
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering the parameters to produce highly reproducible and scalable iron oxide nanoparticles†
Ashish
Avasthi
 a,
Carlos
Caro
a,
María Luisa
Garcia-Martin
*ab and
Manuel
Pernia Leal
*c
a,
Carlos
Caro
a,
María Luisa
Garcia-Martin
*ab and
Manuel
Pernia Leal
*c
aInstituto de Investigación Biomédica de Málaga y Plataforma en Nanomedicina (IBIMA Plataforma BIONAND), Universidad de Málaga, C/Severo Ochoa, 35, 29590 Málaga, Spain. E-mail: mlgarcia@ibima.eu
bBiomedical Research Networking Center in Bioengineering, Biomaterials & Nanomedicine (CIBER-BBN), Málaga, Spain
cDepartamento de Química Orgánica y Farmacéutica, Facultad de Farmacia, Universidad de Sevilla, 41012 Seville, Spain. E-mail: mpernia@us.es
First published on 11th April 2023
Abstract
Nanomedicine has been long hailed as a game changer for treating several ailments, but its translation from bench to bedside is facing some hurdles. Over the past few decades, there have been a plethora of reports regarding the synthesis of nanomaterials and, in particular, of iron oxide nanoparticles. However, very few reports discuss the role of stirring speed, reproducibility, and scalability. This work attempts to comprehensively revisit the most widely used existing protocols and discuss how the particle size or shape varies when certain parameters are altered and different precursors and solvents are used. It also discusses the probability of reproducing and scaling up the reactions while deciphering the effect of the ramp rate on size and shape. Lastly, it upgrades the existing methods and suggests a modification to produce highly reproducible and scalable nanoparticles of ∼4 nm, which can be further tuned to ∼2 nm by merely modifying the stirring speed.
1. Introduction
Over the last decade, nanomedicine has shown exceedingly promising results, which, if translated to be used as common clinical practices, hold the capability of revolutionizing the healthcare industry in the future. This successful translation from bench to bedside requires, in the first place, a very efficient synthesis process. There are numerous reports on nanoparticles (NPs) synthesis, but rarely do they mention the scalability and/or reproducibility to make large-scale synthesis plausible, making these two factors the main roadblocks in bringing nanomedicine to the patient. Iron oxide, in particular its nanosized counterpart, is one such material that has been revolutionizing several different industries such as sensor components,1–3 contrast/imaging agents,4–10 targeted drug delivery,11–14 magnetic nanoprobes,15 nanocatalysts16,17 and magnetically responsive materials.18,19In the field of medicine, iron oxide nanoparticles (IONPs) hold an advantageous place due to their non-toxic nature, biocompatibility, relative ease of functionalization, and, in some cases, their ability to act as magnetic hyperthermia mediators. Up until mid-2022, close to 7000 studies (PubMed database) had been published on magnetic NPs, showcasing IONPs as potent contrast agents (CAs) for magnetic resonance imaging (MRI), but extremely low clinical translation has been achieved so far.20 Some of the reasons for this low number include the possible generation of ROS species upon the interaction between bare IONPs and cellular structures,21–24 and, in the absence of proper coating, it could also show increased toxicity, or it could lead to oxidation of IONPs, thereafter forming a more toxic oxide of iron (Fe2O3), which would lead to a reduction in viability of cells upon prolonged exposure.25–28
Despite the abundant use and applications of IONPs, their mechanism of synthesis is still in debate after decades of research. This complexity in defining the mechanism is illustrated by recent reports challenging the pre-established mechanism by LaMer while other studies continue to support the mechanism favoring nucleation.29–34 The continuous efforts placed in decoding the synthesis mechanism imply the importance of its knowledge to aptly tailor the NPs to desired size and shape according to the need of the application. Most properties, such as saturation magnetization, biodistribution, tumor targeting, or cell uptake, depend on the size and shape of the NPs.27,35–40 Moreover, in some reports, it has also been observed that shape is even more relevant than size.41–48 The importance of controlling size and shape is further highlighted by the fact that magnetic properties such as magnetic anisotropy and magnetic susceptibility are particularly important for IONPs to be used as MRI CAs.
The low translation of IONPs in clinical products can also be attributed to the failure to reproduce the results obtained inside the laboratory on an industrial scale. It also calls for a need to re-evaluate our reporting of such application-oriented NPs.49,50 In this work, we evaluate how size and shape are affected when using different precursors and solvents. The thermal decomposition method was chosen for the synthesis as it is the most commonly used method for the preparation of IONPs for biomedical applications due to its several advantages, including narrow size distribution, good crystallinity, and high saturation magnetization values.4 In addition to using different solvents (hexadecene, octadecene, benzyl ether, or diphenyl ether), different temperature ramp rates were also tested since the size and shape of the NPs are shown to be affected by them as well.51 Finally, magnetic relaxivity was determined at clinical magnetic fields since one of the main applications of the NPs synthesized here is as MRI CAs. The results from this extensive study revealed a few loopholes in different synthesis methods, which are discussed in detail throughout the article. In addition to discussing these different results based on parametric differences, a modification to the synthesis method is introduced that helps yield highly reproducible and scalable NPs of ∼4 nm. These NPs have high relaxivity (the highest among all the work presented throughout this article), making them ideal as MRI contrast agents, particularly for difficult-to-reach tumors, e.g., glioblastoma multiforme.52 This size can further be reduced to ∼2 nm.
2. Experimental
2.1 Materials
Iron(III) chloride hexahydrate (CAS – 10025-77-1), hexadecene (CAS – 629-73-2), and diphenyl ether (CAS – 101-84-8) were purchased from Merck. Oleic acid (CAS – 112-80-1), iron acetylacetonate (CAS – 14024-18-1), 1,2-hexadecanediol (CAS – 6920-24-7), oleyl alcohol (CAS – 143-28-2), oleylamine (CAS – 112-90-3), benzyl ether (CAS – 103-50-4), and octadecene (CAS – 112-88-9) were procured from Sigma Aldrich, and sodium oleate (CAS – 143-19-1) from Sigma Life Science. Ethanol (99.9% purity) (CAS – 64-17-5) and hexane (99% purity) (CAS – 110-54-3) were purchased from PanReac AppliChem, acetone (99.5% purity) (CAS – 67-64-1) from Arcos Organics, and toluene (99% purity) (CAS – 108-88-3) from Alfa Aesar. All the chemicals were used without further purification.2.2 Synthesis of IONPs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to precipitate the particles and centrifuging them thrice (6000 rpm). Finally, they were redispersed in toluene.
:acetone mixture (1:1), and collected by centrifugation. Particles were redispersed in toluene.
:1) to precipitate the particles and centrifuging them thrice (6000 rpm). Finally, they were redispersed in toluene.
:acetone mixture (1:1), and collected by centrifugation. Particles were redispersed in toluene.
Further, the method reported by Takahashi et al.57 was also used and modified. 0.17 g of Fe(acac) was mixed along with 1.04 g of 1,2-hexadecandiol, 2.25 g of oleic acid, 0.53 g of oleyl amine, and 10 mL of benzyl ether. The reaction mixture was heated to 110 °C for 1 h with N2 flow. The temperature was then raised to 200 °C and kept at that temperature for 30 min. Under N2 blanket, the mixture was refluxed with a heating rate of 15 °C min−1 and kept refluxing for 1 h. The solution mixture was cooled down to room temperature, and the NPs were precipitated out by adding ethanol. The product was washed with ethanol and resuspended in the mixture of toluene and hexane.
1 g of Fe(acac)3 was mixed in phenyl ether (20 mL) along with 2.58 g of 1,2-hexadecanediol, 1.69 g of oleic acid, 1 g of sodium oleate, 0.9 g of oleyl alcohol and 5 mL of oleylamine under N2, and heated to reflux for 1 h. The mixture was allowed to cool down to room temperature. The NPs were precipitated using ethanol:acetone mixture (1:1), and then collected by centrifugation. The NPs were redispersed in toluene.
2.3 Characterization
3. Results
3.1 Synthesis of IONPs
 | ||
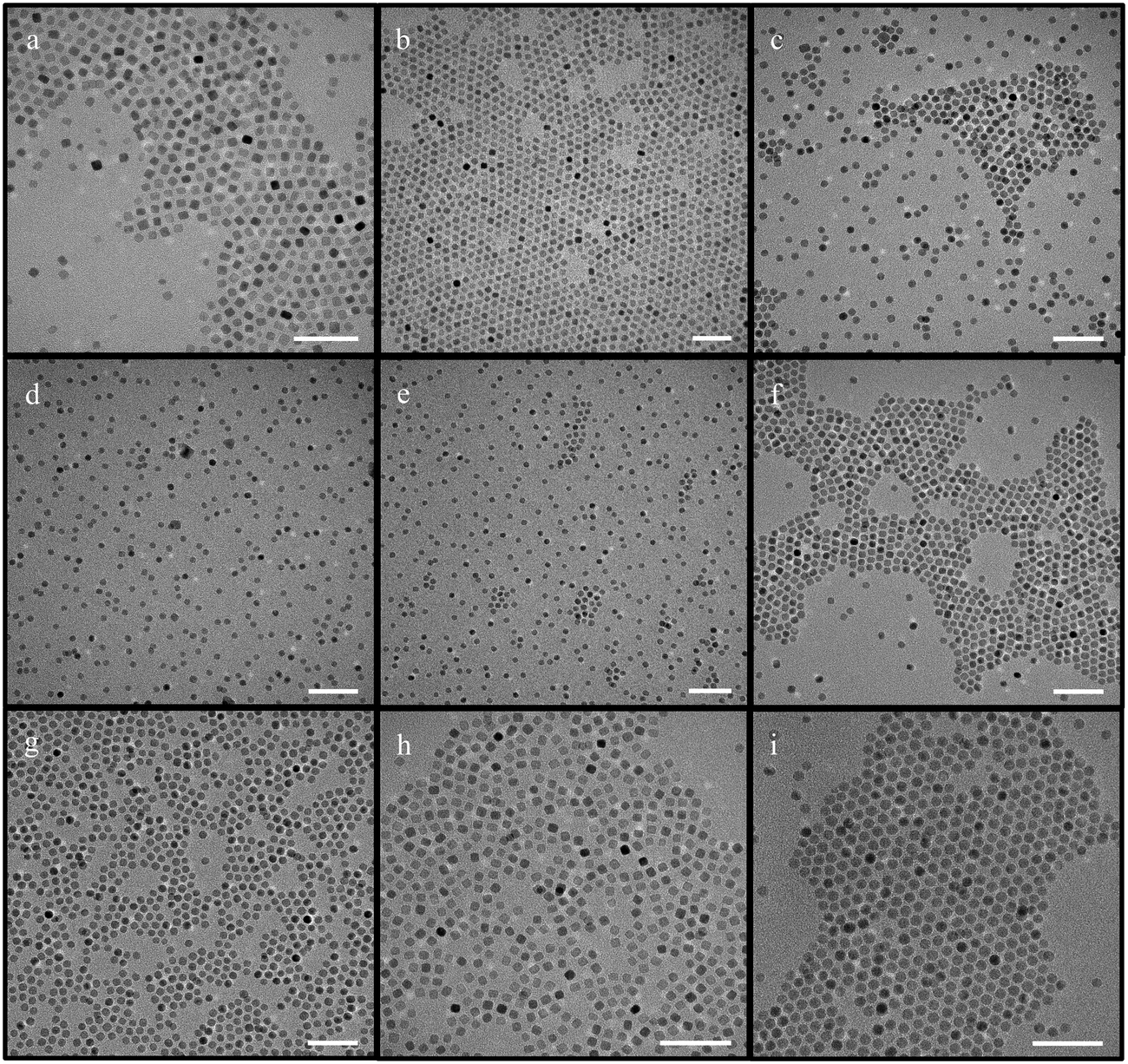
| Fig. 1 IONPs synthesized using iron oleate with octadecene (scale bar – 50 nm) [a–d – protocol from ref. 44 was followed; e–g – concentration of iron oleate was varied; h – synthesis in the absence of oleic acid; i and j – synthesis with increased and decreased ramp rate respectively; k and l reproducibility at low ramp rates; m–o – attempts made with different molar ratios of the reaction with different ramp rates; p – synthesis conducted at increased stirring speed; q–s – synthesis scaled up 5 times; t – synthesis scaled up at a higher stirring speed]. | ||
First, the amount of the precursor added was varied, while the amount of other reactants was kept the same as in the aforementioned protocol. Three different variations to iron oleate quantity were attempted – 1.8 g, 3 g, and 4.5 g. Interestingly, the particle size remained ∼9 nm in all cases, as shown in Fig. 1(e–g).
Next, the amount of the surfactant oleic acid was varied. It was observed that even at a concentration of oleic acid three times that of the original protocol, the NPs were still ∼9 nm (Fig. S1†). Additionally, the synthesis of IONPs in the absence of oleic acid was also tested, but it resulted in high polydispersity (11 ± 3.0 nm) (Fig. 1h).
Since the impact of the ramp rate has been stressed upon, leading to various reports on the inverse relationship between size and ramp rate, changes to it were made.51,59 The ramp rate was initially increased from 3.3 °C min−1 up to 15 °C min−1, but, surprisingly, no discernable change in the size and shape of the particles could be appreciated (Fig. 1i). However, when the ramp rate was decreased to 1 °C min−1, the size increased as predicted to 17.7 ± 1.1 nm (Fig. 1j). Notably, monodisperse NPs of this size were only obtained in 1 out of 7 attempts. The other attempts rendered highly polydisperse NPs, albeit with considerably larger sizes. These results indicate that particles with larger sizes can be formed at low ramp rates but with very low reproducibility (Fig. 1k and l).
Furthermore, when the molar ratio of the whole reaction was varied instead of one reactant (amount of iron oleate, oleic acid, and octadecene were all varied) at the same ramp rate, neither the size nor the shape of the NPs changed noticeably (between 8.4 ± 0.7 nm and 9.2 ± 0.7 nm). On the other hand, changes in the ramp rate from the initial 3.3 °C min−1 to 5 °C min−1, 8 °C min−1, or 15 °C min−1, produced NPs with similar size (∼9.5 ± 1.2 nm) and shape with varying degrees of polydispersity (Fig. 1m–o). There were minor differences in size, which are discussed later. The results achieved are conflicting to the widely accepted postulates that establish a clear relationship between the NPs size and the reaction ramp rate.51,59 Varying the amount of precursor at a lower ramp rate (1 °C) led to some variations in the size and shape of NPs (Fig. S2†). The particles were bimodal, cuboidal or elongated.
Another factor that was evaluated was the stirring speed. Initially, an attempt was made to homogenize the nanoparticles at the lower ramp rate of 1 °C min−1 by increasing the stirring speed. However, it did not help homogenize the particles or develop proper facets (Fig. S3†). Later, the stirring speeds were studied as a variant, with speeds varying from ∼250 rpm up to ∼1600 rpm. Astonishingly, the size of the NPs remained almost invariable (∼8.4 ± 1.1 nm). However, NPs of ∼7 nm were obtained when the stirring speed was increased from ∼800 to ∼1600 rpm in a couple of experiments (Fig. 1p). Additionally, in the reactions where iron oleate concentration was varied, no change was observed in polydispersity.
It is worth noting that the vacuum/N2 cycle was used at the beginning of all experiments, but later some experiments were conducted in the absence of vacuum, with the purging of N2 starting 15 min before the ramp was initiated. However, it did not impact the shape or size of the particles (Fig. S4†).
Finally, the scalability of this synthesis with octadecene was evaluated by performing several scale-up (5×) experiments. The resulting NPs were usually either polydisperse or had bimodal distribution (Fig. 1q–s). On the other hand, increasing the stirring speed to ∼1600 rpm produced bi-pyramidal-like structures for the scale-up experiments (Fig. 1t).
 | ||
| Fig. 2 IONPs synthesized using iron oleate and hexadecene (scale bar – 50 nm) [a and b – synthesis using hexadecene as a solvent following the aforementioned protocol; c–e – synthesis after changing the surfactant/precursor ratio; f – synthesis at increased ramp rate (7 °C min−1); g – synthesis at lower ramp rate (1 °C min−1); h – synthesis at a higher stirring speed (∼1500 rpm); i – synthesis when scaled up to 5 times]. | ||
The size of the NPs remained similar (∼7.5 nm) when the precursor concentration was varied (Fig. 2c–e), suggesting that variation in the precursor/surfactant ratio has no direct effect on the size. As for the effect of the surfactant, the change in just the oleic acid concentration did not produce any change either. Additionally, no remarkable change was observed after increasing the ramp rate from 3 °C min−1 to 5 °C min−1 or 7 °C min−1, resulting in NPs between 7.4–8.5 nm (Fig. 2f). Moreover, no drastic change in size and morphology was observed when the NPs were synthesized in hexadecene at a ramp rate as low as 1 °C min−1, contrary to NPs synthesized using octadecene (Fig. 2g).
Interestingly, an increase in the stirring speed up to ∼1500 rpm at higher ramp rates was suspected of causing a further decrease in size down to ∼6.5 nm (Fig. 2h). However, when the stirring speed was similarly increased at the lower ramp rate, it gave slightly larger NPs, of 9.6 ± 0.8 nm (Fig. S5†). When the stirring speed was decreased to the minimum, polydisperse NPs were formed that were difficult to separate (Fig. S6†).
Reactions conducted using hexadecene showed reasonable scalability, with the particle size being around 7.5 nm. However, these syntheses resulted in mainly spherical NPs instead of the cubic counterparts previously synthesized at a low scale (Fig. 2i). Another important aspect to mention here is that only 1 in 3 scale-up reactions gave the desired product having the same size and shape (Fig. S7†).
Later influenced by the literature, other solvents, such as benzyl ether, a mixture of benzyl ether and octadecene, were also evaluated while using iron oleate as the iron precursor, resulting in highly polydisperse particles39 (Fig. S8†).
The syntheses with benzyl ether, following the protocol by Sun et al.54 and Yang et al.,57 yielded NPs of around 7 nm (6.7 ± 1.4 nm) (Fig. 3a), which were reproducible albeit with some persistent polydispersity. The size of these NPs slightly decreased to ∼5 nm upon further increasing the amount of the surfactant, oleylamine, from 1.6 g to 5 g (Fig. 3b) as previously reported but at the cost of monodispersity.61 As for the ramp rate, the size of the NPs was affected by its increase leading to the synthesis of NPs up to 18.4 ± 4.7 nm at a ramp rate of 15 °C min−1. However, these NPs presented random shapes, showing the lack of reproducibility under these conditions (Fig. 3c). Reactions at ramp rates as low as 1 °C min−1 were not attempted because at the relatively low ramp rate of 4 °C min−1, the size of the NPs had already increased drastically to 37.1 ± 5.5 nm (Fig. 3d).
 | ||
| Fig. 3 IONPs synthesized using iron acetylacetonate as a precursor with benzyl ether and diphenyl ether (scale bar – 50 nm) [a – synthesis using the mentioned protocol with benzyl ether; b – synthesis with increased oleylamine concentration; c – synthesis performed at a ramp rate of 15 °C min−1 (adapted from ref. 46); d – synthesis performed at a lower ramp rate of 5 °C min−1.; e – synthesis when the solvent was diphenyl ether; f – synthesis with diphenyl ether with the addition of sodium oleate]. | ||
Subsequently, diphenyl ether was used as solvent in the synthesis of NPs in place of benzyl ether, resulting in mainly spherical NPs of 4.4 ± 1.9 nm with some polydispersity (Fig. 3e). In this case, no significant change was observed when the ramp rate was increased or decreased. The reproducibility and scalability worked better for reactions with diphenyl ether in comparison to reactions conducted using benzyl ether as solvent; however, a few experiments suffered from large polydispersity issues. Later, in an attempt to reduce the polydispersity, sodium oleate, a well-known shape-directing agent,62 was added to the above reaction, resulting in quasi-cubic NPs with high monodispersity and size of 4.3 ± 0.5 nm (Fig. 3f). However, the particles had somewhat different shapes indicating a lack of control over facet growth.
First, olelyl alcohol was incorporated with octadecene since it has the highest boiling point compared to all the solvents used above. Irrespective of the ramp rate used, the NPs thus produced were highly polydisperse, as well as heterogeneous, with sizes ranging from 5.3 ± 1.6 nm to 7.8 ± 5.8 nm (Fig. 4a and b).
 | ||
| Fig. 4 Synthesis of IONPs with oleyl alcohol as co-solvent (scale bar – 50 nm) [a and b – use of oleyl alcohol with octadecene; c and d – use of oleyl alcohol with hexadecene; e and f – use of oleyl alcohol with benzyl ether; g – use of oleyl alcohol with diphenyl ether; h – synthesis with diphenyl ether and oleyl alcohol at lower stirring speed (∼100 rpm); i – modified protocol with higher stirring speed (∼1500 rpm) showing a size of ∼4 nm; j – modified protocol illustrating a change in size upon decreasing the stirring speed (∼1000 rpm) to ∼2 nm; k and l – synthesis using modified protocol when scaled up to 5 times]. | ||
Then, hexadecene was utilized with oleyl alcohol and exhibited a similar fate, with NPs synthesized between 4.8 ± 1.4 nm and 6.9 ± 2.5 nm (Fig. 4c and d).
Next, oleyl alcohol was used in combination with benzyl ether, which also led to polydispersity and heterogeneity issues, with sizes ranging from 5.7 ± 2.9 nm to 8.5 ± 6 nm (Fig. 4e and f), and a similar trend at different ramp rates or stirring speeds.
In addition, oleyl alcohol was also used in combination with benzyl ether and octadecene, producing very polydisperse NPs (Fig. S9†).
Diphenyl ether was last used as the solvent for the synthesis of the NPs, resulting in sizes of 3.9 ± 0.5 nm (Fig. 4g) and 5.6 ± 2.7 nm (Fig. S10†) in different trials. This variation in size or polydispersity indicates the reproducibility issues with this synthesis. Moreover, the polydispersity of the NPs further increased when the stirring speed was reduced to ∼100 rpm, leading to NPs of 15 ± 7 nm (Fig. 4h).
Eventually, a new synthesis protocol was attempted based on the method recently published by Vangijzegem et al.,58 which combines oleyl alcohol and iron acetylacetonate. This new modification combines iron acetylacetonate along with sodium oleate and oleyl alcohol in order to produce a narrow size distribution, from 4.0 ± 0.6 nm to 4.3 ± 0.7 nm (Fig. 4i). Furthermore, decreasing the stirring speed to ∼1000 rpm elicited a decrease in the size of the NPs, ranging from 2.6 ± 0.3 nm to 2.7 ± 0.5 nm (Fig. 4j) in all experiments conducted, demonstrating the high reproducibility of this protocol.
Notably, the scale-up of this new synthesis protocol rendered NPs of 4.5 ± 1.1 nm, regardless of the stirring speed (Fig. 4k and l). These results clearly demonstrate the superior performance of our new protocol in terms of reproducibility and scalability.
3.2 Characterization of IONPs
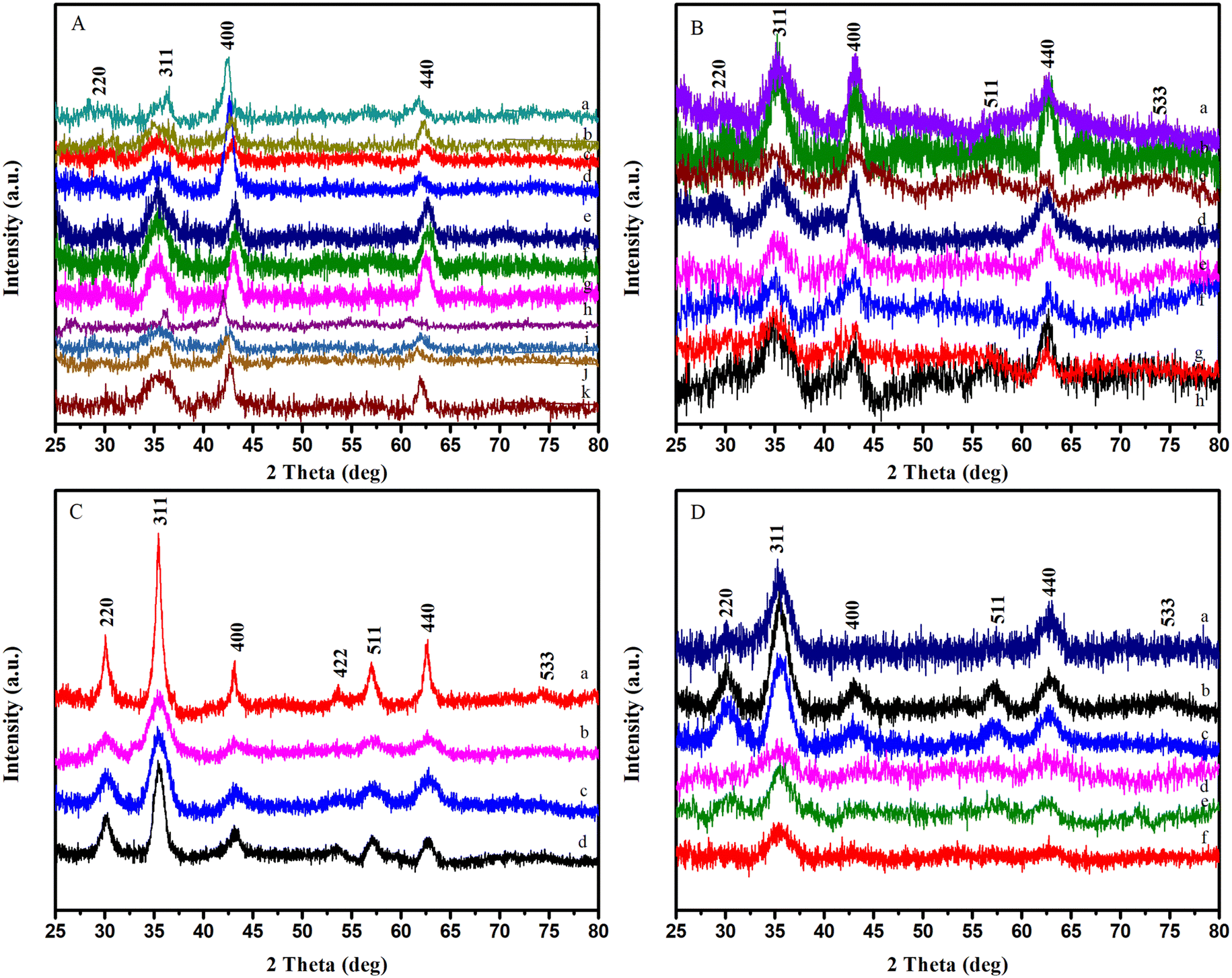 | ||
| Fig. 5 XRD spectra (clockwise) of IONPs synthesized using (A) octadecene with iron oleate, (B) hexadecene with iron oleate, (C) with iron acetylacetonate, (D) with the use of oleyl alcohol (also includes the modified protocol). | ||
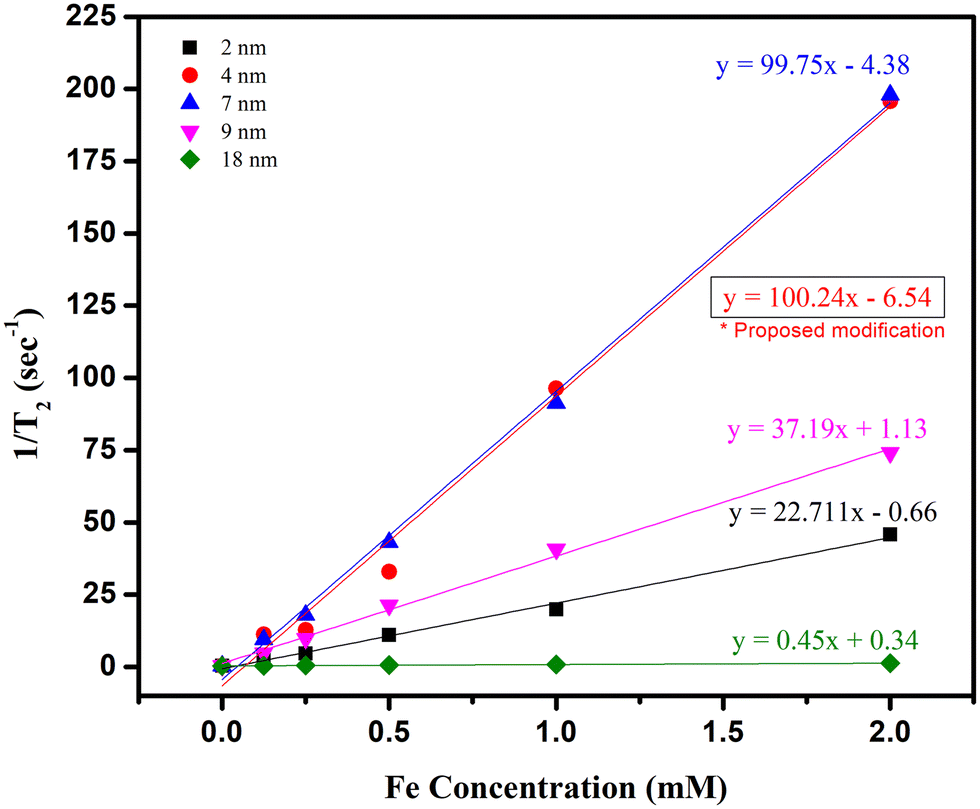 | ||
| Fig. 6 Transverse relaxivity at 1.44 T of NPs with different sizes. r2 values correspond to the slope of the linear fit of 1/T2vs. Fe concentration. | ||
| Sr. no. | TEM size (nm) | HDD in water (nm) | HDD in PBS (nm) | HDD in FBS (nm) |
|---|---|---|---|---|
| 1 | 4.3 ± 0.7 | 24.8 ± 2.1 | 27.5 ± 6.4 | 23.1 ± 4.1 |
| 2 | 7.6 ± 0.7 | 27.8 ± 5.6 | 37.5 ± 4.6 | 25.2 ± 7.5 |
| 3 | 9.3 ± 0.8 | 24.3 ± 2.1 | 31.1 ± 2.9 | 25.9 ± 3.6 |
Table 2 represents the stability of the 3 selected NPs in PBS over 168 h (1 week). As can be observed, all the NPs were very stable and did not show any sign of aggregation.
| Sr. no. | TEM size (nm) | 0 h | 24 h | 48 h | 72 h | 168 h |
|---|---|---|---|---|---|---|
| 1 | 4.3 ± 0.7 | 27.5 ± 6.4 | 28.1 ± 5.5 | 29.4 ± 5.1 | 38.0 ± 4.5 | 25.6 ± 3.3 |
| 2 | 7.6 ± 0.7 | 37.5 ± 4.6 | 29.5 ± 4.8 | 39.9 ± 4.7 | 32.2 ± 7.3 | 29.8 ± 4.3 |
| 3 | 9.3 ± 0.8 | 31.1 ± 2.9 | 31.0 ± 4.4 | 35.9 ± 2.1 | 34.4 ± 2.8 | 33.4 ± 3.3 |
4. Discussion
There is a plethora of articles on the synthesis of IONPs with different shapes and sizes. However, to produce these particles on a large scale for a specific application, it is imperative to choose the correct method. For this purpose, the right chemicals must be chosen to cut down the cost, scale-up must be efficient, reproducibility should be high, and the NPs thus synthesized must be uniform. Unfortunately, many synthesis protocols published to date fail to fulfill all these requirements. The literature indicates that the mechanism of the reaction depends on multiple conditions, and therefore, it becomes difficult to precisely identify, with the current technology, the main responsible variables. Thus, a comprehensive study has been conducted on how different variables, such as the precursor, solvent, temperature, ramp rate, and stirring speed, influence the size, shape, reproducibility, and scalability of IONPs synthesis.All the precursors used for the synthesis of IONPs have different decomposition temperatures as well as different breakdown mechanisms. These different decomposition mechanisms in the precursors have been noted as the prime reason to use different solvents and surfactants with different precursors.63 For instance, the growth of IONPs from iron oleate was initially thought to be directly from iron oleate, but improvements in scientific techniques unveiled the presence of iron-oxo clusters.64 Meanwhile, iron acetylacetonate decomposes slightly differently when compared to iron oleate. It needs to be reduced prior to its decomposition to Fe2+ ions, so 1,2-hexadecanediol was used for this purpose. Moreover, the theories behind the mechanism for the formation of nanoparticles have also evolved from simple LaMer's model of nucleation and growth31 to a continuous process30 and defect-mediated ripening.29
Iron oleate was used in this study with two different solvents, viz. octadecene (b.p. 315 °C) and hexadecene (b.p. 274 °C). In the experiments conducted using the protocol established by Park et al.,53,60 the synthesized particles were monodispersed and homogenous, with a size around 9 nm. They also displayed acceptable reproducibility. However, these NPs showed a cubic shape instead of the predicted spherical one. On the other hand, the experiments conducted with iron oleate and a mixture of solvents, such as octadecene and benzyl ether, yielded a bimodal distribution, with smaller NPs being monodisperse while larger NPs were heterogeneous (Fig. S8†). Further, since it has been noted in the literature that the structure of iron oleate plays an important part in attaining uniform NPs, FTIR spectroscopy was performed prior to all the reactions to assure the stability of the precursor (Fig. S11†).50,65,66 It is worth noting that the reactions with 3 months, 6 months, and 1 year-old iron oleate with octadecene rendered similar sizes and shapes, suggesting that just like its amount, the age of the precursor is not a critical factor once iron oleate has been properly prepared and preserved.
Another important variable in the synthesis that leads to changes in NPs size and shape is the residence time in reflux.54,58 Thus, time-dependent experiments were performed to study this effect using iron oleate as precursor with both solvents – octadecene and hexadecene. Aliquots were taken out after 5, 15, 30, 45, and 60 min and were analyzed by TEM (Fig. S12†). The reaction with octadecene began to show monodisperse particles within the first 5 min, with no change in shape or size after 60 min. However, the cubical facets became slightly more prominent after 15 min, which can be due to the preferential growth of the crystal (110 to 111). The polydispersity stayed below 1 nm in all aliquots. However, when aliquots were taken out of the reaction with hexadecene, the homogenous and monodisperse particles were not formed until after 60 min (Fig. S13†). After the first 5 min, almost no NPs were formed. In subsequent aliquots, very few NPs (9.5 ± 3 nm) were formed, but after 60 min, the NPs population in a similar aliquot increased dramatically, and size became 8.4 ± 0.7 nm, which suggests that the mechanisms of synthesis defined thus far may need to be redefined for every solvent.
Following the study on residence time, a reaction with octadecene as solvent was conducted at 250 °C and it was allowed to stay for 4 h. It produced very monodisperse quasi-cubic NPs of ∼16 nm (Fig. S14†). This suggests that the importance of residence time is not only solvent-dependent but also temperature-dependent, in particular at temperatures closer to the nucleation temperature.
On the other hand, temperature fluctuations during nucleation and growth have also been postulated to cause changes in size and shape.51 Therefore, we performed experiments where the real-time temperature changes were followed while the ramp was set to increase at a constant temperature. The real-time temperature was noted every minute, and the average change was calculated for temperatures ranging from 200 to 250 °C, 250 to 290 °C, and 290 to 320 °C, to determine which part of the reaction – nucleation, continued nucleation or growth – is the driving force for the change in size. However, as can be seen in Table 3 and Fig. 7, no definite conclusion could be drawn from the gathered data.
| Experiment | 200–250 °C (°C min−1) | 250–290 °C (°C min−1) | 290–320 °C (°C min−1) | ∼Size (nm) |
|---|---|---|---|---|
| 1 | 3.8 | 2 | 3.3 | 9.8 |
| 2 | 5 | 8 | 1.1 | 17.8 |
| 3 | 4.1 | 2.1 | 2.3 | 9.3 |
| 4 | 3.1 | 3 | 3 | 8.7 |
| 5 | 3.3 | 2.8 | 2.5 | 9 |
| 6 | 2.9 | 3 | 1.6 | 9.6 |
| 7 | 3.3 | 3 | 2.1 | 8.9 |
| 8 | 3.8 | 4 | 4 | 8.6 |
| 9 | 3.3 | 3.3 | 1.7 | 8.5 |
| 10 | 3.1 | 3 | 2 | 9.3 |
| 11 | 3.1 | 4 | 2.5 | 11.5 |
| 12 | 3.3 | 2.8 | 3.3 | 9.4 |
| 13 | 3.1 | 2.8 | 3 | 7.1 |
| 14 | 2.9 | 3.6 | 1.8 | 18.3 |
| 15 | 20 | 16 | 4 | 8.9 |
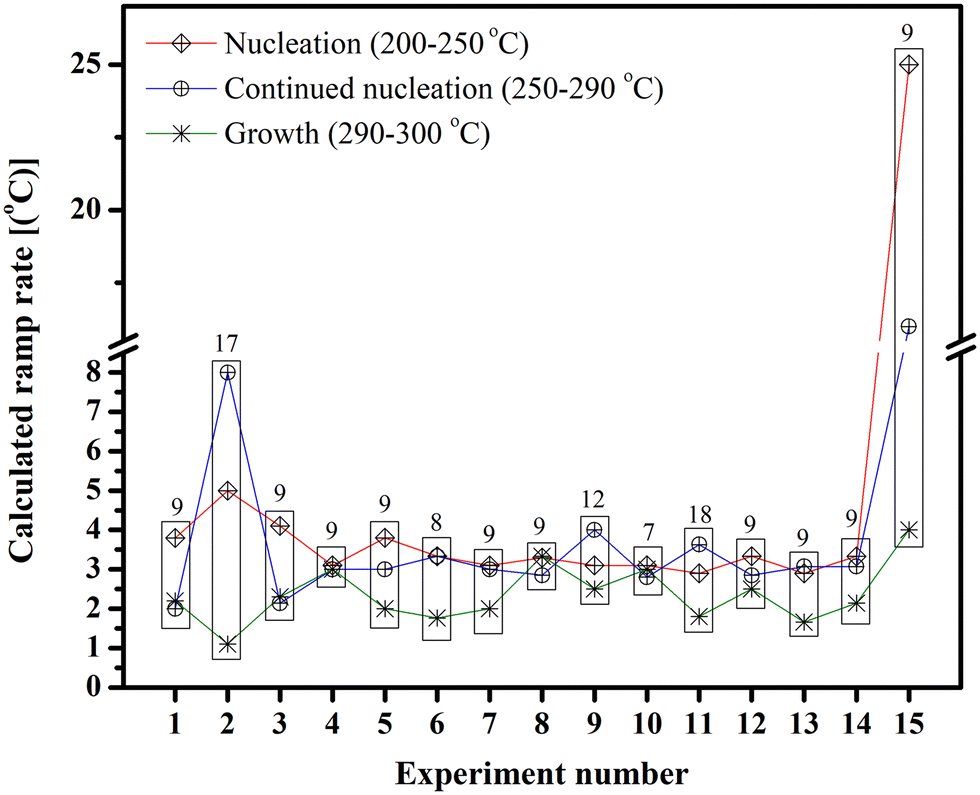 | ||
| Fig. 7 Temperature variation during IONPs synthesis using iron oleate with octadecene at different times (the numbers on top of each rectangle is the size of the NPs formed). | ||
Iron acetylacetonate was used with benzyl ether (b.p. 298 °C) and diphenyl ether (b.p. 258 °C.) as solvents. Particles synthesized with iron acetylacetonate exhibited a size range of 2–7 nm, as mentioned in previous articles.54,63 Moreover, it was observed that the crystallinity of particles was better when synthesized using iron acetylacetonate, as well as their relative reproducibility (Fig. 5). In most experiments, NPs with narrow size distribution were produced, albeit with slight polydispersity. Keeping in mind the difficulty of producing monodisperse NPs below 5 nm with clear facets, it can be concluded that the syntheses worked well. On the other hand, the reproducibility of the reaction with benzyl ether was relatively low if only monodisperse particle synthesis was concerned. In the case of diphenyl ether, the size of NPs was smaller, ∼4.5 nm. This smaller size was expected since the reflux temperature for the reaction was relatively closer to the nucleation temperature due to the lower boiling point of the solvent.
It has been accentuated in the literature that the rate at which temperature is increased directs the size and shape of the NPs. Therefore, we have thoroughly analyzed the effect of this parameter in our experiments. First, it was observed that the starting point of the ramp (room temperature, 100 °C or 200 °C) is not a critical factor in determining the size and shape of NPs as long as it is not beyond the nucleation temperature. During the study, the initiation of the ramp was attempted using all three temperatures, leading to similar results.
Nonetheless, when different ramp rates were attempted, their effect on the size of NPs varied for different precursor and solvent combinations, as evidenced in Fig. 8. The trend lines drawn in the figure illustrate the dependence on the ramp rate. It depicts a very interesting trend demonstrating that the previously reported inverse relationship does exist but only up to a certain range and for a certain combination of precursor and solvent. For instance, in reactions conducted with iron oleate in different solvents (octadecene and hexadecene), the size of NPs was almost the same at the ramp rate of 7 °C min−1, resulting in the smallest NPs. However, the NPs size was increased if the ramp rate was increased further or decreased beyond 7 °C min−1. This size increase was more prominent in octadecene, particularly at the low ramp rate of 1 °C min−1.
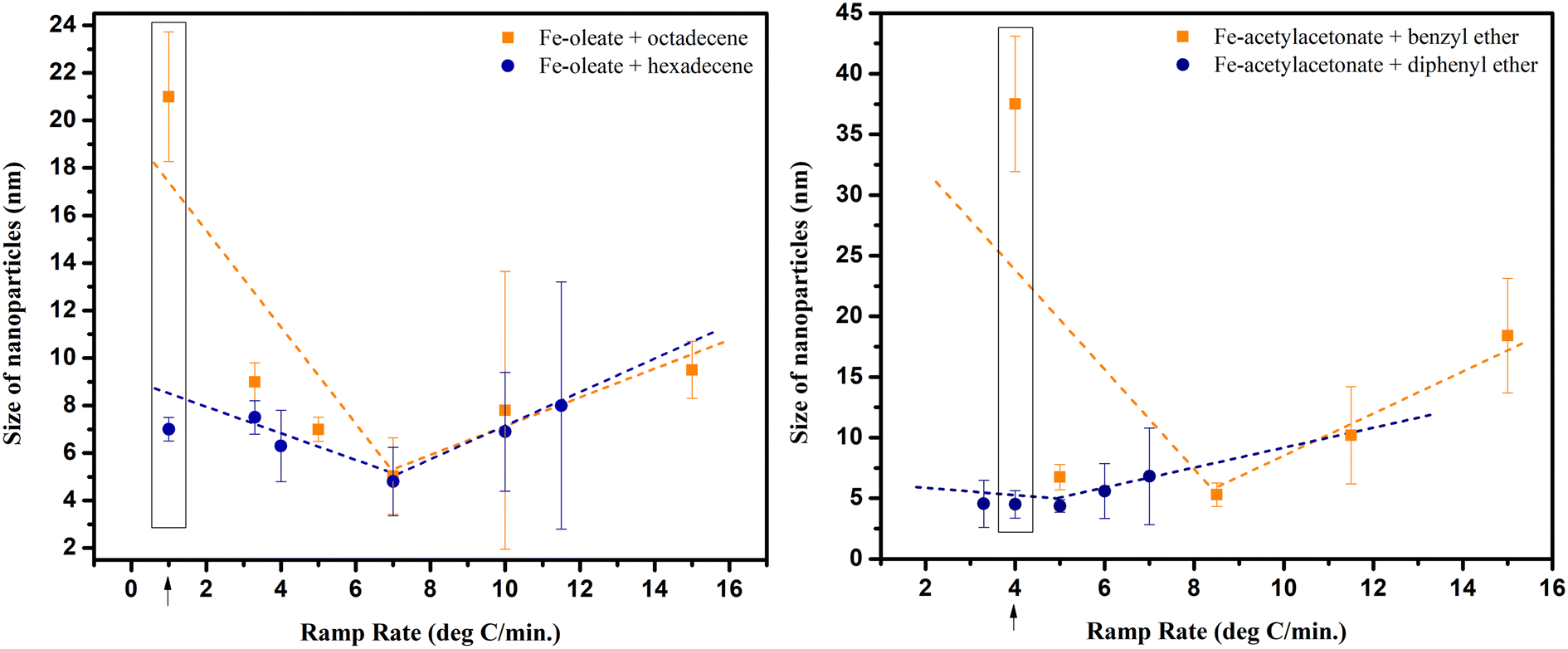 | ||
| Fig. 8 Graphs showing the effect of ramp rate on NPs when using different precursors and different solvents (* shows the ramp rate at which the size of the particles synthesized is the same in both solvents; ↑ shows the difference in size at low ramp rate). | ||
Furthermore, the synthesis with iron acetylacetonate as precursor and benzyl ether as solvent also resulted in a V-shaped trend, but intriguingly, with diphenyl ether, the NPs size was not greatly influenced by the ramp rate. This uniformity in size, irrespective of the ramp rate, was likely due to the low boiling point of diphenyl ether.
Coming to the use of surfactants, the role of oleylamine and oleic acid as capping and reducing agents has been debated over the years with experimental61,67 as well as computational studies.68 However, there have been no concluding remarks except for their influence on the shape and the purification of IONPs from the reaction mixture.
In the experiments performed here, varying the amount of oleic acid influenced the size of IONPs in a ramp rate-dependent manner. Increasing the amount of oleic acid at a high ramp rate (15 °C min−1) resulted in NPs with narrow polydispersity, but at a low ramp rate (5 °C min−1), NPs turned out to be highly polydisperse. This effect could be attributed to the dynamic interaction between the carboxyl group from oleic acid and the surface of NPs, as reported in the literature.68,69 Therefore, at low ramp rates, the growth phase takes longer than at higher ramp rates. Thus, a longer time spent in the growth phase could have allowed the particles to undergo more growth owing to inconsistent passivation of the surface.
The effect of oleylamine on the size of IONPs has been controversial over the years.70 Interestingly, oleylamine has been shown to form complexes with most iron precursors, leading to NPs synthesis.33 A consistent increase in oleylamine concentration resulted in a decrease in NPs size but with high polydispersity. This decrease in the size of NPs could be attributed to changes in electrostatic pressure, steric hindrance, and proton adsorption/desorption play, which occurs due to the addition of 2 surfactants: oleic acid and oleylamine, as previously reported.68 Supposedly, oleylamine acts as a proton acceptor in addition to helping maintain electrostatic pressure. Meanwhile, this electrostatic pressure is responsible for the desorption of oleic acid caused by the increased amount of free protons.
This inconsistency was altered by balancing carboxyl ion and amino ion concentration, which was done by adding sodium oleate at high oleylamine concentrations, resulting in an important reduction in the polydispersity of the particles. This detailed study conducted in this article hints that the particle size depends on the ramp rate, but this dependence is secondary and indirect to the oleic acid/oleylamine ratio (if they are present in the reaction). The importance of this ratio has been studied extensively in the literature, showcasing different theories on ways it influences the size and shape of IONPs. However, the exact role of each component and the mechanism remain elusive.69
Recently, the formation of iron-oxo-oleate species during the synthesis has been demonstrated irrespective of the iron precursor used. These species react in the presence of alcohols such as oleyl alcohol at high temperatures, forming ester groups whose influence on the synthesis is still unclear.30
Although, oleyl alcohol has been shown to influence the synthesis of NPs in multiple ways – its addition to the reaction mixture decreases the boiling point of the solvent, it is shown to reduce iron oleate complex,7 and it is also a known growth inhibitor.71 Synthesis of ultra-small IONPs by addition of oleyl alcohol was suggested by Kim et al.7 Thus, in the studies that were performed, oleyl alcohol was used in combination with different solvents such as hexadecene, octadecene, and benzyl ether. It was observed that it effectively reduced the size of the NPs, but failed to produce uniform particles, along with reproducibility issues, even though it produced good results when used with diphenyl ether. The results achieved demonstrated a reduction in size with low polydispersity and relatively high reproducibility. This reduction in size was expected, but the reduction in polydispersity was surprising. It could perhaps be due to the lower boiling point of the diphenyl ether and the way diphenyl ether interacts with oleyl alcohol under certain temperature conditions.
Stirring speed has been mentioned in previous reports as a variable in nanoparticle synthesis; however, it has seldom been studied in detail.72,73 In this study, the stirring speed appeared to be an important parameter in controlling the shape and size of the NPs. It was observed that an increase in speed up to ∼1500 rpm resulted in a change in the NP size while maintaining the same size of bead, flask, and volume in the reaction. When the speed was decreased to its lowest level of 100 rpm (also for 0 rpm), largely heterogeneous NPs appeared. A speculative explanation could be a change in the strength of the van der Waals strain by altering the time spent nearby to other seeds, leading to changes in Ostwald ripening.
The ability to reproduce a reaction and use the same reaction at a larger scale are both important aspects for the commercialization of NPs. In the case of this study, as can be seen in Fig. 9, when octadecene was used, monodisperse NPs of ∼9 nm were formed, and they were reproducible around 50% of the time. However, if some leeway was taken into account for polydispersity, it rose to around 70%. This reproducibility data does not contain the experiments performed at low ramp rates wherein it plummets down to 20%. The scalability for the reactions with octadecene as solvent had some issues with polydispersity/bimodal distribution. It has been mentioned earlier that increasing the stirring speed in scale-up experiments led to bi-pyramidal structures.
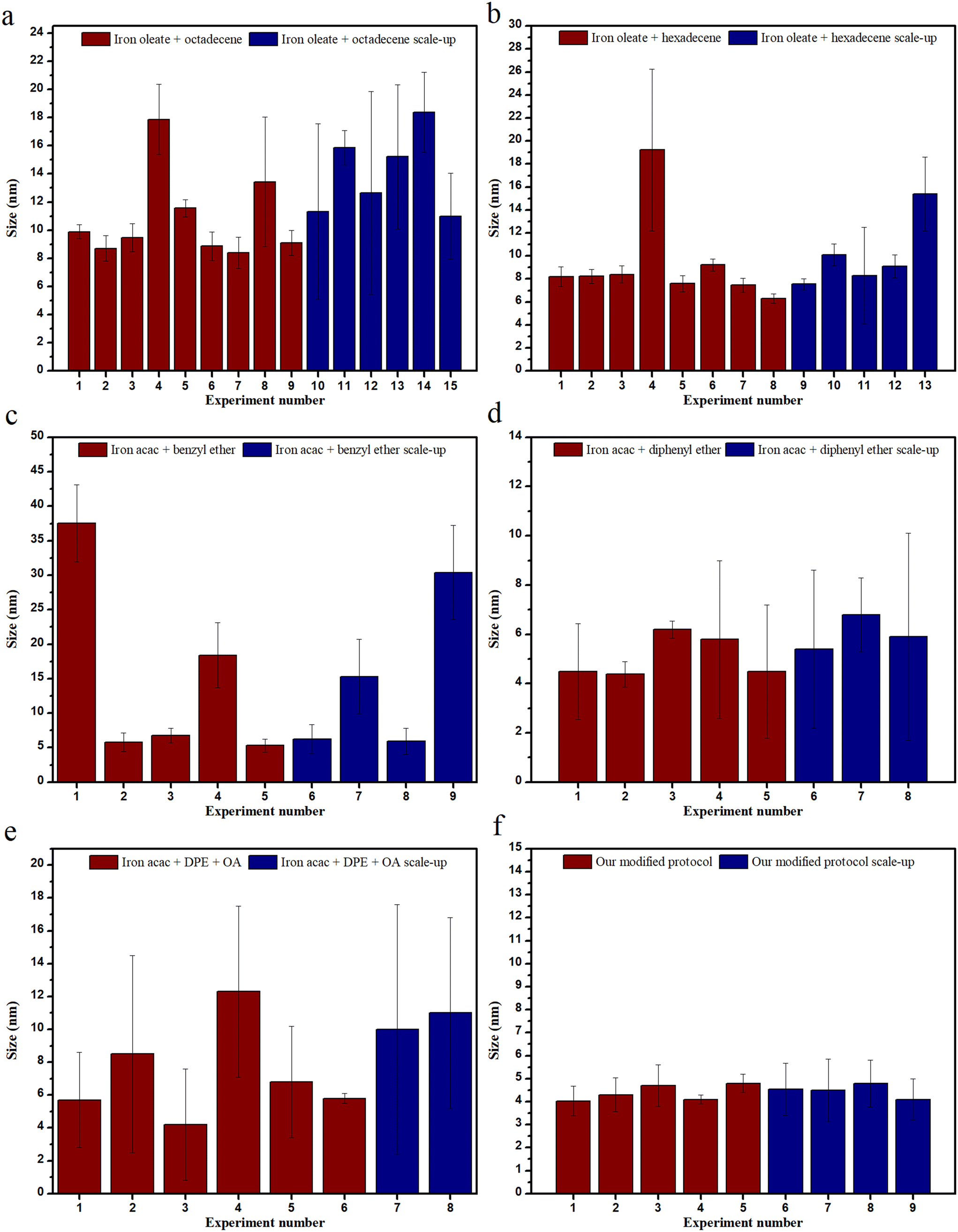 | ||
| Fig. 9 Reproducibility and scalability of IONPs using different precursors and solvents. | ||
Hexadecene, on the other hand, when used as a solvent, had much higher reproducibility and better scalability. Reproducibility, when highly monodisperse particles are considered, was around 60%, which goes up to ∼85% if monodispersity is not an issue, while scalability works ∼50% of the time (∼30–40% if high monodispersity is required). The issue that arose while scaling up the IONPs was that the shape of the particles changed from cubic to spherical.
For the reactions performed with Fe(acac)3 and benzyl ether, the reproducibility for very monodisperse NPs was around 60%, but scalability was an issue here, along with polydispersity. Although, when the solvent was exchanged for diphenyl ether, both reproducibility and scalability increased to ∼75%, albeit with some polydispersity.
The reactions conducted with oleyl alcohol as co-solvent did not bode well for reproducibility or scalability. The only solvent that seemed to perform well with oleyl alcohol was diphenyl ether, although the polydispersity issue persisted.
This issue of polydispersity in scale-up experiments could be attributed to the high temperatures involved during the reactions, along with the larger flasks eliciting a need for improved heating conditions. This hypothesis is also supported by the fact that reproducibility at a smaller scale (1×) performed better than at a larger scale (5×).
All the results from this work indicate that the transition from small-scale to large-scale production will potentially require some changes in the synthesis protocol.
A new protocol has been established by slightly modifying previous protocols. Thus, iron acetylacetonate was used with oleyl alcohol, sodium oleate, oleylamine, oleic acid and mixed with diphenyl ether to attain a size ranging from 2–4.5 nm consistently. Both sizes were highly reproducible (∼95%), but only ∼4 nm NPs were successfully synthesized when the reaction was scaled up. The specific reason for the successful scale-up of 4 nm and not the 2 nm particles remains elusive. However, the heat gradient theory may hold some answers. Multiple surfactants and reducing agents were used here so as to maintain the carboxyl and amine group ratio, in addition to decreasing the energy required for the synthesis of intermediate complexes while increasing the metal ion concentration. Further, at the same time, a solvent with a low boiling point was used in combination with oleyl alcohol.30 It can be seen in Fig. 9 that our modified protocol performs significantly better than others in terms of both reproducibility and scalability.
In summary, this study provides some insights regarding the size range that can be easily formed using iron acetylacetonate and iron oleate using different solvents with relative ease and high reproducibility. For instance, it was observed that for the synthesis of NPs in a range of 5–7 nm, it is recommended to use iron acetylacetonate as the precursor and benzyl ether as the solvent, while particles with sizes below 5 nm can be prepared by using iron acetylacetonate and diphenyl ether. The use of iron oleate as precursor would be indicated for particles between 7 to 12 nm with 7–8.5 nm synthesized using hexadecene in a more facile manner, and from 8.5 nm up to ∼12 nm using octadecene. Depending on the required application, a size different from these mentioned ranges could be attained, but it would require fine-tuning to the reaction conditions. After the successful syntheses, a phase transfer was performed on selected NPs using a gallol-polyethylene glycol (PEG 3000 kDa) ligand. All the IONPs displayed small hydrodynamic diameters (below 40 nm), which is advantageous for in vivo applications, particularly for their use as CAs and tumor targeting.52 The similarities in the hydrodynamic diameter could be due to the orientation of PEG and the quantity bound to the surface of the NPs due to the difference in core size.
5. Conclusion
The main objective of this scientific report was the production of IONPs with a reliable, reproducible, and scalable synthetic method that can compete in the development of new clinical MRI CAs. This study makes it evident that the shift from small-scale to large-scale production is not a straightforward process and, therefore, the industrial manufacture of these NPs will most likely require some changes in the synthesis protocols. The proposed modified protocol showed improved reproducibility and scalability over previously reported protocols. The stability of the PEGylated NPs in PBS buffer was evaluated over 168 h resulting small particle size of 25 nm. Moreover, a high transverse relaxivity value of 100 mM−1 s−1 was obtained for these PEGylated NPs, along with low cytotoxicity. This study made the synthesis of IONPs of certain sizes lucid in terms of ease by using specific precursor and solvent combinations (Fig. 10). Also, it figured out some loopholes and limitations in previously published protocols. Further, this study indicates the essentiality of individually studying the mechanisms of synthesis for every precursor and solvent combination. Finally, it introduces a new modification to synthesize 2 and 4 nm IONPs with relative ease and high reproducibility.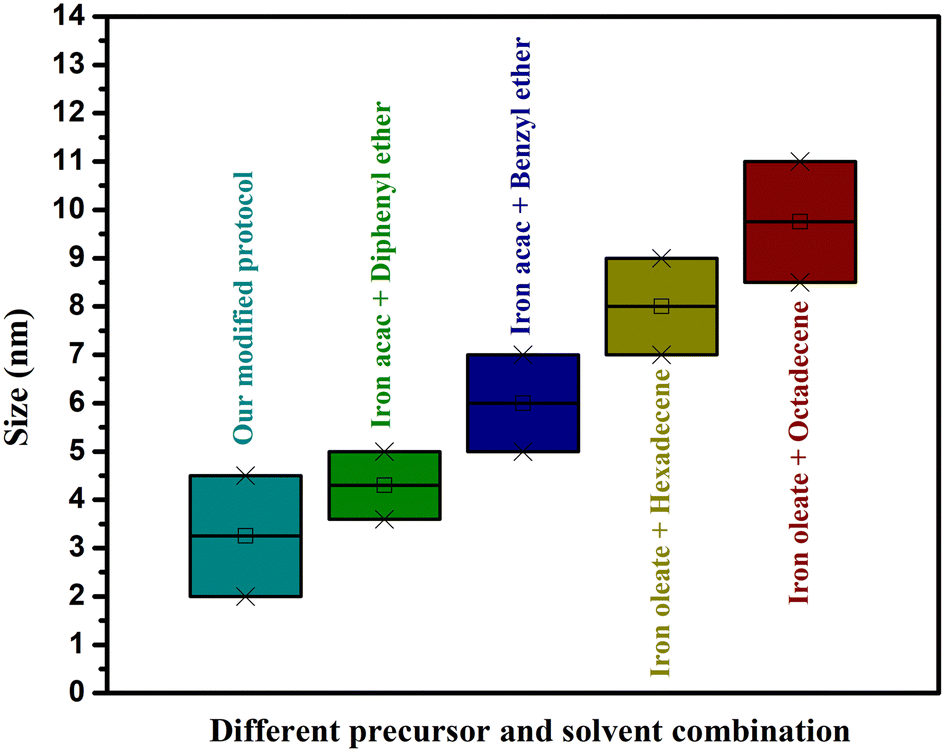 | ||
| Fig. 10 Box plot showing different solvent/precursor combinations and the IONPs size. | ||
Author contributions
A. A.: investigation, writing-original draft. C. C.: investigation. M. L. G. M.: writing-review and editing, fundraising, M. P. L.: writing-review and editing, fundraising.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A. acknowledges Marie Skłodowska-Curie COFUND Programme for his Ph.D. fellowship (NanoMedPhD grant agreement No. 713721, DOI: 10.3030/713721). C. Caro thanks the Consejería de Salud y Familias, Junta de Andalucía, for his senior postdoctoral grant (RH-0040-2021). Financial support was provided by grants PID2020-118448RBC21 and PID2020-118448RBC22, funded by MCIN/AEI/10.13039/501100011033; grant CTQ2017-86655-R funded by the Ministerio de Economia, Industria y Competitividad, Spain; grant P18-RT-1663/PAIDI2020, funded by the Consejería de Economía y Conocimiento, Junta de Andalucía, Spain; and grant P20_00727/PAIDI2020, funded by the Consejería de Transformación Económica, Industria, Conocimiento y Universidades, Junta de Andalucía, Spain. The authors thank Juan F. López for assistance with TEM experiments. TEM, relaxivity and MRI experiments were performed at the ICTS “NANBIOSIS”, more specifically in the Unit 28 at the “Instituto de Investigación Biomédica de Málaga y Plataforma en Nanomedicina (IBIMA Plataforma BIONAND)”.References
- E. Proniewicz, A. Tąta, A. Szkudlarek, J. Świder, M. Molenda, M. Starowicz and Y. Ozaki, Electrochemically synthesized γ-Fe2O3 nanoparticles as peptide carriers and sensitive and reproducible SERS biosensors. Comparison of adsorption on γ-Fe2O3 versus Fe, Appl. Surf. Sci., 2019, 495, 143578 CrossRef CAS.
- L. Khanna, N. Verma and S. Tripathi, Burgeoning tool of biomedical applications-Superparamagnetic nanoparticles, J. Alloys Compd., 2018, 752, 332–353 CrossRef CAS.
- W. Xie, Z. Guo, F. Gao, Q. Gao, D. Wang, B.-S. Liaw, Q. Cai, X. Sun, X. Wang and L. Zhao, Shape-, size-and structure-controlled synthesis and biocompatibility of iron oxide nanoparticles for magnetic theranostics, Theranostics, 2018, 8(12), 3284 CrossRef CAS.
- A. Avasthi, C. Caro, E. Pozo-Torres, M. P. Leal and M. L. García-Martín, Magnetic nanoparticles as MRI contrast agents, Top. Curr. Chem., 2020, 378, 1–43 CrossRef PubMed.
- J. Wang, B. Zhang, G. Yang, L. Su, L. Wang and F. Gao, Transferrin-conjugated superparamagnetic iron oxide nanoparticles as in vivo magnetic resonance imaging contrast agents, J. Nanosci. Nanotechnol., 2020, 20(4), 2018–2024 CrossRef CAS PubMed.
- N. Denora, C. Lee, R. M. Iacobazzi, J. Y. Choi, I. H. Song, J. S. Yoo, Y. Piao, A. Lopalco, F. Leonetti and B. C. Lee, TSPO-targeted NIR-fluorescent ultra-small iron oxide nanoparticles for glioblastoma imaging, Eur. J. Pharm. Sci., 2019, 139, 105047 CrossRef CAS PubMed.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon and H. B. Na, Large-scale synthesis of uniform and extremely small-sized iron oxide nanoparticles for high-resolution T 1 magnetic resonance imaging contrast agents, J. Am. Chem. Soc., 2011, 133(32), 12624–12631 CrossRef CAS PubMed.
- M. P. Leal, C. Muñoz-Hernández, C. C. Berry and M. L. García-Martín, In vivo pharmacokinetics of T 2 contrast agents based on iron oxide nanoparticles: optimization of blood circulation times, RSC Adv., 2015, 5(94), 76883–76891 RSC.
- N. Dhas, R. Kudarha, A. Pandey, A. N. Nikam, S. Sharma, A. Singh, A. Garkal, K. Hariharan, A. Singh, P. Bangar, D. Yadhav, D. Parikh, K. Sawant, S. Mutalik, N. Garg and T. Mehta, Stimuli responsive and receptor targeted iron oxide based nanoplatforms for multimodal therapy and imaging of cancer: Conjugation chemistry and alternative therapeutic strategies, J. Controlled Release, 2021, 333, 188–245 CrossRef CAS PubMed.
- L. Jia, X. Li, H. Liu, J. Xia, X. Shi and M. Shen, Ultrasound-enhanced precision tumor theranostics using cell membrane-coated and pH-responsive nanoclusters assembled from ultrasmall iron oxide nanoparticles, Nano Today, 2021, 36, 101022 CrossRef CAS.
- M. Norouzi, V. Yathindranath, J. A. Thliveris, B. M. Kopec, T. J. Siahaan and D. W. Miller, Doxorubicin-loaded iron oxide nanoparticles for glioblastoma therapy: a combinational approach for enhanced delivery of nanoparticles, Sci. Rep., 2020, 10(1), 11292 CrossRef.
- D. Zhi, T. Yang, J. Yang, S. Fu and S. Zhang, Targeting strategies for superparamagnetic iron oxide nanoparticles in cancer therapy, Acta Biomater., 2020, 102, 13–34 CrossRef CAS.
- P. Hernández, A. Lucero-Acuña, I. E. Moreno-Cortez, R. Esquivel and E. Álvarez-Ramos, Thermo-Magnetic Properties of Fe3O4@ Poly (N-Isopropylacrylamide) Core–Shell Nanoparticles and Their Cytotoxic Effects on HeLa and MDA-MB-231 Cell Lines, J. Nanosci. Nanotechnol., 2020, 20(4), 2063–2071 CrossRef.
- S. Khoee and M. Jalaeian Bashirzadeh, Preparation of Janus-type superparamagnetic iron oxide nanoparticles modified with functionalized PCL/PHEMA via photopolymerization for dual drug delivery, J. Appl. Polym. Sci., 2021, 138(1), 49627 CrossRef CAS.
- Y. Ye, S. Mao, S. He, X. Xu, X. Cao, Z. Wei and S. Gunasekaran, Ultrasensitive electrochemical genosensor for detection of CaMV35S gene with Fe3O4-Au@ Ag nanoprobe, Talanta, 2020, 206, 120205 CrossRef CAS PubMed.
- C. Saengruengrit, A. Sharma, D. Solonenko, P. Thamyongkit, T. Saetan, S. Wacharasindhu, S. Krause, S. Sattayaporn, G. Salvan and D. R. Zahn, Iron oxide nanospheres and nanocubes modified with carboxyphenyl porphyrin and their magnetic, optical properties and photocatalytic activities in room temperature amide synthesis, J. Magn. Magn. Mater., 2021, 521, 167515 CrossRef CAS.
- M. Hammad, P. Fortugno, S. Hardt, C. Kim, S. Salamon, T. C. Schmidt, H. Wende, C. Schulz and H. Wiggers, Large-scale synthesis of iron oxide/graphene hybrid materials as highly efficient photo-Fenton catalyst for water remediation, Environ. Technol. Innovation, 2021, 21, 101239 CrossRef CAS.
- A. S. Gaspar, P. H. Santos, O. Borges, B. F. Costa and L. Durães, Biocompatible and high-magnetically responsive iron oxide nanoparticles for protein loading, J. Phys. Chem. Solids, 2019, 134, 273–285 CrossRef.
- B. P. Nowak, M. Niehues and B. J. Ravoo, Magneto-responsive hydrogels by self-assembly of low molecular weight peptides and crosslinking with iron oxide nanoparticles, Soft Matter, 2021, 17(10), 2857–2864 RSC.
- C. L. Ventola, Progress in nanomedicine: approved and investigational nanodrugs, Pharm. Ther., 2017, 42(12), 742 Search PubMed.
- G. Liu, J. Gao, H. Ai and X. Chen, Applications and potential toxicity of magnetic iron oxide nanoparticles, Small, 2013, 9(9–10), 1533–1545 CrossRef CAS PubMed.
- D. Ling and T. Hyeon, Iron Oxide Nanoparticles: chemical design of biocompatible iron oxide nanoparticles for medical applications (Small 9–10/2013), Small, 2013, 9(9–10), 1449–1449 CrossRef CAS.
- Z. Yarjanli, K. Ghaedi, A. Esmaeili, S. Rahgozar and A. Zarrabi, Iron oxide nanoparticles may damage to the neural tissue through iron accumulation, oxidative stress, and protein aggregation, BMC Neurosci., 2017, 18, 1–12 CrossRef PubMed.
- L. Wu, W. Wen, X. Wang, D. Huang, J. Cao, X. Qi and S. Shen, Ultrasmall iron oxide nanoparticles cause significant toxicity by specifically inducing acute oxidative stress to multiple organs, Part. Fibre Toxicol., 2022, 19(1), 24 CrossRef CAS PubMed.
- A. K. Gupta and S. Wells, Surface-modified superparamagnetic nanoparticles for drug delivery: preparation, characterization, and cytotoxicity studies, IEEE Trans. Nanobioscience, 2004, 3(1), 66–73 CrossRef PubMed.
- T. J. Brunner, P. Wick, P. Manser, P. Spohn, R. N. Grass, L. K. Limbach, A. Bruinink and W. J. Stark, In vitro cytotoxicity of oxide nanoparticles: comparison to asbestos, silica, and the effect of particle solubility, Environ. Sci. Technol., 2006, 40(14), 4374–4381 CrossRef CAS.
- Q. Feng, Y. Liu, J. Huang, K. Chen, J. Huang and K. Xiao, Uptake, distribution, clearance, and toxicity of iron oxide nanoparticles with different sizes and coatings, Sci. Rep., 2018, 8(1), 2082 CrossRef PubMed.
- N. Malhotra, J. S. Lee, R. A. D. Liman, J. M. S. Ruallo, O. B. Villaflores, T. R. Ger and C. D. Hsiao, Potential Toxicity of Iron Oxide Magnetic Nanoparticles: A Review, Molecules, 2020, 25(14), 3159 CrossRef CAS PubMed.
- Q. Zhang, X. Peng, Y. Nie, Q. Zheng, J. Shangguan, C. Zhu, K. C. Bustillo, P. Ercius, L. Wang and D. T. Limmer, Defect-mediated ripening of core-shell nanostructures, Nat. Commun., 2022, 13(1), 1–10 Search PubMed.
- H. Chang, B. H. Kim, H. Y. Jeong, J. H. Moon, M. Park, K. Shin, S. I. Chae, J. Lee, T. Kang and B. K. Choi, Molecular-level understanding of continuous growth from iron-oxo clusters to iron oxide nanoparticles, J. Am. Chem. Soc., 2019, 141(17), 7037–7045 CrossRef CAS.
- V. K. LaMer and R. H. Dinegar, Theory, production and mechanism of formation of monodispersed hydrosols, J. Am. Chem. Soc., 1950, 72(11), 4847–4854 CrossRef CAS.
- L. Grote, M. Seyrich, R. Döhrmann, S. Y. Harouna-Mayer, F. Mancini, E. Kaziukenas, I. Fernandez-Cuesta, C. A. Zito, O. Vasylieva and F. Wittwer, Imaging Cu2O nanocube hollowing in solution by quantitative in situ X-ray ptychography, Nat. Commun., 2022, 13(1), 1–11 Search PubMed.
- M. Strach, V. Mantella, J. R. Pankhurst, P. Iyengar, A. Loiudice, S. Das, C. M. Corminboeuf, W. van Beek and R. Buonsanti, Insights into Reaction Intermediates to Predict Synthetic Pathways for Shape-Controlled Metal Nanocrystals, J. Am. Chem. Soc., 2019, 141(41), 16312–16322 CrossRef CAS PubMed.
- A. Feld, A. Weimer, A. Kornowski, N. Winckelmans, J. P. Merkl, H. Kloust, R. Zierold, C. Schmidtke, T. Schotten, M. Riedner, S. Bals and H. Weller, Chemistry of Shape-Controlled Iron Oxide Nanocrystal Formation, ACS Nano, 2019, 13(1), 152–162 CrossRef CAS PubMed.
- Y. J. Kim, B. C. Park, Y. S. Choi, M. J. Ko and Y. K. Kim, Quantitative Analysis on Cellular Uptake of Clustered Ferrite Magnetic Nanoparticles, Electron. Mater. Lett., 2019, 15(4), 471–480 CrossRef CAS.
- A. A. Narkhede, J. A. Sherwood, A. Antone, K. R. Coogan, M. S. Bolding, S. Deb, Y. Bao and S. S. Rao, Role of Surface Chemistry in Mediating the Uptake of Ultrasmall Iron Oxide Nanoparticles by Cancer Cells, ACS Appl. Mater. Interfaces, 2019, 11(19), 17157–17166 CrossRef CAS PubMed.
- V. Patsula, M. Moskvin, S. Dutz and D. Horák, Size-dependent magnetic properties of iron oxide nanoparticles, J. Phys. Chem. Solids, 2016, 88, 24–30 CrossRef CAS.
- Y.-W. Jun, Y.-M. Huh, J.-S. Choi, J.-H. Lee, H.-T. Song, S. Kim, S. Yoon, K.-S. Kim, J.-S. Shin and J.-S. Suh, Nanoscale size effect of magnetic nanocrystals and their utilization for cancer diagnosis via magnetic resonance imaging, J. Am. Chem. Soc., 2005, 127(16), 5732–5733 CrossRef CAS PubMed.
- A. Demortiere, P. Panissod, B. P. Pichon, G. Pourroy, D. Guillon, B. Donnio and S. Begin-Colin, Size-dependent properties of magnetic iron oxide nanocrystals, Nanoscale, 2011, 3(1), 225–232 RSC.
- S. Kaga, N. P. Truong, L. Esser, D. Senyschyn, A. Sanyal, R. Sanyal, J. F. Quinn, T. P. Davis, L. M. Kaminskas and M. R. Whittaker, Influence of Size and Shape on the Biodistribution of Nanoparticles Prepared by Polymerization-Induced Self-Assembly, Biomacromolecules, 2017, 18(12), 3963–3970 CrossRef CAS PubMed.
- T. Thurn-Albrecht, J. Schotter, G. A. Kastle, N. Emley, T. Shibauchi, L. Krusin-Elbaum, K. Guarini, C. T. Black, M. T. Tuominen and T. P. Russell, Ultrahigh-density nanowire arrays grown in self-assembled diblock copolymer templates, Science, 2000, 290(5499), 2126–2129 CrossRef CAS PubMed.
- H. Zeng, M. Zheng, R. Skomski, D. J. Sellmyer, Y. Liu, L. Menon and S. Bandyopadhyay, Magnetic properties of self-assembled Co nanowires of varying length and diameter, J. Appl. Phys., 2000, 87(9), 4718–4720 CrossRef CAS.
- Q. Song and Z. J. Zhang, Shape control and associated magnetic properties of spinel cobalt ferrite nanocrystals, J. Am. Chem. Soc., 2004, 126(19), 6164–6168 CrossRef CAS PubMed.
- C. Jiang, C. W. Leung and P. W. Pong, Magnetic-field-assisted assembly of anisotropic superstructures by iron oxide nanoparticles and their enhanced magnetism, Nanoscale Res. Lett., 2016, 11(1), 1–12 CrossRef PubMed.
- Z. Nemati, J. Alonso, I. Rodrigo, R. Das, E. Garaio, J. Á. García, I. A. Orue, M.-H. Phan and H. Srikanth, Improving the heating efficiency of iron oxide nanoparticles by tuning their shape and size, J. Phys. Chem. C, 2018, 122(4), 2367–2381 CrossRef CAS.
- D. Vanhecke, F. Crippa, M. Lattuada, S. Balog, B. Rothen-Rutishauser and A. Petri-Fink, Characterization of the Shape Anisotropy of Superparamagnetic Iron Oxide Nanoparticles during Thermal Decomposition, Materials, 2020, 13(9), 2018 CrossRef CAS PubMed.
- J. Lim, S. P. Yeap, C. H. Leow, P. Y. Toh and S. C. Low, Magnetophoresis of iron oxide nanoparticles at low field gradient: The role of shape anisotropy, J. Colloid Interface Sci., 2014, 421, 170–177 CrossRef CAS.
- G. Niraula, J. A. Coaquira, G. Zoppellaro, B. M. Villar, F. Garcia, A. F. Bakuzis, J. O. P. Longo, M. C. Rodrigues, D. Muraca and A. I. Ayesh, Engineering Shape Anisotropy of Fe3O4-γ-Fe2O3 Hollow Nanoparticles for Magnetic Hyperthermia, ACS Appl. Nano Mater., 2021, 4(3), 3148–3158 CrossRef CAS.
- I. Rubia-Rodriguez, A. Santana-Otero, S. Spassov, E. Tombacz, C. Johansson, P. De La Presa, F. J. Teran, M. Del Puerto Morales, S. Veintemillas-Verdaguer, N. T. K. Thanh, M. O. Besenhard, C. Wilhelm, F. Gazeau, Q. Harmer, E. Mayes, B. B. Manshian, S. J. Soenen, Y. Gu, A. Millan, E. K. Efthimiadou, J. Gaudet, P. Goodwill, J. Mansfield, U. Steinhoff, J. Wells, F. Wiekhorst and D. Ortega, Whither Magnetic Hyperthermia? A Tentative Roadmap, Materials, 2021, 14(4), 706 CrossRef CAS.
- K. M. Kirkpatrick, B. H. Zhou, P. C. Bunting and J. D. Rinehart, Size-Tunable Magnetite Nanoparticles from Well-Defined Iron Oleate Precursors, Chem. Mater., 2022, 34(17), 8043–8053 CrossRef CAS PubMed.
- P. Guardia, A. Riedinger, S. Nitti, G. Pugliese, S. Marras, A. Genovese, M. E. Materia, C. Lefevre, L. Manna and T. Pellegrino, One pot synthesis of monodisperse water soluble iron oxide nanocrystals with high values of the specific absorption rate, J. Mater. Chem. B, 2014, 2(28), 4426–4434 RSC.
- C. Caro, A. Avasthi, J. M. Paez-Munoz, M. Pernia Leal and M. L. Garcia-Martin, Passive targeting of high-grade gliomas via the EPR effect: a closed path for metallic nanoparticles?, Biomater. Sci., 2021, 9(23), 7984–7995 RSC.
- J. Park, K. An, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Ultra-large-scale syntheses of monodisperse nanocrystals, Nat. Mater., 2004, 3(12), 891–895 CrossRef CAS PubMed.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, Monodisperse MFe2O4 (M = Fe, Co, Mn) nanoparticles, J. Am. Chem. Soc., 2004, 126(1), 273–279 CrossRef CAS PubMed.
- H. Zeng, P. M. Rice, S. X. Wang and S. Sun, Shape-controlled synthesis and shape-induced texture of MnFe2O4 nanoparticles, J. Am. Chem. Soc., 2004, 126(37), 11458–11459 CrossRef CAS PubMed.
- S. Sun and H. Zeng, Size-controlled synthesis of magnetite nanoparticles, J. Am. Chem. Soc., 2002, 124(28), 8204–8205 CrossRef CAS PubMed.
- H. Yang, T. Ogawa, D. Hasegawa and M. Takahashi, Synthesis and magnetic properties of monodisperse magnetite nanocubes, J. Appl. Phys., 2008, 103(7), 07D526 CrossRef.
- T. Vangijzegem, D. Stanicki, A. Panepinto, V. Socoliuc, L. Vekas, R. N. Muller and S. Laurent, Influence of experimental parameters of a continuous flow process on the properties of very small iron oxide nanoparticles (VSION) designed for T1-weighted magnetic resonance imaging (MRI), Nanomaterials, 2020, 10(4), 757 CrossRef CAS PubMed.
- P. Guardia, R. Di Corato, L. Lartigue, C. Wilhelm, A. Espinosa, M. Garcia-Hernandez, F. Gazeau, L. Manna and T. Pellegrino, Water-soluble iron oxide nanocubes with high values of specific absorption rate for cancer cell hyperthermia treatment, ACS Nano, 2012, 6(4), 3080–3091 CrossRef CAS.
- J. Park, E. Lee, N. M. Hwang, M. Kang, S. C. Kim, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park and T. Hyeon, One-nanometer-scale size-controlled synthesis of monodisperse magnetic iron oxide nanoparticles, Angew. Chem., Int. Ed., 2005, 44(19), 2873–2877 CrossRef PubMed.
- Z. Xu, C. Shen, Y. Hou, H. Gao and S. Sun, Oleylamine as both reducing agent and stabilizer in a facile synthesis of magnetite nanoparticles, Chem. Mater., 2009, 21(9), 1778–1780 CrossRef CAS.
- M. V. Kovalenko, M. I. Bodnarchuk, R. T. Lechner, G. Hesser, F. Schäffler and W. Heiss, Fatty acid salts as stabilizers in size-and shape-controlled nanocrystal synthesis: the case of inverse spinel iron oxide, J. Am. Chem. Soc., 2007, 129(20), 6352–6353 CrossRef CAS PubMed.
- A. Roca, M. Morales and C. Serna, Synthesis of monodispersed magnetite particles from different organometallic precursors, IEEE Trans. Magn., 2006, 42(10), 3025–3029 Search PubMed.
- S. G. Kwon, Y. Piao, J. Park, S. Angappane, Y. Jo, N. M. Hwang, J. G. Park and T. Hyeon, Kinetics of monodisperse iron oxide nanocrystal formation by “heating-up” process, J. Am. Chem. Soc., 2007, 129(41), 12571–12584 CrossRef CAS PubMed.
- E. Wetterskog, M. Agthe, A. Mayence, J. Grins, D. Wang, S. Rana, A. Ahniyaz, G. Salazar-Alvarez and L. Bergström, Precise control over shape and size of iron oxide nanocrystals suitable for assembly into ordered particle arrays, Sci. Technol. Adv. Mater., 2014, 15(5), 055010 CrossRef CAS PubMed.
- L. M. Bronstein, X. Huang, J. Retrum, A. Schmucker, M. Pink, B. D. Stein and B. Dragnea, Influence of iron oleate complex structure on iron oxide nanoparticle formation, Chem. Mater., 2007, 19(15), 3624–3632 CrossRef CAS.
- D. Wilson and M. Langell, XPS analysis of oleylamine/oleic acid capped Fe3O4 nanoparticles as a function of temperature, Appl. Surf. Sci., 2014, 303, 6–13 CrossRef CAS.
- R. A. Harris, P. M. Shumbula and H. T. van der Walt, Analysis of the interaction of surfactants oleic acid and oleylamine with iron oxide nanoparticles through molecular mechanics modeling, Langmuir, 2015, 31(13), 3934–3943 CrossRef CAS PubMed.
- S. Mourdikoudis, M. Menelaou, N. Fiuza-Maneiro, G. Zheng, S. Wei, J. Perez-Juste, L. Polavarapu and Z. Sofer, Oleic acid/oleylamine ligand pair: a versatile combination in the synthesis of colloidal nanoparticles, Nanoscale Horiz., 2022, 7(9), 941–1015 RSC.
- G. Salas, C. Casado, F. J. Teran, R. Miranda, C. J. Serna and M. P. Morales, Controlled synthesis of uniform magnetite nanocrystals with high-quality properties for biomedical applications, J. Mater. Chem., 2012, 22(39), 21065–21075 RSC.
- Y. Bao, J. Sherwood and Z. Sun, Magnetic iron oxide nanoparticles as T 1 contrast agents for magnetic resonance imaging, J. Mater. Chem. C, 2018, 6(6), 1280–1290 RSC.
- S. Ali, S. A. Khan, Z. H. Yamani, M. T. Qamar, M. A. Morsy and S. Sarfraz, Shape-and size-controlled superparamagnetic iron oxide nanoparticles using various reducing agents and their relaxometric properties by Xigo acorn area, Appl. Nanosci., 2019, 9(4), 479–489 CrossRef CAS.
- W. Wu, Q. He and C. Jiang, Magnetic iron oxide nanoparticles: synthesis and surface functionalization strategies, Nanoscale Res. Lett., 2008, 3(11), 397–415 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00516f |
| This journal is © The Royal Society of Chemistry 2023 |