
In situ copper nanoparticle immobilization on the indigo carmine-functionalized chitosan: a versatile biocatalyst towards CO2 fixation and click reactions in water†
Vida
Khodkari
a,
Mohammad Taghi
Nazeri‡
a,
Siamak
Javanbakht‡
a and
Ahmad
Shaabani
 *ab
*ab
aDepartment of Organic Chemistry, Shahid Beheshti University, Daneshjou Boulevard Street, Tehran, 1983969411, Iran. E-mail: a-shaabani@sbu.ac.ir; Tel: +982129902800
bPeoples' Friendship University of Russia (RUDN University), 6, Miklukho-Maklaya Street, Moscow, 117198, Russian Federation
First published on 29th September 2022
Abstract
For catalyst construction, biopolymeric chitosan has attracted the interest of many researchers due to its biodegradability and economic viability. Accordingly, this study utilized chitosan as a green support to produce a versatile heterogeneous catalytic system. Chitosan was first functionalized with indigo carmine (IC) and then metal nanoparticles (i.e., Cu2O and CuO) were immobilized on the IC-functionalized chitosan (IC-CS). The successful synthesis process was fully confirmed using different techniques such as SEM, FT-IR, EDX, XRD, TGA, and DTG. After ensuring catalyst fabrication, the catalytic activities of Cu2O/IC-CS and CuO/IC-CS were investigated for the oxidation state of copper in the click reaction and CO2 fixation, respectively. The obtained results revealed the application of these biocatalysts in the above-mentioned reactions with water as a green solvent in the shortest time with high efficiency and selectivity for the desired products.
1. Introduction
In synthetic chemistry, catalytic organic transformations as key methods have received great interest.1–3 For example, coupling and click reactions as the key tools have received significant interest.4–6 They include an extensive range of synthetic compounds, ranging from the manufacturing of pharmaceuticals and industrial materials to the construction of natural products and organic building blocks.7,8 Moreover, controlling the amount of greenhouse gases in the atmosphere has become a critical scientific, industrial, and technological priority, and this topic has encouraged the catalytic chemistry research society to develop new ways to capture, use, and store the CO2 gas. Specifically, in recent years, the catalytic fixation of CO2 and the possibility of its transformation into value-added products (substituted urea, formic acid, dimethyl carbonate, etc.) has attracted tremendous attention worldwide. Although different metals are used for catalytic organic transformations, copper is commonly used in this contribution. In fact, copper is economical, less toxic, and more abundant in terms of economic viability and sustainability in comparison with other metals. Thus, the use of copper catalysts is rapidly growing for a variety of catalytic organic reactions.9,10 On the other hand, the productivity and stability of copper catalysts in the presence of biopolymeric ligands could be well enhanced under mild circumstances.11–13Among different biopolymers, chitosan as a naturally appearing polysaccharide, has attracted massive interest in different fields for instance, textile industry,14 water treatment,15 optical and electronic devices,16 drug delivery17 and catalysis.18 The existence of amine functional groups in the chitosan structure provides a special chance for the absorption of metal ions by chelating and accordingly eliminating poisonous metals from aquatic media.19 Despite more investigations on chitosan for heavy metal removal from environmental pollutants, a high number of reports have recently utilized chitosan in efficient support catalysis.20,21 To date, a variety of transition metals for instance silver,22 copper,23 palladium,24 platinum and iridium25,26 were supported on chitosan and applied as a heterogeneous biocatalyst to organize numerous organic transformations. In this regard, quite a few reports provided acceptable results considering green chemistry and environmental principles.20,27–33 Owing to the low catalytic efficiency of pristine chitosan and the absence of some functions in its structure that are essential for powerful metal ion chelation, there is considerable metal ion leaching in the reaction media.34 Subsequently, this causes environmental contamination, which also reduces the recyclability of the catalyst. To solve this problem, the post-modification method has been reported for utilizing the full capability of this biopolymer.34,35
In this contribution, chitosan was typically modified with the great potential coordinative functional groups to effectively anchor different metal ions. In recent years, significant signs of progress have been made on the functionalization of biopolymers through a variety of strategies to improve and enhance the coordination potency of metal ions.36–40 Although considerable development has taken place in this aspect, this opinion still is a great challenge to ideally follow the green chemistry merits, i.e., prevention, less hazardous chemical syntheses, atom economy, harmless solvents, reducing derivatives, designing safer chemicals for energy productivity, design for degradation, use of renewable feedstocks, real-time analysis for preventing contamination, etc.
With this viewpoint and concerning the shortcomings noted for chitosan as an ideal-supported catalyst and also in the continuance of our continuing interest for catalytic synthesis of methodologies;41–44 herein, this work utilized a superior electrostatic interaction for functionalization of chitosan with indigo carmine (IC) through a new simple strategy and accordingly improving its ligation towards metal ions/nanoparticles. IC as a typical recalcitrant dye is commonly used in textile dyeing processes. Metal ligand-able active sites in the structure of IC with the high affinity to immobilize copper nanoparticles, i.e., Cu2O and CuO, enable IC-functionalized chitosan (IC-CS) as a novel supported catalyst. This biocatalyst promoted various organic transformations such as CO2 fixation and click reactions in water, indicating excellent activity under milder and sustainable conditions without metal leaching and considerable loss of activity.
2. Experimental
2.1. Materials and methods
Indigo carmine, chitosan, ascorbic acid, copper(II) chloride formula (CuCl2·2H2O), and other chemicals, reagents, and solvents used in this study were obtained from Merck Co. and utilized without further purification. An Aquinox 55 Bruker Instrument was used for collecting the Fourier Transform Infrared (FT-IR) spectra of the materials. The crystal structure of synthesized materials was studied using X-Ray diffraction (XRD, Siemens diffractometer, Silicon Dirft 2017, USA, Cu-Kα radiation at 35 kV). Scanning electron microscope (SEM) images were captured using the ESEM QUANTA 200 model (USA). Thermogravimetric analysis (TGA) analysis was performed using the STA 1500 device at a heating rate of 10 °C min−1 under a nitrogen atmosphere and a flow rate of 50 mL min−1 using an alumina sample holder. Inductively coupled plasma optical emission spectroscopy (ICP-OES) was used to determine the amount of copper in the catalyst.2.2. Synthesis of catalysts
2.3. General procedure for the catalytic synthesis of 1,2,3-triazole compounds
In a general procedure, sodium azide, alkynes, benzyl bromide derivatives, and the desired amounts of catalyst were added to distilled water. The resultant mixture was heated and treated with an ultrasonic batch at 60 °C. The progress of the catalytic reaction was controlled using thin-layer chromatography (TLC) and n-hexane/ethyl acetate as a chromatogram solvent. At the end of the reaction, the catalyst was separated by filtering after cooling the reaction to ambient temperature, followed by the product being extracted using chloroform to reach a pure product.2.4. General procedure for the catalytic synthesis of cyclic carbonates (method A)
In a general procedure, desired amounts of biocatalyst, tetrabutylammonium bromide (TBAB) as a co-catalyst, and epoxide were added to water as a solvent. Then, CO2 was added to the reaction vessel using a balloon with a pressure of 1 bar and treated at 80 °C. After 2 h, the reaction mixture was cooled down to ambient temperature, and unreacted CO2 was slowly released. Finally, the biocatalyst was washed several times with ethyl acetate and then the supernatant was evaporated under a vacuum. The obtained crude products were purified using column chromatography on silica gel (n-hexane/EtOAc) to reach a cyclic carbonate.2.5. General procedure for the catalytic synthesis of cyclic carbonates (method B)
A stainless-steel reactor (100 mL) was used for the CO2 fixation reactions. In a general procedure, desired amounts of the synthesized catalyst, tetrabutylammonium bromide (TBAB), and epoxide compounds were added to the reactor. After sealing the reactor, CO2 was charged inside it until the pressure reached the desired value (2 and 3 bar) at a suitable temperature (80 and 90 °C). After completing the reaction, the reactor was allowed to cool to ambient temperature, then, the unreacted CO2 was slowly released. The biocatalyst was washed several times with ethyl acetate and then the supernatant was evaporated under a vacuum. The obtained crude product was purified using column chromatography on silica gel (n-hexane/EtOAc) to reach a cyclic carbonate.3. Results and discussion
Immobilizing metal nanoparticles on different supported biopolymers via green and simple methods has recently attracted considerable attention.46 Among different biopolymers, chitosan, the deacetylated form of chitin, is the suitable supported catalyst for metal nanoparticle immobilization. Particularly, the hydroxy and amino functional groups on the chitosan surface trigger a high potential for the adsorption of an extensive range of molecules, including metal ions. However, the significant leaching of metal ions from unfunctionalized chitosan into the catalytic reaction medium encourages us to modify it through a novel strategy with high potential ligand-able functional groups, i.e., indigo carmine molecules for preventing the mentioned disadvantages. The synthesized IC-CS was then used for copper nanoparticle immobilization in two different paths. Cu2O and CuO NPs were in situ synthesized by loading Cu2+ ions onto the IC-CS and followed by treating them with ascorbic acid and NaOH solutions to synthesize Cu2O/IC-CS and CuO/IC-CS, respectively (Scheme 1).47 To study the chemical and physical structure of constructed materials and as well as their catalytic activity towards CO2 fixation and click reactions following characterizations were performed.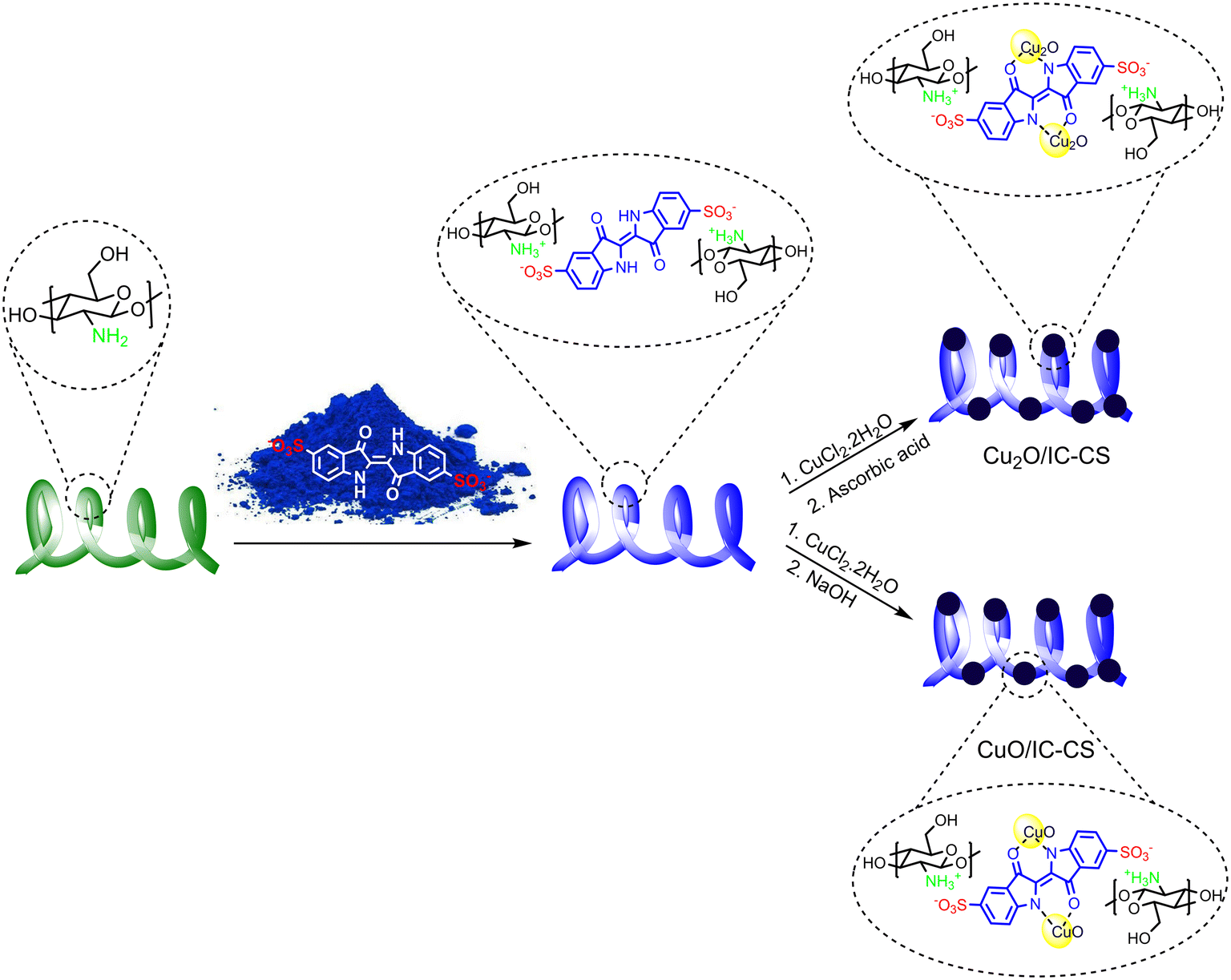 | ||
| Scheme 1 The synthesis routes of Cu2O/IC-CS and CuO/IC-CS. | ||
3.1. FT-IR analysis
To understand the bonding structure of the materials, FT-IR spectra of IC, CS, IC-CS, Cu2O/IC-CS, and CuO/IC-CS were recorded (Fig. 1). The IC FT-IR spectrum showed bands around 3450 cm−1 and 3380–3800 cm−1 are related to the N–H and O–H stretching vibrations with hydrogen bonds of intermolecular and intramolecular, respectively. Also, an absorption spectrum with a less intense peak around 1600 cm−1 and another more intense at 1410 cm−1 were observed and attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC stretching vibrations, respectively.48 In the CS FT-IR spectrum, broad multiple peaks can be seen between 3200–3700 cm−1 related to the stretching of –OH and –NH2 groups with intermolecular and intramolecular hydrogen bonds. The polysaccharide skeleton indicating –C–O– and –C–N– stretching frequencies appeared in the range of 1300–1450 cm−1 and 1000–1200 cm−1, respectively.49 Also, the peak at about 2920 cm−1 was observed due to the C–H stretching. The IC-CS FT-IR spectrum showed all the indicator absorption peaks present in the IC and CS spectra, indicating the formation of an ion-pair composition. Outstandingly, the intensity of the characteristic peaks of C–H stretching at 2800 and 2900 cm−1 was increased after the formation of an ionic bond between CS and IC. The spectra of IC-CS and the Cu2O/IC-CS and CuO/IC-CS heterogeneous biocatalyst were similar with a little dissimilarity. The strong characteristic absorption band at 599 cm−1 due to the Cu-O bond vibration confirmed the successful in situ formation of Cu2O and CuO nanoparticles within the CS polymeric network.
O and CC stretching vibrations, respectively.48 In the CS FT-IR spectrum, broad multiple peaks can be seen between 3200–3700 cm−1 related to the stretching of –OH and –NH2 groups with intermolecular and intramolecular hydrogen bonds. The polysaccharide skeleton indicating –C–O– and –C–N– stretching frequencies appeared in the range of 1300–1450 cm−1 and 1000–1200 cm−1, respectively.49 Also, the peak at about 2920 cm−1 was observed due to the C–H stretching. The IC-CS FT-IR spectrum showed all the indicator absorption peaks present in the IC and CS spectra, indicating the formation of an ion-pair composition. Outstandingly, the intensity of the characteristic peaks of C–H stretching at 2800 and 2900 cm−1 was increased after the formation of an ionic bond between CS and IC. The spectra of IC-CS and the Cu2O/IC-CS and CuO/IC-CS heterogeneous biocatalyst were similar with a little dissimilarity. The strong characteristic absorption band at 599 cm−1 due to the Cu-O bond vibration confirmed the successful in situ formation of Cu2O and CuO nanoparticles within the CS polymeric network.
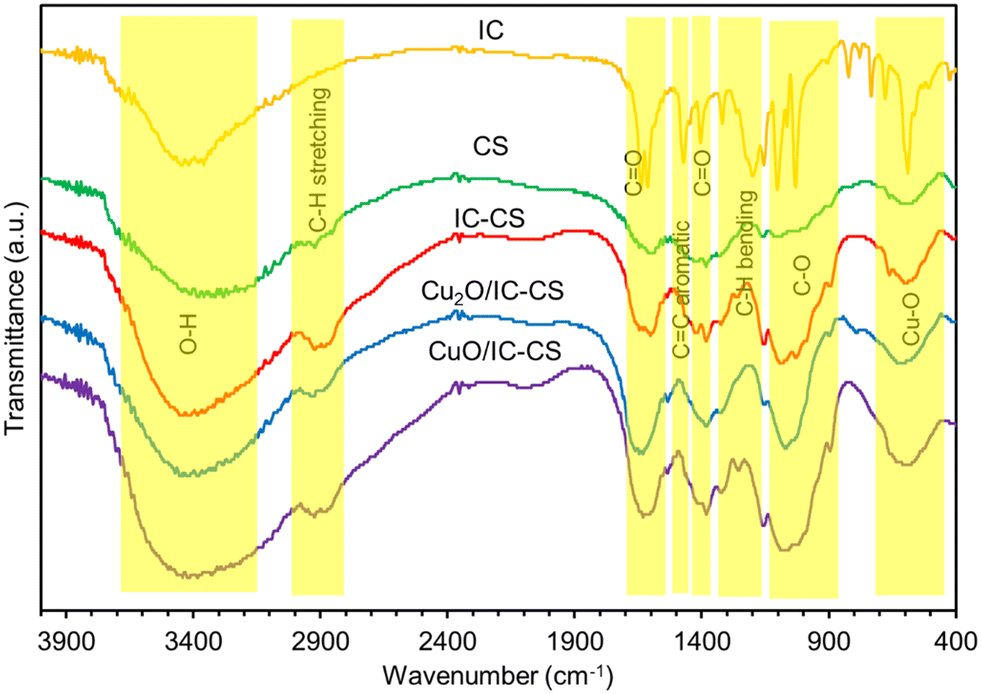 | ||
| Fig. 1 FT-IR spectra for the IC, CS, IC-CS, Cu2O/IC-CS, and CuO/IC-CS. | ||
3.2. Morphological characteristics
Fig. 2 illustrates the SEM images of Cu2O/IC-CS and CuO/IC-CS biocatalysts at the low and high magnification states. The morphologies of the prepared bio-nanocomposites (Cu2O/IC-CS and CuO/IC-CS) exhibited many cavities and wrinkles with tight and smooth levels. This is due to the surface interactions between the IC-CS chains and the Cu2O and CuO nanoparticles, which can be attributed to the intermolecular bonds represented in Scheme 1. As shown in the high magnification SEM images (Fig. 2A′ and B′), the spherical CuO and Cu2O nanoparticles of different sizes are uniformly distributed on the surface of IC-CS. These structures of Cu2O/IC-CS and CuO/IC-CS have a high tendency to adsorb the reagents and accordingly located them next to the catalytic active sites, i.e., Cu2O and CuO nanoparticles.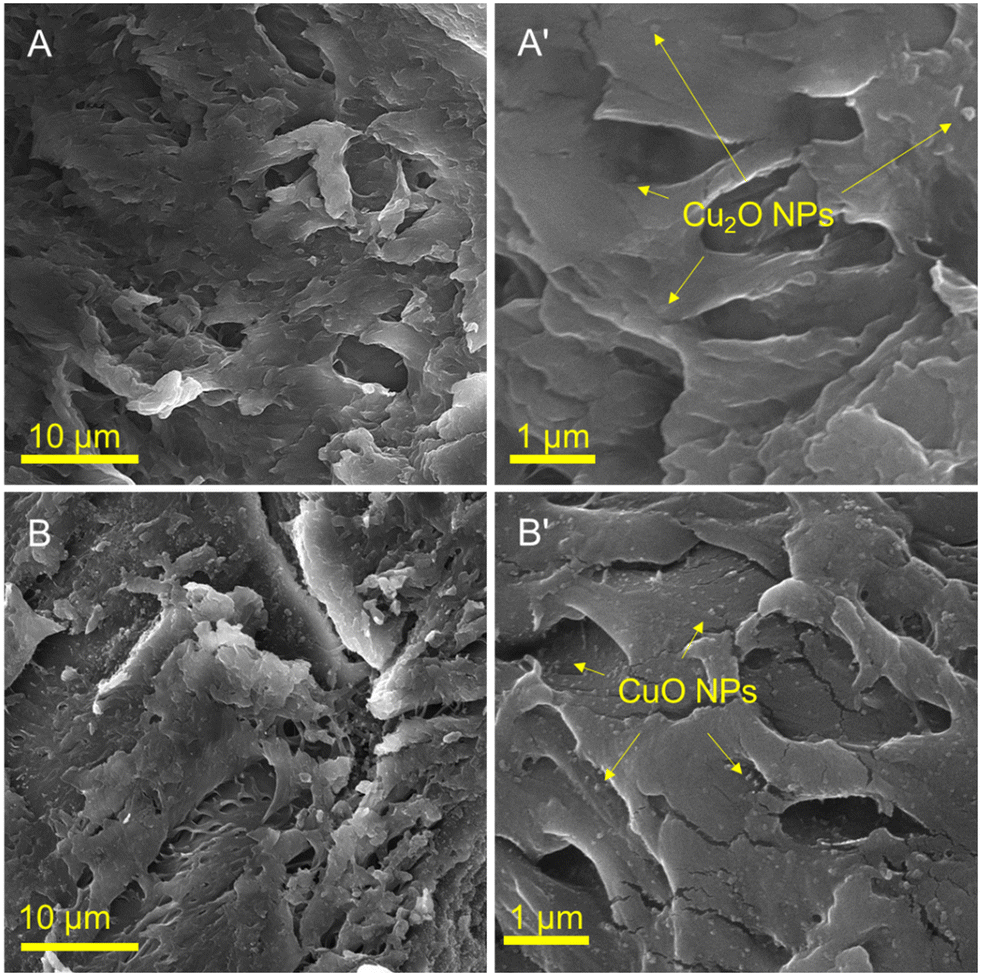 | ||
| Fig. 2 SEM images of Cu2O/IC-CS (A and A′), CuO/IC-CS (B and B′). | ||
The size and morphology of the immobilized Cu2O and CuO nanoparticles on the IC-CS were studied by TEM analysis (Fig. 3). They can be seen as spherical particles in the polymeric structure of the Cu2O/IC-CS and CuO/IC-CS bio-nanocomposite with an average size of about 52 and 92 nm, respectively (Fig. 3A′ and B′, calculated using ImageJ software).57 According to the TEM images, the immobilized Cu2O and CuO nanoparticles were well dispersed without significant aggregation over the biopolymeric chitosan matrix, indicating the efficiency of the construction procedure.
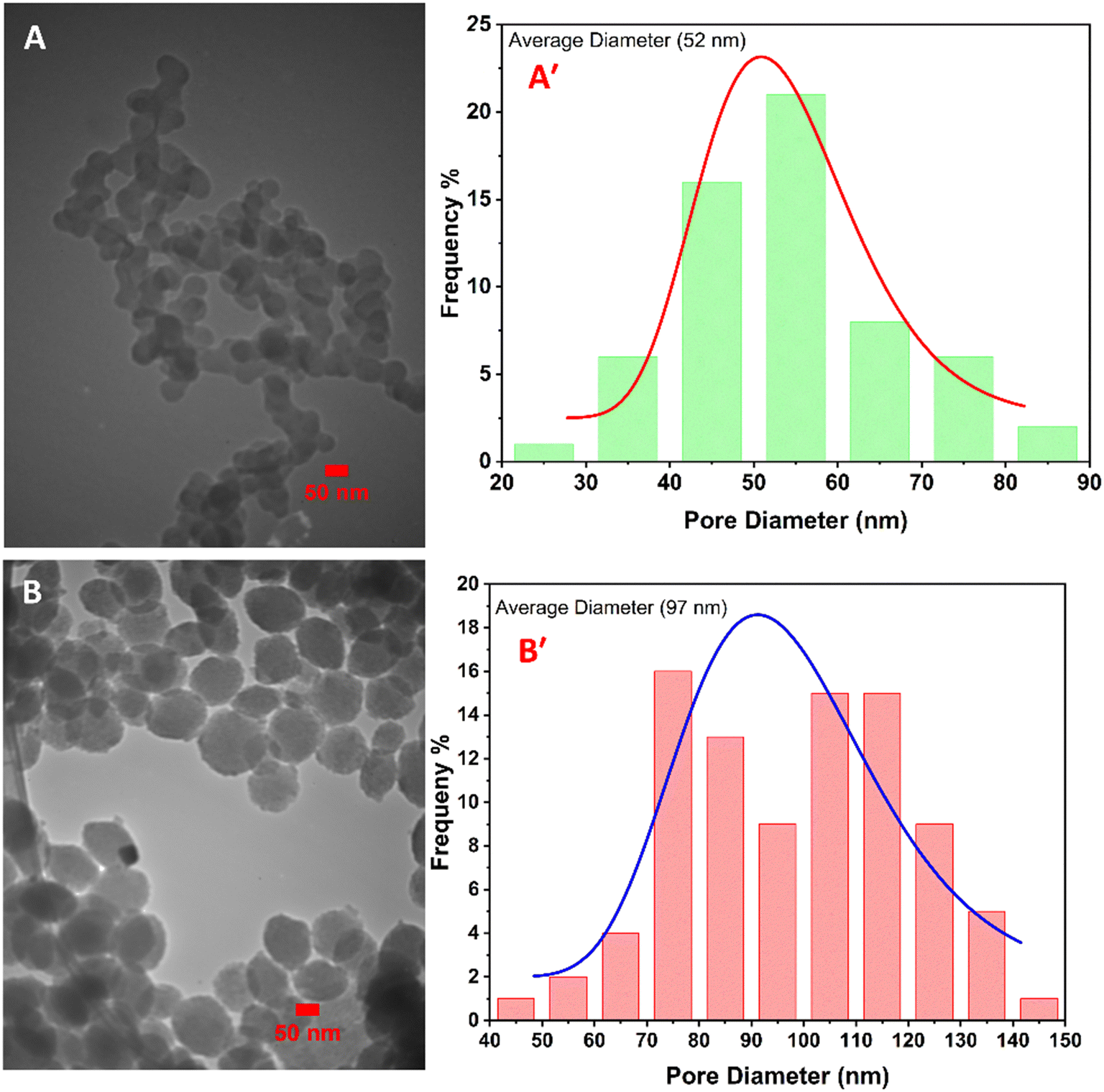 | ||
| Fig. 3 TEM images of Cu2O/IC-CS (A) and CuO/IC-CS (B). The size distribution of immobilized nanoparticles in Cu2O/IC-CS (A′) and CuO/IC-CS (B′). | ||
The element information about Cu2O/IC-CS and CuO/IC-CS biocatalysts was achieved using the EDX analysis (Fig. 4). The presence of only C, N, O, and Cu elements in the biocatalysts well confirmed the successful formation of the Cu2O and CuO nanoparticles within the IC-CS structure and as well as their purity. Furthermore, the uniform distribution of copper in the biocatalyst was confirmed from elemental mapping analysis.
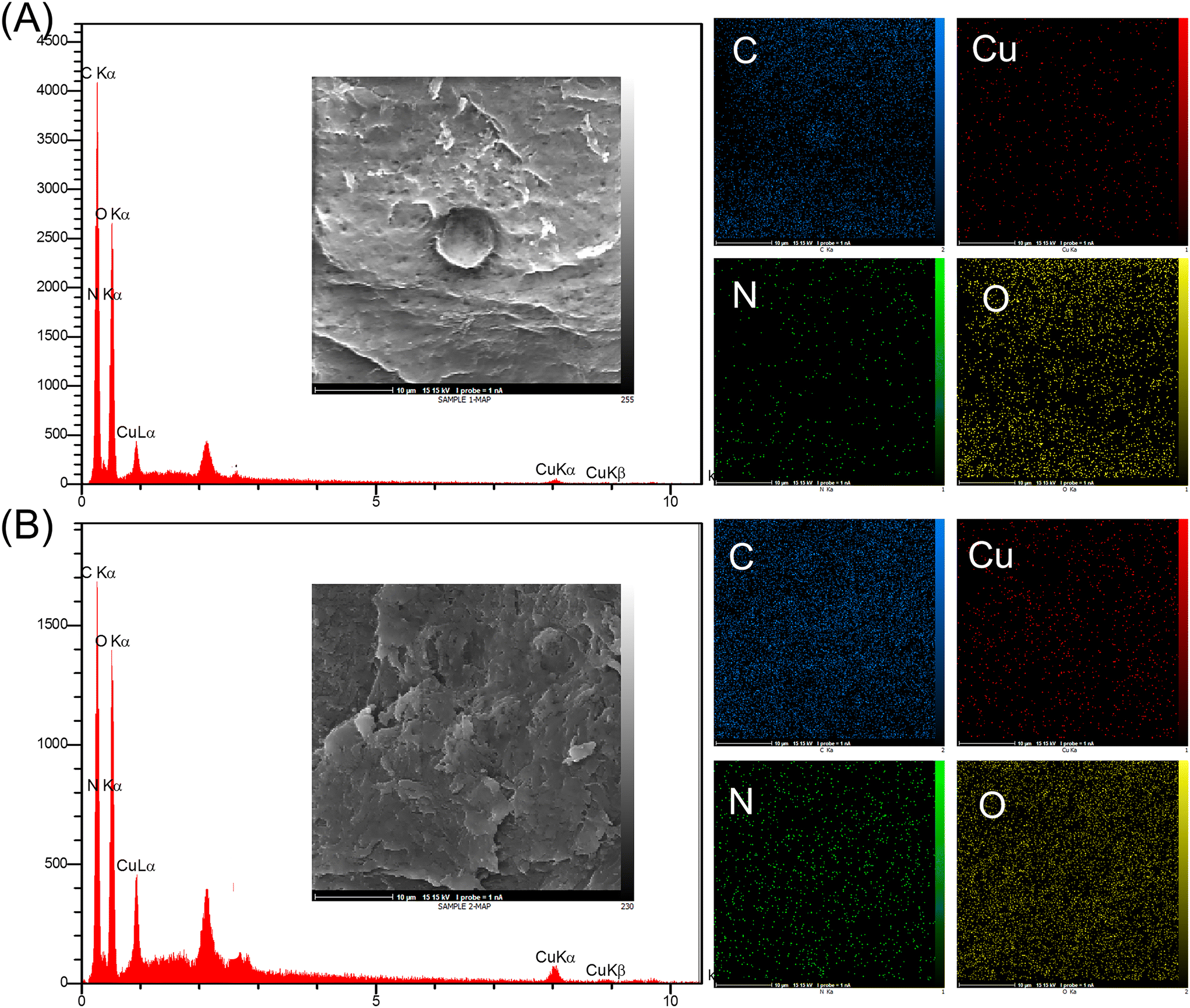 | ||
| Fig. 4 EDS spectra and elemental mapping images of Cu2O/IC-CS (A) and CuO/IC-CS (B). | ||
3.3. XRD analysis
Fig. 5 illustrates the XRD patterns of Cu2O/IC-CS and CuO/IC-CS. According to the literature,50 the pure CS exhibits the typical characteristic diffraction at 2θ values at about 11° and 20° due to the semi crystallinity of CS. In the XRD patterns of Cu2O/IC-CS and CuO/IC-CS, the typical diffraction peaks of Cu2O and CuO nanoparticles did not appear,51,52 which can be related to their homogenous generation and dispersion without aggregation over the biopolymeric chitosan matrix. However, they showed two sharp peaks at 2θ, 44.2°, and 64.6°, probably, these diffractions can be related to the IC or its complex with Cu metal ions. As the focus of this work was on the application of the prepared bio-nanocomposite as a versatile catalyst for organic transformations, we recommended further investigations to fully address this subject.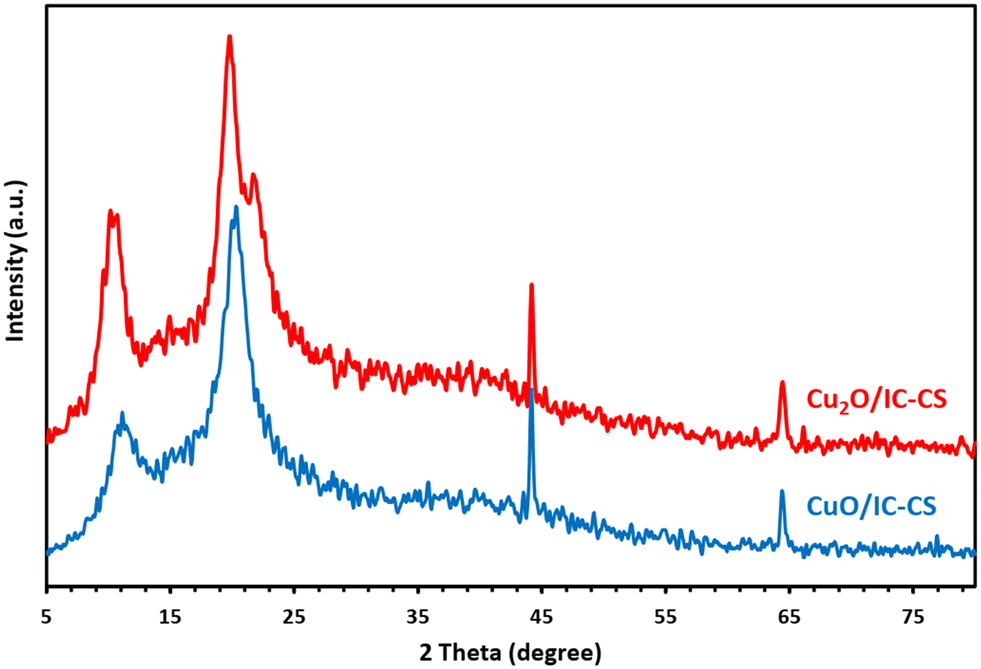 | ||
| Fig. 5 XRD patterns of Cu2O/IC-CS and CuO/IC-CS. | ||
3.4. TGA analysis
TGA studies on Cu2O/IC-CS, CuO/IC-CS, and IC were conducted to determine their thermal stabilities, and the data are shown in Fig. 6. As mentioned in previous reports, the weight loss occurred at three stages for pure CS.53,54 The first one began at 55 °C and was completed at 157 °C with about 10% weight loss due to the loss of absorbed water in the polymeric matrix. The second stage began at 243 °C and ended at 352 °C with about 52% weight loss due to the CS backbone decomposition. The third stage was initiated at 392 °C and completed at 477 °C, which may be due to the removal and vaporization of volatile products. For pure IC, weight loss occurred at three stages (Fig. 6). The first one began at 25 °C and was completed at 235 °C with about 7% weight loss due to the loss of absorbed water in IC. The second stage began at 235 °C and ended at 546 °C with about 28% weight loss due to the IC backbone decomposition. The third stage was initiated at 546 °C and completed at 800 °C with about 46% weight loss due to the removal and vaporization of volatile organic products. For both biocatalysts (Cu2O/IC-CS, CuO/IC-CS), three distinct weight losses were observed under N2 from ambient temperature to 800 °C. The first stage with 20% and 11% weight loss began at 40 °C and was completed at 159 °C, which may be attributed to the removal of water from the Cu2O/IC-CS and CuO/IC-CS, respectively. The next weight losses of 51% and 40% (140–361 °C) were related to the degradation of the polymeric CS. In the final weight loss, the residual weight after 700 °C was usually related to the inorganic content of Cu2O and CuO nanoparticles. The residual weight for the Cu2O/IC-CS and CuO/IC-CS was measured to be about 5% and 35%, respectively. Under thermal conditions, inorganic metal oxide nanomaterials typically do not decompose until their melting and reduction points.10,46,54 The residual weight for the Cu2O/IC-CS is lower compared to CuO/IC-CS, which is probably due to the reduction of Cu2O by the polymer through the thermal reduction process.10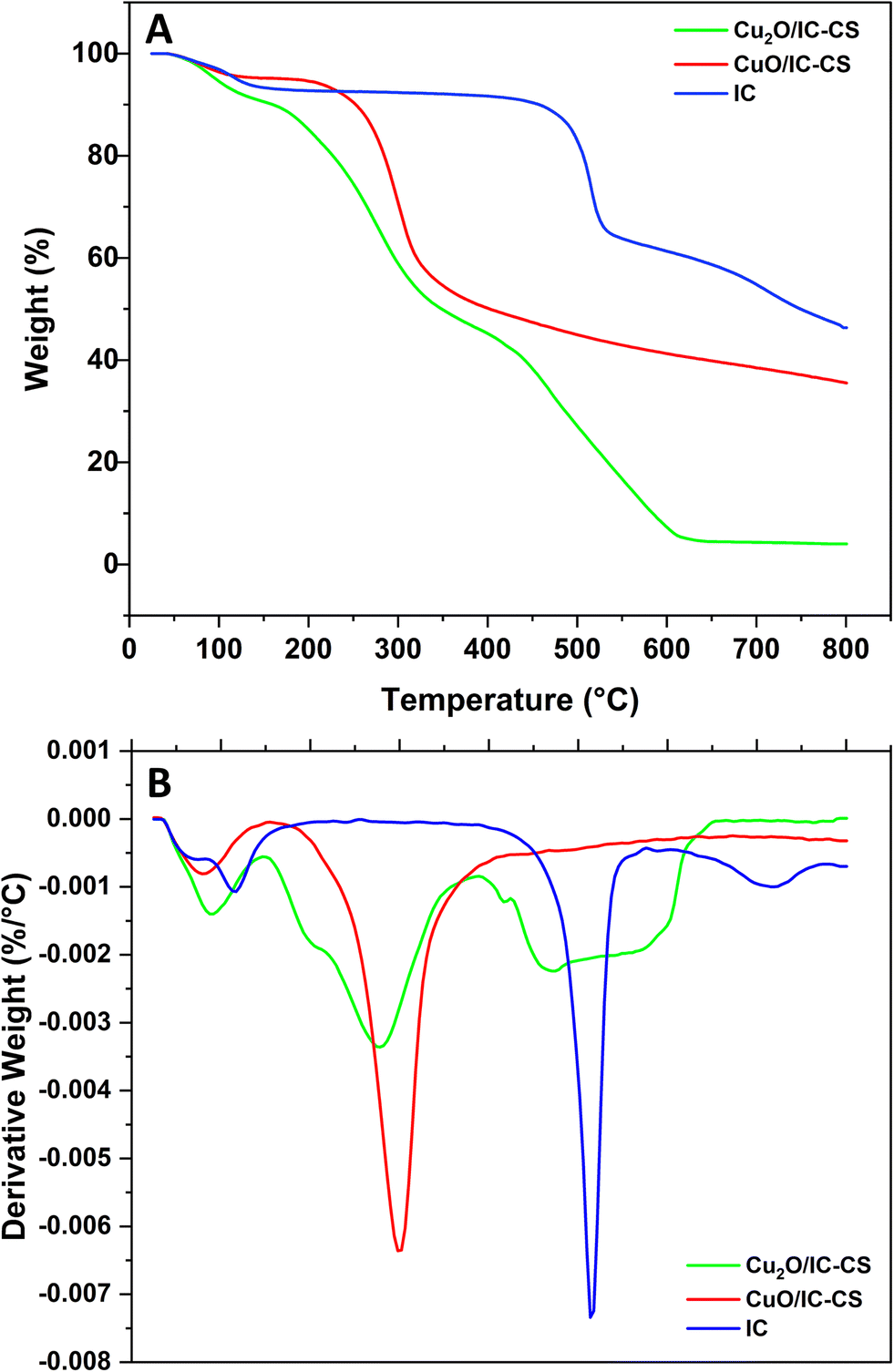 | ||
| Fig. 6 TGA (A) and DTG (B) curves of Cu2O/IC-CS and CuO/IC-CS. | ||
4. The catalytic activity of the synthesized Cu2O/IC-CS and CuO/IC-CS
To evaluate the catalytic activity of the prepared Cu2O/IC-CS biocatalyst, a click reaction was performed for the synthesis of 1,2,3-triazole compounds 3a and 3a′ (Scheme 2, path a). This reaction was selected due to the reported catalytic activity for Cu2O nanoparticles.10 After comprehensive studies of the model reaction under different conditions (Table S1, ESI†), the optimal click reaction conditions were achieved in the presence of 5 mg of Cu2O/IC-CS as a catalyst under ultrasonic irradiation in water as a green solvent at 60 °C. Under these conditions, desired products with high regioisomers and efficiency were obtained in the shortest time under green conditions. The obtained results indicated that the Cu2O/IC-CS biocatalyst was efficient in the click reaction. Also, due to the reported catalytic activity for CuO nanoparticles,46 the catalytic properties of CuO/IC-CS biocatalyst were investigated in the CO2 fixation reaction for the synthesis of cyclic carbonate 5a using epoxide 4a (Scheme 2, path b) and the results of this investigation are provided in Table S2 (ESI†). The optimal reaction conditions were attained in the presence of CuO/IC-CS and tetrabutylammonium bromide (TBAB) as a co-catalyst in water as a green solvent at 80 °C. The remarkable advantages of the current biocatalyst are a great efficiency, high CO2 fixation capacity, operational simplicity, rapid and mild reactions with the green and non-hazardous condition, and desired yields.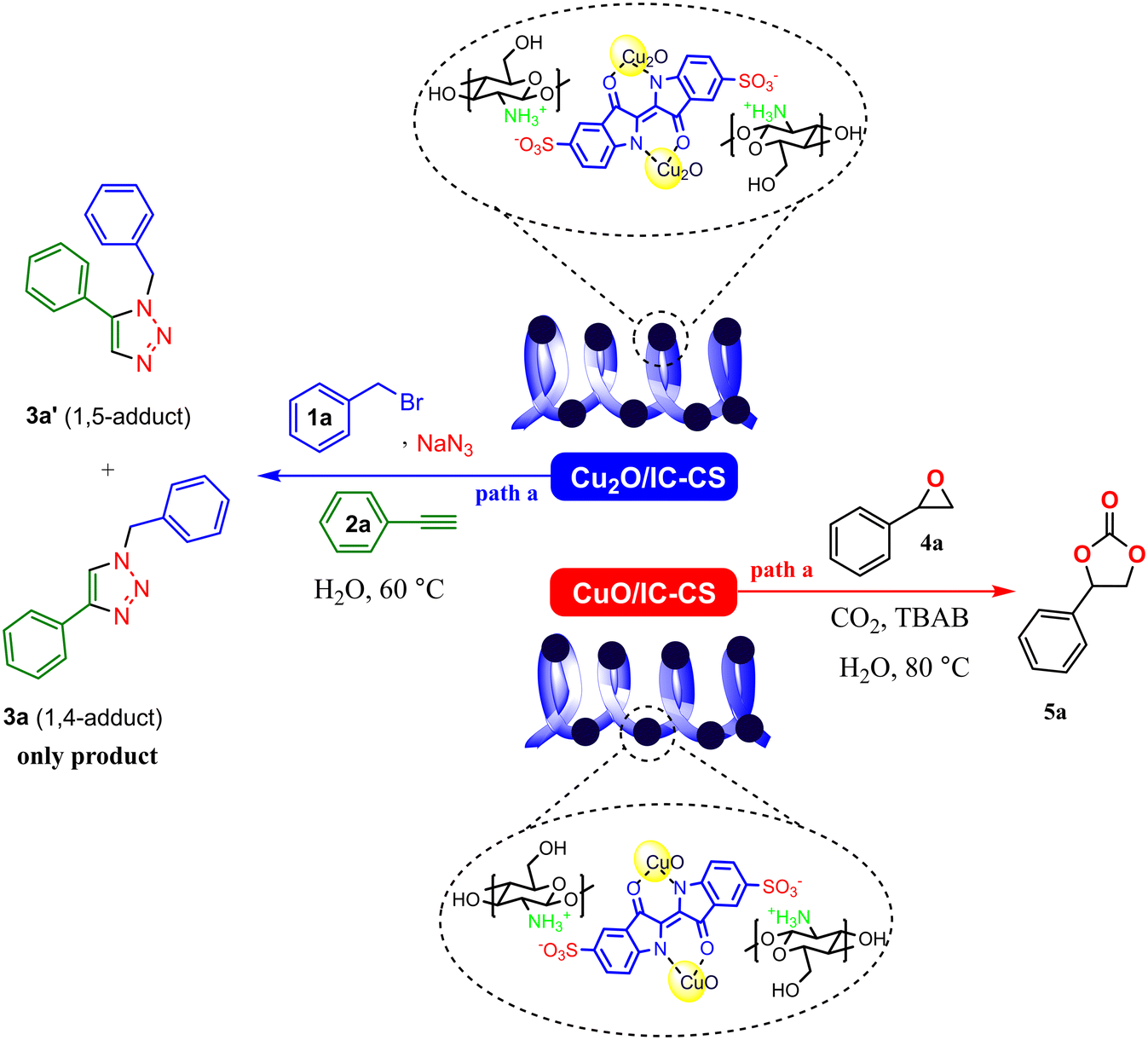 | ||
| Scheme 2 Illustration of the catalytic activities of Cu2O/IC-CS and CuO/IC-CS in the click reaction and CO2 fixation reaction, respectively. | ||
After determining the optimal conditions for the click reaction, various triazole compounds with different types of substituents in the benzyl bromide and acetylene moieties were introduced into the catalytic reaction (Table 1). It is noteworthy that this reaction was performed in the presence of Cu2O/IC-CS as a catalyst with a wide tolerance for various substituents on benzyl bromide and acetylene, which gives the desired product with high regioisomers and yield. On the other hand, when benzyl chloride and iodide were used instead of benzyl bromide, a significant difference in the reaction time was observed (Table 1, Entries 9 and 10). Considering the rate-determining step in this type of reaction, the halogen, which is a better leaving group, will have a direct effect on the time of the reaction.55 These results indicate the high efficiency of the catalyst for selecting an only isomer in the click reaction.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R 1 | R 2 | X | Yieldb (%) | TONb | TOFc (h−1) | Product |
| a Reaction conditions alkyne (1.00 mmol), sodium azide (1.00 mmol), benzyl bromide (1.00 mmol), and 5 mg Cu2O/IC-CS in water, 60 °C, 40 min, under ultrasonic irradiation. b Turn-over number (TON). c Turn-over frequency (TOF). d Reaction time 50 min. e Reaction time 20 min. | |||||||
| 1 | H | Ph | Br | 97 | 0.440 | 0.661 | 3a |
| 2 | Br | Ph | Br | 93 | 0.422 | 0.634 | 3b |
| 3 | H | C(Me)2OH | Br | 91 | 0.413 | 0.620 | 3c |
| 4 | H | MePh | Br | 93 | 0.422 | 0.634 | 3d |
| 5 | H | CH2N(Me)2 | Br | 91 | 0.413 | 0.620 | 3e |
| 6 | Br | MePh | Br | 90 | 0.409 | 0.610 | 3f |
| 7 | Br | CH2N(Me)2 | Br | 90 | 0.409 | 0.610 | 3g |
| 8 | Br | C(Me)2OH | Br | 89 | 0.404 | 0.606 | 3h |
| 9 | H | Ph | Cl | 94 | 0.427 | 0.640 | 3a |
| 10 | H | Ph | I | 97 | 0.440 | 0.661 | 3a |

To determine the productivity of the CuO/IC-CS biocatalyst, a variety of epoxides 4 were used in the chemical fixation reaction of CO2, providing the corresponding cyclic carbonates 5 with high selectivity and yield (Table 2). Both aromatic 4a–b and aliphatic 4c–f epoxides in the presence of this catalyst are good substrates for synthesizing cyclic carbonate 5a–f. 2-(Phenoxymethyl)oxirane 5b is the most reactive epoxide, while 2-butyloxirane 5f showed the lowest activity among the studied epoxies.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Epoxide | Product | Yieldb (%) | TON | TOF (h−1) |
| a Conditions of reaction: epoxide (3 mmol), CO2 (1 bar balloon), TBAB (12 mg), and 30 mg CuO/IC-CS in water, 80 °C, 2 h. b Isolated yield. | |||||
| 1 |
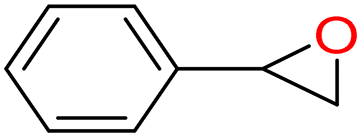 4a
4a
|
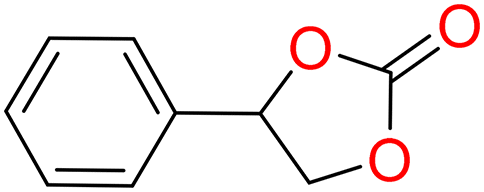 5a
5a
|
96 | 0.269 | 0.134 |
| 2 |
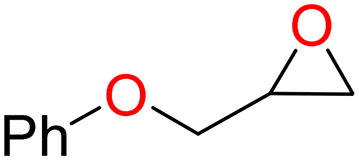 4b
4b
|
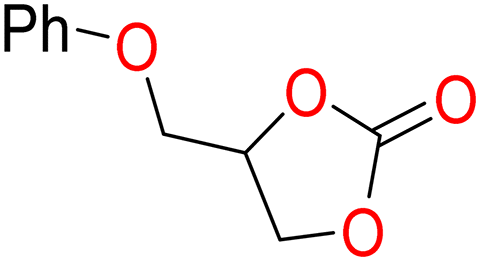 5b
5b
|
97 | 0.271 | 0135 |
| 3 |
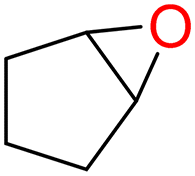 4c
4c
|
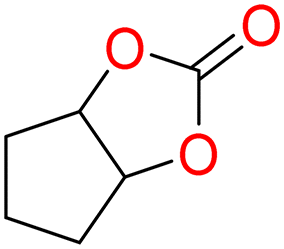 5c
5c
|
93 | 0.260 | 0.130 |
| 4 |
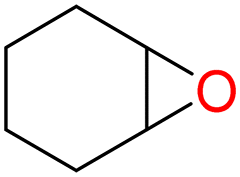 4d
4d
|
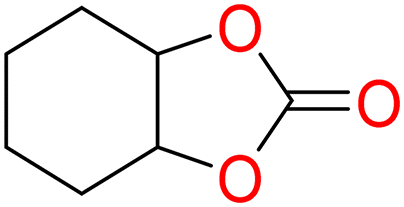 5d
5d
|
90 | 0.252 | 0.126 |
| 5 |
 4e
4e
|
 5e
5e
|
94 | 0.263 | 0.131 |
| 6 |
 4f
4f
|
 5f
5f
|
87 | 0.243 | 0.121 |
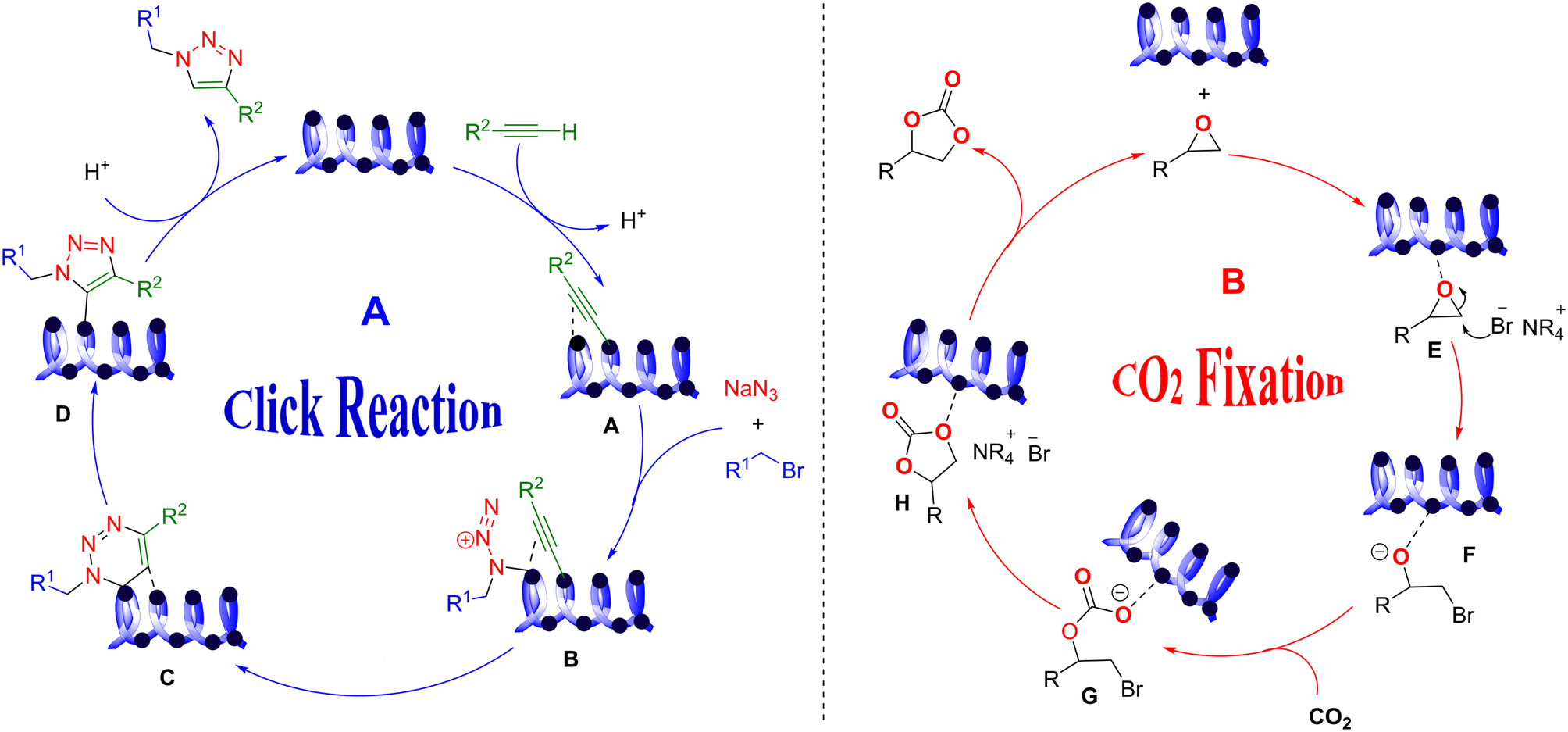
The suggested mechanism for the click reaction is presented in Scheme 3A. In the first step, alkynyl is coordinated to copper in Cu2O/IC-CS and forms a copper(I) acetylide complex that is obtained through the π-coordination of an alkyne with Cu(I) species A. This improves the hydrogen acidity of alkyne, tolerating the construction of the copper acetylide with the already activated alkyne. Then, copper acetylide is a σ-coordinated complex in water solvents. The formed alkyl azide is then activated in coordination with the second copper atom B. The process continues with the formation of the first C–N bond until the final separation of the triazole ring D from the copper. Finally, the desired product 7 is created with high regioisomers.56
 | ||
| Scheme 3 A suggested mechanism for the Click reaction (A) and CO2 fixation reaction (B) in the presence of Cu2O/IC-CS and CuO/IC-CS biocatalysts, respectively. | ||
Moreover, the proposed mechanism for the CO2 fixation reaction can be seen in Scheme 3B. The high catalytic activity of CuO/IC-CS originated from the cooperative effect of the nucleophilic bromine anion (Br−) in TBAB and Cu(II) in the center of the catalyst. Firstly, the Cu–O bond was constructed between the epoxy and immobilized CuO nanoparticles on the IC-CS network. In the next step, the bromide anion from TBAB attacked the less-hindered carbon atom of the epoxide to open the ring to form intermediate F. After interacting with CO2, intermediate G was received via a nucleophilic reaction. Finally, the bromide was removed from the intermediate G to form the carbonate ring. According to the above-mentioned plausible mechanisms, the biocatalysts were recovered and could be utilized in the next catalytic cycle.57,58
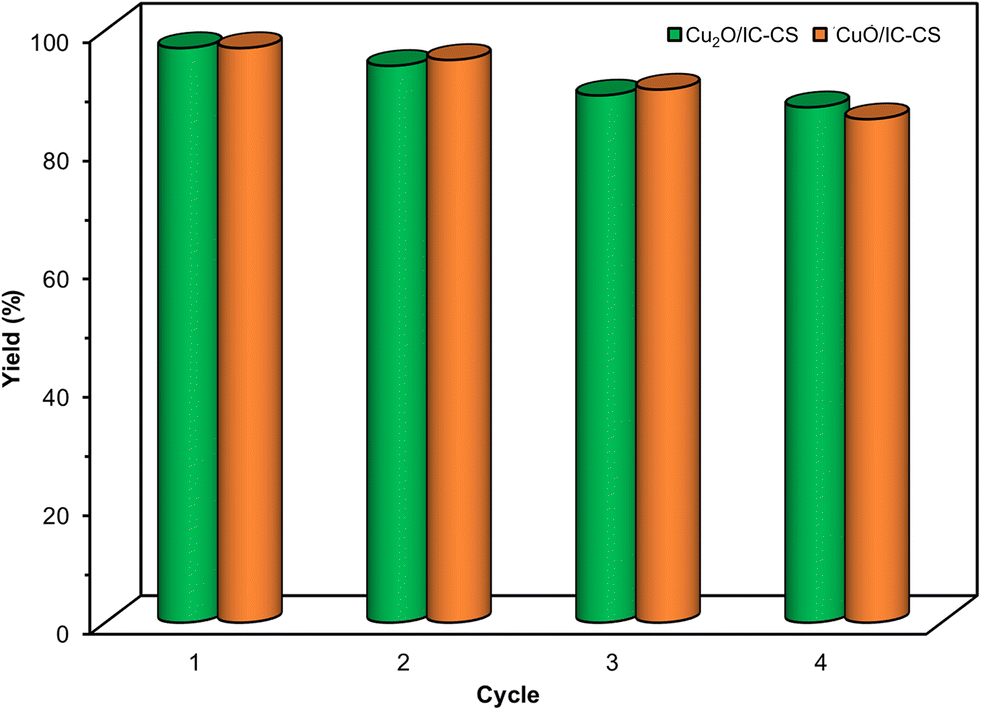
The catalytic stability of the copper nanoparticles immobilized on the indigo carmine-functionalized chitosan (Cu2O/IC-CS and CuO/IC-CS) was considered for exploring their potential activity as reusable biocatalysts (Fig. 7). It is clear that the Cu2O/IC-CS and CuO/IC-CS biocatalysts displayed comparatively stable activities in four runs for the selective cycloaddition reaction under optimal conditions. The conversion percentage of the starting material to five-member heterocycles in click and CO2 fixation reactions showed a slight decrease in the yield, indicating the desired stability of biocatalysts with good reusability without a significant loss of their activity.
 | ||
| Fig. 7 Recycling experiment of the constructed Cu2O/IC-CS and CuO/IC-CS biocatalyst for the model click and CO2 fixation reaction, respectively. | ||
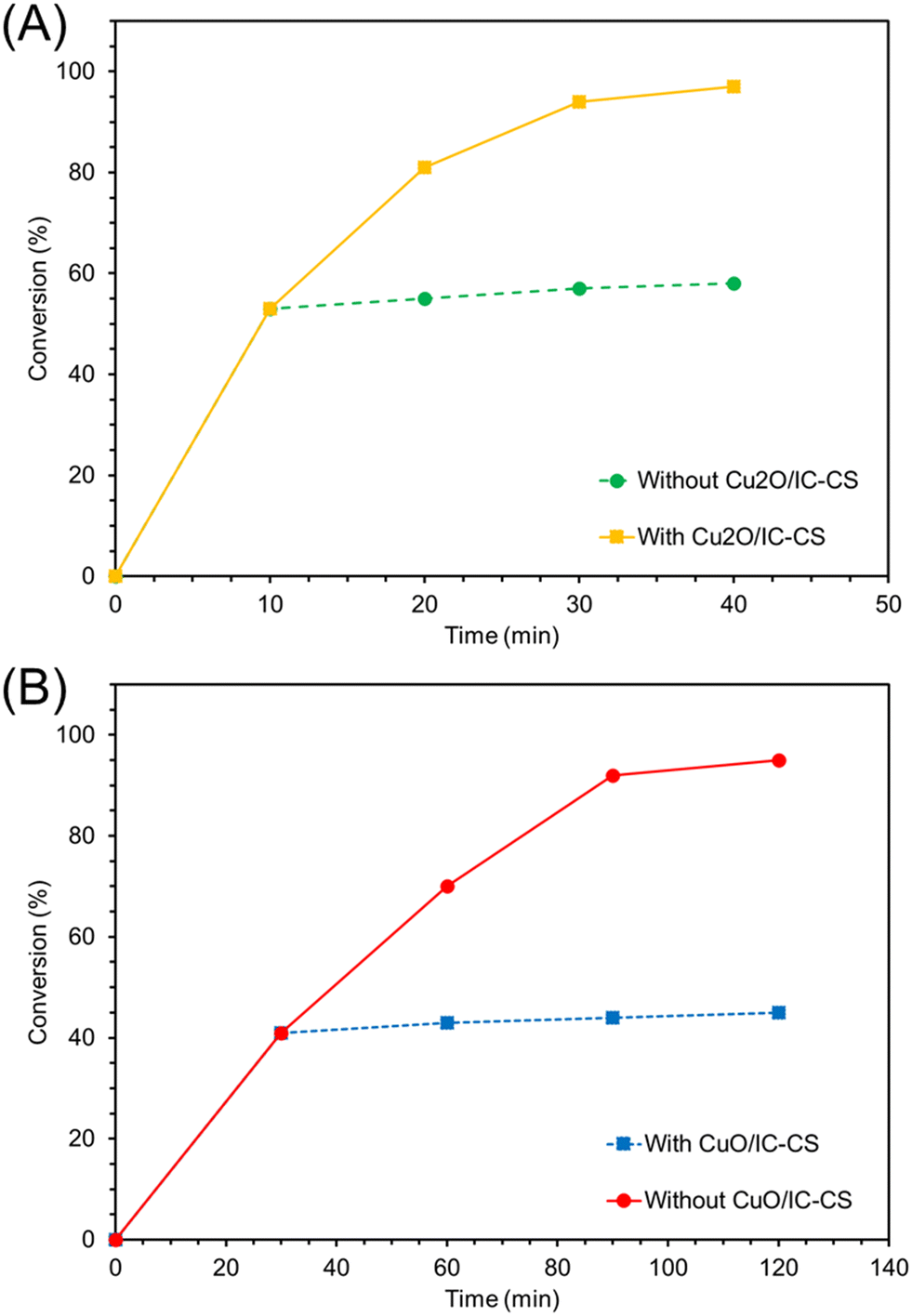
To determine the copper loading in the biocatalyst, the ICP-OES analysis of Cu2O/IC-CS and CuO/IC-CS were obtained and the results indicated 2.9 and 5.7 wt% copper loading in the final biocatalyst. Moreover, the actual heterogeneous feature of the biocatalyst was verified via the hot filtration test. Separating Cu2O/IC-CS and CuO/IC-CS from the mixture of model reactions after 9 min and 30 min only led to insignificant progress in the starting material conversion (Fig. 8A and B), respectively.
 | ||
| Fig. 8 Hot filtration tests of the Cu2O/IC-CS (A) and CuO/IC-CS (B) biocatalysts for the model click and CO2 fixation reaction, respectively. | ||
Ultimately, to compare the catalytic performance of the presented biocatalysts in click and CO2 fixation reactions with other catalytic systems based on copper nanoparticles, conditional factors regarding time, pressure, temperature, solvent, and amount of catalyst were studied and the data are summarized in Table 3. Comparing the click reaction in this investigation with previous reports (Table 3), we can refer to the simple preparation of Cu2O/IC-CS as a green biocatalyst, and alternatively, the synthesized triazole is obtained in the shortest time with high-isomer selectivity in a green process. On the other hand, the use of CuO/IC-CS biocatalyst in the CO2 fixation reaction in comparison with similar reports suggests that, in addition to being a greener biocatalyst, it can be successfully employed in water as a green solvent and has a short catalytic reaction time with high efficiency at low CO2 pressure for the synthesis of carbonates.
| Entry | Catalyst | Catalyst and co- catalyst (mg) | Condition | Time (h) | Yield (%) | Type of catalytic reaction | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Cu2O nanocrystals | 32/− | EtOH, 55 °C | 1 | 96 | Click | 59 |
| 2 | Cu/SiO2 composite | 38/− | H2O, 70 °C, MW (50 W) | 0.6 | 92 | Click | 60 |
| 3 | Cu2O@ARF | 25/− | H2O, 25 °C | 1 | 92 | Click | 61 |
| 4 | Cu-CS NPs/MWCNT@Fe3O4 | 10/− | H2O, 70 °C | 0.5 | 99 | Click | 62 |
| 5 | CS NP/MWCNT@Fe3O4 | 20/− | H2O, 70 °C | 0.5 | 84 | Click | 62 |
| 6 | Cu2O/IC-CS bio-nanocomposite | 5/− | H2O, 60 °C, ultrasonic | 0.4 | 97 | Click | This work |
| 7 | Paper matrices immobilized with CuO nanoflakes | 10/− | Solvent free, 100 °C, CO2 (4 bar) | 4 | 96 | CO2 fixation | 63 |
| 8 | CuAl2O4/TBAI | 90/16 | Solvent free, 85 solvent free, CO2 (3 bar) | 10 | 98 | CO2 fixation | 64 |
| 9 | CuO | 39/16 | Solvent free, 85 solvent free, CO2 (3 bar) | 10 | 85 | CO2 fixation | 64 |
| 10 | CuO/IC-CS/TBAB | 10/4 | H2O, 80 °C, CO2 (1 bar balloon) | 2 | 96 | CO2 fixation | This work |
5. Conclusion
In summary, in this study, we developed chitosan as a green support to build a versatile heterogeneous biocatalytic structure based on functionalization with indigo carmine and subsequent immobilization of copper nanoparticles. This successful biocatalyst fabrication was confirmed by various analyzes such as FT-IR, SEM, EDX, XRD TGA, and DTG. The resulting bio-nanocomposites can act as a versatile biocatalyst for the synthesis of 1,2,3-triazole and cyclic carbonates with good yields in aqueous media as the green solvent. In fact, the contribution of copper metal in the complexation of biopolymeric ligands can improve its catalytic efficiency over organic transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper has been supported by the Research Council of Shahid Beheshti University and the RUDN University Strategic Academic Leadership Program (A. Shaabani).References
- P. P. Nair, R. M. Philip and G. Anilkumar, Org. Biomol. Chem., 2021, 19, 4228–4242 RSC.
- M. Nasrollahzadeh, M. Sajjadi, F. Ghorbannezhad and S. M. Sajadi, Chem. Rec., 2018, 18, 1409–1473 CrossRef CAS PubMed.
- A. Ramazani, A. Dabbaghi and F. Gouranlou, Curr. Org. Chem., 2018, 22, 362–369 CrossRef CAS.
- E. Arefi, A. Khojastehnezhad and A. Shiri, Sci. Rep., 2021, 11, 1–14 CrossRef PubMed.
- A. Khojastehnezhad, M. Bakavoli, A. Javid, M. M. Khakzad Siuki and M. Shahidzadeh, Res. Chem. Intermed., 2019, 45, 4473–4485 CrossRef CAS.
- Z. Ghadamyari, A. Khojastehnezhad, S. M. Seyedi, F. Taghavi and A. Shiri, ChemistrySelect, 2020, 5, 10233–10242 CrossRef CAS.
- G. Kaur, G. Singh and J. Singh, Mater. Today Chem., 2018, 8, 56–84 CrossRef CAS.
- P. Leophairatana, C. C. De Silva and J. T. Koberstein, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 75–84 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337–2364 CrossRef CAS.
- A. Shaabani, M. Shadi, R. Mohammadian, S. Javanbakht, M. T. Nazeri and F. Bahri, Appl. Organomet. Chem., 2019, 33, e5074 Search PubMed.
- F. Bahri, M. Shadi, R. Mohammadian, S. Javanbakht and A. Shaabani, Carbohydr. Polym., 2021, 265, 118067 CrossRef CAS PubMed.
- B. J. Borah, D. Dutta, P. P. Saikia, N. C. Barua and D. K. Dutta, Green Chem., 2011, 13, 3453–3460 RSC.
- H. Naeimi and M. Moradian, Appl. Catal., A, 2013, 467, 400–406 CrossRef CAS.
- M. A. Hassan, T. P. Li and Z. Z. Noor, Journal of Chemical and Natural Resources Engineering, 2009, 4, 43–53 Search PubMed.
- S. Kobayashi, T. Kiyosada and S.-I. Shoda, J. Am. Chem. Soc., 1996, 118, 13113–13114 CrossRef CAS.
- A. Sugunan, C. Thanachayanont, J. Dutta and J. Hilborn, Sci. Technol. Adv. Mater., 2005, 6, 335 CrossRef CAS.
- J. H. Park, G. Saravanakumar, K. Kim and I. C. Kwon, Adv. Drug Delivery Rev., 2010, 62, 28–41 CrossRef CAS PubMed.
- J. Baudoux, K. Perrigaud, P.-J. Madec, A.-C. Gaumont and I. Dez, Green Chem., 2007, 9, 1346–1351 RSC.
- E. Guibal, Prog. Polym. Sci., 2005, 30, 71–109 CrossRef CAS.
- M. Chtchigrovsky, A. Primo, P. Gonzalez, K. Molvinger, M. Robitzer, F. Quignard and F. Taran, Angew. Chem., Int. Ed., 2009, 48, 5916–5920 CrossRef CAS.
- J. J. Hardy, S. Hubert, D. J. Macquarrie and A. J. Wilson, Green Chem., 2004, 6, 53–56 RSC.
- D. Wei, W. Sun, W. Qian, Y. Ye and X. Ma, Carbohydr. Res., 2009, 344, 2375–2382 CrossRef CAS PubMed.
- L. Qi, Z. Xu, X. Jiang, Y. Li and M. Wang, Bioorg. Med. Chem. Lett., 2005, 15, 1397–1399 CrossRef CAS PubMed.
- V. Calò, A. Nacci, A. Monopoli, A. Fornaro, L. Sabbatini, N. Cioffi and N. Ditaranto, Organometal, 2004, 23, 5154–5158 CrossRef.
- M. Adlim, M. A. Bakar, K. Y. Liew and J. Ismail, J. Mol. Catal. A: Chem., 2004, 212, 141–149 CrossRef CAS.
- B. J. Arena, Chitin-and chitosan-based immobilized metal catalysts, US Pat., 4274980, 1981 Search PubMed.
- T. Baran, E. Açıksöz and A. Menteş, Carbohydr. Polym., 2016, 142, 189–198 CrossRef CAS PubMed.
- J. Sun, J. Wang, W. Cheng, J. Zhang, X. Li, S. Zhang and Y. She, Green Chem., 2012, 14, 654–660 RSC.
- Z. Ghadamyari, A. Khojastehnezhad, S. M. Seyedi and A. Shiri, ChemistrySelect, 2019, 4, 10920–10927 CrossRef CAS.
- A. Khojastehnezhad, M. Bakavoli, A. Javid, M. M. Khakzad Siuki and F. Moeinpour, Catal. Lett., 2019, 149, 713–722 CrossRef CAS.
- S. Allameh, A. Davoodnia and A. Khojastehnezhad, Chin. Chem. Lett., 2012, 23, 17–20 CrossRef CAS.
- A. Shaabani, V. Khodkari, M. T. Nazeri, S. Ghasemi, R. Mohammadian and S. Shaabani, J. Iran. Chem. Soc., 2019, 16, 1793–1800 CrossRef CAS.
- F. Moeinpour and A. Khojastehnezhad, Eur. J. Chem., 2012, 9, 504–509 CAS.
- V. K. Thakur and M. K. Thakur, ACS Sustainable Chem. Eng., 2014, 2, 2637–2652 CrossRef CAS.
- M. Lee, B.-Y. Chen and W. Den, Appl. Sci., 2015, 5, 1272–1283 CrossRef CAS.
- C. Camacho, J. C. Matías, D. García, B. K. Simpson and R. Villalonga, Electrochem. Commun., 2007, 9, 1655–1660 CrossRef CAS.
- C. Fritzmann, J. Löwenberg, T. Wintgens and T. Melin, Desalination, 2007, 216, 1–76 CrossRef CAS.
- B. Ganem, Acc. Chem. Res., 2009, 42, 463–472 CrossRef CAS PubMed.
- R. Afshari and A. Shaabani, ACS Comb. Sci., 2018, 20, 499–528 CrossRef CAS PubMed.
- R. Kakuchi, Angew. Chem., Int. Ed., 2014, 53, 46–48 CrossRef CAS PubMed.
- M. Mahyari, M. S. Laeini and A. Shaabani, Chem. Commun., 2014, 50, 7855–7857 RSC.
- A. Shaabani, R. Mohammadian, S. E. Hooshmand, A. Hashemzadeh and M. M. Amini, ChemistrySelect, 2017, 2, 11906–11911 CrossRef CAS.
- A. Shaabani, A. Rashidi Vahid, S. Shaabani, R. Mohammadian, M. T. Nazeri and M. Keramati Nejad, Appl. Organomet. Chem., 2018, 32, e4510 CrossRef.
- A. Shaabani, R. Mohammadian, A. Hashemzadeh, R. Afshari and M. M. Amini, New J. Chem., 2018, 42, 4167–4174 RSC.
- M. Yadollahi, I. Gholamali, H. Namazi and M. Aghazadeh, Int. J. Biol. Macromol., 2015, 73, 109–114 CrossRef CAS PubMed.
- S. Javanbakht, V. Khodkari, M. T. Nazeri and A. Shaabani, React. Chem. Eng., 2022, 7, 1210–1218 RSC.
- M. Radetić and D. Marković, Cellulose, 2019, 26, 8971–8991 CrossRef.
- Y. Raji, I. Mechnou, W. Yassine, Z. Kadri, K. Oumghar, O. Cherkaoui and S. Zyade, 2020.
- S. Javanbakht and A. Shaabani, Int. J. Biol. Macromol., 2019, 123, 389–397 CrossRef CAS PubMed.
- S. Farhoudian, M. Yadollahi and H. Namazi, Int. J. Biol. Macromol., 2016, 82, 837–843 CrossRef CAS PubMed.
- M. A. Bhosale, K. D. Bhatte and B. M. Bhanage, Powder Technol., 2013, 235, 516–519 CrossRef CAS.
- C. Tamuly, I. Saikia, M. Hazarika and M. R. Das, RSC Adv., 2014, 4, 53229–53236 RSC.
- C. D. T. Neto, J. A. Giacometti, A. E. Job, F. C. Ferreira, J. L. C. Fonseca and M. R. Pereira, Carbohydr. Polym., 2005, 62, 97–103 CrossRef CAS.
- P. S. Kumar, M. Selvakumar, S. G. Babu, S. K. Jaganathan, S. Karuthapandian and S. Chattopadhyay, RSC Adv., 2015, 5, 57493–57501 RSC.
- C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS.
- K. Ladomenou, V. Nikolaou, G. Charalambidis and A. G. Coutsolelos, Coord. Chem. Rev., 2016, 306, 1–42 CrossRef CAS.
- M. T. Nazeri, S. Javanbakht, M. Nabi and A. Shaabani, Carbohydr. Polym., 2022, 119144 CrossRef CAS PubMed.
- M. T. Nazeri, S. Javanbakht, M. Ramezani and A. Shaabani, J. Taiwan Inst. Chem. Eng., 2022, 136, 104428 CrossRef CAS.
- K. Chanda, S. Rej and M. H. Huang, Chem. – Eur. J., 2013, 19, 16036–16043 CrossRef CAS PubMed.
- C. S. Radatz, L. do Amaral Soares, E. R. Vieira, D. Alves, D. Russowsky and P. H. Schneider, New J. Chem., 2014, 38, 1410–1417 RSC.
- S. Ghosh, S. Saha, D. Sengupta, S. Chattopadhyay, G. De and B. Basu, Ind. Eng. Chem. Res., 2017, 56, 11726–11733 CrossRef CAS.
- A. Shaabani, R. Afshari and S. E. Hooshmand, New J. Chem., 2017, 41, 8469–8481 RSC.
- S. Aggrawal, R. Sharma and P. Mohanty, J. CO2 Util., 2021, 46, 101466 CrossRef CAS.
- R. Khalifeh, M. Karimi, M. Rajabzadeh, A. Hafizi and F. S. Nogorani, J. CO2 Util., 2020, 41, 101233 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00195k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |