
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid catalysis for enantioselective Baeyer–Villiger oxidation and stereoselective epoxidation: a Cp*Ir complex to fuel FMN and FAD reduction for flavoprotein monooxygenase modules†
Robert
Röllig
 *a,
Caroline E.
Paul
b,
Pierre
Rousselot-Pailley
a,
Selin
Kara
*cd and
Véronique
Alphand
*a
*a,
Caroline E.
Paul
b,
Pierre
Rousselot-Pailley
a,
Selin
Kara
*cd and
Véronique
Alphand
*a
aAix Marseille Univ, CNRS, Centrale Marseille, iSm2 UMR 7313, Marseille, France. E-mail: robert.rollig@inrae.fr; v.alphand@univ-amu.fr
bBiocatalysis section, Department of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629HZ Delft, The Netherlands
cBiocatalysis and Bioprocessing Group, Department of Biological and Chemical Engineering, Aarhus University, Gustav Wieds Vej 10, 8000 Aarhus, Denmark
dInstitute of Technical Chemistry, Leibniz University Hannover, Callinstr. 5, 30167 Hannover, Germany
First published on 4th August 2023
Abstract
Taking advantage of the unique properties of two-component flavo-monooxygenases and the ability of [Cp*Ir(bpy-OMe)H]+ to transfer hydrides to reduce flavins, we extended the scope of the pH- and oxygen–robust iridium(III)-complex to drive the enzymatic reaction of a FMN-dependent Baeyer–Villiger monooxygenase and a FAD-dependent styrene monooxygenase (respectively FPMO Group C and E), using formic acid as H-donor for NADH recycling.
Introduction
Harnessing electron transfer to reveal the unique catalytic properties of enzymes is the wrench to expand biocatalytic reaction systems. Redox platforms adopted by Nature include flavins, which are ubiquitous organic cofactors and mostly occur as flavin mononucleotide, FMN(H2), or flavin adenine dinucleotide, FAD(H2). Flavins are reduced by a stoichiometric supply of organic molecules as sacrificial electron donors, typically via enzymatic electron transfer by other biological mediators such as NAD(P)H or ferredoxins. The efficiency of such flavin-dependent enzymatic systems is demonstrated by numerous chemo-, regio- and enantioselective reactions.1 Despite its broad scope, the industrial challenge for flavin-mediated enzymatic synthesis lies in the cost of its electron donors and the amount of generated waste, as underlined in several reviews.2–4 To enlarge opportunities and allow easier control of the reaction, simplifications of the system aiming for a direct reduction of flavins5 to regenerate the operative form of the enzyme are desired.In this context, certain flavoprotein monooxygenases (FPMOs)1 display an interesting structural feature. In contrast to the single-component FPMOs (group A, B, G and H), groups C to F are two-component enzymes1,6 with a potential for a (direct) chemical hydride transfer. The enzymatic activity originates from an individual protein, the monooxygenase module. In the natural system, a reductase provides the required FMN(H2) or FAD(H2) through NAD(P)H oxidation, followed by the free diffusion of the flavin to the FPMO's active site. Previously, applied with the well-known styrene monooxygenase StyA (group E FPMO), this reductase/nicotinamide system was replaced by a rhodium catalyst allowing the direct FAD reduction in enzymatic epoxidations. However, the system never reached full conversion and stopped at a moderate turnover-number (TON) of eleven for the catalyst.7,8
Baeyer–Villiger (BV) oxidations (oxygen atom insertion adjacent to a carbonyl group) are also among the most applied oxyfunctonalisation in organic synthesis. Despite their high enantioselectivity, the industrial exploitation of enzymatic BV oxidations by type I Baeyer–Villiger Monooxygenases (BVMOs) is hampered by the classic limitations of the enzymatic processes such as low TON,9 while the search for efficient enantioselective chemical catalysts is still ongoing.10
To address this issue, we wished to take advantage of the type II BVMO specificity, which are two component enzymes. Combining both previous concepts in a chemoenzymatic process, we applied an hybrid catalysis strategy to the oxidising module of 2,5-diketocamphane-monooxygenase I (2,5-DKCMO).11–15 This enzyme is one of the two known representatives of the mechanistically intriguing family of type II BVMOs, belonging to the strictly FMN-dependent group C of FPMOs.1,16,17 Due to its outstanding stability,14,15 we combined the enzyme with the cyclopentadienyl type iridium complex 1–(H+),18 a pH- and O2–robust electron mediator, with conspicuous catalytic properties for the acetic acid hydrogenation.19 Then, to expand the scope of 1–(H+) for hybrid catalysis, we associated this catalyst with the highly stereoselective styrene monooxygenase, SfStyA.20 The principle of the hybrid system, combining 1–(H+) with a two-component flavin-dependent monooxygenase, is shown Fig. 1.
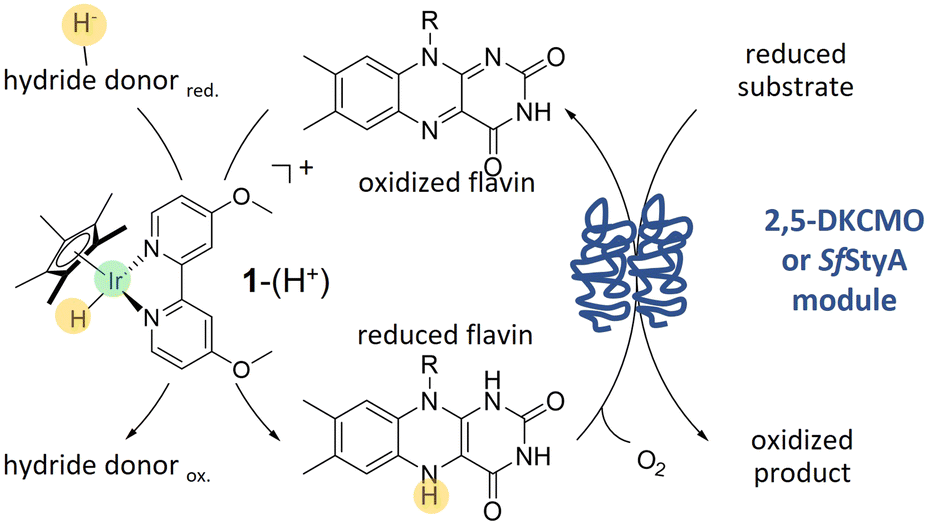 | ||
| Fig. 1 Flavin reduction mediated by 1–(H+) for hydroquinone-dependent monooxygenases. Flavin is reduced in solution by the iridium complex-mediated trans-hydrogenation and (re)oxidized enzymatically through the formation of a flavin C4a-hydroperoxide, which inserts the single oxygen into the (reduced) substrate. A hydride donor (re)generates the reactive 1–(H+) for another cycle. | ||
Experimental
Chemicals
All chemicals were utilized as supplied without further purification. Ketone 2a and lactone (1R,5S)-2c were obtained from Fisher Scientific and Merck respectively. The other authentic lactone samples were synthesized by microbiological biotransformation according to described procedures.21,22 Styrene derivatives and products of the epoxidation were synthesized as described in the literature.23Synthesis of [Cp*Ir(bpy-OMe)SO4]
[Cp*Ir(4,4′-dimethoxy-2,2′-bipyridine)SO4] or [Cp*Ir(bpy-OMe)SO4], named here 1–(SO4), was synthesized with a method adapted to the literature.23,24 The precursor of 1–(SO4), [Cp*Ir(4,4′-dimethoxy-2,2′-bipyridine)Cl2], was synthesized from 20 μmol of dichloro(pentamethylcyclo-pentadienyl)–iridium(III) dimer and two equivalents of 4,4′-dimethoxy-2,2′-bipyridine in 2 mL methanol. The mixture was stirred at room temperature until dissolution and concentrated under reduced pressure. The residue was solubilized in 0.1 mL of methylene chloride before a dropwise addition of cyclohexane. The formed precipitate was filtrated, washed (3 × 1 mL cyclohexane) and dried. Without further purification, 27 μmol of the chloro complex and an equivalent of Ag2SO4 were stirred in 2 mL H2O at room temperature overnight. The precipitated AgCl was washed (3 × 1 mL H2O), the aqueous phases were pooled and concentrated under reduced pressure. 1–(SO4) was obtained as a yellow solid (see Fig. S1† for 1H NMR spectrum). The catalytically active [Cp*Ir(bpy-OMe)H]+, 1–(H+), is formed in (aqueous) solution in the presence of formate,24 as displayed in Fig. S2.†Preparation of the enzymes
The cloning, recombinant expression, and purification of the synthetic 2,5-diketocamphane monooxygenase I from Pseudomonas putida ATCC 17453 (2,5-DKCMO, accession number: AAR21560.1) is described elsewhere.14 The enzyme was desalted, directly lyophilized without cryoprotectants and stored at −20 °C without any further preparation from purification. Styrene monooxygenase from Sphingopyxis fribergensis Kp.5.2 (SfStyA, accession number: AJA07151)25 was produced via recombinant expression in E. coli and purified as previously described.20Substrate and products of 2,5-DKCMO biotransformation
2,5-DKCMO accepts only a few number of substrates.14,15 Among them, bicyclo[3.2.0]hept-2-en-6-one 2a, a well-known substrate of BVMOs, was chosen for its high activity against the enzyme in spite of moderate enantioselectivity and regioselectivity. Two regioisomeric lactones, called respectively “normal” and “abnormal” lactones, can be produced in various ratio as displayed Fig. 2. “Abnormal” lactones are formed almost exclusively by enzymatic BV oxidation in contrast to “normal” lactones that also can be formed by chemical oxidation. With 2,5-DKCMO, as already described,14 the (−) enantiomer of the ketone 2a reacted preferentially to give the (+)-normal lactone, (+)-(1R,5S)-2-oxabicyclo[3.3.0]oct-6-en-3-one 2b. Meanwhile, the (+)-ketone enantiomer of 2a was transformed more slowly into both regioisomeric lactones (−)-(1S,5R)-2b and (+)-(1S,5R)-2c (for details, see Fig. 2 and S3† and for comparison with the “natural” system, see ref. 14).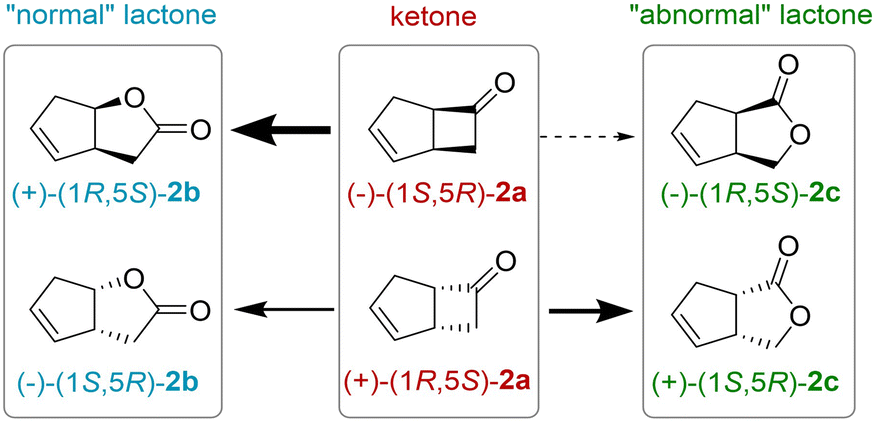 | ||
| Fig. 2 Enantioselective BV oxidation of rac-bicyclo[3.2.0]hept-2-en-6-one 2a. The thickness of the arrows indicates the preference of the enzyme 2,5-DKCMO. | ||
The experiments were described using the “total lactone yield”, defined as the sum the individual lactone yields and by the enantiomeric excess (ee) of the ketone. These data were used for the calculation of E, the enantiomeric ratio, using the non-linear least square analysis of the experimental data set based on Sih's formula:26
| E = ln[1 − (1 + ees)]/ln[1 − c(1 − ees)] |
Biotransformations
Experiments were performed in closed 2 mL glass vials with a total volume of 0.5 mL in K2HPO4-solutions of various molarities. The pH of the solutions was regulated by the addition of formic acid. Exemplary, the obtain pH 6.7, 100 mM formic acid was added in 225 mM K2HPO4-solutions, or 1 M in 450 mM, respectively. All reactions took place in an orbital shaker at 400 rpm and 20 °C. The final concentration of 2,5-DKCMO and SfStyA were respectively 7.3 μM and 5.2 μM.rac-bicyclo[3.2.0]hept-2-en-6-one (rac-2a) was supplied from a 100 mM stock solution in ethanol (to a final ethanol content of 5% (v/v)). Styrene 3a, or its derivates 4a–6a, respectively, were added similarly from a solution in DMSO. Further details on the reaction conditions are listed in the caption of the figures and tables.
Samples were taken from the reaction medium and extracted in ethyl acetate solution of 1.1 mM tridecane as the internal standard for reactions with rac-2a, and 2 mM dodecane for reactions with styrene 3a and styrene derivatives (4a–6a), in a volumetric ratio of 1-to-2. All experiments were performed duplicates or triplicates. Organic phases were analysed by Gas Chromatography according to the conditions described in the ESI.† Compound assignments were determined by comparison with authentic samples. The two enantiomers of 2a and the four lactone isomers 2b and 2c were separated using a 25 m × 0.25 mm CP-Chirasil Dex CB capillary column (Agilent Technologies). Styrene, styrene oxide and their derivatives were separated with a 25 m × 0.25 mm β-Dex120 column (Sigma-Aldrich). Yields were calculated respectively using tridecane and dodecane as internal standard. The parameters of GC analyses are reported in ESI† and Table S1.
Results and discussion
A promising approach for recycling nicotinamide cofactors is the use of a hybrid photocatalyst–enzyme system with a light-harvesting photocatalyst to provide reducing equivalents.27,28 However, efficient photocatalysts have yet to be discovered, so research into simple catalyst–enzyme hybrid systems is still relevant, especially as some of the difficulties to be overcome are similar.Changing the redox state of flavin is demanding, especially in its dissolved, unbound form, as the reduced semiquinones are unstable (single-electron) radicals. To avoid this radical formation, the oxidized quinone should be directly reduced to the hydroquinone in a hydride transfer (two-electron reduction), accomplished efficiently by flavin reductases, or specific protein domains in Nature.1,6,16 Chemically, this reaction is exigent, as catalysts often lack selectivity, and/or stability, especially in the presence of dioxygen.29 Moreover, chemical approaches often operate at reaction conditions unsuitable for most enzymes.
Transition metal complexes catalyse hydride transfers and have been demonstrated to support enzymes to reduce NAD+ and analogues,2,4,18 but have been rarely applied to flavin cofactors directly.7,8,30,31 The d9 transition metal complex [Cp*Rh(bpy)(H)]+ proved successful, as it catalysed the stereospecific in situ regeneration of reduced nicotinamides and flavins from cathodes, phosphite, or formate, and hence has been combined with many enzymes.2–4,32,33 However, as previously mentioned, the direct regeneration of FADH2 by this complex for an enzymatic epoxidation gave moderate outcomes.8
On the other hand, iridium-catalysed transfer hydrogenation was reported,4,34 whereas biotinylated d6 piano-stool Ir complexes were used for artificial (metallo)enzymes.35 Moreover, simpler Ir(III) organometallic complexes, such as [Cp*Ir(H)]+,36 [Cp*Ir(bpy)(H)]+,37 and [Cp*Ir(bpy-OMe)H]+,38 referred here as 1–(H+), drew attention as they are water-soluble, O2-resistant and have been demonstrated to perform the acid-catalysed transfer hydrogenations of alkenes, carbonyl compounds,37 imines38 and also nicotinamides.34,39,40
The latter hydride 1–(H+) offers an unusually broad pH range (pH 4–10),41 making it a promising candidate for chemoenzymatic redox cascades, especially for the (bio)catalytically versatile class of FPMOs. Formic acid was chosen as a readily available, environmentally friendly, and inexpensive hydride source.
Reduction of FMN and FAD by formic acid as hydride donor
To investigate the feasibility of a hybride catalysis with 1–(H+) we tested its ability to reduced FMN and FAD to their hydroquinone forms. The reduction spectra of the flavins are displayed in Fig. 3. Neither FMNH2 nor FADH2 formation was detected during the initial 14 min due to fast aerobic reoxidation.31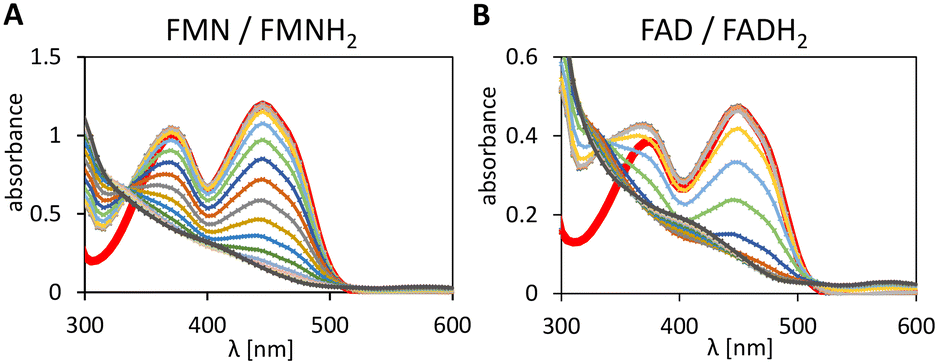 | ||
| Fig. 3 Reduction of FMN (A) and FAD (B) mediated by 1–(H+) with formic acid as the hydride donor. Experimental conditions: K2HPO4 solution [0.23 M, pH 6.7] with 100 mM formic acid, flavin [50 μM], 1–(SO4) [25 μM], 1.0 mL, 20 °C, 30 min in the presence of dioxygen. Spectra were recorded every minute. The red line shows the spectrum before the addition of 1–(SO4). Experiments were performed in triplicates. | ||
The characteristic absorption pattern was not observed in control experiments (no formic acid, or no Ir-complex, data not shown), which demonstrates the flavin reduction is mediated by 1–(H+) and driven by the hydride donation from formic acid.
Mechanism of flavin reduction
The proposed mechanism through the hydrido complex 1–(H+) as the catalytically active species for the flavin (FMN or FAD) reduction is shown Fig. S2.†The flavin hydroquinone is formed in solution, and can diffuse to the active side of the flavoprotein. Its formation empowers the transition of two electrons from the hydride donor to the enzymatic part. Coupling the reactions, however, requires maintaining conditions favourable to both reactions, and also, as we previously demonstrated,14,15 balancing the flavin reduction and oxidation rates to minimize the instability of the reduced flavin in the presence of dioxygen. Indeed, a reduction rate similar or lower than the oxidation rate must be favoured.
Enantioselective BV oxidation by 2,5-DKCMO
Various set-ups were investigated to combine the i) 1–(H+) mediated, formic acid-driven flavin reduction with ii) the regio- and enantioselective BV oxidation of rac-bicyclo[3.2.0]hept-2-en-6-one 2a by the model enzyme.To discriminate whether pH or formic acid concentration are the key parameter, further experiments were carried out at formic acid concentrations of 100 and 200 mM as well as at pH 6.7 and 7.4. The results confirmed i) the strong pH effect with twice the lactone yield at pH 6.7 compared to pH 7.4 at 100 mM formic acid, and revealed ii) an effect of the formic acid concentration with an optimum at 100 mM HCO2H at pH 6.7 as shown in Fig. 4, 5B and C (also see Table S3, Fig. S4†).
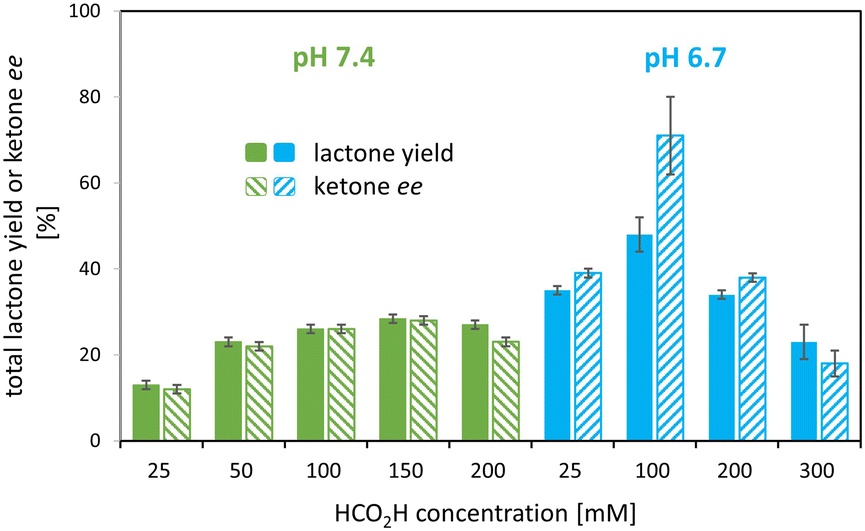 | ||
| Fig. 4 Effect of pH and HCO2H concentration on total lactone yield and ketone ee after 24 h. Experimental conditions: rac-2a [5 mM], oxygenase [312 μg mL−1] (lyophilized), catalase [400–1000 units mL−1], FMN [10 μM], 1–(SO4) [100 μM]. pH was adjusted with K2HPO4 solutions of various molarities. Experiments in duplicates. | ||
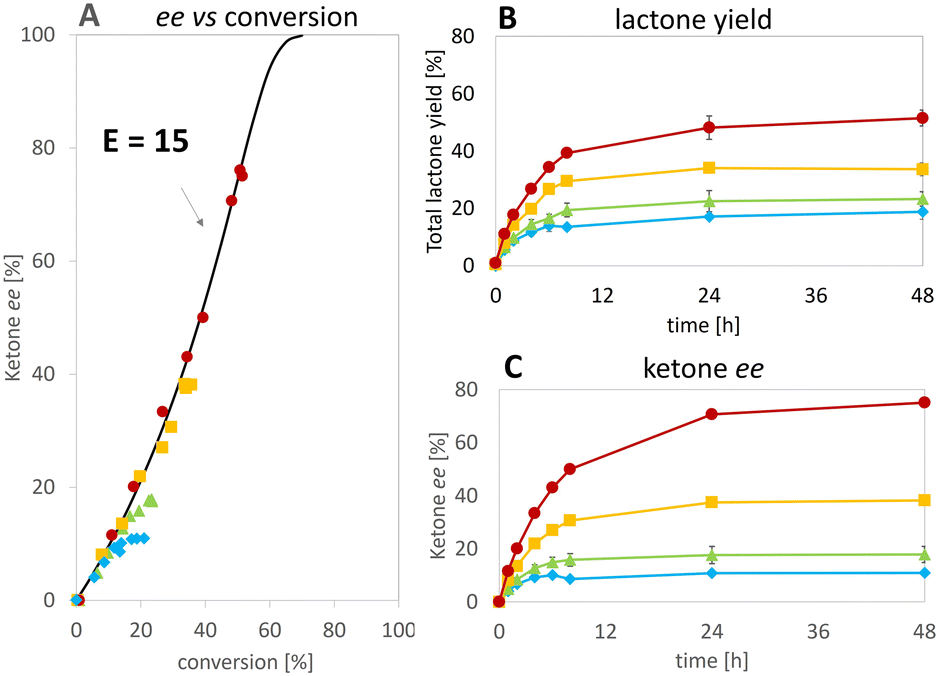 | ||
Fig. 5 Effect of the hydride donor HCO2H concentration at pH 6.7:  100 mM, 100 mM,  200 mM, 200 mM,  300 mM, 300 mM,  400 mM. ees of the ketone 2a over conversion (A); yields of lactones (B) and ees of 2a (C) over time. Experimental conditions: rac-2a [5 mM], oxygenase [312 μg mL−1] (lyophilized), catalase [400–1000 units mL−1], FMN [10 μM], 1–(SO4) [100 μM], pH 6.7. Experiments in duplicates. 400 mM. ees of the ketone 2a over conversion (A); yields of lactones (B) and ees of 2a (C) over time. Experimental conditions: rac-2a [5 mM], oxygenase [312 μg mL−1] (lyophilized), catalase [400–1000 units mL−1], FMN [10 μM], 1–(SO4) [100 μM], pH 6.7. Experiments in duplicates. | ||
At the best, applying the hydride donor HCO2H in 20-fold excess to the substrate, we obtained an analytical yield of 48 ± 4% (TON of 1–(H+) = 24, TON of 2,5-DKCMO = 331, see Table 1). The enantiomeric ratio (E value)26 was 15 (see Fig. 5A), which is in the expected range for this biotransformation. No alcohol side-products (potentially formed by the reduction of the ketone or hydrolysis of the lactones) were observed.
| Systema | Rate | Metal catalyst | Enzyme | Ref. | ||
|---|---|---|---|---|---|---|
| [mM h−1] | TOF | TON | TOF | TON | ||
| a Metal-catalyst/enzyme/H− donor/substrate. b Rate based on the substrate consumption. c Determined after 2 hour. d Determined after 15 min. | ||||||
| Ir/2,5-DKCMO/NADH/2a | 2.9 | 29 | 42 | 400 | 580 | This study |
| Ir/2,5-DKCMO/HCO2H/2a | 0.5c | 5 | 24 | 70 | 330 | |
| Ir/SfStyA/HCO2H/3a | 1.3b | 14 | 66 | 250 | 1140 | |
| Rh/StyA/HCO2H/3a | 1.9d | 10 | 11 | 600 | 670 | 8 |
| SfStyA/BNAH/3a | 3.9 | — | — | 1300 | 1700 | 20 |
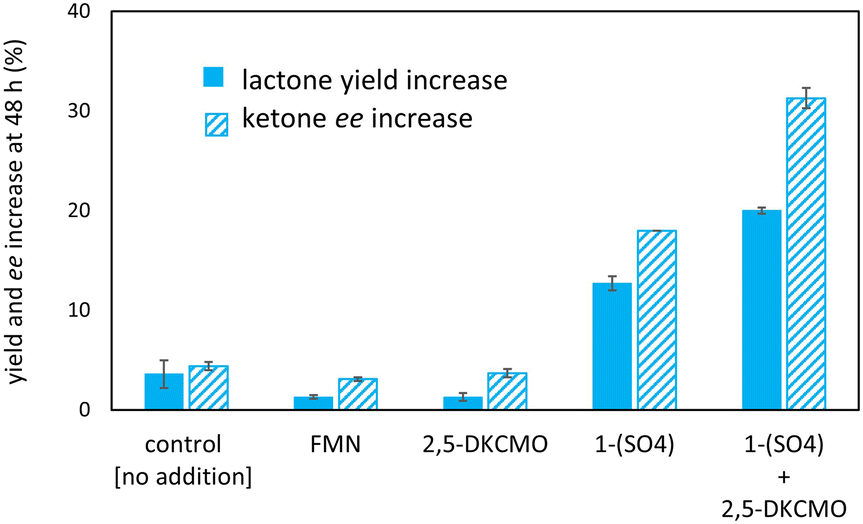 | ||
| Fig. 6 Effect of the independent addition of FMN, 1–(SO4), and 2,5-DKCMO after 24 hours [respectively 10 μM, 50 mM and 7.3 μM]. A total lactone yield of 15 ± 1% and ketone ee of 15 ± 0% were determined at 24 h (prior to the addition of each system component in the concentrations specified here). The control remained untreated. The ordinate displays the differences between 24 h and 48 h. Initial experimental conditions: rac-2a [5 mM], lyophilized oxygenase [312 μg mL−1, 7.3 μM], catalase [400–1000 units mL−1], FMN [10 μM], 1–(SO4) [50 μM], HCO2H [25 mM], K2HPO4 buffer [100 mM, pH 6.7]. Experiments in duplicates. | ||
Consequently, under the conditions tested, deactivation of the Ir 1–(H+) complex occurred before that of the enzyme, unlike the “mutual enzyme/complex deactivation” frequently mentioned in other hybrid systems.7 Nevertheless, the integrity of the enzyme seems to be rapidly affected at the highest concentration of formic acid, as indicated by the decrease in conversion and, to a lesser extent, enantioselectivity in experiments at 300 and 400 mM of HCO2H (see Fig. 5A). In contrast, the experimental data gained at 100 and 200 mM of formic acid fit with the theoretical curve for an E value of 15.
The privation of hydride donor due to H2-formation was not observed, and is highly unlikely, as the reaction requires the excitation of 1–(H+) by the light of distinct wavelengths.42 However, the reaction of the hydrido complex, or the formed flavin hydroquinone, with dioxygen may occur, as already reported for [Cp*Rh(bpy)H]+,41,43 diminishing hydride donor for the enzyme coupled reaction. Interestingly, the increase in enzyme stability and/or decrease in hydrogen peroxide formation (oxygen dilemma) was promoted in photobiocatalysis by Gonçalves et al., who used MOPS buffer as a sacrificial electron donor and FMN and FAD stabilizers.44,45
Applying various concentrations of the Ir complex and NADH (see Fig. S5†), we selected as best conditions 100 μM of 1–(SO4) and 25 mM of NADH for 5 mM substrate, the yield was increased to 80% achieving initial rates >90-fold higher than in the reaction without Ir complex. Enantioselectivity ratio (E = 12) was similar to that calculated for the experiment with 100 mM of HCO2H (see Fig. 7). This observation contrasts with that of de Gonzalo et al.46 who combined a Rh catalyst with FAD-dependent type I BVMOs (FPMO group B, single-component enzyme with integrated cofactor reductase activity) in the presence and absence of NADPH. In this experiment, they reported the decrease of enantioselectivity, which was ascribed to the absence of the native nicotinamide cofactor as it participates to the structural shaping of the active site in the natural system,30,47,48 thus disqualifying this type of enzyme in for the hybrid catalysis we aimed for. In our case, comparing the E values with formic acid and NADH (see Fig. 7A), we demonstrate that the hydride donor does not affect the enantioselectivity of the flavoprotein monooxygenase module.
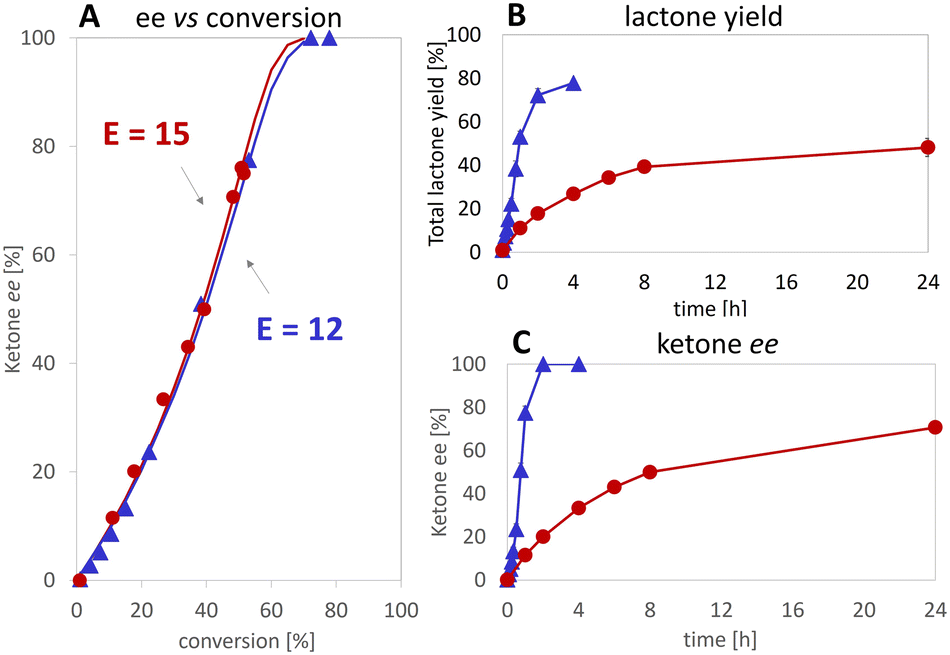 | ||
Fig. 7 Comparison of HCO2H ( ) and NADH ( ) and NADH ( ) as hydride donors. ees of the ketone 2a over conversion (A); yields of lactones (B) and ees of 2a (C) over time. Experimental conditions: lyophilized 2,5-DKCMO [312 μg mL−1], 1–(SO4) [100 μM], FMN [10 μM], catalase [400–1000 U mL−1], HCO2H [100 mM] or NADH [25 mM]. Experiments were performed in duplicates. ) as hydride donors. ees of the ketone 2a over conversion (A); yields of lactones (B) and ees of 2a (C) over time. Experimental conditions: lyophilized 2,5-DKCMO [312 μg mL−1], 1–(SO4) [100 μM], FMN [10 μM], catalase [400–1000 U mL−1], HCO2H [100 mM] or NADH [25 mM]. Experiments were performed in duplicates. | ||
Indeed, we even managed to accelerate the FMN reduction to the level at which the enzymatic reaction (BV oxidation) became the rate-limiting step14,15 as we observed a 4.9-fold higher initial rate when the oxygenase concentration was quintupled (see Fig. S6†). These results likewise demonstrate the roles of NADH as i) hydride acceptor from 1–(H+), and ii) hydride donor for 1–(H2O).
Extention of 1–(SO4) scope: enantioselective epoxidation by SfStyA
The reduction of FAD by the Ir complex 1 shown in Fig. 3B prompted us to investigate the enlargement towards the other flavin cofactor. We selected the highly stereoselective epoxidation of styrene 3a and the styrene derivatives 4a–6a (see Fig. 8) by SfStyA,20 as the model reaction to investigate the compatibility of the iridium complex with another enzyme.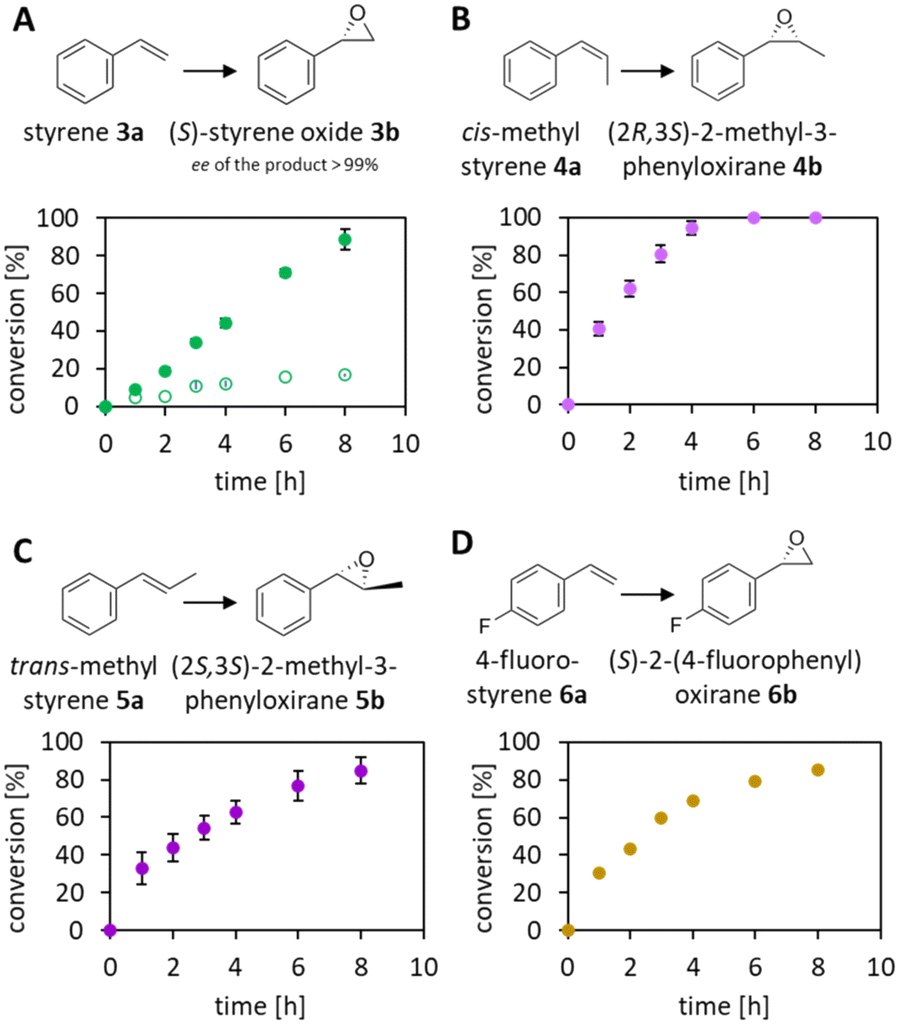 | ||
| Fig. 8 Stereoselective epoxidation of styrene (A) and derivatives (B–D) catalysed by SfStyA and 1–(H+)-mediated FAD reduction. Conversions were based on substrate disappearance. A ● 100 mM and ○ 450 mM of HCO2H, as the hydride donor at pH 6.7. Reaction conditions: substrate [6 mM], SfStyA [247 μg mL−1], FAD [10 μM], 1–(SO4) [91 μM], catalase [400–1000 U mL−1], HCO2H [100 mM]. | ||
Herein, we observed full conversion of all tested substrates with the formation of the corresponding (S)-epoxides (Fig. 8) and their hydrolysis products (not pictured). Similarly to the 2,5-DKCMO system, the trend towards the best performance at 100 mM of formic acid was observed (see Fig. 4, 5 and 8A). Table 1 summarizes the hybrid catalysis of this study and compares kinetic and catalytic parameters with previously reported approaches applied to styrene. The TON of 1–(H+) in the SfStyA/HCO2H system (TON = 66) was higher than in the 2,5-DKCMO/HCO2H system (TON = 24), and likewise higher than reported in a similar StyA/HCO2H approach (TON = 11).8 The values from styrene derivatives (not reported) were in the same range as for styrene with the best activity for cis-methylstyrene as reported in literature.20
We ascribe this increase to the overall faster enzymatic reaction, lessening the interplay of the Ir centre with nucleophilic residues of SfStyA. Here, the substrate was converted before the total inactivation of the catalysts. The difference in the performance of the three systems is, inter alia, due to the structural variations of the enzymes, particularly the number and accessibility of peripheral functional groups, which make them more or less sensitive to deactivation. Our system proved to be significantly more stable than the [Cp*Rh(bpy)–H+]/StyA combination,8 outstretching the catalytic cycles by six-fold while reaching full conversion of the styrene substrate.
Conclusion
The potential contribution of type II BVMOs for further enantioselective chemoenzymatic BV oxidations was highlighted. We showed that 1–(H+) mediates hydride transfers to fuel two-component flavin dependent monooxygenases driven by the oxidation of inexpensive formic acid. The proof of functionality for FMN(H2)-dependent 2,5-DKCMO, as well as FAD(H2)-dependent SfStyA, confirmed the flexibility of the organometallic complex for flavin reductions in aqueous solutions. Nevertheless, the performance still needs to be improved. Immobilized or encapsulated metal catalyst technology32,33,49 may increase the resilience of the combined catalysts, and likewise boost further hybrid catalytic approaches with FMN or FAD dependent enzymes.Author contributions
Conceptualisation: R. Röllig and V. Alphand; data curation: R. Röllig, S. Kara and V. Alphand; investigation: R. Röllig, C. E. Paul, P. Rousselot-Pailley; writing – original draft: R. Röllig and V. Alphand; writing – review & editing: R. Röllig, C. E. Paul, S. Kara and V. Alphand.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the European Union's Horizon 2020 MSCA ITN-EJD program under grant agreement No. 764920. They thank Prof. Dr D. Tischler for the plasmid bearing the SfStyA gene and Dr K. Duquesne for the plasmid bearing the 2,5-DKCMO gene.References
- C. E. Paul, D. Eggerichs, A. H. Westphal, D. Tischler and W. J. H. van Berkel, Biotechnol. Adv., 2021, 107712 CrossRef CAS PubMed.
- F. Hollmann, I. W. C. E. Arends and K. Buehler, ChemCatChem, 2010, 2, 762–782 CrossRef CAS.
- X. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. Shi, Chem, 2017, 2, 621–654 CAS.
- S. Fukuzumi, Y.-M. Lee and W. Nam, J. Inorg. Biochem., 2019, 199, 110777 CrossRef CAS PubMed.
- M. Ismail, L. Schroeder, M. Frese, T. Kottke, F. Hollmann, C. E. Paul and N. Sewald, ACS Catal., 2019, 9, 1389–1395 CrossRef CAS PubMed.
- J. Sucharitakul, R. Tinikul and P. Chaiyen, Arch. Biochem. Biophys., 2014, 555–556, 33–46 CrossRef CAS PubMed.
- F. Hollmann, B. Witholt and A. Schmid, J. Mol. Catal. B: Enzym., 2002, 19–20, 167–176 CrossRef CAS.
- F. Hollmann, P.-C. Lin, B. Witholt and A. Schmid, J. Am. Chem. Soc., 2003, 125, 8209–8217 CrossRef CAS PubMed.
- S. Schmidt and U. T. Bornscheuer, in The Enzymes, Elsevier, 2020, vol. 47, pp. 231–281 Search PubMed.
- C. Liu, K. Wen, X. Zeng and Y. Peng, Adv. Synth. Catal., 2020, 362, 1015–1031 CrossRef CAS.
- D. G. Taylor and P. W. Trudgill, J. Bacteriol., 1986, 165, 489–497 CrossRef CAS PubMed.
- H. Iwaki, S. Grosse, H. Bergeron, H. Leisch, K. Morley, Y. Hasegawa and P. C. K. Lau, Appl. Environ. Microbiol., 2013, 79, 3282–3293 CrossRef CAS PubMed.
- M. Kadow, K. Balke, A. Willetts, U. T. Bornscheuer and J.-E. Bäckvall, Appl. Microbiol. Biotechnol., 2014, 98, 3975–3986 CrossRef CAS PubMed.
- R. Röllig, C. E. Paul, M. Claeys-Bruno, K. Duquesne, S. Kara and V. Alphand, Org. Biomol. Chem., 2021, 19, 3441–3450 RSC.
- R. Röllig, C. E. Paul, K. Duquesne, S. Kara and V. Alphand, ChemBioChem, 2022, 23, e202200293 CrossRef PubMed.
- W. Van Berkel, N. Kamerbeek and M. Fraaije, J. Biotechnol., 2006, 124, 670–689 CrossRef CAS PubMed.
- A. Willetts, Microorganisms, 2018, 7, 1 CrossRef PubMed.
- J. Canivet, G. Süss-Fink and P. Štěpnička, Eur. J. Inorg. Chem., 2007, 2007, 4736–4742 CrossRef.
- T. P. Brewster, A. J. M. Miller, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2013, 135, 16022–16025 CrossRef CAS PubMed.
- L. Martínez-Montero, D. Tischler, P. Süss, A. Schallmey, M. C. R. Franssen, F. Hollmann and C. E. Paul, Catal. Sci. Technol., 2021, 11, 5077–5085 RSC.
- I. Hilker, M. C. Gutiérrez, R. Furstoss, J. Ward, R. Wohlgemuth and V. Alphand, Nat. Protoc., 2008, 3, 546–554 CrossRef CAS PubMed.
- L. Butinar, M. Mohorčič, V. Deyris, K. Duquesne, G. Iacazio, M. Claeys-Bruno, J. Friedrich and V. Alphand, Phytochemistry, 2015, 117, 144–153 CrossRef CAS PubMed.
- L. Dadci, H. Elias, U. Frey, A. Hoernig, U. Koelle, A. E. Merbach, H. Paulus and J. S. Schneider, Inorg. Chem., 1995, 34, 306–315 CrossRef CAS.
- J. M. McFarland and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 13490–13491 CrossRef CAS PubMed.
- M. Oelschlägel, C. Rückert, J. Kalinowski, G. Schmidt, M. Schlömann and D. Tischler, Int. J. Syst. Evol. Microbiol., 2015, 65, 3008–3015 CrossRef PubMed.
- C. S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294–7299 CrossRef CAS.
- N. Yang, Y. Tian, M. Zhang, X. Peng, F. Li, J. Li, Y. Li, B. Fan, F. Wang and H. Song, Biotechnol. Adv., 2022, 54, 107808 CrossRef CAS PubMed.
- V. Alphand, W. J. H. Van Berkel, V. Jurkaš, S. Kara, R. Kourist, W. Kroutil, F. Mascia, M. M. Nowaczyk, C. E. Paul, S. Schmidt, J. Spasic, P. Tamagnini and C. K. Winkler, ChemPhotoChem, 2023, e202200325 CrossRef CAS.
- C. E. Paul, D. Tischler, A. Riedel, T. Heine, N. Itoh and F. Hollmann, ACS Catal., 2015, 5, 2961–2965 CrossRef CAS.
- A. Taglieber, F. Schulz, F. Hollmann, M. Rusek and M. T. Reetz, ChemBioChem, 2008, 9, 565–572 CrossRef CAS PubMed.
- M. M. Grau, M. Poizat, I. W. C. E. Arends and F. Hollmann, Appl. Organomet. Chem., 2010, 24, 380–385 CAS.
- T. Himiyama, M. Waki, Y. Maegawa and S. Inagaki, Angew. Chem., Int. Ed., 2019, 58, 9150–9154 CrossRef CAS PubMed.
- Y. Deng, M. Odziomek, C. Sanchez, O. Back, V. Mougel and M. Fontecave, ChemCatChem, 2020, 12, 1236–1243 CrossRef CAS.
- Y. Okamoto and T. R. Ward, Biochemistry, 2017, 56, 5223–5224 CrossRef CAS PubMed.
- A. D. Liang, J. Serrano-Plana, R. L. Peterson and T. R. Ward, Acc. Chem. Res., 2019, 52, 585–595 CrossRef CAS PubMed.
- N. Makihara, S. Ogo and Y. Watanabe, Organometallics, 2001, 20, 497–500 CrossRef CAS.
- T. Abura, S. Ogo, Y. Watanabe and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 4149–4154 CrossRef CAS PubMed.
- J. M. McFarland and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 13490–13491 CrossRef CAS PubMed.
- S. Bose, A. H. Ngo and L. H. Do, J. Am. Chem. Soc., 2017, 139, 8792–8795 CrossRef CAS PubMed.
- S. Betanzos-Lara, Z. Liu, A. Habtemariam, A. M. Pizarro, B. Qamar and P. J. Sadler, Angew. Chem., Int. Ed., 2012, 4 Search PubMed.
- S. M. Barrett, S. A. Slattery and A. J. M. Miller, ACS Catal., 2015, 5, 6320–6327 CrossRef CAS.
- S. M. Barrett, B. M. Stratakes, M. B. Chambers, D. A. Kurtz, C. L. Pitman, J. L. Dempsey and A. J. M. Miller, Chem. Sci., 2020, 11, 6442–6449 RSC.
- F. Hollmann, A. Schmid and E. Steckhan, Angew. Chem., Int. Ed., 2001, 40, 169–171 CrossRef CAS.
- L. C. P. Gonçalves, H. R. Mansouri, E. L. Bastos, M. Abdellah, B. S. Fadiga, J. Sá, F. Rudroff and M. D. Mihovilovic, Catal. Sci. Technol., 2019, 9, 1365–1371 RSC.
- L. C. P. Gonçalves, H. R. Mansouri, S. PourMehdi, M. Abdellah, B. S. Fadiga, E. L. Bastos, J. Sá, M. D. Mihovilovic and F. Rudroff, Catal. Sci. Technol., 2019, 9, 2682–2688 RSC.
- G. de Gonzalo, G. Ottolina, G. Carrea and M. W. Fraaije, Chem. Commun., 2005, 3724 RSC.
- F. Hollmann, A. Taglieber, F. Schulz and M. T. Reetz, Angew. Chem., Int. Ed., 2007, 46, 2903–2906 CrossRef CAS PubMed.
- A. Alfieri, E. Malito, R. Orru, M. W. Fraaije and A. Mattevi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6572–6577 CrossRef CAS PubMed.
- Y. Okamoto, V. Köhler, C. E. Paul, F. Hollmann and T. R. Ward, ACS Catal., 2016, 6, 3553–3557 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00411b |
| This journal is © The Royal Society of Chemistry 2023 |