
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA combined crystallographic and theoretical investigation of noncovalent interactions in 1,3,4-oxadiazole-2-thione-N-Mannich derivatives: in vitro bioactivity and molecular docking†
Lamya H. Al-Wahaibi a,
Kowsalya Alagappanb,
Rosa M. Gomilac,
Olivier Blacqued,
Antonio Frontera*c,
M. Judith Percinoe,
Ali A. El-Emam*f and
Subbiah Thamotharan*b
a,
Kowsalya Alagappanb,
Rosa M. Gomilac,
Olivier Blacqued,
Antonio Frontera*c,
M. Judith Percinoe,
Ali A. El-Emam*f and
Subbiah Thamotharan*b
aDepartment of Chemistry, College of Sciences, Princess Nourah bint Abdulrahman University, Riyadh 11671, Saudi Arabia
bBiomolecular Crystallography Laboratory and DBT-Bioinformatics Center, School of Chemical and Biotechnology, SASTRA Deemed University, Thanjavur 613 401, India. E-mail: thamu@scbt.sastra.edu
cDepartament de Química, Universitat de les Illes Balears, Ctra. de Valldemossa km 7.5, Baleares, 07122 Palma de Mallorca, Spain. E-mail: toni.frontera@uib.es
dDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland
eUnidad de Polímeros y Electrónica Orgánica, Instituto de Ciencias, Benemérita Universidad Autónoma de Puebla, Val3-Ecocampus Valsequillo, Independencia O2 Sur 50, San Pedro Zacachimalpa, Puebla 72960, CP, México
fDepartment of Medicinal Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt. E-mail: elemam@mans.edu.eg
First published on 21st November 2023
Abstract
Two 1,3,4-oxadiazole-2-thione-N-Mannich derivatives, specifically 5-(4-chlorophenyl)-3-[(2-trifluoromethylphenylamino)methyl]-1,3,4-oxadiazole-2(3H)-thione (1) and 5-(4-chlorophenyl)-3-[(2,5-difluorophenylamino)methyl]-1,3,4-oxadiazole-2(3H)-thione (2), were synthesized and then characterized by elemental analysis and NMR (1H and 13C) spectroscopy and the single crystal X-ray diffraction method. The formed weak intermolecular interactions in the solid-state structures of these derivatives were thoroughly investigated utilizing a variety of theoretical tools such as Hirshfeld surface analysis and quantum theory of atoms in molecules (QTAIM). Furthermore, the CLP-PIXEL and density functional theory calculations were used to study the energetics of molecular dimers. Numerous weak intermolecular interactions such as C–H⋯S/Cl/F/π interactions, a directional C–Cl⋯Cl halogen bond, π-stacking, type C–F⋯F–C contact and a short F⋯O interaction, help to stabilize the crystal structure of 1. Crystal structure 2 also stabilizes with several weak intermolecular contacts, including N–H⋯S, C–H⋯N//Cl/F interactions, a highly directional C1–Cl1⋯C(π) halogen bond and C(π)⋯C(π) interaction. In vitro antimicrobial potency of compounds 1 and 2 was assessed against various Gram-positive and Gram-negative bacterial strains and the pathogenic yeast-like Candida albicans. Both compounds showed marked activity against all tested Gram-positive bacteria and weak activity against Escherichia coli and lacked inhibitory activity against Pseudomonas aeruginosa. In addition, compounds 1 and 2 displayed good in vitro anti-proliferative activity against hepatocellular carcinoma (HepG-2) and mammary gland breast cancer (MCF-7) cancer cell lines. Molecular docking studies revealed the binding modes of title compounds at the active sites of prospective therapeutic targets.
1. Introduction
1,3,4-Oxadiazoles are an imperative class of heterocyclic compounds with diverse pharmacological properties.1 1,3,4-Oxadiazole is a valuable structural core in currently employed chemotherapeutic agents such as the broad spectrum antibacterial drug furamizole,2 the anticancer agent zibotentan,3 the herbicidal and fungicidal agent oxadiazon4 and the antiretroviral drug raltegravir.5 The chemotherapeutic efficacy of anticancer 1,3,4-oxadiazoles is attributed to targeting thymidylate synthase,6 epidermal growth factor receptors (EGFR),7 vascular endothelial growth factor receptors (VEGF),8 histone deacetylases (HDAC),9 tubulin polymerase,10 methionine aminopeptidase (MetAP),11 focal-adhesion kinase (FAK),12 telomerase,13 and nuclear factor κB.14 It has been reported that several solid tumors overexpress thymidine phosphorylase, which plays a crucial role in angiogenesis.15 In addition, the antifungal activity of several 1,3,4-oxadiazole derivatives is attributed to potent succinate dehydrogenase inhibitory activity.16 Furthermore, in the last two decades, organo-fluorine compounds represent one of the most fast-growing classes of bioactive compounds.17In view of the previously reported observations and as a continuation of an interest in developing chemotherapeutic agents,18 and structural characterization of 1,3,4-oxadiazole-based derivatives,19 we report herein the synthesis, characterization, supramolecular features, nature of intermolecular interactions, preliminary antimicrobial and anti-proliferative activities of two 3-fluorophenylaminomethyl-1,3,4-oxadiazole-2(3H)-thione derivatives. The antioxidant activity of the N-Mannich base of 1,3,4-oxadiazole derivatives bearing 1,4,-benzodioxan was reported.20 Similarly, the crystal structures of the adamantyl group attached to the central 1,3,4-oxadiazole moiety were reported and these compounds were similar to those of the title compounds.21 Non-covalent interactions (NCI), particularly hydrogen bonds, have become one of the most important topics not only in synthetic chemistry, but also in biological and material systems.22 The occurrence and importance of C–H groups as donors in hydrogen bond formation in biological macromolecules such as proteins and nucleic acids were described.23 In contrast, σ-holes, another type of NCI, possess electron-deficient regions that arise when electron density is anisotropically distributed on group 14 elements (tetrels), 15 elements (pnictogens), 16 elements (chalcogens) and 17 elements (halogens) elements when they are covalently bonded to electron-withdrawing groups.24 The title compounds exhibit some unique intermolecular interactions such as halogen bond (Cl⋯Cl and Cl⋯π), and C–F⋯F–C and F⋯O noncovalent interactions compared to the closely related reported structures.
In this work, we present structural analysis of two closely related 1,3,4-oxadiazole-2-thione N-Mannich base derivatives to investigate the intermolecular interactions formed in these solid-state structures, the energetics, nature of the intermolecular interactions and in vitro bioactivities. Furthermore, a molecular docking analysis was performed to delineate the favorable orientation of these compounds at the active sites of four different drug targets. The title compounds exhibit a variety of conventional and non-conventional intermolecular contacts. The PIXEL energy calculation, Hirshfeld surface analysis, a DFT study, QTAIM, molecular electrostatic potential, and NCI plot analysis were carried out to characterize the intermolecular interactions observed in these compounds. Some of these computational tools have been successfully applied to various solid-state structures and modeled complexes to analyze various non-covalent interactions with respect to their geometrical preferences, strength and nature.25
2. Materials and methods
2.1. Synthesis and crystallization
To a solution of 5-(4-chlorophenyl)-1,3,4-oxadiazole-2(3H)-thione C (1.06 g, 5.0 mmol) in ethanol (10 mL), 37% formaldehyde solution (1.0 mL) and 2-trifluoromethylaniline (0.81 g, 5.0 mmol) or 2,5-difluoroaniline (0.65 g, 5.0 mmol) were added and the reaction mixture was stirred at room temperature for 5 hours and allowed to stand overnight. The precipitated crude products were filtered, washed with water, dried and crystallized from aqueous ethanol to yield the target compounds 1 and 2. Suitable single crystals for X-ray diffraction were obtained by slow evaporation of a solution of the title compound in ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :chloroform (1:1, v/v) at room temperature.
:chloroform (1:1, v/v) at room temperature.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S).S).
S).S).2.2. Single crystal X-ray analysis
For crystals 1 and 2, the X-ray intensity data were collected at 160 K on a Rigaku OD SuperNova/Atlas area-detector diffractometer using Cu Kα radiation (λ = 1.54184 Å) from a micro-focus X-ray source and an Oxford Instruments Cryojet XL cooler. The suitable single crystal was mounted using polybutene oil on a flexible loop fixed on a goniometer head and immediately transferred to the diffractometer. Pre-experiment, data collection, data reduction and analytical absorption correction26 were performed with the program CrysAlisPro (CrysAlis, Version 1.171.40.16c, Rigaku Oxford Diffraction, 2018). Using Olex2,27 the structure was solved with the SHELXT28 small molecule structure solution program and refined with the SHELXL2018/3 program package29 by full-matrix least-squares minimization on F2. The position of amine H atom was located from a difference Fourier map and refined freely along with its isotropic displacement parameters. All the remaining H atoms were placed in calculated positions (C–H = 0.95–0.99 Å) and constrained to ride on their parent atoms, with the Uiso(H) = 1.2Ueq(C). PLATON was used to check the result of the X-ray analysis.30 Mercury program was used to generate crystal packing and molecular dimers.312.3. Computational methods
The crystal structure geometry with the normalized H positions according to the typical neutron diffraction values (C–H = 1.083 and N–H = 1.009 Å) was used for all calculation. The nature of intermolecular interactions and their contributions were studied using the Hirshfeld surface32 and 2D-fingerprint plots33 with the CrystalExplorer 17.5 program.34 The calculations reported herein were performed using the Turbomole 7.0 program.35 The crystallographic coordinates were used for the calculations of the supramolecular assemblies. We have used the crystallographic coordinates for the assemblies because we are interested in evaluating the interactions as they stand in the solid state since the purpose of this manuscript is not finding the most favourable orientation of the molecules in the isolated dimers in the gas phase. The level of theory used for the calculations was RI-BP86-D3/def2-TZVP.36 The molecular electrostatic potential (MEP) surface plots were generated using the wavefunctions obtained at the same level of theory and the 0.001 a.u. isosurface to simulate the van der Waals envelope. The topological analysis of the electron density was carried out according to the QTAIM and noncovalent interaction plot index (NCIplot) methods proposed by Bader37 and W. Yang et al.,38 respectively. Both were represented using the VMD program39 using the following settings for the NCI plot index representation: s = 0.5 a.u.; cut-off ρ = 0.04 a.u. and colour scale −0.03 ≤ sign(λ2) ρ ≤ 0.03 a.u. The electron localization function (ELF)40 analysis was performed using the MultiWFN program41 at the PB86-D3/def2-TZVP level of theory.2.4. In vitro antimicrobial activity
The in vitro antimicrobial activity of the 1,3,4-oxadiazole derivatives 1 and 2 towards a panel of standard pathogenic bacterial and fungal strains namely; Staphylococcus aureus ATCC 6571, Bacillus subtilis ATCC 5256, Micrococcus luteus ATCC 27141 (Gram-positive bacteria), Escherichia coli ATCC 8726, Pseudomonas aeruginosa ATCC 27853 (Gram-negative bacteria), and the yeast-like pathogenic fungus Candida albicans MTCC 227 was evaluated. The preliminary screening was carried out following the semi-quantitative agar-disc diffusion method with Müller-Hinton agar medium.42 The minimal inhibitory concentration (MIC) of compounds 1 and 2 was assessed against the same microorganism used in the preliminary screening using the micro-dilution susceptibility method in Müller-Hinton Broth.432.5. In vitro anti-proliferative activity
The in vitro anti-proliferative activity of compounds 1 and 2 was evaluated against HepG-2 (hepatocellular carcinoma), PC-3 (human prostate cancer), HCT-116 (colorectal carcinoma) and MCF-7 (mammary gland breast cancer) tumor cell lines using the 3-[4,5-dimethylthiazoyl-2-yl]-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay.442.6. Molecular docking simulations
Molecular docking analysis was carried out for compounds 1 and 2 and co-crystallized ligands in the respective protein-inhibitor complexes using the Schrödinger suite (Schrödinger suite, LLC, New York, NY, 2021). In this study, we used two cancer targets namely, human thymidine phosphorylase (PDB ID: 1UOU; co-crystallized ligand ID: CMU),45 human thymidylate synthase (PDB ID: 6QXG; co-crystallized ligand ID: UFP)46 and a bacterial target namely, thymidine phosphorylase from Escherichia coli K12 strain (PDB ID: 4EAD; co-crystallized ligand ID: 0NP) and pyrimidine-nucleoside phosphorylase from Staphylococcus aureus strain COL (PDB ID: 3H5Q; co-crystallized ligand ID: THM). In all four cases, one subunit (chain A) was retrieved from the protein data bank (http://www.rcsb.org). The co-crystallized ligands in these targets and the compounds 1 and 2 were prepared using the ligprep module and the target proteins were prepared using the protein preparation wizard. For both ligands and proteins, OPLS4 force field was applied.47 The glide XP docking score was used to predict the most favourable orientation of the ligand at the protein active site.48 The protein-ligand interactions for the best predicted poses were analyzed using the PLIP Web server.493. Results and discussion
3.1. Chemical synthesis
4-Chlorobenzohydrazide B was prepared from the commercially-available methyl 4-chlorobenzoate A via treatment with hydrazine in ethanol.50 1,3,4-Oxadiazole-2(3H)-thione C was obtained via reaction of the carbohydrazide B with carbon disulfide in ethanolic potassium hydroxide as previously described.6 1,3,4-Oxadiazole-2(3H)-thiones were reported to undergo aminomethylation through reaction with primary aromatic amines and formaldehyde to yield the corresponding N-Mannich bases.18 Consequently, treatment of C with 2-trifluoromethylaniline or 2,5-difluoroaniline and formaldehyde solution and various primary aromatic amines in ethanol at room temperature yielded the target 3-arylaminomethyl N-Mannich bases 1 and 2 in good yields (Scheme 1). The structures of compounds 1 and 2 were confirmed by elemental analyses, 1H NMR and 13C NMR spectral data in addition to single crystal X-ray analysis.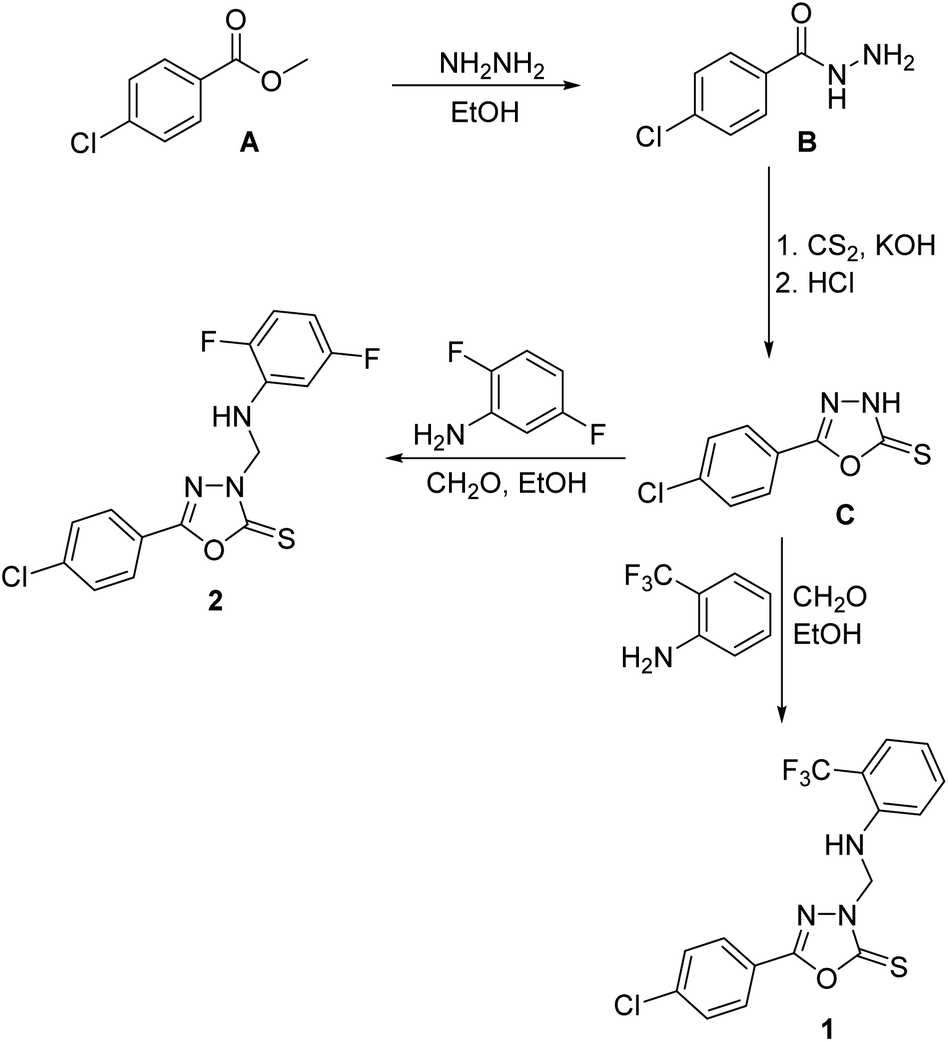 | ||
| Scheme 1 The synthetic pathway for compounds 1 and 2. | ||
3.2. Molecular and crystal structures
In order to gain insights into how the substituents affect molecular conformation, crystal packing, and intermolecular interactions, the single crystal X-ray analyses of compounds 1 and 2 were determined at 160 K. The X-ray analysis revealed that both compounds crystallize in the orthorhombic crystal system with the non-centrosymmetric space groups P212121 (compound 1) and Iba2 (compound 2). A summary of X-ray crystallographic data and refinement parameters is presented in Table 1. As shown in Fig. 1, both compounds contain a 4-chlorophenyl ring (ring A) attached to an oxadiazole ring (ring B). There is a difference between these two compounds in their substituted ring C. In compound 1, the trifluoromethyl group is present at the ortho position. In compound 2, the fluorine substitutions occur at the ortho and meta positions. The amino methylene group bridges the rings B and C in both compounds.| Compound 1 | Compound 2 | |
|---|---|---|
| Empirical formula | C16H11ClF3N3OS | C15H10ClF2N3OS |
| Formula weight | 385.79 | 353.77 |
| Temperature (K) | 160 (1) | 160 (1) |
| Crystal system | Orthorhombic | Orthorhombic |
| Space group | P212121 | Iba2 |
| a (Å) | 4.7906 (2) | 28.8781 (9) |
| b (Å) | 13.1169 (6) | 13.8936 (4) |
| c (Å) | 25.4767 (12) | 7.7325 (3) |
| α (°) | 90 | 90 |
| β (°) | 90 | 90 |
| γ (°) | 90 | 90 |
| Volume (Å3) | 1600.90 (12) | 3102.43 (17) |
| Z | 4 | 8 |
| ρcalc (g cm−3) | 1.601 | 1.515 |
| μ (mm−1) | 3.741 | 3.709 |
| F(000) | 784.0 | 1440.0 |
| Crystal size (mm3) | 0.27 × 0.03 × 0.02 | 0.11 × 0.05 × 0.02 |
| Radiation | Cu Kα (λ = 1.54184) | Cu Kα (λ = 1.54184) |
| 2θ range for data collection/° | 6.94 to 148.798 | 6.122 to 148.86 |
| Index ranges | −5 ≤ h ≤ 5, −14 ≤ k ≤ 16, −31 ≤ l ≤ 31 | −36 ≤ h ≤ 36, −13 ≤ k ≤ 17, −9 ≤ l ≤ 9 |
| Reflections collected | 15968 |
15474 |
| Independent reflections | 3244 [Rint = 0.0711, Rsigma = 0.0460] | 3163 [Rint = 0.0466, Rsigma = 0.0326] |
| Data/restraints/parameters | 3244/0/230 | 3163/1/212 |
| Goodness-of-fit on F2 | 1.035 | 1.061 |
| Final R indexes [I> = 2σ (I)] | R1 = 0.0399, wR2 = 0.0994 | R1 = 0.0270, wR2 = 0.0581 |
| Final R indexes [all data] | R1 = 0.0473, wR2 = 0.1042 | R1 = 0.0334, wR2 = 0.0607 |
| Largest diff. peak/hole (e Å−3) | 0.38/−0.32 | 0.18/−0.16 |
| Flack parameter | 0.013(17) | −0.004(10) |
| CCDC no. | 2302018 | 2302019 |
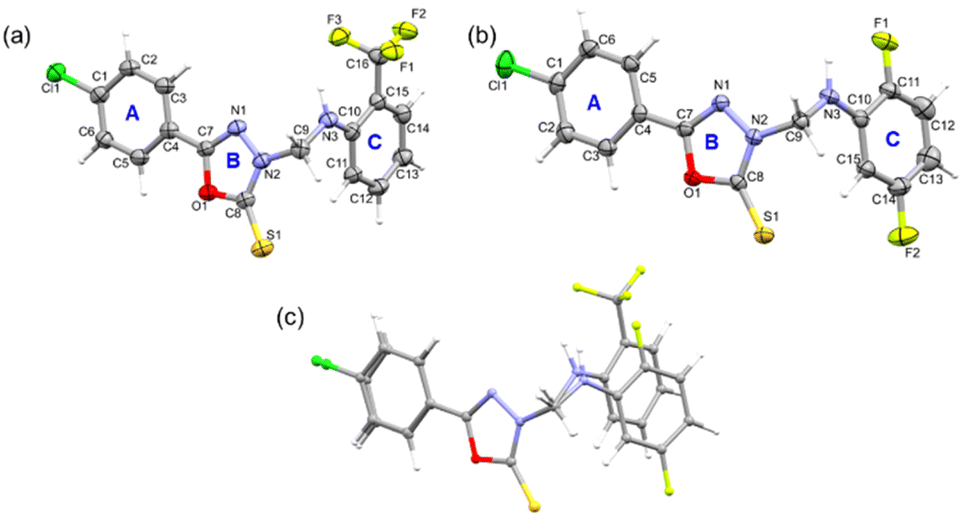 | ||
| Fig. 1 The ORTEP representations of compounds (a) 1 and (b) 2 are drawn at the 50% probability level, and (c) a structural overlay illustrates the deviation around ring C between compounds 1 and 2. | ||
Molecular geometry from the CSD database is used as knowledge base in an algorithm to verify molecular geometry using the mogul geometry check utility.51 We performed the mogul geometry check for compounds 1 and 2 and the results showed unusual bond length and valence angles involving the oxadiazole ring for 1. The length of the oxadiazole ring's C8–N2 bond (1.349 Å) is slightly higher than the mean value of 1.337 Å, and the four valence angles of the oxadiazole ring also deviate from the respective mean value. In addition to the valence angles of the oxadiazole, one of the valence angles (C11–C10–C15 = 117.44°; mean value = 119.87°) of ring C is also affected. It is to be noted that no such deviated geometrical parameters are observed for structure 2. The deviation of geometrical parameters could indicate the effect due to the trifluoromethyl group in ring C of 1.
Furthermore, in both structures, rings A and B are nearly coplanar (5.88° in 1 and 5.64° in 2) and the dihedral angle between mean planes of A/B and C rings is 84.64° in 1 and 71.46° in 2. We also note that both molecules adopt a L-shaped molecular conformation which is very similar to that of the structures, namely 2-(4-fluorobenzylthio)-5-(5-bromothiophen-2-yl)-1,3,4-oxadiazole and 4-(4-chlorophenyl)-3-[(4-fluorobenzyl)sulfanyl]-5-(thiophen-2-yl)-4H-1,2,4-triazole reported earlier.19,52 One of the arms of L comprises rings A and B, while the other arm of L consists of ring C. Fig. 1c shows the structural deviation around ring C, whereas rings A and B are overlaid very well. This structural deviation could be due to the substituted ring C mentioned above.
In the solid state, both compounds exhibit different types of weak hydrogen bonds and other unorthodox contacts because of the presence of suitable donors, acceptor groups, and aromatic rings. However, we are particularly interested in the ability of the chlorophenyl ring to establish halogen bonding interactions as a σ-hole donor. It is well known that chlorine is a very weak σ-hole donor and its ability to form halogen bonds has been an issue of discussion in the literature.53 Several nonclassical noncovalent interactions observed in molecular solids were described in the literature to highlight their cooperativity with conventional noncovalent interactions and the role of individual interaction in supramolecular assemblies.54
3.3. Qualitative analysis of intermolecular interactions: Hirshfeld surface and 2D-fingerprint plots
With a fingerprint plot, HS analysis has become an invaluable tool for exploring the nature of intermolecular interactions within crystal structures. In this way, characteristic interactions within structures and surfaces around molecules can be easily identified. In a dnorm surface, intermolecular contacts can be easily compared in terms of van der Waals radii via a simple red-white-blue colour scheme.In compound 1, the intense red regions represent close inter-contacts such as C(π)⋯C(π), C–F⋯F–C, and three-centered intermolecular C–H⋯F interactions (Fig. 2a). These short contacts have a significant impact on crystal packing. In compound 2, six different intermolecular interactions such as N–H⋯S, C–H⋯F, C(π)⋯C(π), S(lp)⋯C(π), F⋯O contacts and a σ-hole of type C–Cl⋯C(π) halogen bond with varying degrees of red spots on the HS (Fig. 2b). It is thought that these contacts are essential for the stabilization of crystal structure 2.
 | ||
| Fig. 2 Red spots on the Hirshfeld surfaces with two different orientations show the close inter-contacts observed in structures (a) 1 and (b) 2. | ||
The 2D fingerprint plots obtained from the Hirshfeld surface calculation show how various intermolecular interactions contribute to crystal packing and how substituents affect the contribution of these interactions. The selected 2D fingerprint plots for compounds 1 and 2 are shown in Fig. S1 (ESI†). The main contributions come from H⋯H contacts, which range from 20.6 to 21% and whose contributions to the crystal packing of 1 and 2 are very similar. The intermolecular H⋯F contacts significantly contribute to the crystal packing of both compounds. Since structure 2 has only two F atoms, the contribution of this contact is expected to be less compared to structure 1. Moreover, the fingerprint plots show that this contact is distributed differently between the two structures. For example, double sharp tips have a tip distance (de + di) of ∼2.3 Å in 2, while blunt tips have a slightly longer tip distance of 2.5 Å in 1. We also note a slight difference (2.8%) in the contribution of H⋯C contacts between the two structures. The observed close contact appears around 2.80 Å and can form intermolecular C–H⋯C(π) interactions in 1, while the corresponding contact in 2 appears to be longer than 3.0 Å, suggesting that this contact is not essential for stabilization.
Another significant contribution comes from the H⋯Cl interactions, and the tip distance is about 2.9 Å in the fingerprint plots in both structures and has a slightly higher contribution to the total HS area in 2 compared to 1. Although the relative contribution of H⋯S contact differs between these two structures (10.1% in 1 and 7.0% in 2), the fingerprint plots also show differences in the dnorm spacing. The intermolecular H⋯S contacts appear at 2.5 Å in 2, while the corresponding short contact distance is 3.0 Å in 1. The distribution of H⋯S contact confirms the presence of the N–H⋯S interaction, and the same type of intermolecular interaction is not possible in 1. It is, however, possible to establish a C–H⋯S interaction. It is interesting to note that the intermolecular F⋯F contact in 1 is established with a tip distance of 2.8 Å and contributes about 4.0% to crystal packing. However, in 2, this contact disappears. The relative contribution of the H⋯N contact is almost the same in both structures. However, this contact is relatively shorter in 2 (∼2.7 Å) than in structure 1 (over 2.8 Å). This feature indicates that structure 2 tends to establish a C–H⋯N interaction, whereas this contact is weak in structure 1 due to the longer separation between the H and N atoms. Similarly, the C⋯C contact, representing the π-stacking interaction, contributes 4.7% to the crystal packing of 1, while the corresponding contact contributes only about 1.7% in 2. Red-blue triangles on the surfaces of rings A and B in the shape index diagram and green dots at 3.6 Å in the 2D-FP plots indicate that these two rings can stack on each other (Fig. S2, S3, ESI†).
The halogen bonds show an interesting feature in the 2D fingerprint plots. In 1, a homo-halogen bond of the type Cl⋯Cl interaction contributes about 1.7% to the crystal packing and is located as short as 3.5 Å, indicating its short nature and found important for stabilization. The corresponding contact is in 2 beyond 3.9 Å, indicating its weak nature. The relative contribution of another potential halogen bond of the type Cl⋯C(π) is almost the same in both structures. However, this contact is relatively shorter in 2 (∼3.4 Å) than in structure 1 (3.6 Å). This observation suggests that the Cl⋯Cl halogen bond in 1 and the Cl⋯C(π) halogen bond in 2 play an important role in stabilizing the respective crystal structure. We also note that the F atom establishes a noncovalent interaction with the oxadiazole O atom. The 2D-FP plots show that the F⋯O interaction in 2 is relatively shorter than in structure 1 although its contribution to crystal packing is comparable between the two structures. Furthermore, there is a short S⋯C contact at 3.5 Å with a contribution of 3.8% in structure 2 and the corresponding contact at slightly over 3.5 Å with a contribution of 1.1% in 1. The 2D-FP plots for the other possible inter-contacts are relatively longer than the sum of the vdW radii of the interacting atoms in these structures.
3.4. Supramolecular self-assembly in 1
In the solid state, molecule 1 is packed columnar along the crystallographic bc and ac planes, as shown in Fig. 3a and b. The energetically significant dimers in this structure were identified by CLP-PIXEL calculations (Fig. 3c–f). The results showed that five important dimers contribute to stabilizing the crystal structure. Various types of intermolecular interactions are formed in these dimers, including C–H⋯F, C–H⋯Cl, C–H⋯S, C–H⋯π, π-stacking, F⋯O, C–F⋯F–C contacts, and a homo halogen bonding type of C–Cl⋯Cl interaction. The intermolecular interaction energies for these dimers were calculated using the CLP-PIXEL method and ranged from −13.3 to −1.3 kcal mol−1. Accurate dimerization energies were calculated using the DFT method at the B97-D3/def2-TZVP level of theory to compare these energies with those calculated using the CLP-PIXEL method. As shown in Table 2, the dimerization energies calculated with both methods are comparable except for dimer D1. The CLP-PIXEL method underestimates the energy of the D1 dimer, which could be due to the dispersion effect of the observed intermolecular interactions in D1.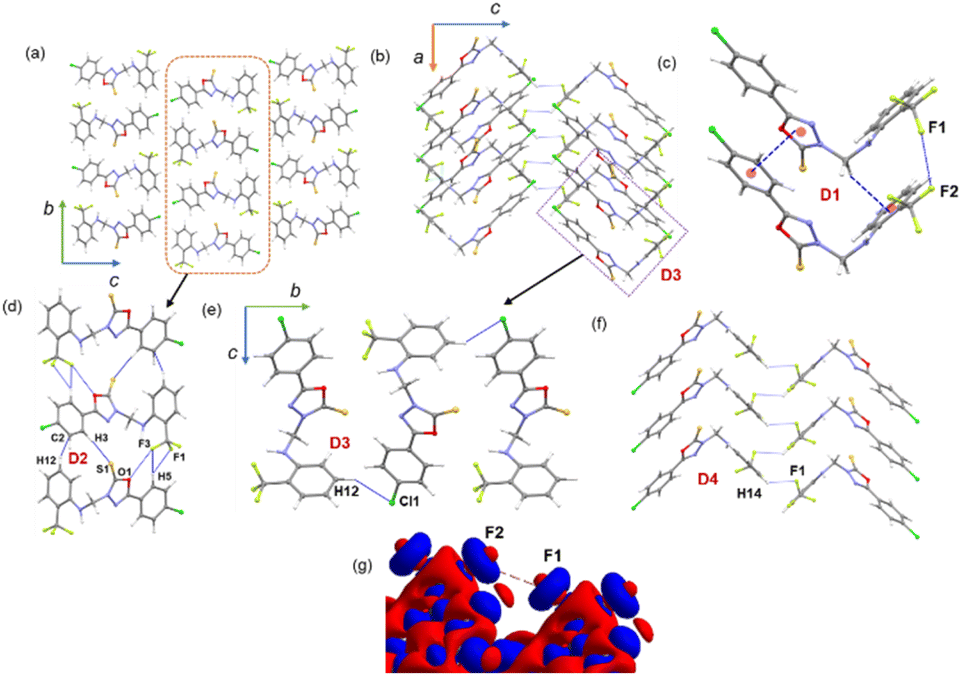 | ||
| Fig. 3 Crystal packing of 1 projected onto the crystallographic (a) bc plane, (b) ac plane, and (c–f) dimers observed in 1, supramolecular self-assembly mediated by various intermolecular contacts, and (g) deformation density showing charge depletion (red) and charge concentrated region (blue) around interacting fluorine atoms. | ||
| Dimer | CD | Symmetry | Important interactions | Geometry H⋯A (Å), ∠D–H⋯A (°) | PIXEL/MP2/6-31G** | B97D3/def2-tzvp | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ecoul | Epol | Edisp | Erep | Etot | ΔEcp | |||||
| D1 | 4.791 | x − 1, y, z | Cg1⋯Cg2 | 3.484 (2) | −4.8 | −2.5 | −20.6 | 14.6 | −13.3 | −17.7 |
| C9–H9B⋯Cg3 | 2.80, 139 | |||||||||
| C16–F1⋯F2–C16 | 2.832 (4), 149.2 (2) | |||||||||
| D2 | 6.647 | −x + 1, y − 1/2, −z + 3/2 | C5–H5⋯F1 | 2.44, 134 | −2.9 | −1.8 | −8.2 | 6.1 | −6.8 | −5.7 |
| C5–H5⋯F3 | 2.43, 169 | |||||||||
| C3–H3⋯S1 | 2.98, 126 | |||||||||
| C12–H12⋯C2(π) | 2.82, 134 | |||||||||
| C16–F3⋯O1 | 3.049 (4), 151.7 (2) | |||||||||
| D3 | 7.965 | −x, y − 1/2, −z + 3/2 | C12–H12⋯Cl1 | 2.91, 136 | −1.6 | −0.7 | −4.4 | 2.5 | −4.3 | −4.0 |
| D4 | 12.479 | x − 1/2, −y + 1/2, −z + 2 | C14–H14⋯F1 | 2.60, 136 | −0.5 | −0.1 | −1.8 | 0.7 | −1.7 | −1.4 |
| D5 | 14.397 | x + 1/2, −y + 1/2, −z + 1 | C1–Cl1⋯Cl1 | 3.523 (1), 162.5 (1) | −0.6 | −0.3 | −2.2 | 1.7 | −1.3 | −1.2 |
A highly stable dimer D1 consists of two adjacent L-shaped molecules held together by intermolecular π-stacking, C–H⋯π and C–F⋯F–C interactions (Fig. 3c). The π-stacking interaction is formed between rings A and B (centroid-to-centroid distance = 3.484 (2) Å), while the bridging methylene group participates in an intermolecular C–H⋯π interaction with the π-system of ring C. The C–F⋯F–C interaction is formed between adjacent CF3 groups. The components of electrostatic and dispersion energies contribute 26 and 74%, respectively, to stabilizing D1. The higher contribution of dispersion energies could be due to the effect of the π-stacking and C–H⋯π and C–F⋯F–C interactions.
Four different types of halogen–halogen bonding (involving Cl, Br and I) geometries were described in the literature as shown in Scheme 2.55 The homo fluorine (F⋯F) interactions are also shown as one of the four types of geometries. To gain more insight into the geometrical preference of C–F⋯F–C contacts from trifluoromethyl moiety, we searched the Cambridge Structural Database (CSD, version 2022.3.0) using a template containing trifluoromethylphenyl residues rather than Ph-F residues, and the search yielded 584 hits with 970 C–F⋯F–C contacts. The separation of F⋯F contacts ranges from 2.503 to 2.940 Å, and the shortest one was observed for the entry with refcode HEWFAQ (d = 2.503 Å), and the longest separation was observed for the entry with refcode DIDQAK (d = 2.940 Å). More than 80% of contacts fall between 2.803 and 2.940 Å (Fig. S4, ESI†). In this study, we observed the F⋯F distance is 2.832 (4) Å, which lies in the most preferred region. Tothadi et al. described the classification of homo-halogen bonding based on the difference in angles (0° ≤ |θ1 − θ2| ≤ 15°: type I; 30° ≤ |θ1 − θ2|: type II and 15° ≤ |θ1 − θ2| ≤ 30°: quasi type I/II).56 According to this classification, 52.4% of the contacts exhibit type I geometry, similar to Cl⋯Cl contacts surveyed earlier. 16.1% of the contacts belong to quasi-type I/II geometry (Fig. S5, ESI†). In the present study, F⋯F contact belongs to type I/II geometry with the |θ1 − θ2| = 29.85°. The remaining contacts belong to type II geometry. The CSD search also indicates that some F⋯F contacts adopt a Z-shaped geometry. Furthermore, we calculated the deformation density for the dimer, showing the C–F⋯F–C interaction (Fig. 3g).
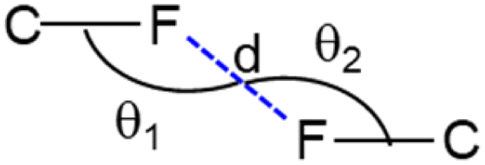 | ||
| Scheme 2 Geometrical parameters of C–F⋯F–C interactions involving trifluoromethyl moieties found in CSD database. Different types geometries are defined as: type I (90° < θ1 ≈ θ2 < 180°; parallel displaced geometry), type II (θ1 ∼ 90°; θ2 ∼ 180°; L-shaped geometry) and type III (θ1 ≈ θ2 = 180°; linear geometry) and type IV (θ1 ≈ θ2 = 90°; Z-shaped geometry). | ||
The neighbouring molecules stacked along the crystallographic b axis in a column are connected by a large number of intermolecular interactions, including intermolecular C–H⋯F/S/C(π) interactions and unconventional F⋯O contacts. These interactions lead to the formation of the dimer D2, and the contribution of the electrostatic and dispersion energies to the stabilization is 36% and 64%, respectively. There are rectangular molecular arrangements in the repetitive structural motif of 1 (dimer D3) (see Fig. 3, dashed rectangular box). This motif is stabilized by an intermolecular C–H⋯Cl interaction and links the molecules into a supramolecular C(14) chain that runs parallel to the crystallographic b axis. Notably, the dispersion energy contributes more (75%) towards stabilizing dimer D3. A weak molecular dimer (D5) is stabilized by a directional σ-hole bonding namely C–Cl⋯Cl halogen bonding interaction. This dimer is also predominantly driven by the dispersion energy component, contributing 71% towards the total stabilization energy. This halogen bond exhibits an L-shaped geometry (type II geometry with |θ1 − θ2| = 70.37°). To investigate the existence of σ-hole contact in 1, we carried out molecular electrostatic potential surface analysis, and the results are presented in a separate section.
3.5. Supramolecular self-assembly in 2
In the crystalline state, molecule 2 is arranged in columnar along the crystallographic ab plane, as shown in Fig. 4a. From the CLP-PIXEL calculation, one can extract the energetically significant dimers in this crystal structure (Table 3). There are five such dimers (D1–D5) stabilized by varieties of intermolecular interactions such as N–H⋯S, C–H⋯NF/Cl, C(π)⋯C(π) contact, S(lp)⋯π contact (Fig. 4a–e). Furthermore, a directional halogen bond of type C–Cl⋯C(π) and a fluorine bond of type C–F⋯O interaction (Fig. 4b). It is important to emphasize that some of the intermolecular interactions such as N–H⋯S, C–H⋯N, lp⋯π, C–Cl⋯C(π) and C–F⋯O contacts are specific to compound 2. These specific interactions play a major role in stabilizing the solid-state structure. Furthermore, the intermolecular interaction energies for these dimers are calculated using the CLP-PIXEL method, ranging from −12.5 to −1.7 kcal mol−1. The dimerization energies calculated by the DFT and CLP-PIXEL methods are comparable (Table 3). Slight deviations in energies in some dimers occur due to the effect of dispersive forces and the types of interactions present in them.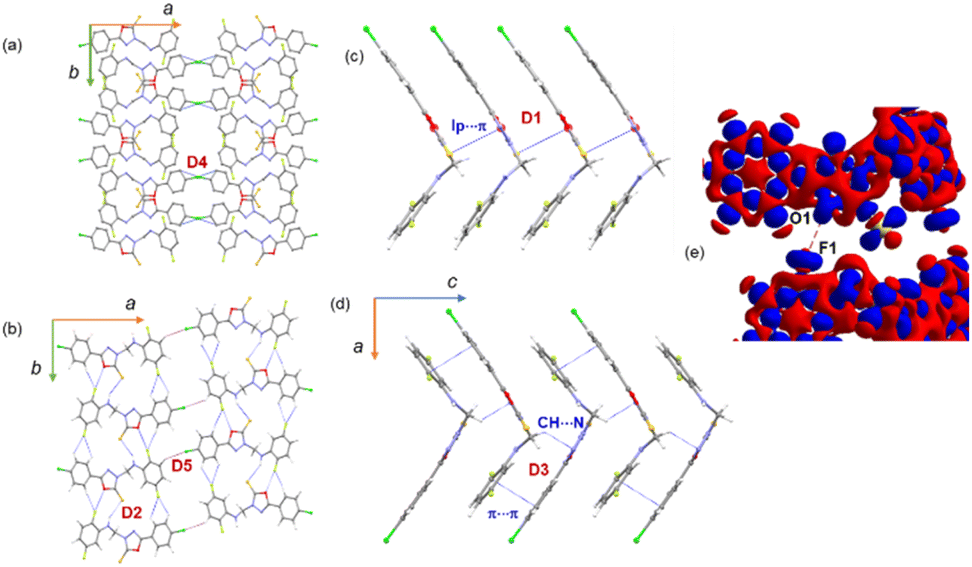 | ||
| Fig. 4 (a) Adjacent columns are interlinked by directional C–Cl⋯C(π) halogen bond, (b) supramolecular ribbon formed by motif D2 and adjacent supramolecular ribbons are interlinked by weak C–H⋯Cl hydrogen bonds, (c) L-shaped molecular layer held together by S(lp)⋯π interaction, and (d) molecular dimer D3 is formed by intermolecular C–H⋯N interaction and supported by a weak C(π)⋯C(π) interaction, (d) 3D deformation electron density shows intermolecular F⋯O contact (red: charge depletion region and blue: charge concentrated region). | ||
| Dimer | CD | Symmetry | Important interactions | Geometry H⋯A (Å), ∠D–H⋯A (°) | PIXEL/MP2/6-31G** | B97D3/def2-tzvp | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ecoul | Epol | Edisp | Erep | Etot | ΔEcp | |||||
| D1 | 5.089 | x, –y + 1, z − 1/2 | S1⋯Cg1 | 3.343 (1) | −6.2 | −3.2 | −14.2 | 11.0 | −12.5 | −14.7 |
| D2 | 7.089 | −x + 3/2, y − 1/2, z | N3–H3A⋯S1 | 2.50, 149 | −8.4 | −4.4 | −7.2 | 9.8 | −10.2 | −8.3 |
| C5–H5⋯F2 | 2.56, 122 | |||||||||
| C6–H6⋯F2 | 2.62, 119 | |||||||||
| C3–H3⋯F1 | 2.23, 173 | |||||||||
| C11–F1⋯O1 | 2.923 (1), 160.36(1) | |||||||||
| D3 | 5.493 | −x + 3/2, −y + 3/2, z − 1/2 | C9–H9A⋯N1 | 2.73, 137 | −3.1 | −1.4 | −12.6 | 7.6 | −9.4 | −12.1 |
| C11⋯C6 | 3.303 (4) | |||||||||
| D4 | 13.587 | −x + 1, y, z − 1/2 | C1–Cl1⋯C6(π) | 3.390(3), 170.9(1) | −0.9 | −0.5 | −3.8 | 3.2 | −2.1 | −2.4 |
| D5 | 14.890 | x − 1/2, −y + 3/2, z | C12–H12⋯Cl1 | 2.90, 153 | −0.7 | −0.3 | −1.6 | 1.0 | −1.7 | −1.5 |
In this structure, the strongest dimer (D1) stabilizes with the intermolecular lp⋯π interaction involving S1 and the π-system of ring B (Fig. 4c). This interaction links the molecules into a chain parallel to the crystallographic c axis. It is also noted that electrostatic energy contributes 40% towards the total stabilization energy for this dimer. In the solid state, dimer D2 extends along the b axis to generate a molecular ribbon, and adjacent ribbons formed by dimer D2 are interconnected by dimer D5 (Fig. 4b). These two dimers collectively generate a supramolecular chain along the ab plane. The former dimer D2 stabilizes with five intermolecular interactions. The N–H⋯S interaction is formed between the amine group and the S atom of the oxadiazole moiety. Both fluorine atoms (F1 and F2) of ring C have been involved in intermolecular C–H⋯F interactions. One of the fluorine atoms (F1) is engaged in an unorthodox fluorine bond of type F⋯O contact. The 3D deformation density calculated from theoretical charge density analysis for the F⋯O contact shows the small polarizability of electron density around the fluorine atom (Fig. 4e). The charge depletion region of fluorine (red) pointed towards the charge concentrated region on the O1 atom (blue). The CSD search reveals a very small number of F⋯O contacts exist. The CLP-PIXEL calculation reveals that the contribution of electrostatic and dispersion energies is 64 and 36% towards the stabilization of dimer D2, whereas the dispersion energy contributes (62%) more to the stabilization of dimer D5 than the electrostatic energy.
As illustrated in Fig. 4d, a supramolecular chain is generated by the dimer D3 which stabilizes with two intermolecular interactions. In this dimer, the bridge methylene group acts as a donor and one of the N atoms of the oxadiazole ring is involved as an acceptor for the intermolecular C–H⋯N interaction. The stability of D3 is supported by a C(π)⋯C(π) contact (3.303 (4) Å) between rings A and C in an offset mode and the dispersion energy contributes 74% towards stabilization. Dimer D4 stabilizes with a weak C1–Cl1⋯C6(π) halogen bond. As shown in Fig. 4a, this halogen bond interlinks the adjacent molecular sheets formed on the crystallographic ab plane.
3.6. Nature of halogen bonding interactions in 1 and 2
Initially, we used molecular electrostatic potential surface calculations to investigate the existence of σ-hole at the Cl atom in compounds 1 and 2. The surfaces are represented in Fig. 5, showing that in both compounds, the MEP maximum is located at the NH bond (+38.3 and +41.4 kcal mol−1 for 1 and 2, respectively), and the MEP minimum is located at the S-atom of the thione group (−22.6 and −23.2 kcal mol−1, respectively). The MEP is also large and positive over the five-membered ring (+21.9 kcal mol−1 in both compounds) disclosing that the five-membered ring is electron deficient and exerts a significant electron withdrawing effect on the chlorophenyl ring. This increases the MEP value at the chlorine's sigma-hole and reduces the nucleophilicity of the negative belt (MEP value is more positive). In Fig. 5 (top-center), an amplification of the MEP around the chlorine atom is provided. In both compounds the MEP surface and anisotropy at the Cl-atom are equivalent: +11.3 kcal mol−1 at the σ-hole and −3.7 kcal mol−1 at the negative belt.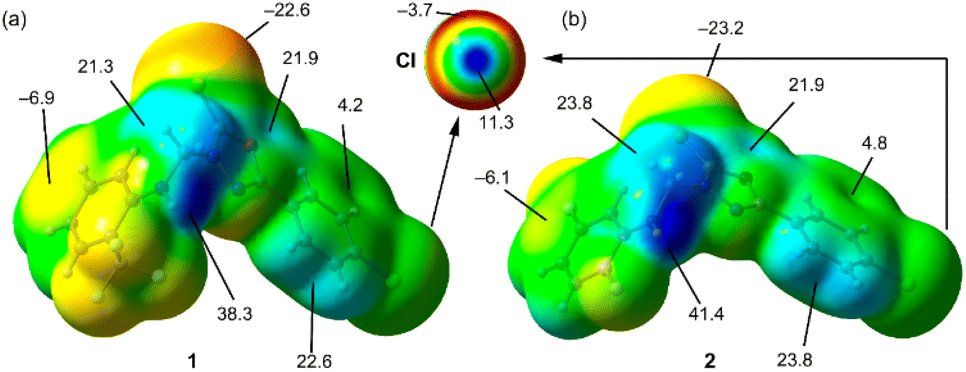 | ||
| Fig. 5 MEP surfaces of compounds 1 (a) and 2 (b) and the RI-BP86-D3/def2-TZVP level of theory. Some MEP values are indicated in kcal mol−1. An amplification of the MEP around the Cl-atom is also provided. | ||
In Fig. 6, we have represented two selected dimers of compounds 1 and 2, where the Cl-atom is involved in halogen bonding interactions. In the case of compound 1, the C–Cl bond points to the negative belt of the adjacent Cl-atom and in the case of compound 2, to one C-atom of the aromatic ring. The dimerization energies are modest (−1.2 kcal mol−1 and −2.4 kcal mol−1) but favourable in both cases. It should be emphasized that the MEP over the chlorophenyl ring is positive, thus indicating that in compound 2, the halogen bond is likely not dominated by electrostatic effects and that other forces like dispersion or polarization likely compensate for the small electrostatic repulsion. The directionality of the HaB in compound 2 is better than in compound 1, in line with the stronger interaction. Fig. 6 also shows the combined QTAIM and NCI plot analysis of both HaB dimers. The QTAIM analysis shows one bond critical point (CP, represented as a small red sphere) and the bond path (orange line) interconnecting both Cl-atoms in the dimer of 1, thus confirming the existence of the HaB contact. Moreover, the interaction is further characterized by a green RDG isosurface coincident with the location of bond CP, which supports the attractive nature of the interaction. For compound 2, the interaction is characterized a bond CP and a bond path connecting the Cl-atom to one C-atom of the aromatic ring. The NCIplot shows the presence of a green RDG isosurface that emerges upon dimerization and embraces one of the CC bonds of the aromatic ring, suggesting that both C-atoms are involved in the interaction, as further studied below.
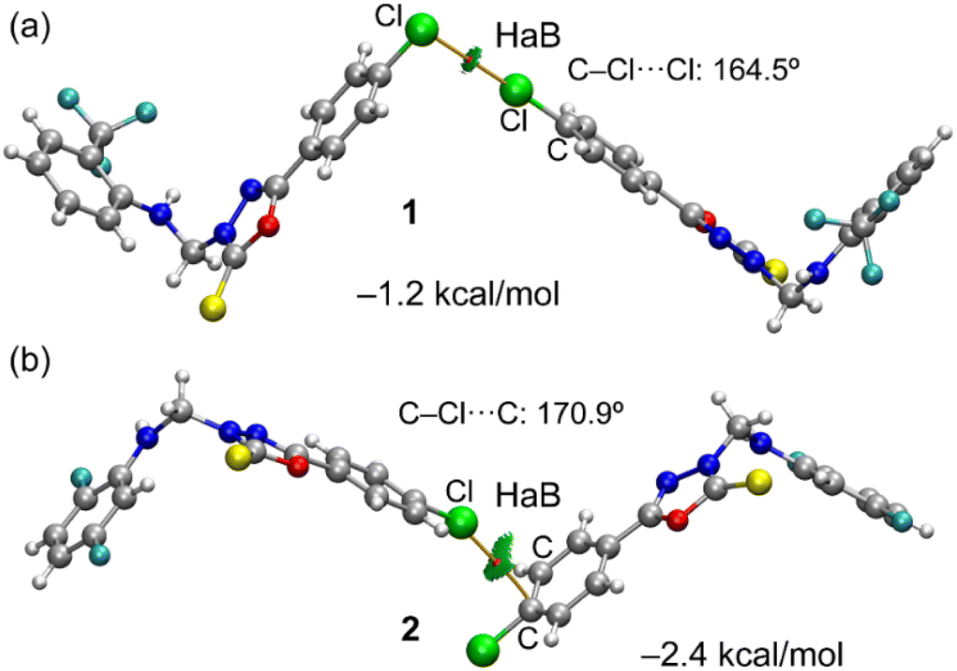 | ||
| Fig. 6 QTAIM and NCIplot analyses of the HaB dimers observed in the X-ray structures of compounds 1 (a) and 2 (b). Only intermolecular bond CP (red spheres) and bond paths (orange lines) are represented. For the NCI plot settings, see computational methods. | ||
To further investigate the halogen bonding nature of the contacts, especially in compound 2 where the MEP analysis suggests that the chlorophenyl π-system is electron deficient, we have used the electron localization function (ELF). This computational tool is useful to identify LPs and σ-holes.57 The 2D ELF maps are represented in Fig. 7, where we have used the 2D plane defined by the atoms involved in the HaB (these atoms are also indicated in Fig. 6). It can be observed that in compound 1, the σ-hole at the Cl-atom is pointing to the belt of the electron donor Cl-atom of the adjacent molecule, as expected for a true halogen bonding interaction. In fact, the bond path that connects both Cl-atoms crosses very close to the maximum electron depletion (σ-hole). For compound 2, the σ-hole points to the π-system of the CC bond, thus also confirming the HaB nature, in spite of the positive MEP.
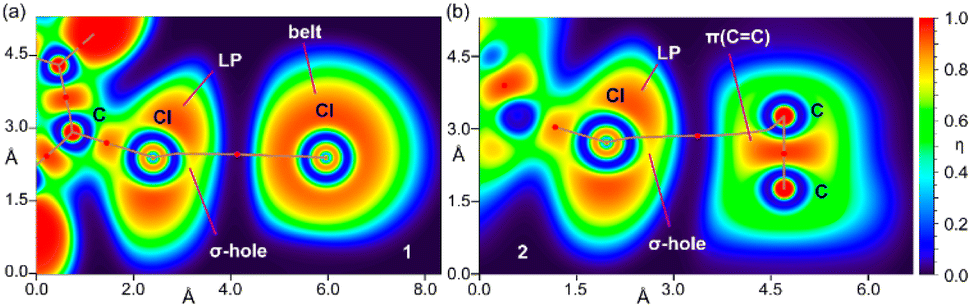 | ||
| Fig. 7 2D maps of η(r) function for the dimers of 1 (a) and 2 (b). The molecular planes correspond to the C-atoms labelled in Fig. 1. | ||
3.7. Topological analysis of intermolecular interactions
To assess the nature and strength of the intermolecular interactions observed in various molecular dimers of 1 and 2, we calculated the topological properties of these interactions mentioned in the experimental section. The molecular graphs of dimers show the bond critical points (BCPs) for the intermolecular interactions illustrated in Fig. S6 and S7 (ESI†), and the topological parameters for the intermolecular interactions are summarized in Table S1 (ESI†). All the observed interactions are closed-shell interactions according to the value of |–V(r)/G(r)| < 1, ∇2ρ(r) > 0 and H(r) > 1.58 In 1, the dissociation energies (De) for two of the C–H⋯F interactions are relatively higher compared to other interactions observed. It is of interest to note that the dissociation energy of C16–F1⋯F2–C16 interaction is comparable to the strength of two of the C–H⋯F interactions. The experimental charge density values were reported for the F⋯F interactions with type I, type II and, quasi-I/II geometries.59 The electron density (ρ(r)) is in the range of 0.02–0.07 e Å−3 and the Laplacian of the electron density (∇2ρ(r)) falls in the range of 0.5–1.3 e Å−5. In the present study, these values are comparable to experimental charge density values. We also noted that the strength of the C–Cl⋯Cl halogen bond and C⋯C and C–H⋯π, C–H⋯S, and one of the C–H⋯F interactions is relatively weaker than the F⋯F interaction.In 2, the strength of N–H⋯S (De = 2.2 kcal mol−1) and one of the C–H⋯F interactions (De = 2.1 kcal mol−1) is comparable, and they are relatively higher compared to other interactions. We also noted that the strength of a halogen bond (C–Cl⋯π) is comparable to that of C–H⋯N/F. The dissociation energy of the unconventional fluorine bond of type F1⋯O1 contact is the same as that of a Cl⋯π halogen bond and unconventional S⋯π contact. The intermolecular C–H⋯Cl interaction is relatively weaker than other interactions observed in 2.
3.8. In vitro antimicrobial activity
The results of the initial antimicrobial screening of compounds 1 and 2 (200 μg/disc), the antibacterial antibiotics gentamicin sulfate, ampicillin trihydrate, and the antifungal drug clotrimazole (100 μg/disc) are outlined in Table 4. As evident from Table 4, compounds 1 and 2 showed marked antibacterial activity against all tested Gram-positive bacterial strains, marginal inhibitory activity against Escherichia coli, and lacked inhibitory activity against Pseudomonas aeruginosa. Compound 1 displayed weak inhibitory activity against the pathogenic fungus Candida albicans, while compound 2 was totally inactive.| Compound | Diameter of growth inhibition zonea (mm) | |||||
|---|---|---|---|---|---|---|
| SA | BS | ML | EC | PA | CA | |
| a Figures shown in parentheses represent the MIC values (μg mL−1), NT: not tested, (−): inactive (inhibition zone < 10 mm. | ||||||
| 1 | 21 (4) | 18 (16) | 16 | 12 | — | 11 |
| 2 | 22 (8) | 24 (2) | 18 (16) | 11 | — | — |
| Gentamicin sulfate | 27 (1) | 26 (2) | 20 (2) | 22 (0.5) | 21 (0.5) | NT |
| Ampicillin trihydrate | 22 (2) | 23 (1) | 20 (2) | 16 (8) | 18 (8) | NT |
| Clotrimazole | NT | NT | NT | NT | NT | 21 (4) |
The minimal inhibitory concentration (MIC) of compounds 1 and 2 was assessed against the same microorganism used in the preliminary screening using the micro-dilution susceptibility method in Müller-Hinton Broth.60 The values of the MIC for compounds 1, 2 and the reference antimicrobial drugs gentamicin sulfate, ampicillin trihydrate, and clotrimazole (Table 4) were almost consistent with their growth inhibition zones.
3.9. In vitro anti-proliferative activity
The results of in vitro anti-proliferative activity of compounds 1 and 2 and the anticancer drug doxorubicin are shown in Table 5. The anti-proliferative activity outcomes showed that 2,5-difluorphenyl analogue 2 displayed marked anti-proliferative activity with IC50 < 25 μM against HepG-2 and MCF-7 cell lines and retained weak activity against PC-3 and HCT-116 cell lines. Meanwhile, the 2-(trifluoromethyl)phenyl analogue 1 displayed good anti-proliferative activity against HepG-2 and moderate activity against MCF-7 cell lines, and its activity against PC-3 and HCT-116 cell lines was almost similar to compound 1.3.10. Molecular docking analysis
In order to corroborate the experimental in vitro data, we performed a molecular docking simulation for four important drug targets, two of which are expressed in many tumor cells (human thymidine phosphorylase and human thymidylate synthase) and one target (thymidine phosphorylase) taken from the E.coli K12 strain and a drug target namely pyrimidine-nucleoside phosphorylase from Staphylococcus aureus strain COL was considered. The extra precision (XP) glide scoring scheme was used to predict the favourable pose at the protein active site and a score for the most favourable pose. Table 6 summarizes the glide XP docking scores for compounds 1 and 2 against these targets. For the control experiment, the co-crystallized ligand bound to these targets was docked and the score was compared with compounds 1 and 2. The co-crystallized ligands showed a higher binding affinity in all four cases toward the respective target. However, compounds 1 and 2 also displayed a negative glide XP score, suggesting that they could also bind to the active site of the respective target. The intermolecular interactions between compounds 1 and 2 and the critical active site residues in the respective protein targets are illustrated in Fig. 8.| Compound code | Glide XP docking score |
|---|---|
| Human thymidine phosphorylase | |
| CMU | −11.578 |
| 1 | −4.917 |
| 2 | −6.748 |
|
|
| E.coli K12 strain thymidine phosphorylase | |
| 0NP | −10.487 |
| 1 | −6.987 |
| 2 | −5.926 |
|
|
| Human thymidylate synthase | |
| UFP | −9.271 |
| 1 | −4.519 |
| 2 | −4.024 |
|
|
| Staphylococcus aureus strain COL pyrimidine-nucleoside phosphorylase | |
| THM | −7.599 |
| 1 | −3.812 |
| 2 | −4.278 |
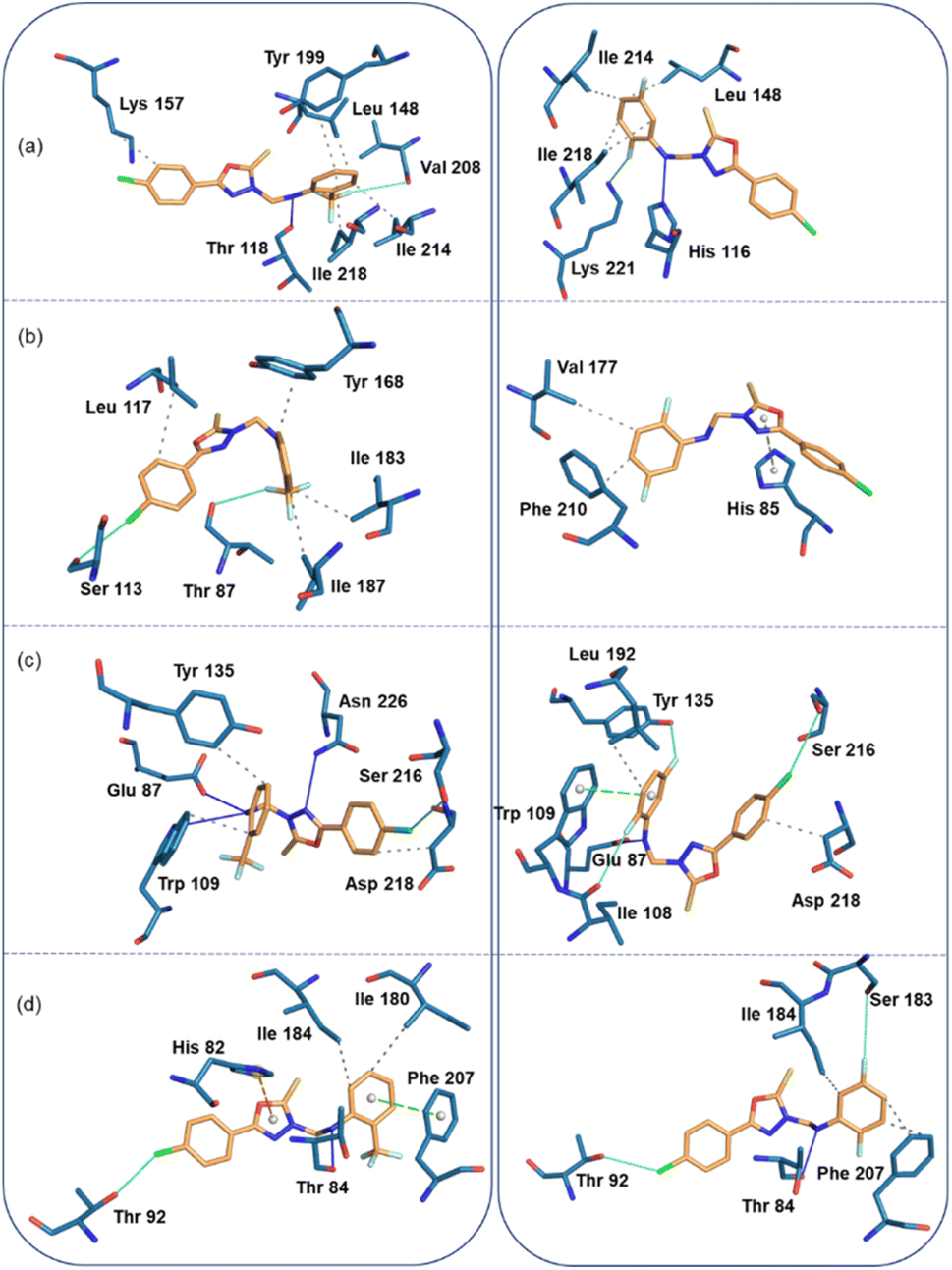 | ||
| Fig. 8 Protein-ligand interactions (left panel: compound 1 and right panel: compound 2). (a) Human thymidine phosphorylase, (b) E. coli thymidine phosphorylase, (c) Human thymidylate synthase, and (d) S. aureus pyrimidine-nucleoside phosphorylase. | ||
In the human thymidine phosphorylase complex, the binding affinity of compound 2 is slightly better than compound 1. An analysis of protein-ligand interaction reveals that in the human thymidine phosphorylase complex, residues Leu 148, Lys 157, Tyr 199, Ile 214, and Ile 218 establish hydrophobic interactions with the terminal aromatic rings of compound 1. The amine group of 1 is involved in a hydrogen bond (N–H⋯O) with the backbone O atom of the Thr 118, and one of the F atoms of compound 1 has established a linear F⋯O contact (3.94 Å and 165°) with the backbone O atom of the Val 208. Compound 2 also establishes similar types of hydrophobic interactions with active site residues in human thymidine phosphorylase complex. Instead of an N–H⋯O hydrogen bond, an N–H⋯N hydrogen bond is formed between compound 2 and His 116. Similarly, instead of a linear F⋯O contact, an intermolecular F⋯N contact (3.14 Å, 156°) is formed between 2 and side chain N atom of the Lys 221 (Fig. 8a).
In the E.coli thymidine phosphorylase complex, compound 1 shows a slightly better affinity for the enzyme than compound 2, as shown in Table 6. Fig. 8b shows different modes for compounds 1 and 2 at the active site, as seen from the interacting residues. We also see from this figure that compound 1 is slightly folded when it binds to the enzyme, whereas compound 2 exhibits a relatively extended conformation at the active site. The differences in affinity could be understood as the preferable binding conformations of these compounds. In addition to the hydrophobic interactions formed by residues Leu 117, Tyr 168, Ile 183, and Ile 187, a highly directional σ-hole (halogen bond) interaction is formed between the Cl atom of 1 and the O atom of the backbone Ser 113 (Cl⋯O = 3.68 Å, ∠C–Cl⋯O = 175°). One of the F atoms of 1 is involved in an interaction with the backbone O atom of Thr 87 residue (F⋯O = 3.17 Å, ∠C–F⋯O = 147°). The strong affinity could arise from forming halogen contacts between compound 1 and the active site residues. Unlike compound 1, compound 2 establishes only two hydrophobic interactions with the active site residues Val 177 and Phe 210. In addition, the residue His 85 forms a π-stacking interaction (with a T-shaped configuration) with the oxadiazole ring.
In the thymidylate synthase complex, both compounds exhibit folded conformations at the active site and show comparable affinity towards the enzyme. As shown in Fig. 8c, compound 1 establishes three hydrogen bonds with the active site residues Glu 87, Trp 109, and Asn 226. In addition, the residues Trp 109, Tyr 135, and Asp 218 have hydrophobic interactions with compound 1. There is a σ-hole interaction is formed between the Cl atom and the side chain O atom of Ser 216 (Cl⋯O = 3.44 Å, ∠C–Cl⋯O = 148°). The number of hydrogen bonds (Glu 87) and hydrophobic interactions (Leu 192 and Asp 218) is reduced in enzyme-compound 2 compared to the enzyme-compound 1 complex. However, three halogens (F and Cl) involved non-bonded interactions formed with the O atoms of the residues Ile 108, Tyr 135 and Ser 216 residues. The F⋯O (Ile 108) distance is 3.31 Å and ∠C–F⋯O = 162°. For another F⋯O (Tyr 135), separation is 2.90 Å and ∠C–F⋯O = 144°. The C–Cl⋯O (3.74 Å, 161°) halogen bond is highly directional and helps stabilize the complex.
In the S. aureus pyrimidine-nucleoside phosphorylase complex, both compounds show comparable affinity for the enzyme. Compound 1 establishes a hydrogen bonding interaction with the Thr 84 residue (Fig. 8d). The active site residues Ile 180 and 184 are involved in hydrophobic interactions. The active site residue Phe 207 stacks against ring C of compound 1 and His 82 establishes a cation-π interaction with the central oxadiazole ring. It is interesting to observe that the A ring of the Cl atom participates in the halogen bonding interaction with the Thr 92 residue (C–Cl⋯O = 3.4 Å, and ∠C–Cl⋯O = 153°). All these active site residues are involved in intermolecular interactions except for residues His 82 and Ile 180 in 2. Ser 183 is in contact with one of the F atoms of the ring C. The distance between F and O is 3.9 Å and ∠C–F⋯O = 157°).
4. Conclusions
This effort involved the synthesis of two 3-arylaminomethyl N-Mannich bases and their structural characterization by a single-crystal X-ray diffraction approach. A thorough examination of the nature and strength of many types of intermolecular interactions, including hydrogen bonds, halogen bonding, unconventional F⋯O fluorine bond, and C–F⋯F–C contact. Further, the C–F⋯F–C contact exhibits a quasi-type I/II geometry. Based on the CSD analysis, the geometrical preference (type I) of C–F⋯F–C contact is quite comparable to the Cl⋯Cl contact. Moreover, the σ-hole on the Cl atom is revealed by the electrostatic potential map, and it functions as a donor for two distinct kinds of halogen bonds (Cl⋯Cl in compound 1 and Cl⋯π in compound 2). The former contact exhibits L-shaped type II geometry, while the latter contact adopts type III linear geometry. Cl⋯π halogen bond-mediated dimer is two-fold stronger than one generated by a Cl⋯Cl halogen bond, according to dimerization energy calculations. Cooperativity with other interactions, such as C–H⋯S, N–H⋯S interactions, and unconventional F⋯O fluorine bond, is one of the common characteristics shared by C–H⋯F hydrogen bonds. Furthermore, the in vitro experiments showed that compounds 1 and 2 displayed antibacterial activity against all tested Gram-positive bacterial strains and marginal activity against E. coli. In addition, compound 1 showed weak inhibition activity against Candida albicans, while compound 2 was completely inactive. This result suggests the role of substituent groups, especially trifluorophenyl and di-substituted fluorophenyl, in the observed biological activity. We also showed that the title compounds had antiproliferative activities against different cancer cell lines. Molecular docking analysis indicated that both compounds could establish intermolecular interactions with active site residues of bacterial and cancer protein targets. We also noted that halogen atoms (Cl and F) are found to be involved in the intermolecular interactions with the active site residues, indicating that these halogens play a significant role in the observed bioactivity. Solid-state interactions and their strength and bioactivity help optimize the ligand molecules to achieve better antibacterial and anticancer activity while retaining the triazole core.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project No. (PNURSP2023R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. ST acknowledges the Department of Biotechnology (DBT), Government of India (grant number: BT/PR40144/BTIS/137/46/2022) and SASTRA Deemed University for the financial support towards the purchase of the Schrödinger software suite.References
- (a) K. Rana and J. K. Sahu, Curr. Drug Res. Rev., 2021, 13, 90–100 CrossRef CAS; (b) N. Desai, J. Monapara, A. Jethawa, V. Khedkar and B. Shingate, Arch. Pharm., 2022, 355, 2200123 CrossRef CAS; (c) J.-J. Wang, W. Sun, W.-D. Jia, M. Bian and L.-J. Yu, J. Enzyme Inhib. Med. Chem., 2022, 37, 2304–2319 CrossRef.
- M. Ogata, H. Atobe, H. Kushida and K. Yamamoto, J. Antibiot., 1971, 24, 443–451 CrossRef CAS.
- D. R. Shepard and R. Dreicer, Expert Opin. Invest. Drugs, 2010, 19, 899–908 CrossRef CAS.
- K. C. Rees, Z. Dou and D. C. Whitehead, ACS Infect. Dis., 2022, 8, 911–917 CrossRef CAS.
- V. Summa, A. Petrocchi, F. Bonelli, B. Crescenzi, M. Donghi, M. Ferrara, F. Fiore, C. Gardelli, O. Gonzalez Paz, D. J. Hazuda, P. Jones, O. Kinzel, R. Laufer, E. Monteagudo, E. Muraglia, E. Nizi, F. Orvieto, P. Pace, G. Pescatore, R. Scarpelli, K. Stillmock, M. V Witmer and M. Rowley, J. Med. Chem., 2008, 51, 58435855 CrossRef.
- (a) S. Bajaj, M. S. Kumar, H. Tinwala and M. YC, Bioorg. Chem., 2021, 111, 104873 CrossRef CAS; (b) Z. M. M. Alzhrani, M. M. Alam, T. Neamatallah and S. Nazreen, J. Enzyme Inhib. Med. Chem., 2020, 35, 1116–1123 CrossRef CAS; (c) S. A. Shahzad, M. Yar, M. Bajda, L. Shahzadi, Z. A. Khan, S. A. R. Naqvi, S. Mutahir, N. Mahmood and K. M. Khan, Bioorg. Chem., 2015, 60, 37–41 CrossRef CAS.
- M. M. Alam, S. Nazreen, A. S. A. Almalki, A. A. Elhenawy, N. I. Alsenani, S. E. I. Elbehairi, A. M. Malebari, M. Y. Alfaifi, M. A. Alsharif and S. Y. M. Alfaifi, Pharmaceuticals, 2021, 14, 870 CrossRef.
- M. Hagras, M. A. Saleh, R. R. Ezz Eldin, A. A. Abuelkhir, E. G. Khidr, A. A. El-Husseiny, H. A. El-Mahdy, E. B. Elkaeed and I. H. Eissa, J. Enzyme Inhib. Med. Chem., 2022, 37, 380–396 Search PubMed.
- (a) S. Valente, D. Trisciuoglio, T. De Luca, A. Nebbioso, D. Labella, A. Lenoci, C. Bigogno, G. Dondio, M. Miceli, G. Brosch, D. Del Bufalo, L. Altucci and A. Mai, J. Med. Chem., 2014, 57, 6259–6265 CrossRef CAS; (b) V. R. Pidugu, N. S. Yarla, A. Bishayee, A. M. Kalle and A. K. Satya, Apoptosis, 2017, 22, 1394–1403 CrossRef.
- M. C. Tuma, A. Malikzay, X. Ouyang, D. Surguladze, J. Fleming, S. Mitelman, M. Camara, B. Finnerty, J. Doody, E. L. P. Chekler, P. Kussie and J. R. Tonra, Transl. Oncol., 2010, 3, 318–325 CrossRef.
- J. Sun, M.-H. Li, S.-S. Qian, F.-J. Guo, X.-F. Dang, X.-M. Wang, Y.-R. Xue and H.-L. Zhu, Bioorg. Med. Chem. Lett., 2013, 23, 2876–2879 CrossRef CAS.
- M. D. Altıntop, B. Sever, G. Akalın Çiftçi, G. Turan-Zitouni, Z. A. Kaplancıklı and A. Özdemir, Eur. J. Med. Chem., 2018, 155, 905–924 CrossRef.
- (a) S. Bajaj, P. P. Roy and J. Singh, Adv. Anticancer Agents Med. Chem., 2017, 17, 1869–1883 CAS; (b) J. Sun, H. Zhu, Z.-M. Yang and H.-L. Zhu, Eur. J. Med. Chem., 2013, 60, 23–28 CrossRef CAS.
- C. D. Mohan, N. C. Anilkumar, S. Rangappa, M. K. Shanmugam, S. Mishra, A. Chinnathambi, S. A. Alharbi, A. Bhattacharjee, G. Sethi, A. P. Kumar, Basappa and K. S. Rangappa, Front. Oncol., 2018, 8, 42 CrossRef.
- E. Mitsiki, A. C. Papageorgiou, S. Iyer, N. Thiyagarajan, S. H. Prior, D. Sleep, C. Finnis and K. R. Acharya, Biochem. Biophys. Res. Commun., 2009, 386, 666–670 CrossRef CAS.
- (a) Y.-Y. Wu, W.-B. Shao, J.-J. Zhu, Z.-Q. Long, L.-W. Liu, P.-Y. Wang, Z. Li and S. Yang, J. Agric. Food Chem., 2019, 67, 13892–13903 CrossRef CAS; (b) Z. Yang, Y. Sun, Q. Liu, A. Li, W. Wang and W. Gu, J. Agric. Food Chem., 2021, 69, 13373–13385 CrossRef CAS.
- (a) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS; (b) J. Han, L. Kiss, H. Mei, A. M. Remete, M. Ponikvar-Svet, D. M. Sedgwick, R. Roman, S. Fustero, H. Moriwaki and V. A. Soloshonok, Chem. Rev., 2021, 121, 4678–4742 CrossRef CAS; (c) J. He, Z. Li, G. Dhawan, W. Zhang, A. E. Sorochinsky, G. Butler, V. A. Soloshonok and J. Han, Chinese Chem. Lett., 2023, 34, 107578 CrossRef CAS.
- (a) L. H. Al-Wahaibi, A. A. B. Mohamed, S. S. Tawfik, H. M. Hassan and A. A. El-Emam, Molecules, 2021, 26, 2110 CrossRef CAS; (b) A. A. El-Emam, O. A. Al-Deeb, M. Al-Omar and J. Lehmann, Bioorg. Med. Chem., 2004, 12, 5107–5113 CrossRef CAS.
- (a) L. H. Al-Wahaibi, A. Alsfouk, A. A. El-Emam and O. Blacque, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 611–615 CrossRef CAS; (b) L. H. Al-Wahaibi, N. S. Kumar, A. A. El-Emam, N. S. Venkataramanan, H. A. Ghabbour, A.-M. S. Al-Tamimi, J. Percino and S. Thamotharan, J. Mol. Struct., 2019, 1175, 230–240 CrossRef CAS.
- L. Ma, Y. Xiao, C. Li, Z.-L. Xie, D.-D. Li, Y.-T. Wang, H.-T. Ma, H.-L. Zhu, M.-H. Wang and Y.-H. Ye, Bioorg. Med. Chem., 2013, 21, 6763–6770 CrossRef CAS.
- (a) A.-M. S. Al-Tamimi, O. A. Al-Deeb, A. A. El-Emam, S. W. Ng and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o729 CrossRef CAS; (b) A.-M. S. Al-Tamimi, A. M. Alafeefy, A. A. El-Emam, S. W. Ng and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o730 CrossRef CAS; (c) M. A. Al-Alshaikh, H. A. Ghabbour, A.-M. S. Al-Tamimi, M. S. M. Abdelbaky, S. Garcia-Granda and A. A. El-Emam, Z. Kristallogr.–New Cryst. Struct., 2016, 231, 301–303 CrossRef CAS; (d) R. I. Al-Wabli, N. A. El-Emam, H. A. Ghabbour, N. G. Haress and A. A. El-Emam, Z. Kristallogr.–New Cryst. Struct., 2016, 231, 815–817 CrossRef CAS; (e) L. H. Al-Wahaibi, H. A. Ghabbour, F. A. M. Al-Omary, E. R. T. Tiekink and A. A. El-Emam, Z. Kristallogr.–New Cryst. Struct., 2022, 237, 587–591 CrossRef CAS.
- (a) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS; (b) G. R. Desiraju, Chem. Commun., 2005, 2005, 2995–3001 RSC; (c) G. Bulusu and G. R. Desiraju, J. Indian Inst. Sci., 2020, 100, 31–41 CrossRef.
- Z. S. Derewenda, Int. J. Mol. Sci., 2023, 24, 13165 CrossRef CAS.
- (a) C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef CAS; (b) G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS; (c) A. Varadwaj, P. R. Varadwaj, H. M. Marques and K. Yamashita, Inorganics, 2022, 10, 149 CrossRef CAS; (d) P. R. Varadwaj, A. Varadwaj, H. M. Marques and K. Yamashita, CrystEngComm, 2023, 25, 1411–1423 RSC.
- (a) J. Badorrek and M. Walter, J. Comput. Chem., 2022, 43, 340–348 CrossRef CAS; (b) M. Hajji, N. Abad, M. A. Habib, S. M. H. Elmgirhi and T. Guerfel, J. Indian Chem. Soc., 2021, 98, 100208 CrossRef CAS; (c) M. Hajji, H. Mtiraoui, N. Amiri, M. Msaddek and T. Guerfel, Int. J. Quantum Chem., 2019, 119, e26000 CrossRef.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef.
- O. V Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef.
- G. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, 2017.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef CAS; (b) H. L. Schmider and A. D. Becke, J. Chem. Phys., 1998, 108, 9624–9631 CrossRef CAS; (c) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14(27–28), 33–38 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- G. L. Woods and J. A. Washington, Antibacterial susceptibility tests: dilution and disk diffusion methods, in Manual of Clinical Microbiology, ed. P. R. Murray, E. J. Baron, M. A. Pfaller, F. C. Tenover and R. H. Yolken, American Society of Microbiology, Washington, DC, USA, 1995 Search PubMed.
- I. Wiegand, K. Hilpert and R. E. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- R. A. Norman, S. T. Barry, M. Bate, J. Breed, J. G. Colls, R. J. Ernill, R. W. A. Luke, C. A. Minshull, M. S. B. McAlister, E. J. McCall, H. H. J. McMiken, D. S. Paterson, D. Timms, J. A. Tucker and R. A. Pauptit, Structure, 2004, 12, 75–84 CrossRef CAS.
- C. Pozzi, S. Ferrari, R. Luciani, G. Tassone, M. P. Costi and S. Mangani, Molecules, 2019, 24, 1257 CrossRef.
- C. Lu, C. Wu, D. Ghoreishi, W. Chen, L. Wang, W. Damm, G. A. Ross, M. K. Dahlgren, E. Russell, C. D. Von Bargen, R. Abel, R. A. Friesner and E. D. Harder, J. Chem. Theory Comput., 2021, 17, 4291–4300 CrossRef CAS.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS.
- M. F. Adasme, K. L. Linnemann, S. N. Bolz, F. Kaiser, S. Salentin, V. J. Haupt and M. Schroeder, Nucleic Acids Res., 2021, 49, W530–W534 CrossRef CAS.
- I. Saraogi, B. H. M. Mruthyunjayaswamy, O. B. Ijare, Y. Jadegoud and T. N. Guru Row, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, o1341–o1342 CrossRef CAS.
- I. J. Bruno, J. C. Cole, M. Kessler, J. Luo, W. D. S. Motherwell, L. H. Purkis, B. R. Smith, R. Taylor, R. I. Cooper, S. E. Harris and A. G. Orpen, J. Chem. Inf. Comput. Sci., 2004, 44, 2133–2144 CrossRef CAS.
- L. H. Al-Wahaibi, B. Rahul, A. A. B. Mohamed, M. S. M. Abdelbaky, S. Garcia-Granda, A. A. El-Emam, M. J. Percino and S. Thamotharan, ACS Omega, 2021, 6, 6996–7007 CrossRef CAS.
- P. R. Varadwaj, A. Varadwaj and H. M. Marques, Crystals, 2020, 10, 146 CrossRef CAS.
- (a) M. Hajji, J. S. Al-Otaibi, M. Belkhiria, S. Dhifaoui, M. A. Habib, S. M. H. Elmgirhi, H. Mtiraoui, R. Bel-Hadj-Tahar, M. Msaddek and T. Guerfel, J. Mol. Struct., 2021, 1223, 128998 CrossRef CAS; (b) M. J. Calhorda, Chem. Commun., 2000, 801–809 RSC; (c) M. Hajji, A. Haouas, N. Abad and T. Guerfel, ChemistrySelect, 2023, 8, e202302624 CrossRef CAS.
- M. A. A. Ibrahim, R. R. A. Saeed, M. N. I. Shehata, M. N. Ahmed, A. M. Shawky, M. M. Khowdiary, E. B. Elkaeed, M. E. S. Soliman and N. A. M. Moussa, Int. J. Mol. Sci., 2022, 23, 3114 CrossRef CAS.
- S. Tothadi, S. Joseph and G. R. Desiraju, Cryst. Growth Des., 2013, 13, 3242–3254 CrossRef CAS.
- D. F. Mertsalov, R. M. Gomila, V. P. Zaytsev, M. S. Grigoriev, E. V Nikitina, F. I. Zubkov and A. Frontera, Crystals, 2021, 11, 1406 CrossRef CAS.
- C. Gatti, Z. Kristallogr. - Cryst. Mater., 2005, 220, 399–457 CrossRef CAS.
- (a) L. Singla, A. Kumar, C. M. Robertson, P. Munshi and A. Roy Choudhury, Cryst. Growth Des., 2023, 23, 853–861 CrossRef CAS; (b) D. Chopra, T. S. Cameron, J. D. Ferrara and T. N. Guru Row, J. Phys. Chem. A, 2006, 110, 10465–10477 CrossRef CAS; (c) V. R. Hathwar and T. N. Guru Row, Cryst. Growth Des., 2011, 11, 1338–1346 CrossRef CAS; (d) M. S. Pavan, K. Durga Prasad and T. N. Guru Row, Chem. Commun., 2013, 49, 7558–7560 RSC; (e) D. E. Hibbs, J. Overgaard, J. A. Platts, M. P. Waller and M. B. Hursthouse, J. Phys. Chem. B, 2004, 108, 3663–3672 CrossRef CAS.
- I. Wiegand, K. Hilpert and R. E. W. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2302018 and 2302019. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra07169c |
| This journal is © The Royal Society of Chemistry 2023 |