews:
(a) S. Ghibu, C. Richard, C. Vergely, M. Zeller, Y. Cottin and L. Rochette, Antioxidant Properties of an Endogenous Thiol: Alpha-lipoic Acid, Useful in the Prevention of Cardiovascular Diseases, J. Cardiovasc. Pharmacol., 2009, 54, 391–398 CrossRef CAS PubMed;
(b) K. P. Shay, R. F. Moreau, E. J. Smith, A. R. Smith and T. M. Hagen, Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential, Biochim. Biophys. Acta, 2009, 1790, 1149–1160 CrossRef CAS PubMed;
(c) P. M. Bingham, S. D. Stuart and Z. Zachar, Lipoic acid and lipoic acid analogs in cancer metabolism and chemotherapy, Expert Rev. Clin. Pharmacol., 2014, 7, 837–846 CrossRef CAS PubMed;
(d) F. A. Moura, K. Q. de Andrade, J. C. F. dos Santos and M. O. F. Goulart, Lipoic Acid: Its Antioxidant and Anti-Inflammatory Role and Clinical Applications, Curr. Top. Med. Chem., 2015, 15, 458–483 CrossRef CAS PubMed;
(e) B. Salehi, Y. B. Yılmaz, G. Antika, T. B. Tumer, M. F. Mahomoodally, D. Lobine, M. Akram, M. Riaz, E. Capanoglu, F. Sharopov, N. Martins, W. C. Cho and J. Sharifi-Rad, Insights on the Use of α-Lipoic Acid for Therapeutic Purposes, Biomolecules, 2019, 9, 356 CrossRef CAS PubMed;
(f) S. Jeffrey, P. I. Samraj and B. S. Raj, Curr. Diabetes Rev., 2021, 17, e011821190404 CrossRef CAS PubMed;
(g) D. Kaur, T. Behl, A. Sehgal, S. Singh, N. Sharma, S. Chigurupati, A. Alhowail, A. Abdeen, S. F. Ibrahim, C. Vargas-De-La-Cruz, M. Sachdeva, S. Bhatia, A. Al-Harrasi and S. Bungau, Decrypting the potential role of α-lipoic acid in Alzheimer's disease, Life Sci., 2021, 284, 119899 CrossRef CAS PubMed;
(h) D. P. Theodosis-Nobelos, G. Papagiouvannis, P. Tziona and E. A. Rekka, Lipoic acid. Kinetics and pluripotent biological properties and derivatives, Mol. Biol. Rep., 2021, 48, 6539–6550 CrossRef PubMed;
(i) H. Xie, X. Yang, Y. Cao, X. Long, H. Shang and Z. Jia, Role of lipoic acid in multiple sclerosis, CNS Neurosci. Ther., 2022, 28, 319–331 CrossRef CAS PubMed;
(j) B. Fasipe, A. Faria and I. Laher, Potential for Novel Therapeutic Uses of Alpha Lipoic Acid, Curr. Med. Chem., 2023, 30, 3942–3954 CrossRef CAS PubMed.
- M. H. Brookes, B. T. Golding, D. A. Howes and A. T. Hudson, Proof that the absolute configuration of natural α-lipoic acid is R by the synthesis of its enantiomer [(S)-(–)-α-lipoic acid] from (S)-malic acid, J. Chem. Soc. Chem. Commun., 1983, 19, 1051–1053 RSC.
-
(a) A. Marshall, R. Graul, M. Y. Morgan and S. Sherlock, Treatment of alcohol-related liver disease with thioctic acid: a six month randomised double-blind trial, Gut, 1982, 23, 1088–1093 CrossRef CAS PubMed;
(b) N. Ramakrishnan, W. W. Wolfe and G. N. Catravas, Radioprotection of Hematopoietic Tissues in Mice by Lipoic Acid, Radiat. Res., 1992, 130, 360–365 CrossRef CAS PubMed;
(c) S. Stoll, H. Hartmann, S. Cohen and W. Müller, The potent free radical scavenger α-lipoic acid improves memory in aged mice: putative relationship to NMDA receptor deficits, Pharmacol., Biochem. Behav., 1993, 46, 799–805 CrossRef CAS PubMed;
(d) A. Faust, V. Burkart, H. Ulrich, C. H. Weischer and H. Kolb, Effect of lipoic acid on cyclophosphamide-induced diabetes and insulitis in non-obese diabetic mice, Int. J. Immunopharmacol., 1994, 16, 61–66 CrossRef CAS PubMed;
(e) P. Ou, H. J. Tritschler and S. P. Wolff, Thioctic (lipoic) acid: a therapeutic metal-chelating antioxidant?, Biochem. Pharmacol., 1995, 50, 123–126 CrossRef CAS PubMed;
(f) S. Golbidi, M. Badran and I. Laher, Diabetes and alpha lipoic acid, Front. Pharmacol, 2011, 2, 69 Search PubMed.
- J. S. Yadav, S. V. Mysorekar and K. Garyali, Synthesis of α-lipoic acid-A highly useful biologically active compound, J. Sci. Ind. Res., 1990, 49, 400–409 CAS.
- R. Ramachanderan and B. Schaefer, Lipoic acid, ChemTexts, 2019, 5, 18 CrossRef.
- M. W. Bullock, J. A. Brockman Jr, E. L. Patterson, J. V. Pierce, H. vonSultza, F. Sanders and E. L. R. Stokstad, J. Am. Chem. Soc., 1952, 74, 3455 CrossRef CAS.
- M. W. Bullock, J. A. Brockman Jr, E. L. Patterson, J. V. Pierce, H. vonSultza, F. Sanders and E. L. R. Stokstad, Syntheses in the Thioctic Acid Series, J. Am. Chem. Soc., 1954, 76, 1828–1832 CrossRef CAS.
-
(a) Q. F. Soper, W. E. Buting Jr, J. E. Cochrau and A. Pohland, Syntheses of DL-α-Lipoic Acid, J. Am. Chem. Soc., 1954, 76, 4109–4112 CrossRef CAS;
(b) D. S. Acker and W. J. Wayn, Synthesis of Racemic, Optically Active and Radioactive α-Lipoic Acids, J. Am. Chem. Soc., 1957, 79, 6483–6484 CrossRef CAS.
-
(a) L. J. Reed and C. Niu, Syntheses of DL-α-Lipoic Acid, J. Am. Chem. Soc., 1955, 77, 416–419 CrossRef CAS;
(b) R. C. Thomas and L. J. Reed, Synthesis and Properties of High Specific Activity DL-α-Lipoic Acid-S235, J. Am. Chem. Soc., 1955, 77, 5446–5448 CrossRef CAS.
-
(a) D. S. Acker and C. W. Todd, US Pat. US2752374, 1956;
(b) L. J. Reed, US Pat. US2980716, 1961;
(c) H. Ose and K. Yoshida, US Pat. US3223712, 1965.
- D. S. Acker and W. J. Wayne, Synthesis of Racemic, Optically Active and Radioactive α-Lipoic Acids, J. Am. Chem. Soc., 1957, 79, 6483–6487 CrossRef CAS.
- E. Walton, A. F. Wagner, F. W. Bachelor, L. H. Peterson, F. W. Holly and K. Folkers, Synthesis of (+)-α-Lipoic Acid and its Optical Antipode, J. Am. Chem. Soc., 1955, 77, 5144–5149 CrossRef CAS.
- A. V. R. Rao, S. V Mysorekar and J. S. Yadav, Synthesis of 1,3-Diols: Its Application to the Synthesis of α-Lipoic Acid, Synth. Commun., 1987, 17, 1339–1347 CrossRef CAS.
- E. A. Braude, R. P. Linstead and K. R. H. Wooldridge, 594. A new synthesis of 6:8-thioctic acid, J. Chem. Soc., 1956, 3074–3078 RSC.
- J. Tsuji, H. Yasuda and T. Mandai, Synthesis of dl-.alpha.-lipoic acid from a butadiene telomer, J. Org. Chem., 1978, 43, 3606–3607 CrossRef CAS.
- P. Dhar, N. Chidambaram and S. Chandrasekaran, Piperidinium tetrathiotungstate as sulfur transfer reagent: synthesis of cyclic disulfides, J. Org. Chem., 1992, 57, 1699–1702 CrossRef CAS.
- S. P. Chavan, R. R. Kale and K. Pasupathy, A Facile Regioselective Decomposition of Tosylhydrazones: An Application towards the Synthesis of α-Lipoic Acid, Synlett, 2005, 7, 1129–1132 CrossRef.
- A. N. Purude, K. P. Pawar, N. B. Patil, U. R. Kalkote and S. P. Chavan, Total synthesis of (R)-lipoic acid and (S)-lipoic acid via an Mn (III)-salen-catalyzed oxidative kinetic resolution, Tetrahedron: Asymmetry, 2015, 26, 281–287 CrossRef CAS.
- A. F. Wagner, E. Walton, C. H. Hoffman, L. H. Peterson, F. W. Holly and K. Folkers, Synthesis of DL-Dimethyldihydro-α-lipoic Acid, J. Am. Chem. Soc., 1955, 77, 5140–5143 CrossRef CAS.
- B. V. Rao and A. S. Rao, A Common Strategy for the Synthesis of (+)-Hepialone and (±)-α-Lipoic Acid, Synth. Commun., 1995, 25, 1531–1543 CrossRef CAS.
- A. Segre, R. Viterbo and G. Parisi, A New Synthesis of 6-Thioctic Acid (DL-α-Lipoic Acid), J. Am. Chem. Soc., 1957, 79, 3503–3505 CrossRef CAS.
- F. Balkenhohl and J. Paust, A Short and Productive Synthesis of Racemic α-Lipoic Acid, Z. Naturforsch., 1999, 54, 649–654 CrossRef CAS.
- S. P. Chavan, K. Shivsankar and K. Pasupathy, Simplistic Expedient and Practical Synthesis of (±)-α-Lipoic Acid, Synthesis, 2005, 8, 1297–1300 CrossRef.
-
(a) Y. Zhang, W. Zhang, G. Zheng and J. Xu, Identification of an ε-Keto Ester Reductase for the Efficient Synthesis of an (R)-α-Lipoic Acid Precursor, Adv. Synth. Catal., 2015, 357, 1697–1702 CrossRef CAS;
(b) B. Zehnpfennig, P. Wiriyasermkul, D. A. Carlson and M. Quick, Interaction of α-Lipoic Acid with the Human Na+/Multivitamin Transporter (hSMVT), J. Biol. Chem., 2015, 290, 16372–16382 CrossRef CAS PubMed.
- Y. Numbu and T. Endo, Stereoselective Reduction of Racemic N-hydroxyamino Acid by Optically Active Thiols-iron(II), Chem. Lett., 1985, 14, 999–1002 CrossRef.
-
(a) T. Tsunoda, H. Kaku, M. Nagaku and E. Okuyama, Deracemization of 2-Alkylcyclohexanones Utilizing Host-Guest Molecular Association with Optically Active Host Compounds in Basic Suspension Media, Tetrahedron Lett., 1997, 38, 7759–7760 CrossRef CAS;
(b) H. Kaku, N. Okamoto, T. Nishii, M. Horikawa and T. Tsunoda, Enantioselective Total Synthesis of (R)-α-Lipoic Acid: An Application of Thermodynamically Controlled Deracemization of (±)-2-(2-Methoxyethyl) Cyclohexanone, Synthesis, 2010, 17, 2931–2934 CrossRef.
- L. S. Bose, L. Fatima and S. Rajender, An Efficient, Highly Enantioenriched Route to L-Carnitine and α-Lipoic Acid via Hydrolytic Kinetic Resolution, Synthesis, 2006, 11, 1863–1867 CrossRef.
- N. W. Fadnavis and K. Koteshwar, Remote control of stereoselectivity: lipase catalyzed enantioselective esterification of racemic α-lipoic acid, Tetrahedron: Asymmetry, 1997, 8, 337–339 CrossRef CAS.
- H. Yan, Z. Wang and L. Chen, Kinetic resolution of α-lipoic acid via enzymatic differentiation of a remote stereocenter, J. Ind. Microbiol. Biotechnol., 2009, 36, 643–648 CrossRef CAS PubMed.
- N. Liu, L. Wang, Z. Wang, L. Jiang, Z. Wu, H. Yue and X. Xie, Microwave-Assisted Resolution of α-Lipoic Acid Catalyzed by an Ionic Liquid Co-Lyophilized Lipase, Molecules, 2015, 20, 9949–9960 CrossRef CAS PubMed.
- Y. R. S. Laxmi and D. S. Iyengar, Chemoenzymatic Synthesis of Methyl(6S)-(-)-6,8-Dihydroxyoctanoate: A Precursor to (R)-(+)-α-Lipoic Acid, Synthesis, 1996, 5, 594–596 CrossRef.
- W. Zhou, Y. Ni, G. Zheng, H. Chen, Z. Zhu and J. Xu, Enzymatic resolution of a chiral chlorohydrin precursor for (R)-α-lipoic acid synthesis via lipase catalyzed enantioselective transacylation with vinyl acetate, J. Mol. Catal. B: Enzym., 2014, 99, 102–107 CrossRef CAS.
- N. W. Fadnavis, R. L. Babu, S. K. Vadivel, A. A. Deshpande and U. T. Bhalerao, Lipase catalyzed regio- and stereospecific hydrolysis: chemoenzymatic synthesis of both (R)- and (S)-enantiomers of α-lipoic acid, Tetrahedron: Asymmetry, 1998, 9, 4109–4112 CrossRef CAS.
-
(a) A. V. R. Rao, M. K. Gurjar, K. Garyau and T. Ravindranatha, Enantiospecific synthesis of (R-(+)-α-lipoic acid from d-glucose, Carbohydr. Res., 1986, 148, 51–55 CrossRef CAS;
(b) A. V. R. Rao, A. V. Purandare, E. R. Reddy and M. K. Curjar, Synthesis of R-(+)-α-lipoic acid, Synth. Commun., 1987, 17, 1095–1102 CrossRef CAS.
- M. H. Brookes and B. T. Golding, Syntheses of α-(R)- and α-(S)-Lipoic Acid from (S)-Malic Acid, J. Chem. Soc., Perkin Trans. 1, 1988, 9–12 RSC.
- J. S. Yadav, S. V. Mysorekar, S. M. Pawar and M. K. Gurjar, Synthesis of (3R, 4R)-1, 2-Divinylglycol and its Unsymmetrical Derivatives: An Application to the Synthesis of R-(+)-α-Lipoic Acid, J. Carbohydr. Chem., 1990, 9, 307–316 CrossRef CAS.
- C. Crévisy, B. Herbage, M. Marrel, L. Toupet and R. Grée, A New Iron-Mediated Strategy for the Synthesis of α-Lipoic Acid and Analogues, Eur. J. Org Chem., 1998, 1949–1954 CrossRef.
- Z. Wei, H. Lan, J. Zheng and P. Huang, New and Concise Approach to (R)-α-Lipoic Acid, Synth. Commun., 2009, 39, 691–701 CrossRef CAS.
- S. P. Chavan, K. P. Pawar, C. Praveen and N. B. Patil, Chirality induction and chiron approaches to enantioselective total synthesis of α-lipoic acid, Tetrahedron, 2015, 71, 4213–4218 CrossRef CAS.
- W. S. Johnson, C. Edington, J. D. Elliott and I. R. Silverman, Asymmetric Synthesis via Acetal Templates. 10. 1 Aldol-Type Reactions. Preparation of a Nonactic Acid Intermediate, J. Am. Chem. Soc., 1984, 106, 7588–7591 CrossRef CAS.
- J. D. Elliott, J. Steele and W. S. Johnson, Asymmetric synthesis via acetal templates. 12. Highly diastereoselective coupling reactions with a ketene acetal. An efficient, asymmetric synthesis of R-(+)-α-lipoic acid, Tetrahedron Lett., 1985, 26, 2535–2538 CrossRef CAS.
- R. B. Menon, M. A. Kumar and T. Ravindranathan, Stereospecific synthesis of α-lipoic acid, Tetrahedron Lett., 1987, 28, 5313–5314 CrossRef CAS.
- P. C. B. Page, C. M. Rayner and I. Sutherland, Enantioselective Synthesis of R-(+)-α-Lipoic Acid, J. Chem. Soc., Chem. Commun., 1986, 18, 1408–1409 RSC.
- R. Zimmer, U. Hain, M. Berndt, R. Gewald and H. Reissig, Enantioselective synthesis of (S)- and (R)-6-hydroxy-8-nonene-carboxylates by asymmetric catalysis: a formal synthesis of (R)-α-lipoic acid and its (S)-antipode, Tetrahedron: Asymmetry, 2000, 11, 879–887 CrossRef CAS.
- T. T. Upadhya, M. D. Nikalje and A. Sudalai, Asymmetric dihydroxylation and hydrogenation approaches to the enantioselective synthesis of R-(+)-α-lipoic acid, Tetrahedron Lett., 2001, 42, 4891–4893 CrossRef CAS.
- S. P. Chavan, C. Praveen, G. Ramakrishna and U. R. Kalkote, Enantioselective synthesis of R-(+)-α and S-(-)-α-lipoic acid, Tetrahedron Lett., 2004, 45, 6027–6028 CrossRef CAS.
- R. Zimmer, A. Peritz, R. Czerwonka, L. Schefzig and H. Reißig, Optimisation, Scope and Limitations of Enantioselective Aldol Reactions of an S-Ketene Silyl Acetal with Aliphatic Aldehydes under (R)-BINOL-Titanium(IV) Catalysis Conditions, Eur. J. Org Chem., 2002, 3419–3428 CrossRef CAS.
- S. Zhang, X. Chen, J. Zhang, W. Wang and W. Duan, An Enantioselective Formal Synthesis of (+)-(R)-α-Lipoic Acid by an L-Proline-Catalyzed Aldol Reaction, Synthesis, 2008, 3, 383–386 Search PubMed.
- S. P. Panchgalle, G. F. Jogdand, S. P. Chavan and U. R. Kalkote, Enantioselective synthesis of (R)-(+)-α-lipoic acid via proline-catalyzed sequential α-aminoxylation and HWE olefination of aldehyde, Tetrahedron Lett., 2010, 51, 3587–3589 CrossRef CAS.
- A.
S. Gopalan and H. K. Jacobs, Stereochemical control of yeast reductions: synthesis of R-(+)-α-lipoic acid, Tetrahedron Lett., 1989, 30, 5705–5708 CrossRef CAS.
- A. S. Gopalan and H. K. Jacobs, Bakers' Yeast Reduction of Alkyl6-Chloro-3-oxohexanoates: Synthesis of (R)-(+)-α-Lipoic Acid, J. Chem. Soc., Perkin Trans. 1, 1990, 1897–1990 RSC.
- L. Dasaradhi, N. W. Fadnavis and U. T. Bhalerao, A Novel Enantiospecific Synthesis of (S)-(-)-Methyl 6,8-Dihydroxyoctanoate, a Precursor of (R)-(+)-α-Lipoic Acid, J. Chem. Soc., Chem. Commun., 1990, 10, 729–730 RSC.
- M. Bezbarua, A. K. Saikia, N. C. Barua, D. Kalita and A. C. Ghosh, A Short Enantioselective Formal Synthesis of Methyl (S)-(-)-6,8-Dihydroxyoctanoate: A Key Intermediate for the Synthesis of R-(+)-α-Lipoic Acid, Synthesis, 1996, 11, 1289–1290 CrossRef.
- G. Bringmann, D. Herzberg, G. Adam, F. Balkenhohl and J. Paust, A Short and Productive Synthesis of (R)-α-Lipoic Acid, Z. Naturforsch., 1999, 54, 655–661 CrossRef CAS.
- Y. Xu, Q. Chen, Z. Zhang, J. Xu and G. Zheng, Co-evolving the Activity and Thermostability of an ε-Ketoester Reductase for Better Synthesis of (R)-α-Lipoic Acid Precursor, ChemBioChem, 2020, 21, 1341–1346 CrossRef CAS PubMed.
- B. Adger, M. T. Bes, G. Grogan, R. McCague, S. Pedragosa-Moreau, S. M. Roberts, R. Villa, P. W. H. Wan and A. J. Willetts, Application of Enzymic Baeyer-Villiger Oxidations of 2-Substituted Cycloalkanones to the Total Synthesis of (R)-(+)-Lipoic Acid, J. Chem. Soc., Chem. Commun., 1995, 15, 1563–1564 RSC.
-
(a) M. T. Bes, R. Villa, S. M. Roberts, P. W. H. Wan and A. Willetts, Oxidative biotransformations by microorganisms: production of chiral synthons by cyclopentanone monooxygenase from Pseudomonas sp. NCIMB 9872, J. Mol. Catal. B: Enzym., 1996, 1, 127–134 CrossRef CAS;
(b) B. Adger, M. T. Bes, G. Grogan, R. McCague, S. PedragosaMoreau, S. M. Roberts, R. Villa, P. W. H. Wan and A. J. Willetts, The Synthesis of (R)-(+)-Lipoic Acid using a Monooxygenase-Catalysed Biotransformation as the Key Step, Bioorg. Med. Chem., 1997, 5, 253–261 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ac
*ac