
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh order assembly of multiple protein cages with homogeneous sizes and shapes via limited cage surface engineering†
Hyeok Jin
Oh
and
Yongwon
Jung
 *
*
Department of Chemistry, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: ywjung@kaist.ac.kr; Fax: +82-42-350-2810; Tel: +82-42-350-2817
First published on 30th December 2022
Abstract
Protein cages are attractive building blocks to build high order materials such as 3D cage lattices, which offer accurately ordered bio-templates. However, controlling the size or valency of these cage-to-cage assemblies is extremely difficult due to highly multivalent and symmetric cage structures. Here, various high order cage assemblies with homogeneous sizes and geometries are constructed by developing an anisotropic ferritin cage with limitedly exposed binding modules, leucine zipper. The anisotropic ferritin is produced as expressed in cells without the need of complex in vitro cage fabrication by careful subunit manipulation. Ferritin cages with limitedly exposed zippers are assembled around a core ferritin with fully exposed opposing zippers, generating homogeneous high order structures, whereas two fully exposed ferritins are assembled into heterogeneous cage aggregates. Diverse fully exposed core cages are prepared by varying the zipper-ferritin fusion geometries and even by using larger cage structures. With these core cages and the anisotropic ferritin, a range of high order cage assemblies with diverse ferritin valencies (3 to over 12) and sizes (over 40 nm) are created. Cell surface binding and internalization of cage structures are greatly varied by assembly sizes, where high order ferritins are clearly more effective than monomeric ferritin.
Introduction
Most natural processes occur by precise assemblies of multiple biomolecules. In particular, proteins are major assembly components due to their diverse structures and functions. These supramolecular assemblies with their collective (multivalent) properties enable dynamic biochemical and physical activities, which individual molecules cannot offer.1 A wide range of engineered supramolecular protein nanostructures accordingly have been developed as versatile templates for biosensors, cargo delivery, and biocatalysts.2 The most widely investigated examples of such protein supramolecular assemblies are protein cages.3 These protein shell structures have hollow inside cavities that can stably confine diverse foreign cargo molecules, and cage structures can be precisely modified by other biomolecules or synthetics moieties with near atomic-level structural accuracy. Various natural as well as artificial protein cage structures have been reported as multifunctional nanomaterials for diverse applications.3–6 For example, engineered protein cages have been successfully used as vehicles for drug delivery,7,8 reactors for bio/chemical reactions,9,10 and new scaffolds of vaccines.11,12Protein cages are also excellent building blocks to construct even higher order protein structures such as 3D protein lattices.13 These lattices allow highly accurate structural organization of multiple protein cages, offering attractive materials with ordered cage functionalities such as reaction centers. As a lattice building block, protein cages exhibit valuable features including (1) uniform, fairly rigid, and symmetric structures, (2) inside cavities for functional cargo encapsulation, and (3) the availability of facile surface modification to induce cage-to-cage assembly. Various cage proteins such as virus capsids and cage-like proteins14–20 have been assembled into high order structures and used as ordered templates for special arrangement of biochemical reactions.21–25 Ferritin, a ubiquitous iron storage protein, is the most extensively investigated protein cage, and many different strategies to engineer inside and outside ferritin cages have been reported.26 Accordingly, ferritins have also been a popular choice for high order cage array construction. Diverse 2D and 3D ferritin arrays with regular packing patterns have been reported.27–31 In these examples, various cage assembly forces such as electrostatic interactions, aromatic stacking, and metal coordination were symmetrically introduced into 24 meric ferritin cages to build ferritin lattices.
Despite well-ordered structures, self-assembled cage lattices grow continuously in size until most cages are exhausted, similar to the high order assembly of many other nanoparticles (Scheme 1a).32–34 The size or dimension of high order cage assemblies is thereby generally heterogeneous and uncontrollable. Assembling protein cages into defined geometries and sizes will greatly expand the applicability of high order protein cage structures.35 For instance, the size and shape of therapeutic nanoparticles are critical factors for effective tumor targeting.36 However, only a few strategies to manipulate the size distribution of cage-to-cage assemblies by restricting continuous growth have been reported. Biotinylated 24-meric ferritin cages were assembled via biotin-binding tetrameric streptavidin, where the size of ferritin-streptavidin aggregates could be varied by the ferritin/streptavidin ratios.37 In another example, ferritin cages were linearly linked by poly-lysine, and the assembly length distribution could be varied again by the stoichiometric ratios or the cultivation time.38 Recently, more discrete high order cage assemblies were prepared by gel-based purification of heterogeneous assembly mixtures.39 Stable chemical linking and subsequent gel purification produced discrete assemblies containing between one to four ferritin cages. This study also demonstrated that ferritin oligomers provided greatly enhanced anti-tumor therapeutic efficacy compared to a single ferritin cage, again highlighting the need for high order cage assemblies with defined sizes and shapes.
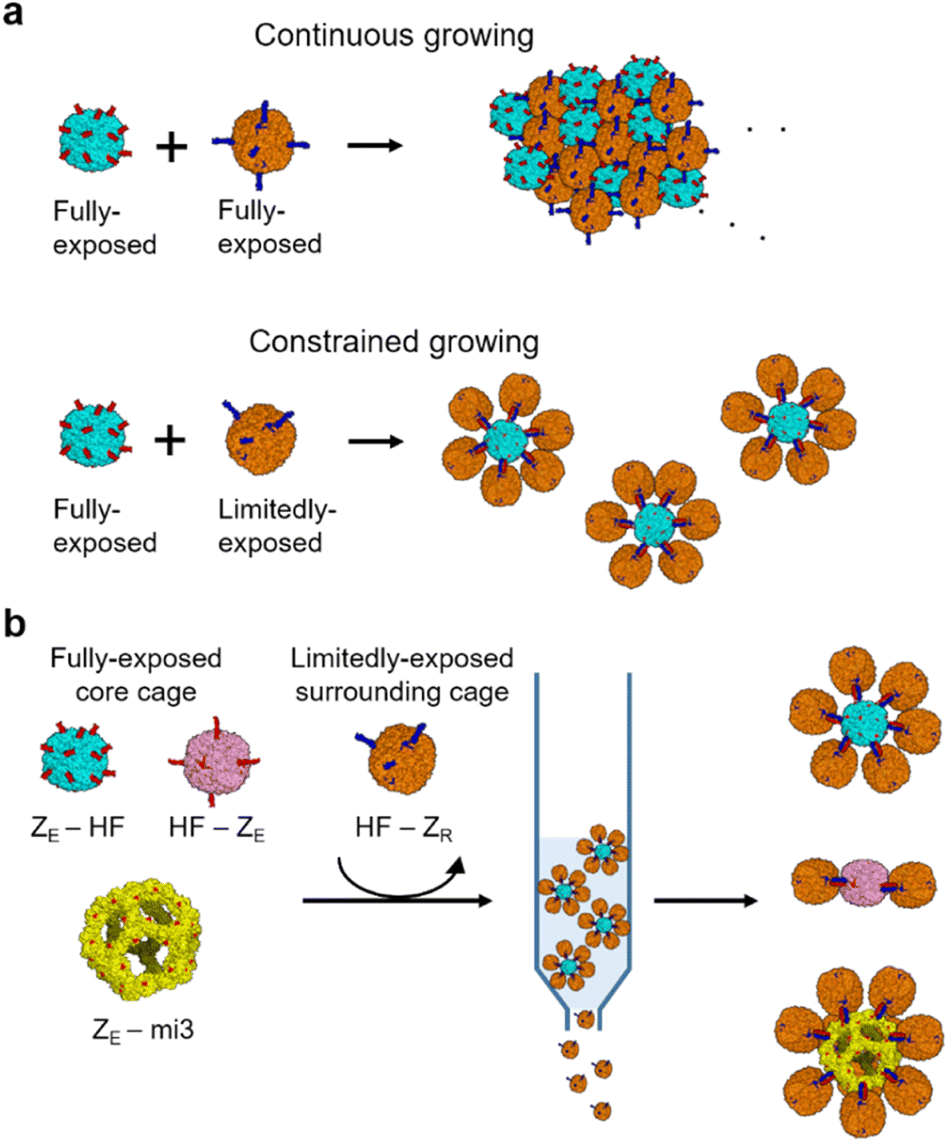 | ||
| Scheme 1 Fabrication of homogeneous high order ferritin assemblies. (a) Schematic diagrams of continuous assembly growth between fully exposed ferritin cages and constrained assembly growth between fully and limitedly exposed ferritin cages. (b) Isolation of high order cage assemblies by removing un-assembled cages with affinity chromatography. | ||
Here, we developed a reliable and facile strategy to build high order ferritin assemblies with homogeneous sizes and shapes by designing a limitedly activated nanocage for constrained assembly (Scheme 1a). We demonstrated that strong binding pair leucine zippers could be fully or limitedly exposed on a ferritin surface with different orientations by systematically manipulating ferritin-zipper fusion. Ferritin cages with limitedly exposed zippers could be assembled on a fully activated cage with minimal continuous growth to form defined cage assemblies with a core cage and surrounding cages (Scheme 1a). Constructed high order structures could be isolated from unassembled cage monomers by simple affinity chromatography, and thereby preparation could be easily scaled up (Scheme 1b). Cage-to-cage assemblies consisting of seven or three ferritin nanocages were constructed through this constrained assembly. In addition, a larger protein cage than ferritin could also be used as a core cage to produce a spherical high order cage structure with over ten surrounding ferritin cages. More ellipsoidal cage assemblies were also constructed by using dimeric ferritin as a core. Lastly, we examined constructed cage assemblies for their cellular binding and uptake, which were clearly more effective compared to a single ferritin cage.
Results and discussion
Fabrication of a ferritin variant with limited surface modification for constrained cage assembly
Diverse high order colloidal structures with precise architectures have been assembled by using synthetic particle building blocks with anisotropically exposed functional groups on their surfaces.33,34,40 We envisioned that growth-constrained high order cage assemblies could be similarly fabricated by confining the activated surface of the protein cage building blocks. However, assembling anisotropic protein cages is difficult and often requires complicated engineering processes including co-assembly of different cage subunits. Ferritin cages can be dis-assembled and re-assembled by controlling pH, and several studies reported partial surface activation of ferritin nanocages by scrambling unmodified and modified subunits during this dis-/re-assembly process.41–46 However, structurally defected ferritins could be generated during the dis-/re-assembly processes,47 and more importantly, we observed that proper ferritin re-assembly was hampered by genetic fusion of foreign proteins such as stable binding modules for cage-to-cage assembly (Fig. S1†). It would be ideal if ferritin building blocks with anisotropically exposed binding modules could be constructed by simple self-assembly of a single kind of subunits as expressed in cells. This approach would minimize ferritin variant exposure to harsh conditions and enable reliable cage production even on a large scale.The human heavy-chain ferritin (HF) subunits form a 24-meric cage structure with the N-termini exposed to the exterior surface, whereas the C-termini (at the end of the E-helix) are facing the inner cavity (Fig. 1a).26 It has been reported that these inward-facing E-helices can be pulled out from the cage (flopping) by fusing proteins exceeding certain sizes to the ferritin C-terminus.48 We envisioned that if 24 E-helices were designed to be only partially flopped, anisotropically activated ferritin cages could be formed from identical subunits rather than co-assembly of different subunits. Sizes, shapes, and charges of fused proteins will likely govern the flopping process. We fused a strongly interacting leucine zipper pair, ZE and ZR (Kd ∼10−15 M) (Fig. S2†), which form a parallel coiled-coil,49 to ferritin for stable and well-defined cage-to-cage assembly. More importantly, these zipper peptides possess small sizes (∼40 amino acids) and distinct charges. Upon C-terminal fusion, many small zipper peptides might stay inside the cavity with only a small portion of peptides exposed to the outside. Moreover, negative ZE and positive ZR might have different flopping tendencies since the ferritin cavity is overall negatively charged.50 ZE and ZR were fused to both the N- and C-termini of ferritin with 15 residue linkers (all protein sequences are given in the ESI†). N-terminal fusion will generate cages with 24 exposed zipper peptides, which are evenly distributed around the cage (Fig. 1a).51,52 On the other hand, for C-terminal fused and fully flopped cages, six sets of four closely clustered zipper peptides will be exposed with an octahedral geometry on a cage surface (Fig. 1a and b). Wild-type ferritin (HF), ZE-fused ferritins (N-terminal fusion: ZE-HF, C-terminal fusion: HF-ZE), and C-terminal ZR-fused ferritin (HF-ZR) were all expressed well in cells into ∼12 nm spherical cage structures, as shown in their transmission electron microscopy (TEM) images (Fig. 1c), except for N-terminal ZR-fused ferritin (ZR-HF). ZR-HF could not properly assemble into the cage structure (Fig. S3†).
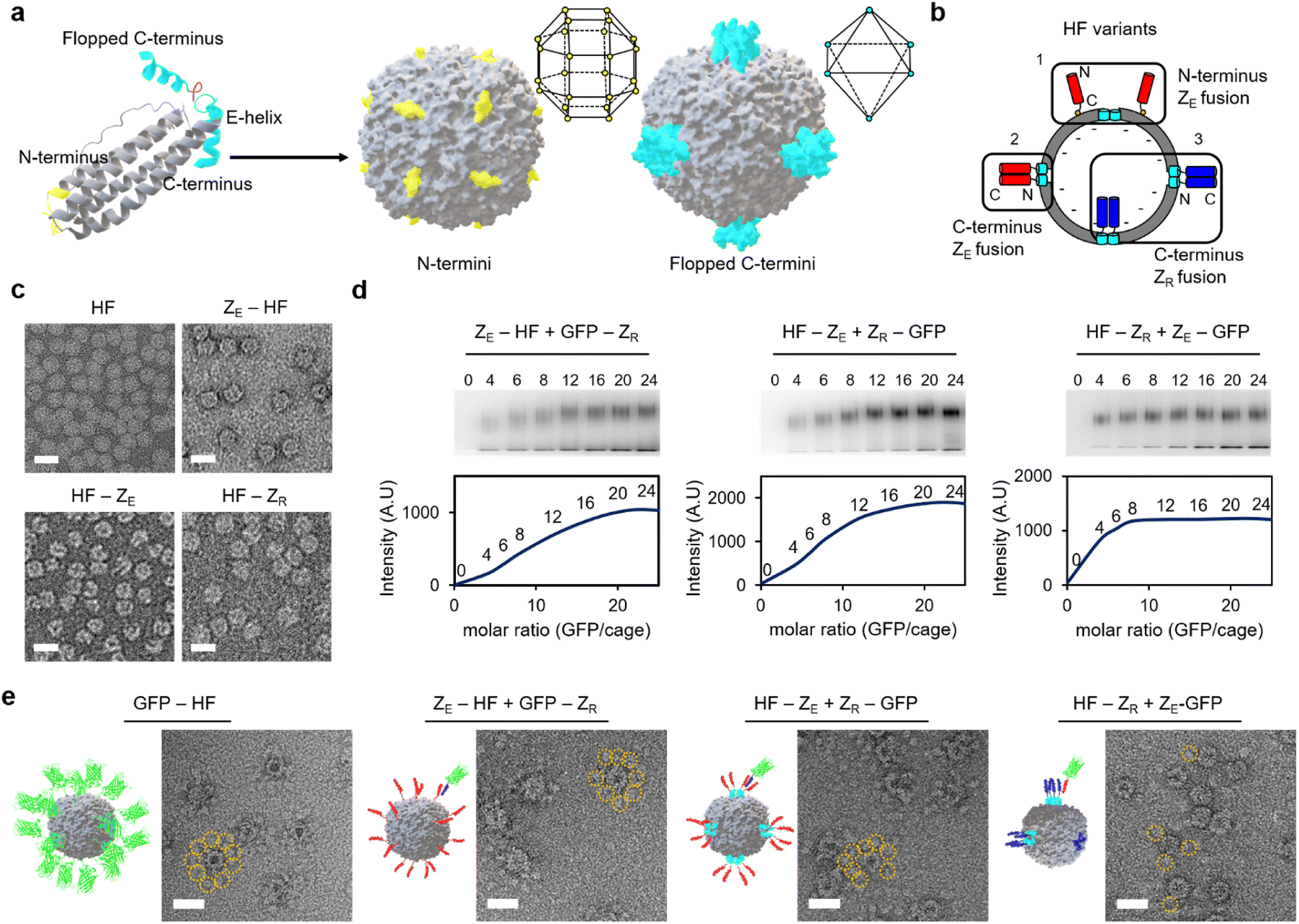 | ||
| Fig. 1 Fabrication of ferritin variants with fully or limitedly exposed zipper peptides. (a) Structures of the ferritin subunit and assembled 24-meric cages. The location of N-terminus (yellow) and C-terminus at the E-helix (cyan) is indicated. Surface exposed N-termini and flopped C-termini on the cage are depicted, and their surface distributions are described with polyhedra. (b) Schematic diagram of zipper exposure on a ferritin surface. ZE peptides (red) fused to both of the N-termini and C-termini of ferritin are fully exposed, whereas C-terminal fused ZR peptides (blue) are only partially exposed. (c) Negative-stained TEM images of HF, ZE-HF, HF-ZE, and HF-ZR. Scale bars are 20 nm. (d) Gel shift assays of zipper-fused ferritin variants binding to their complementary zipper-fused GFPs with increased GFP per cage ratios (0 to 24). Native gel fluorescence (GFP) images (top) and relative intensities of HF-GFP complexes (bottom) are shown. (e) Negative-stained TEM images and schematic structures of GFP-fused HF (GFP-HF) and zipper-fused ferritin variants bound with their complementary zipper-fused GFPs. Stained GFPs around a few HF variants are indicated with dotted yellow circles. Scale bars are 20 nm. | ||
Binding titration of surface exposed zippers on ferritin variants
The exposure degree of zipper peptides on the ferritin outer surface was investigated by titrating binding activities. ZE- and ZR-fused GFPs were prepared, and their binding to zipper ferritin variants at various ratios was examined with an electrophoretic mobility shift assay (Fig. 1d). GFP-ZR (ZR fusion to the GFP C-terminus) was treated with ZE-HF, while ZR-GFP (ZR fusion to the GFP N-terminus) was treated with HF-ZE to ensure an identical ferritin-GFP complex geometry for different variants. When more GFP was added, bands for the ferritin-GFP complexes were gradually shifted and intensified, indicating increased numbers of bound GFPs. The shifted band intensity for ZE-HF, which has fully exposed ZE on the ferritin N-termini, was increased until approximately 20 to 24 GFPs were added to a single ferritin (Fig. 1d). The GFP binding pattern of HF-ZE was similar to that of ZE-HF, indicating that C-terminal fused ZE peptides were also fully exposed (flopped) from the cage. Interestingly, however, the complex band intensity of HF-ZR was saturated after only 6 to 8 GFPs were added. It is clear that only a portion of ZR peptides flopped from the cage, unlike ZE. While negative ZE easily flopped from the negatively charged cavity, positive ZR might preferentially remain in the cavity, and only several ZR peptides were flopped, likely due to the limited cavity space. In fact, when the 15 residue linker between HF and ZR was removed to reduce the C-terminal fused protein size, most ZR peptides remained inside the cage, showing no complex formation with ZE-GFP (Fig. S4†). On the other hand, linker flexibility did not significantly affect this C-terminal flopping (Fig. S4b†). The linker length was further varied from 15 residues to 5 or 30 residues. Flopped ZR peptides were clearly fewer for the shorter linker variant and more for the longer linker variant compared to the original HF-ZR with the 15 residue linker (Fig. S4c†).Full and limited exposure of zipper peptides on ferritin variants were further verified with negative stained TEM images of GFP bound ferritins. For comparison, GFP was genetically fused to the ferritin N-terminus (GFP-HF), which represents a ferritin cage with 24 GFPs that are fully and evenly exposed on the cage surface. Surrounding GFPs along the cage boundary were clearly observed in GFP-HF TEM images (Fig. 1e and S5†). GFP bound zipper ferritin variants were next prepared by adding excess zipper-GFPs to zipper ferritins, and unbound GFPs were removed by size exclusion chromatography (SEC). For both ZE-HF and HF-ZE, many surface bound GFPs were clearly observed in TEM images, similar to GFP-HF, verifying full surface exposure of fused ZE. On the other hand, only a few (one to three) GFPs were observed on ZE-GFP treated HF-ZR, again supporting limited ZR exposure on HF-ZR (Fig. S5†). Dynamic light scattering (DLS) was also used to examine the sizes of HF-ZE and HF-ZR before and after GFP binding (Fig. S6†). Before GFP binding, both variants showed nearly the same size (∼13.5 nm). After GFP binding, however, fully flopped HF-ZE showed a larger size (∼18.2 nm) than limitedly flopped HF-ZR (∼15.7 nm), likely due to a much higher number of GFPs on the fully flopped ferritin. All binding titration data indicate that ferritin variants with fully and limitedly exposed zipper peptides can be constructed by careful genetic fusion of zippers to ferritins.
Constrained high order ferritin assembly with limitedly exposed zipper ferritin
Constructed surface activated ferritin variants with fully or partially exposed zippers were next used to induce constrained ferritin assembly. To properly evaluate the roles of the limitedly activated ferritin (HF-ZR) for constrained assembly, we also prepared fully exposed ZR-ferritin for comparison. It has been reported that the C-terminal E-helix of ferritin can be removed without disrupting the cage structure, and newly generated C-termini of this truncated ferritin (F160) are fully exposed from the cage (Fig. S7a†).53,54 We fused ZR to the F160 C-terminus (F160-ZR) to prepare fully exposed ZR-ferritin. The fused subunits assembled into the ferritin cage structure (Fig. S7b†), and more importantly, gel-based binding titration and TEM images of F160-ZR with bound ZE-GFP confirmed that ZR peptides were fully exposed, similar to ZE-fused ferritins (Fig. S7c†). When fully exposed ZE-ferritins were mixed with also fully exposed F160-ZR, highly heterogeneous and large ferritin assemblages were formed (Fig. 2a), likely through continuous growth of cage-to-cage assemblies. We also added excess F160-ZR to ZE-HF since uneven building block concentrations often hinder continuous high order assembly. However, even with six times more F160-ZR than ZE-HF, these zipper ferritins were still assembled into large heterogeneous assemblages (Fig. 2a and S8a†). The high valency (24) of the surface binding modules, which are also spherically distributed on a ∼13 nm diameter cage, is likely responsible for this strong (uncontrollable) cage assembly growth.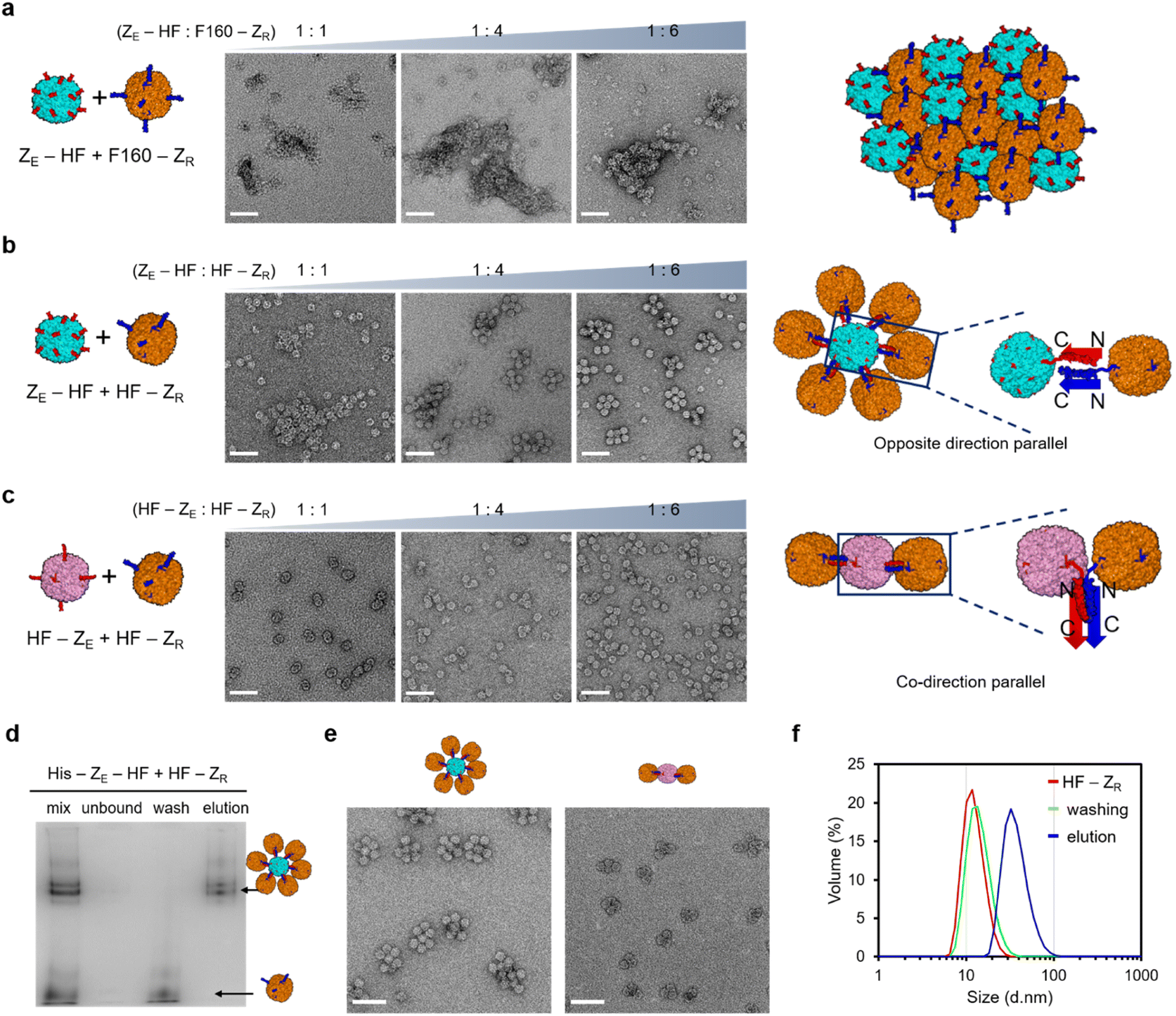 | ||
Fig. 2 Construction of homogeneous high order ferritin assemblies. (a) TEM images of ZE-HF and F160-ZR mixtures with increased F160-ZR. (ZE-HF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :F160-ZR = 1:1, 1:4, and 1:6) Assembled cage structures are schematically expressed in the right. (b) TEM images of ZE-HF and HF-ZR mixtures with increased HF-ZR. Assembled cage structures with zipper binding orientations (arrows) are schematically expressed in the right. (c) TEM images of HF-ZE and HF-ZR mixtures with increased HF-ZR. (d) High order ferritin (ZE-HF:HF-ZR = 1:10) purification with His affinity chromatography. A native fluorescence gel image of cage mixture (mix), resin-unbound flow through (unbound), washed un-assembled HF-ZR, and eluted high order structure is shown. (e) TEM images of purified high order ZE-HF/HF-ZR (left) and HF-ZE/HF-ZR (right). (f) Dynamic light scattering data of HF-ZR (red), un-assembled HF-ZR (green), and eluted high order structure (blue). All scale bars are 50 nm. :F160-ZR = 1:1, 1:4, and 1:6) Assembled cage structures are schematically expressed in the right. (b) TEM images of ZE-HF and HF-ZR mixtures with increased HF-ZR. Assembled cage structures with zipper binding orientations (arrows) are schematically expressed in the right. (c) TEM images of HF-ZE and HF-ZR mixtures with increased HF-ZR. (d) High order ferritin (ZE-HF:HF-ZR = 1:10) purification with His affinity chromatography. A native fluorescence gel image of cage mixture (mix), resin-unbound flow through (unbound), washed un-assembled HF-ZR, and eluted high order structure is shown. (e) TEM images of purified high order ZE-HF/HF-ZR (left) and HF-ZE/HF-ZR (right). (f) Dynamic light scattering data of HF-ZR (red), un-assembled HF-ZR (green), and eluted high order structure (blue). All scale bars are 50 nm. | ||
On the other hand, when anisotropic HF-ZR was added to ZE-HF, continuous assembly growth was clearly limited by increased HF-ZR (Fig. 2b). In particular, at a mixing ratio of 1:6 (ZE-HF:HF-ZR), most observed high order assemblies showed a finite architecture, where a single central cage (likely ZE-HF) was wrapped around once by other cages (likely HF-ZR). With only a limited number of exposed ZR on HF-ZR, after HF-ZR binds to the first ZE-HF, HF-ZR binding to the second ZE-HF for continuous growth could be heavily prevented. We also expect that multiple ZE/ZR interactions are involved in a single HF–HF assembly, which also limits the binding of HF-ZR (with a few exposed ZR) to multiple ZE-HF. Anisotropic HF-ZR was also added to another fully activated HF-ZE. Interestingly, only a few (one to three) HF-ZR cages were bound to HF-ZE at all mixing ratios as shown in the TEM images (Fig. 2c) and band shift gel data (Fig. S8†). Unlike ZE-HF, HF-ZE has an octahedral surface distribution of clustered ZE peptides, offering fewer binding locations for HF-ZR cages. More importantly, the same zipper fusion orientation for HF-ZE and HF-ZR (both C-terminal fusion) demands tight contact between two ferritins for the parallel ZE/ZR binding-based cage assembly (Fig. 2c). In addition, this close ferritin proximity might also hamper HF-ZR wrapping around HF-ZE, whereas ZE-HF was effectively surrounded by HF-ZR through a more spaced ZE/ZR assembly (Fig. 2b). These data indicate that different high order ferritin assemblies with homogeneous sizes and shapes could be constructed by varying the zipper fusion orientations with asymmetrically-flopped HF-ZR. We also examined high order cage assembly with the HF-ZR linker variants (Fig. S4†). The longer linker variant with more exposed ZR was not effective for constrained assembly, showing large cage assemblages at most mixing ratios as demonstrated in TEM (Fig. S9†) and gel shift (Fig. S10†) analyses. On the other hand, the shorter linker variant with fewer surface ZR was highly effective for constrained assembly, further supporting that highly limited binding zipper exposure is critical for constrained cage assembly.
To remove unbound HF-ZR from high order ferritin assemblies, a 6His tag was introduced only into the ZE-fused ferritins (ZE-HF and HF-ZE). His tag-fused ZE-ferritins were mixed with excess (10 fold for ZE-HF, 6 fold for HF-ZE) HF-ZR, and ZE-ferritins with bound HF-ZR were isolated from unbound HF-ZR by using His tag affinity columns. Unbound HF-ZR ferritin monomers were effectively removed by this simple affinity chromatography (Fig. 2d and S11†). SEC analyses also confirmed that high order assemblies were effectively separated from unbound HF-ZR by affinity purification (Fig. S12†). The resulting purified ferritin assemblies showed uniform sizes and shapes in their TEM images (Fig. 2e and S13†). A DLS analysis also confirmed the effective isolation of high order assemblies by removing unbound HF-ZR (Fig. 2f). The DLS size distribution of the high order structure was maintained even after 20 days incubation at 4 °C, and native gel and TEM images of these structures were also unaffected by high salt (500 mM NaCl) or high temperature (42 °C) incubation (Fig. S14†).
Different high order assemblies could be separated in a 3% native gel according to the number of bound HF-ZR (Fig. 2d, S8 and S10†), whereas they were undistinguishable by DLS (Fig. 2f) and SEC (Fig. S12†) analyses. To determine the exact number of bound HF-ZR, assemblies on each native gel band were extracted and analyzed by TEM (Fig. S15†). TEM images showed that isolated ZE-HF/HF-ZR assemblies contained mostly four to six HF-ZR cages surrounding a single ZE-HF with a size of ∼38 nm. Relative band intensities indicated that over 90% of isolated assemblies were the intended ferritin assemblies with a single ZE-HF cage and surrounding HF-ZR cages (Fig. S15a†). The ZE-HF/HF-ZR assemblies were also examined by atomic force microscopy (AFM). These assemblies appeared as rounded particles with sizes of 40–50 nm, while continuous assembly structures between ZE-HF and F160-ZR appeared as highly irregular shapes with over 200 nm sizes (Fig. S16†). The number of bound HF-ZR cages could be decreased by lowering the ZE-HF:HF-ZR mixing ratios, but lowering the mixing ratios also stimulated the formation of polydisperse large assemblages (Fig. 2b and S17†). Mostly two or three HF-ZR cages were bound to HF-ZE in isolated HF-ZE/HF-ZR assemblies (Fig. 2e and S15b†). We also examined another fully exposed ZE-fused ferritin, F160-ZE, which behaved similarly to fully exposed HF-ZE (Fig. S18†). Interestingly, one or two more HF-ZR cages were bound to F160-ZE than to HF-ZE (Fig. S18d†). E-helix truncated F160-ZE might provide better spacing and orientation of exposed ZE than HF-ZE for HF-ZR assembly.
High order ferritin assembly with a bigger core cage
Constrained ferritin assembly with limitedly activated ferritin was conducted with a larger core protein cage. An artificially designed protein cage mi3 contains 60 subunits and has an icosahedron structure with a diameter of ∼25 nm.55 A His tag and ZE were fused to mi3 at the N-terminus (ZE-mi3), which is exposed to the cage exterior. Expressed ZE-mi3 assembled into the native spherical structure, likely with 60 fully exposed ZE peptides (Fig. 3a). A gel band shift assay suggested that nearly 12 anisotropic HF-ZR could be assembled onto this large 60-meric protein cage (Fig. S19†). Excess (20 fold) HF-ZR was mixed with ZE-mi3, and unbound HF-ZR was removed again with His tag affinity columns (Fig. 3b and S20a†). At a low ZE-mi3:HF-ZR ratio (1:6), polydisperse cage assemblages were formed (Fig. 3c). However, with more HF-ZR (ZE-mi3:HF-ZR ratios = 1:12 and 1:24), nearly 50 nm size high order cage assemblies were uniformly formed. TEM images suggest that potentially ∼12 HF-ZR could be surrounding a core mi3 cage (Fig. S20b†). AFM images of ZE-mi3/HF-ZR showed 70–80 nm size particles, and these assemblies were also stable against high salt and high temperature (Fig. S20c and d†). With the present anisotropic HF-ZR, diverse cage-to-cage assemblies can be designed by varying the core template proteins with fused ZE. About 36% to 56% of initially mixed core cage proteins were recovered as high order assemblies after affinity purification and dialysis (Fig. S21†).
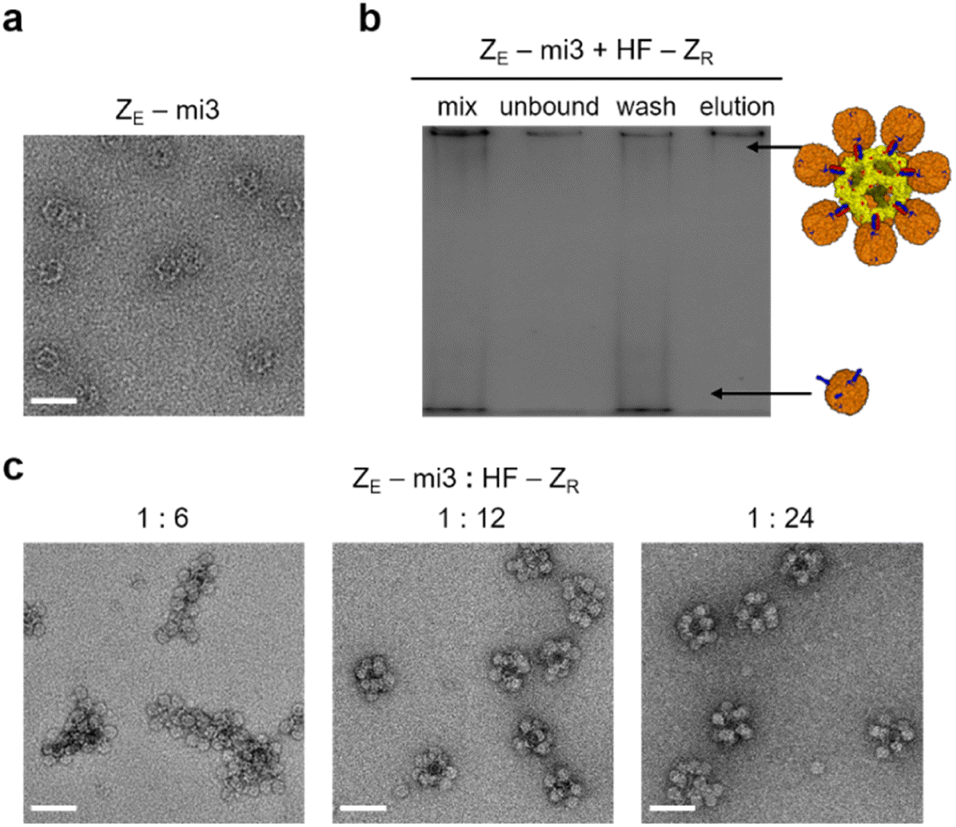 | ||
| Fig. 3 High order ferritin assembly with mi3. (a) TEM image of ZE-mi3. (b) Fluorescence gel image for affinity purification of ZE-mi3/HF-ZR assembly (ZE-mi3:HF-ZR = 1:12). (c) TEM images of purified high order ZE-mi3/HF-ZR assemblies with varying cage ratios (ZE-mi3:HF-ZR = 1:6, 1:12, and 1:24). All scale bars are 50 nm. | ||
High order ferritin assembly with multimeric ferritin cages
To further demonstrate how different high order ferritins can be assembled with different core proteins, particularly with non-spherical structures, we constructed multimeric ferritin structures. Ferritin subunits assemble into a 24-meric cage through a stable dimer intermediate.56 We therefore tandemly fused two subunits with a flexible linker, where the resulting repeat protein can form a single chain dimer intermediate (di-subunit). The ferritin cage structure can be assembled with 12 of these di-subunits (Fig. 4a), and this cage will have 12 exposed N-termini instead of 24. We also envisioned that two monomeric subunits of a di-subunit can individually participate in two separate cage formations, and then two ferritin cages can be linked by this inter-cage assembly, producing a stable ferritin dimer. The di-subunit was expressed and assembled in cells, and purified ferritin proteins were analyzed with a native gel. Interestingly, the assembled ferritin proteins showed a ladder shape band pattern, indicating the formation of multimeric ferritin cage assembly mixtures (Fig. 4b). This indicates that the inter-cage assembly by the di-subunit can also repeatedly occur to link multiple ferritins. Three fast migrating proteins (likely mono-, di-, and tri-meric ferritins) were isolated by gel-based separation, followed by electro-elution, as previously reported.57 TEM images clearly showed the monomeric and multimeric cage structures of these isolated ferritins (Fig. 4c).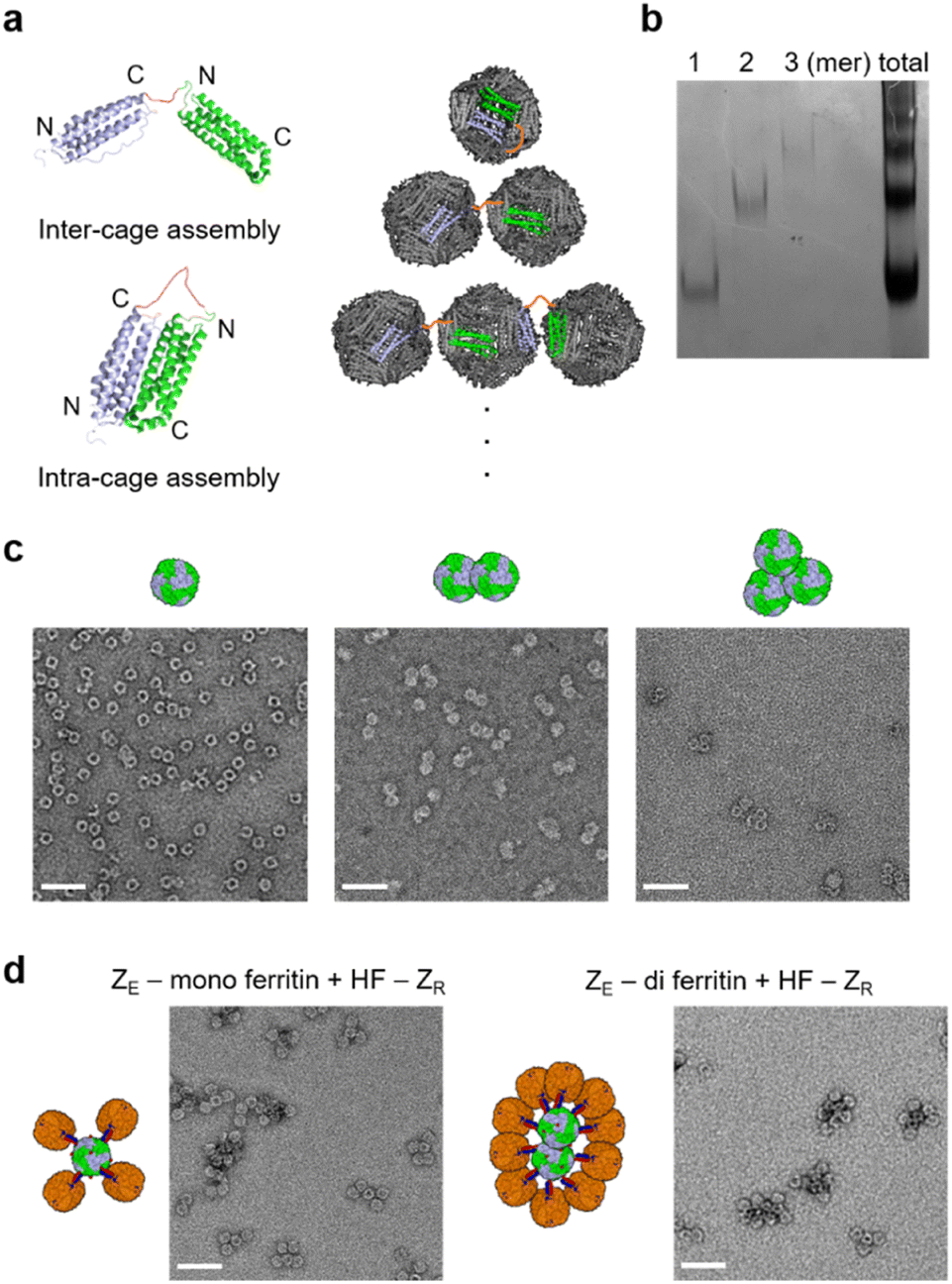 | ||
| Fig. 4 High order ferritin assembly with multimeric ferritins. (a) Schematic diagrams of multimeric ferritin formation from the tandemly repeated ferritin subunits (di-subunit). Ferritin di-subunits assembled into multimeric ferritins via inter-cage assembly (link two cages). (b) Native gel image of the expressed multimeric ferritin mixture (total) and gel-purified discrete ferritin multimers (1, 2, 3 mer). (c) TEM images of the gel-purified ferritin monomer, dimer, and trimer. (d) TEM images of purified high order assemblies between the ZE-fused ferritin monomer (left) or dimer (right) and HF-ZR. All scale bars are 50 nm. | ||
Monomeric and dimeric ferritins consisting of di-subunits with a N-terminal His tag and ZE peptide were prepared. Again, excess HF-ZR was added to these ferritins, and unbound HF-ZR was removed from the high order assemblies. Only three or four HF-ZR cages were bound to the monomeric ferritin (Fig. 4d), likely due to there being only 12 exposed ZE rather than 24 for ZE-HF. For the dimeric (di-) ferritin, more ellipsoidal high order ferritin assemblies were obtained by surrounding two linked cages with HF-ZR, demonstrating that cage-to-cage assembly shapes can be varied by applying different shapes of core proteins.
Cellular binding and uptake of fabricated high order ferritins
Lastly, to examine the influence of increased sizes and valencies of high order cage assemblies on their behaviors, we investigated cellular binding and internalization of fabricated ferritin structures. As discussed earlier, the size and shape of carrier materials are key factors that dictate their pharmaceutical properties.36,58 Since human ferritin has shown an intrinsic targeting ability to the TfR1 receptor, which is overexpressed on tumor cell surfaces,59 ferritin assemblies (ZE-HF/HF-ZR and ZE-mi3/HF-ZR) were directly treated with HeLa cells. Monomeric ferritin was also treated with cells for comparison. ZE-HF and ZE-mi3 were assembled with dye-labeled HF-ZR, and we estimated that 6 and 12 HF-ZR were bound to ZE-HF and ZE-mi3, respectively. All ferritin samples were treated with cells with a final concentration of 100 nM for a monomeric cage (i.e. assembly concentration: 100/7 nM for ZE-HF/HF-ZR with total ∼7 HF and 100/12 nM for ZE-mi3/HF-ZR with total ∼12 HF). First, the ferritin binding efficiency to cell surfaces was investigated at 4 °C, where ferritin internalization will be inhibited.60 Surface bound ferritin signals were clearly higher for the high order assemblies than for the monomeric ferritin (Fig. 5a and b), likely due to the ferritin multivalency of the assemblies. In particular, ZE-HF/HF-ZR showed the strongest signal, which was even two-fold higher than the signal of larger ZE-mi3/HF-ZR. ZE-HF/HF-ZR with ∼six surrounding HF-ZR might have a sufficiently high ferritin multivalency for strong cell surface binding and also a high assembly concentration (∼100/7 nM), compared to ZE-mi3/HF-ZR, which has a high multivalency with ∼12 surrounding HF-ZR but with a low assembly concentration (∼100/12 nM). Next, cellular internalization of the ferritin assemblies was monitored over time at 37 °C. Again, the high order assemblies showed stronger internalization than the monomeric ferritin (Fig. 5c and d). In addition, internalization signals also increased more rapidly for the high order structure.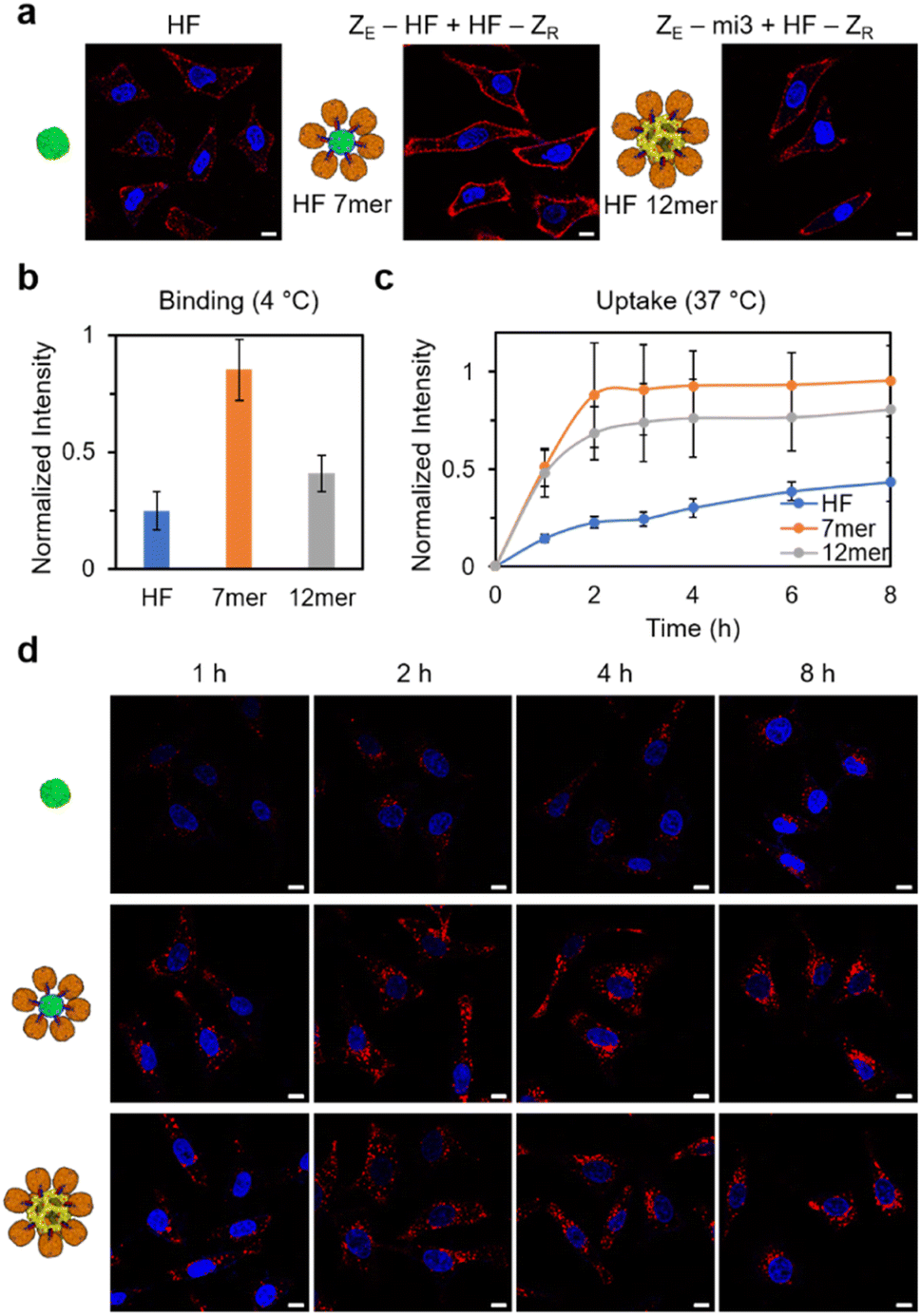 | ||
| Fig. 5 Cellular uptake of high order ferritin assemblies. (a) Confocal images of HeLa cells (blue, DAPI staining) treated with dye-labeled ferritin and high order ferritin assemblies (red) at 4 °C to examine cage surface binding. (b) Relative cell surface binding intensities of ferritin (1 mer), ZE-HF/HF-ZR (total 7 HF, 7 mer), and ZE-mi3/HF-ZR (total 12 HF, 12 mer). (c) Relative cellular uptake signals of ferritin variants at different time points. (d) Confocal images of HeLa cells treated with dye-labeled ferritin variants (red) at 37 °C to examine cage cell internalization. All scale bars are 10 μm. | ||
Conclusions
Various high order ferritin assemblies with defined sizes and shapes were constructed by developing an anisotropic ferritin with limitedly exposed leucine zipper peptides. The anisotropically flopped ferritin was produced by single component cellular expression without applying conventional in vitro co-assembly of different components, making this method highly reliable and suitable for mass production. This limitedly activated ferritin allowed constrained cage-to-cage assembly processes, which were not possible with fully activated ferritins. In addition, this constrained assembly did not require precise mixing concentration control, which is often challenging with different protein cages. We also inserted zipper peptides with different orientations to ferritin variants (HF and F160), which widened the pool of fully activated core proteins for the construction of high order ferritins with varied sizes and shapes. Due to the rigid parallel interaction structure of zipper peptides, different zipper orientations generated different steric hindrance, leading to assembly variation.We also constructed larger and even ellipsoidal cage assemblies by using mi3 and di-ferritin core cages. Increasing the size of protein cage structures is one of the major ongoing goals in the field of protein cage design. The expanded range of attainable cage sizes provided in our work will be highly valuable, particularly for bio-medical applications. In fact, we showed that the ZE-HF/HF-ZR assembly with about seven ferritin cages showed a vastly stronger cell internalization ability than the monomeric HF and the ZE-mi3/HF-ZR assembly with over twelve cages. Our modular building block cages can be individually functionalized and assembled, and these multi-function assemblies can be used in diverse applications such as synergistic drug delivery or linked reaction center design. For future studies, we will develop various limitedly activated ferritins with binding moieties other than ZR by controlling the size and charge of fused moieties. Varying the binding pairs will further diversify attainable structures of high order ferritin structures. Moreover, anisotropic HF will also be assembled with synthetic nanostructures to generate hybrid cage materials.
Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
H. Oh conducted all experiments. H. Oh and Y. Jung designed the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant (NRF-2019R1A2C2008558) and by the Basic Research Laboratory (BRL) Program (2022R1A4A1033471) funded by the Korea government (MSIT).References
- G. P. Whyburn, Y. Li and Y. Huang, J. Mater. Chem., 2008, 18, 3755–3762 RSC.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS.
- M. Uchida, M. T. Klem, M. Allen, P. Suci, M. Flenniken, E. Gillitzer, Z. Varpness, L. O. Liepold, M. Young and T. Douglas, Adv. Mater., 2007, 19, 1025–1042 CrossRef CAS.
- M. Rother, M. G. Nussbaumer, K. Renggli and N. Bruns, Chem. Soc. Rev., 2016, 45, 6213–6249 RSC.
- Y. Zhang, M. S. Ardejani and B. P. Orner, Chem.–Asian J., 2016, 11, 2814–2828 CrossRef CAS.
- M. L. Flenniken, M. Uchida, L. O. Liepold, S. Kang, M. J. Young and T. Douglas, Curr. Top. Microbiol. Immunol., 2009, 327, 71–93 CAS.
- M. Liang, K. Fan, M. Zhou, D. Duan, J. Zheng, D. Yang, J. Feng and X. Yan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14900–14905 CrossRef CAS PubMed.
- L. Li, M. Munoz-Culla, U. Carmona, M. P. Lopez, F. Yang, C. Trigueros, D. Otaegui, L. Zhang and M. Knez, Biomaterials, 2016, 98, 143–151 CrossRef CAS.
- T. Ueno, M. Abe, K. Hirata, S. Abe, M. Suzuki, N. Shimizu, M. Yamamoto, M. Takata and Y. Watanabe, J. Am. Chem. Soc., 2009, 131, 5094–5100 CrossRef.
- A. Liu, C. H. H. Traulsen and J. J. L. M. Cornelissen, ACS Catal., 2016, 6, 3084–3091 CrossRef.
- M. Kanekiyo, C. J. Wei, H. M. Yassine, P. M. McTamney, J. C. Boyington, J. R. Whittle, S. S. Rao, W. P. Kong, L. Wang and G. J. Nabel, Nature, 2013, 499, 102–106 CrossRef PubMed.
- W. Wang, X. Zhou, Y. Bian, S. Wang, Q. Chai, Z. Guo, Z. Wang, P. Zhu, H. Peng, X. Yan, W. Li, Y. X. Fu and M. Zhu, Nat. Nanotechnol., 2020, 15, 406–416 CrossRef.
- W. M. Aumiller, M. Uchida and T. Douglas, Chem. Soc. Rev., 2018, 47, 3433–3469 RSC.
- M. A. Kostiainen, O. Kasyutich, J. J. Cornelissen and R. J. Nolte, Nat. Chem., 2010, 2, 394–399 CrossRef PubMed.
- M. A. Kostiainen, P. Hiekkataipale, A. Laiho, V. Lemieux, J. Seitsonen, J. Ruokolainen and P. Ceci, Nat. Nanotechnol., 2013, 8, 52–56 CrossRef PubMed.
- M. A. Kostiainen, P. Hiekkataipale, J. Á. de la Torre, R. J. M. Nolte and J. J. L. M. Cornelissen, J. Mater. Chem., 2011, 21, 2112–2117 RSC.
- H. Yoshimura, E. Edwards, M. Uchida, K. McCoy, R. Roychoudhury, B. Schwarz, D. Patterson and T. Douglas, J. Phys. Chem. B, 2016, 120, 5938–5944 CrossRef PubMed.
- M. Uchida, B. LaFrance, C. C. Broomell, P. E. Prevelige, Jr. and T. Douglas, Small, 2015, 11, 1562–1570 CrossRef PubMed.
- V. Liljestrom, J. Seitsonen and M. A. Kostiainen, ACS Nano, 2015, 9, 11278–11285 CrossRef.
- P. A. Sontz, J. B. Bailey, S. Ahn and F. A. Tezcan, J. Am. Chem. Soc., 2015, 137, 11598–11601 CrossRef.
- V. Liljestrom, J. Mikkila and M. A. Kostiainen, Nat. Commun., 2014, 5, 4445 CrossRef.
- M. Lach, M. Kunzle and T. Beck, Chem.–Eur. J., 2017, 23, 17482–17486 CrossRef PubMed.
- M. Uchida, K. McCoy, M. Fukuto, L. Yang, H. Yoshimura, H. M. Miettinen, B. LaFrance, D. P. Patterson, B. Schwarz, J. A. Karty, P. E. Prevelige, Jr., B. Lee and T. Douglas, ACS Nano, 2018, 12, 942–953 CrossRef PubMed.
- H. Choi, B. Choi, G. J. Kim, H. U. Kim, H. Kim, H. S. Jung and S. Kang, Small, 2018, 14, e1801488 CrossRef PubMed.
- S. Chakraborti, A. Korpi, M. Kumar, P. Stepien, M. A. Kostiainen and J. G. Heddle, Nano Lett., 2019, 19, 3918–3924 CrossRef.
- G. Jutz, P. van Rijn, B. Santos Miranda and A. Boker, Chem. Rev., 2015, 115, 1653–1701 CrossRef.
- J. Mikkila, E. Anaya-Plaza, V. Liljestrom, J. R. Caston, T. Torres, L. Escosura Ade and M. A. Kostiainen, ACS Nano, 2016, 10, 1565–1571 CrossRef PubMed.
- M. Kunzle, T. Eckert and T. Beck, J. Am. Chem. Soc., 2016, 138, 12731–12734 CrossRef.
- K. Zhou, J. Zang, H. Chen, W. Wang, H. Wang and G. Zhao, ACS Nano, 2018, 12, 11323–11332 CrossRef.
- K. Zhou, H. Chen, S. Zhang, Y. Wang and G. Zhao, Chem. Commun., 2019, 55, 7510–7513 RSC.
- B. Zheng, K. Zhou, T. Zhang, C. Lv and G. Zhao, Nano Lett., 2019, 19, 4023–4028 CrossRef.
- Y. Xia, T. D. Nguyen, M. Yang, B. Lee, A. Santos, P. Podsiadlo, Z. Tang, S. C. Glotzer and N. A. Kotov, Nat. Nanotechnol., 2011, 6, 580–587 CrossRef.
- R. Mérindol, E. Duguet and S. Ravaine, Chem.–Asian J., 2019, 14, 3232–3239 CrossRef.
- E. Elacqua, X. Zheng, C. Shillingford, M. Liu and M. Weck, Acc. Chem. Res., 2017, 50, 2756–2766 CrossRef PubMed.
- A. Korpi, E. Anaya-Plaza, S. Valimaki and M. Kostiainen, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1578 Search PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef PubMed.
- D. Men, T. T. Zhang, L. W. Hou, J. Zhou, Z. P. Zhang, Y. Y. Shi, J. L. Zhang, Z. Q. Cui, J. Y. Deng, D. B. Wang and X. E. Zhang, ACS Nano, 2015, 9, 10852–10860 CrossRef PubMed.
- R. Yang, L. Chen, T. Zhang, S. Yang, X. Leng and G. Zhao, Chem. Commun., 2014, 50, 481–483 RSC.
- Q. Liu, J. Tian, J. Liu, M. Zhu, Z. Gao, X. Hu, A. C. Midgley, J. Wu, X. Wang, D. Kong, J. Zhuang, J. Liu, X. Yan and X. Huang, Adv. Mater., 2021, 33, e2103128 CrossRef PubMed.
- T. S. Skelhon, Y. Chen and S. A. Bon, Soft Matter, 2014, 10, 7730–7735 RSC.
- X. Huang, J. Chisholm, J. Zhuang, Y. Xiao, G. Duncan, X. Chen, J. S. Suk and J. Hanes, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6595–E6602 Search PubMed.
- X. Huang, J. Zhuang, S. W. Chung, B. Huang, G. Halpert, K. Negron, X. Sun, J. Yang, Y. Oh, P. M. Hwang, J. Hanes and J. S. Suk, ACS Nano, 2019, 13, 236–247 CrossRef CAS.
- X. Lin, J. Xie, G. Niu, F. Zhang, H. Gao, M. Yang, Q. Quan, M. A. Aronova, G. Zhang, S. Lee, R. Leapman and X. Chen, Nano Lett., 2011, 11, 814–819 CrossRef CAS PubMed.
- L. Ma, F. Li, T. Fang, J. Zhang and Q. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 11024–11031 CrossRef CAS.
- S. Kang, P. A. Suci, C. C. Broomell, K. Iwahori, M. Kobayashi, I. Yamashita, M. Young and T. Douglas, Nano Lett., 2009, 9, 2360–2366 CrossRef CAS.
- P. A. Suci, S. Kang, M. Young and T. Douglas, J. Am. Chem. Soc., 2009, 131, 9164–9165 CrossRef CAS.
- M. Kim, Y. Rho, K. S. Jin, B. Ahn, S. Jung, H. Kim and M. Ree, Biomacromolecules, 2011, 12, 1629–1640 CrossRef CAS PubMed.
- G. C. A. Luzzago, EMBO J., 1989, 8, 569–576 CrossRef.
- J. R. Moll, S. B. Ruvinov, I. Pastan and C. Vinson, Protein Sci., 2001, 10, 649–655 CrossRef CAS PubMed.
- T. Douglas and D. R. Ripoll, Protein Sci., 1998, 7, 1083–1091 CrossRef CAS PubMed.
- I. S. Jeon, J. D. Yoo, S. Gurung, M. Kim, C. Lee, E. J. Park, R. W. Park, B. Lee and S. Kim, Biomaterials, 2021, 270, 120685 CrossRef CAS PubMed.
- S. Kim, J. O. Jeon, E. Jun, J. Jee, H. K. Jung, B. H. Lee, I. S. Kim and S. Kim, Biomacromolecules, 2016, 17, 1150–1159 CrossRef CAS PubMed.
- M. A. Baraibar, B. B. Muhoberac, H. J. Garringer, T. D. Hurley and R. Vidal, J. Biol. Chem., 2010, 285, 1950–1956 CrossRef CAS PubMed.
- B. Ahn, S. G. Lee, H. R. Yoon, J. M. Lee, H. J. Oh, H. M. Kim and Y. Jung, Angew. Chem., Int. Ed. Engl., 2018, 57, 2909–2913 CrossRef CAS PubMed.
- Y. Hsia, J. B. Bale, S. Gonen, D. Shi, W. Sheffler, K. K. Fong, U. Nattermann, C. Xu, P. S. Huang, R. Ravichandran, S. Yi, T. N. Davis, T. Gonen, N. P. King and D. Baker, Nature, 2016, 535, 136–139 CrossRef CAS PubMed.
- D. Sato, H. Ohtomo, Y. Yamada, T. Hikima, A. Kurobe, K. Fujiwara and M. Ikeguchi, Biochemistry, 2016, 55, 287–293 CrossRef CAS PubMed.
- Y. E. Kim, Y. N. Kim, J. A. Kim, H. M. Kim and Y. Jung, Nat. Commun., 2015, 6, 7134 CrossRef CAS PubMed.
- P. Foroozandeh and A. A. Aziz, Nanoscale Res. Lett., 2018, 13, 339 CrossRef PubMed.
- L. Li, C. J. Fang, J. C. Ryan, E. C. Niemi, J. A. Lebron, P. J. Bjorkman, H. Arase, F. M. Torti, S. V. Torti, M. C. Nakamura and W. E. Seaman, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3505–3510 CrossRef CAS.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Aberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc02772k |
| This journal is © The Royal Society of Chemistry 2023 |