
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple purification of small-molecule-labelled peptides via palladium enolate formation from β-ketoamide tags†
Kenji
Hayamizu
a,
Kota
Koike
 ab,
Kosuke
Dodo
*ab,
Miwako
Asanuma
ab,
Hiromichi
Egami
a and
Mikiko
Sodeoka
*ab
ab,
Kosuke
Dodo
*ab,
Miwako
Asanuma
ab,
Hiromichi
Egami
a and
Mikiko
Sodeoka
*ab
aSynthetic Organic Chemistry Laboratory, RIKEN Cluster for Pioneering Research, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: dodo@riken.jp; sodeoka@riken.jp
bRIKEN Center for Sustainable Resource Science, 2-1, Hirosawa, Wako, Saitama 351-0198, Japan
First published on 14th July 2023
Abstract
Palladium enolates derived from β-ketocarbonyl compounds serve as key intermediates in various catalytic asymmetric reactions. We found that the palladium enolate formed from β-ketoamide is stable in air and moisture and we applied this property to develop a peptide purification system using β-ketoamide as a small affinity tag in aqueous media. A solid-supported palladium complex successfully captured β-ketoamide-tagged molecules as palladium enolates and released them in high yield upon acid treatment. Optimum conditions for the catch and release of tagged peptides from a mixture of untagged peptides were established. To demonstrate the value of this methodology in identifying the binding site of a ligand to its target protein, we purified and identified a peptide containing the ligand-binding site from the tryptic digest of cathepsin B labelled with a covalent cathepsin B inhibitor containing a β-ketoamide tag.
Introduction
In chemical biology research, the binding modes of bioactive compounds provide key information for both mechanistic studies and the design of relatively more potent compounds. X-ray crystallographic and nuclear magnetic resonance (NMR) analyses are generally used to reveal the structure of protein–compound complexes.1,2 However, these methods require a large amount of protein and are often limited by the availability of target proteins. Moreover, the physical properties of the target protein, especially in the case of membrane proteins, can affect efficient crystallization for X-ray analysis or solubilisation for NMR analysis. In contrast, a chemical approach using affinity labelling and mass analysis is applicable to endogenous and small amounts of protein.3–6 Affinity labelling can form a covalent bond between the bioactive compound and its target protein by introducing a reactive group into the target compound. Photoactivatable groups,6 such as aryl azide,7 diazirine,4,8 and benzophenone,9 have been used to label the binding protein upon photoirradiation. Electrophilic groups, such as halomethyl ketone10 or epoxide,11 can react with nucleophilic amino acids, such as cysteine or lysine, and label the binding protein depending on the proximity to the reactive amino acid. Recently, several additional reactive groups have been developed and used for various applications, such as proteome analysis12–14 or live cell imaging of target protein.15–17 However, irrespective of the reactive group, the binding site can be identified via tandem mass spectrometry (MS/MS) analysis of the labelled peptides after enzymatic digestion of the labelled protein. In this method, purification of a labelled peptide is important for efficient analysis.Affinity purification is used to purify molecules of interest via specific interactions between affinity tags and capture molecules, such as antigens and antibodies, or enzymes and substrates. Biotin is often used as an affinity tag for bioactive compounds, and biotin-tagged peptides are purified using avidin-immobilized beads (Fig. 1a, upper panel). Although the avidin–biotin system is one of the most popular methods, it has two major problems. One problem is that the biotin tag is sometimes larger than the original compounds and tends to decrease their biological activity. Another problem is that the high affinity between avidin and biotin often disturbs the efficient elution from the affinity matrix. To overcome these limitations, alkyne or azide tags are introduced into bioactive compounds, and click chemistry is used to install biotin tags after affinity labelling. Various cleavable linkers have also been developed to improve elution efficiency,18,19 and many reports have used click reactions and cleavable linkers (Fig. 1a, lower panel).20–22 In addition, desthiobiotin is used in place of biotin owing to its lower affinity to avidin, which improves the elution efficiency.23 Other affinity tags,24 such as the FLAG tag, can be also installed and used for the purification of the labelled protein. However, multistep and complex procedures including click reaction sometimes result in low purification efficiency of the labelled peptides, as previously reported.25,26
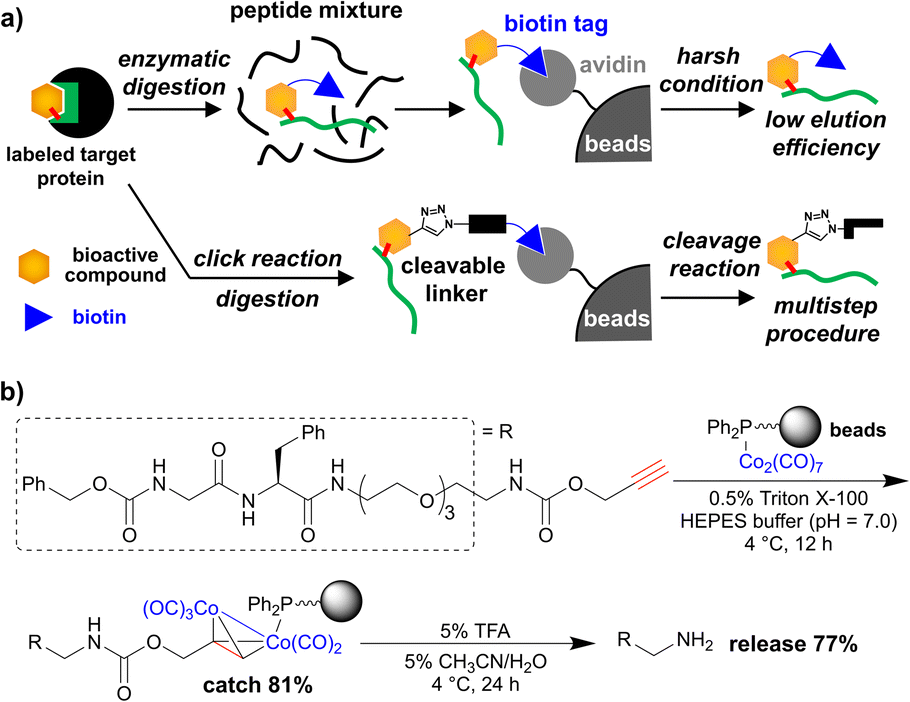 | ||
| Fig. 1 (a) Affinity purification using biotin tag for the small-molecule-labelled peptide; upper panel: biotin tag is pre-installed into the target compound, lower panel: biotin tag is post-installed via click chemistry and a cleavable linker is used for efficient elution. (b) Purification of propargyl-carbamate-tagged dipeptide using cobalt-complex-immobilized beads (previous work). | ||
To overcome the limitations, we have developed a simple and efficient purification method using metal-complex-immobilized beads. We previously applied cobalt–phosphine-complex-immobilized beads for the purification of propargyl-carbamate-tagged molecules including dipeptide (Fig. 1b).27,28 However, to achieve the identification of small-molecule-labelled peptides, a relatively more efficient purification method is needed.
In this study, we focused on palladium enolate29,30 derived from β-ketocarbonyl compounds and the Pd aqua complex, which was originally developed as a reactive species for various asymmetric catalytic reactions (Fig. 2a).31–35 We previously succeeded in isolating palladium enolates formed from β-ketoamide (Fig. 2a, lower left panel),36 showing high stability in air and moisture. Based on this result, we evaluated palladium enolate from different viewpoints and speculated on the potential of the palladium complex to capture β-ketocarbonyl compounds on beads and release them via simple acidic treatment (Fig. 2b). The β-ketoamide structure is small and contained in bacterial signaling molecules,37 suggesting that it may function as a bio-orthogonal tag for bioactive compounds. Therefore, we applied the unique properties of Pd enolates to develop a novel purification method for β-ketoamide-tagged molecules.
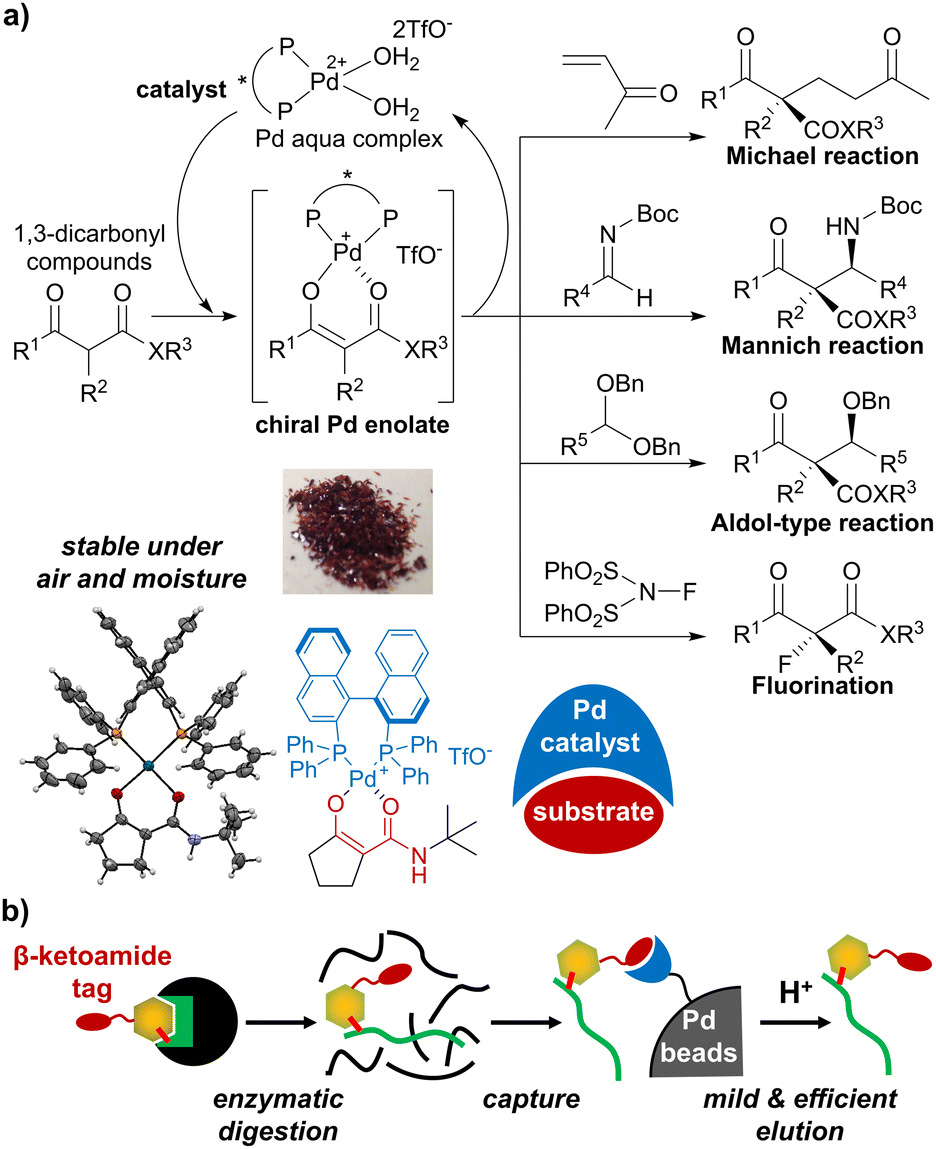 | ||
| Fig. 2 (a) Catalytic asymmetric reactions via palladium enolate. (b) Application of palladium enolate chemistry to affinity purification of peptides. | ||
Results and discussion
First, we prepared a BINAP-Pd-aqua complex immobilized on polymer beads according to our previously reported procedure.38 In this study, we selected TentaGel grafted copolymer beads consisting of hydrophobic regions of polystyrene and hydrophilic regions of polyethylene glycol because its amphiphilic properties were considered suitable for the purification of peptides in aqueous media. We prepared rac-BINAP-carboxylic acid from rac-BINOL in eight steps according to a previously reported procedure (Scheme S1†).39 BINAP-carboxylic acid was immobilized via condensation with TentaGel-NH2 (Scheme 1). The completion of the reaction was confirmed using the Kaiser test. Using the BINAP-immobilized TentaGel, the Pd complex was generated using [Pd(CN3CN)4]2+(TfO−)2. The formation of the Pd aqua complex was confirmed via gel-phase 31P-NMR in CDCl3 as previously reported.38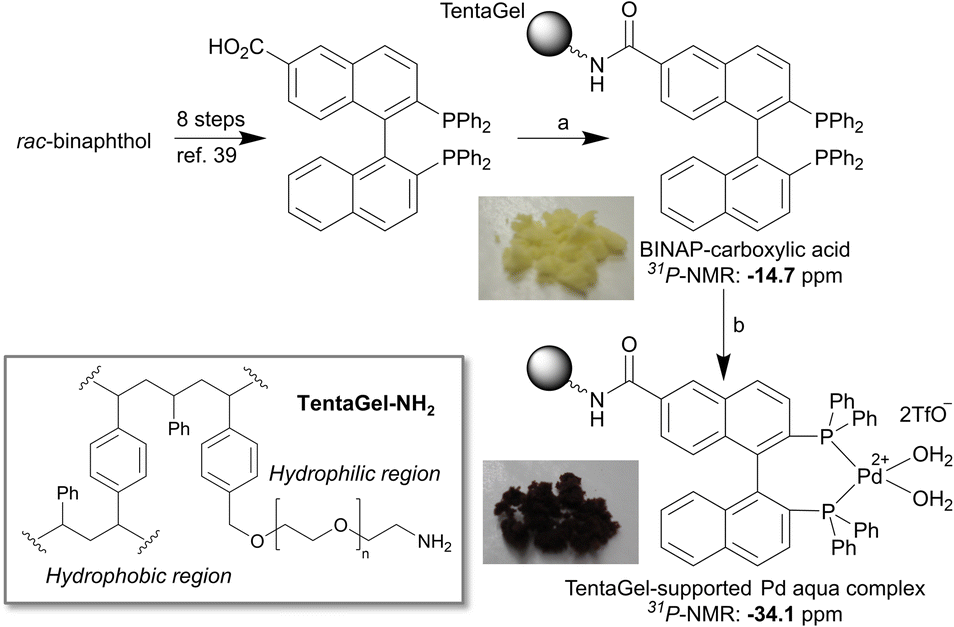 | ||
| Scheme 1 Reagents and conditions: (a) TentaGel-NH2, EDCI, HOBt, DMF, retention time of 13 h. (b) [Pd(CH3CN)4]2+(TfO−)2, wet acetone, retention time of 7 h, 96% in 2 steps. | ||
We have shown that Pd enolate is rapidly formed from β-ketoamide in solution,36 as monitored via31P-NMR. Therefore, we confirmed the formation of Pd enolate on TentaGel in aqueous media using 31P-NMR. The TentaGel-supported Pd aqua complex was mixed with three equivalents of β-ketoamide in 5% CH3CN/H2O and the mixture was incubated at room temperature for 12 h, filtered, washed with 50% CH3CN/H2O and CHCl3, and analysed via31P-NMR (Fig. 3). The signals of BINAP phosphines were shifted from 34.1 ppm to 27.8 and 34.5 ppm, which was in good agreement with those of the isolated Pd enolate complex36 (27.7 and 34.3 ppm, Fig. S1†). These results indicate the quantitative formation of a stable Pd enolate on TentaGel in aqueous media, suggesting that the TentaGel-supported Pd complex could successfully capture β-ketoamide-tagged molecules.
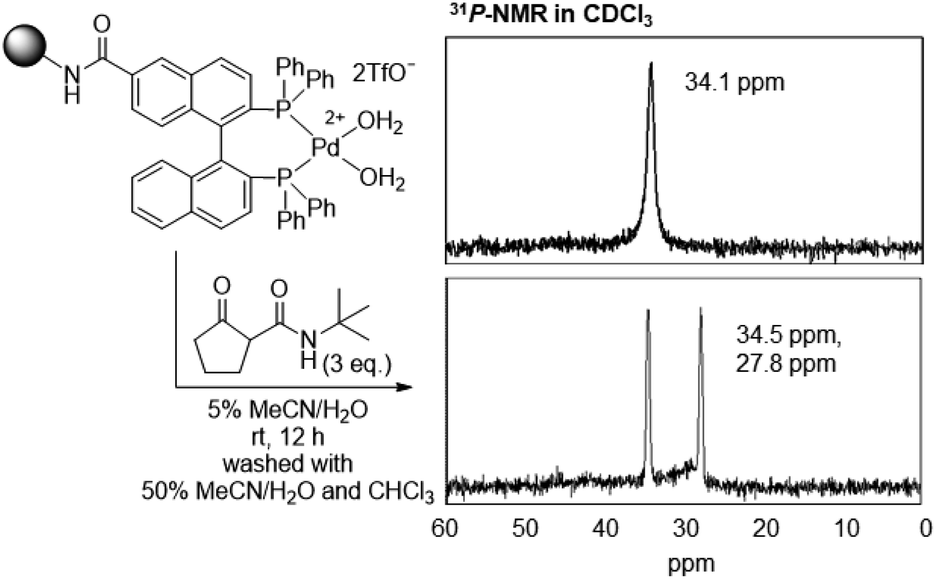 | ||
| Fig. 3 31P-NMR analysis of TentaGel-supported Pd aqua complexes before and after mixing with β-ketoamide. | ||
Based on the successful formation of Pd enolate in aqueous media, we next applied this methodology to the purification of β-ketoamide-tagged molecules. We tested several β-ketoamides and found that the optimum conditions varied depending on the structure (Fig. S2 and S3, Scheme S2†). Thus, we focused on benzoylacetamide (Bza) in further experiments (Fig. S8 and S9†). We prepared a Bza-tagged model peptide with a sequence corresponding to BSA161–167 (Bza-peptide) and investigated the catch and release conditions (Fig. 4 and S4†). The Bza-peptide was mixed with the TentaGel-supported Pd complex and residual peptides in the supernatant and wash fraction were quantified via high-performance liquid chromatography (HPLC) analysis (Fig. S5 and S6†). The catch efficiency was quantified based on the residual amount. We also assessed the compatibility with various buffer components. In the phosphate buffer, the reaction was complete within 1 h at room temperature with a 97% catch efficiency (Fig. 4b, entries 1 and 2). Phosphate-buffered saline (PBS) was also applicable, although a relatively larger amount of Pd complex was required (Fig. 4b, entries 3–5). These results indicate that our system can function in high salt concentrations (PBS: 137 mM NaCl and 2.7 mM KCl). Moreover, 0.1% trifluoroacetic acid (TFA) in 5% CH3CN/H2O, a standard acidic solvent for peptides, was compatible and yielded a catch efficiency of 90% (Fig. 4b, entry 9). We also found that peptides at much lower concentrations could be efficiently captured and eluted by using sufficient amounts of Pd complex (Fig. S10–S12†). Next, we examined the elution conditions required to release the captured peptide. Treatment with 0.3% TFA in 50% CH3CN/H2O for 30 min was sufficient to elute the Bza-peptide quantitatively (98%) (Fig. 4c, entry 3; and S7†). In addition to the increase in acidity after using 0.1–0.3% TFA, the increase in CH3CN content from 5% to 50% appeared to enhance elution via the coordination of CH3CN to the Pd complex. Compared with previous results using a cobalt complex (Fig. 1b),27,28 a relatively more efficient catch and release with shorter incubation time, milder acid treatment, and a longer model peptide were achieved in the present study.
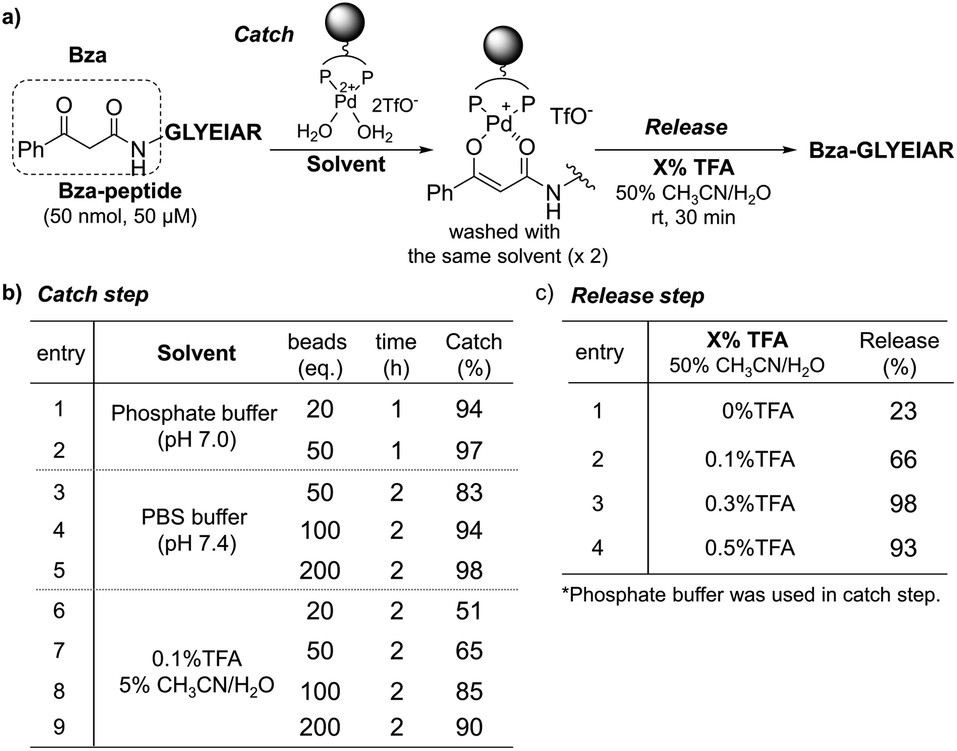 | ||
| Fig. 4 Catch and release of Bza-peptide. (a) Procedure. (b) Catch and (c) release efficiencies were quantified by HPLC analysis. | ||
After establishing the optimized conditions, we attempted the affinity purification of the Bza-peptide from a complex peptide mixture (Fig. 5). We added Bza-peptide to a tryptic digest of bovine serum albumin (BSA), which is expected to contain 55 peptides with a molecular weight over 500 Da (Fig. 5a). The catch and release protocol was applied to the peptide mixture. The initial solution, supernatant after the catch step, washing solution, and eluate were analysed using matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) MS (Fig. 5b and c). The Bza-peptide was selectively detected in the eluate, while the original peptide (BSA161–167) was mainly detected in the wash fraction. Although many other peptides contain functional groups that could potentially interact with the cationic metal, the other peptide captured by the TentaGel-supported Pd complex was solely BSA347–359. Both BSA161–167 and BSA347–359 contain acidic amino acids (Asp or Glu) and a basic amino acid (Arg) (Fig. 5c S13†); however, the latter was bound to Pd beads, whereas the former was not, implying that the amino acid sequence of BSA347–359 may contribute to the binding affinity. The results also indicated the compatibility of the TentaGel-supported Pd complex with functional groups in amino acids, except for cysteine. Because thiol group interacts with metal beads as previously reported,28 our method is unsuitable for a sample containing free thiol of cysteine residue. However, the cysteine thiol group is generally blocked with iodoacetamide before tryptic digestion. Therefore, our purification method is expected to be useful for the purification of affinity-labelled peptides.
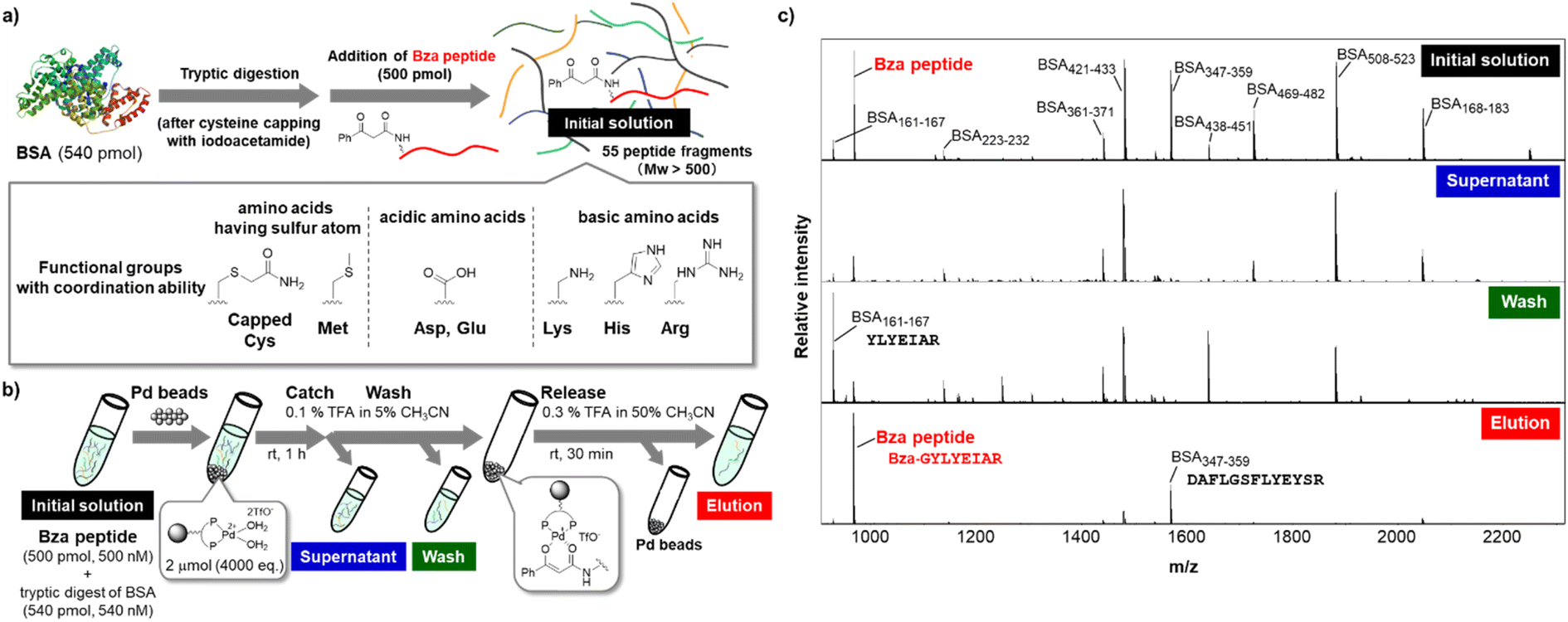 | ||
| Fig. 5 Purification of Bza-peptide (500 pmol, 500 nM) from a complex mixture of peptides generated via tryptic digestion of BSA (540 pmol, 540 nM). (a) Preparation of the mixture of peptides. Functional groups believed to coordinate to Pd complex are shown. (b) Procedure of affinity purification using TentaGel-supported Pd complex. (c) MALDI-TOF MS analysis of the initial solution, supernatant after the catch step, wash fraction, and eluate. The amino acid sequences of BSA161–167, BSA347–359, and Bza-peptide are shown. | ||
Finally, to demonstrate the utility of our methodology for the determination of binding sites in bioactive molecules, we performed affinity labelling of an enzyme with a Bza-tagged inhibitor and applied our system to purify the labelled peptide (Fig. 6). Z-FG-AOMK was previously developed as a specific cathepsin B (CatB) inhibitor,40 which forms a covalent bond with the catalytic cysteine residue.26 We designed Bza-FG-AOMK using a “built-in” strategy, in which the β-ketoamide structure was introduced into Z-FG-AOMK with minimum modification of the structure. Bza-FG-AOMK was synthesized (Scheme S3†) and found to show stronger CatB-inhibitory activity than that of Z-FG-AOMK, indicating the compatibility of the Bza tag (Fig. 6a and S14†). CatB was treated with Bza-FG-AOMK and then digested using trypsin after iodoacetamide treatment. A labelled peptide was purified from the tryptic digest according to the procedure presented in Fig. 6 (Fig. 6b, S15, and S17†), and a specific peptide corresponding to the binding site of Z-FG-AOMK was obtained (Fig. 6c). The labelled amino acid was confirmed to be the catalytic Cys29 via LC-MS/MS analysis (Fig. 6d). In addition to the labelled peptide, CatB246–263 was purified even in the absence of Bza-FG-AOMK (Fig. S16 and S17†), confirming the affinity of this peptide for Pd beads. Although certain non-target peptides may show specific binding to Pd beads, the binding peptide labelled with a Bza-tagged compound can be easily identified by comparison with the control experiment. Furthermore, to compare this approach to the standard purification method using the biotin–avidin system with click reaction, we performed an affinity labelling experiment using alkyne-tagged Bza-FG-AOMK. Purification using the Pd beads was successful, whereas that using the biotin–avidin system was unsuccessful due to loss of the biotinylated peptide after the click reaction and/or its low ionization efficiency (Fig. S18–S20 and Scheme S4†). In the previous study, we applied alkyne-tag Raman screening (ATRaS) method to identify the binding site of Z-FG-AOMK.26 We labelled CatB with Alt-FG-AOMK, an alkyne-tagged Z-FG-AOMK. After tryptic digestion, we fractionated peptides with HPLC and analysed each fraction with Raman microscope26 or Raman plate reader.41 Finally, the labelled peptide was identified by MS analysis of the fractions showing Raman signal from the alkyne-tag. In contrast, this new affinity purification method does not require HPLC fractionation and screening processes, and the labelled peptide was successfully identified by the direct MS analysis of the purified peptide solution.
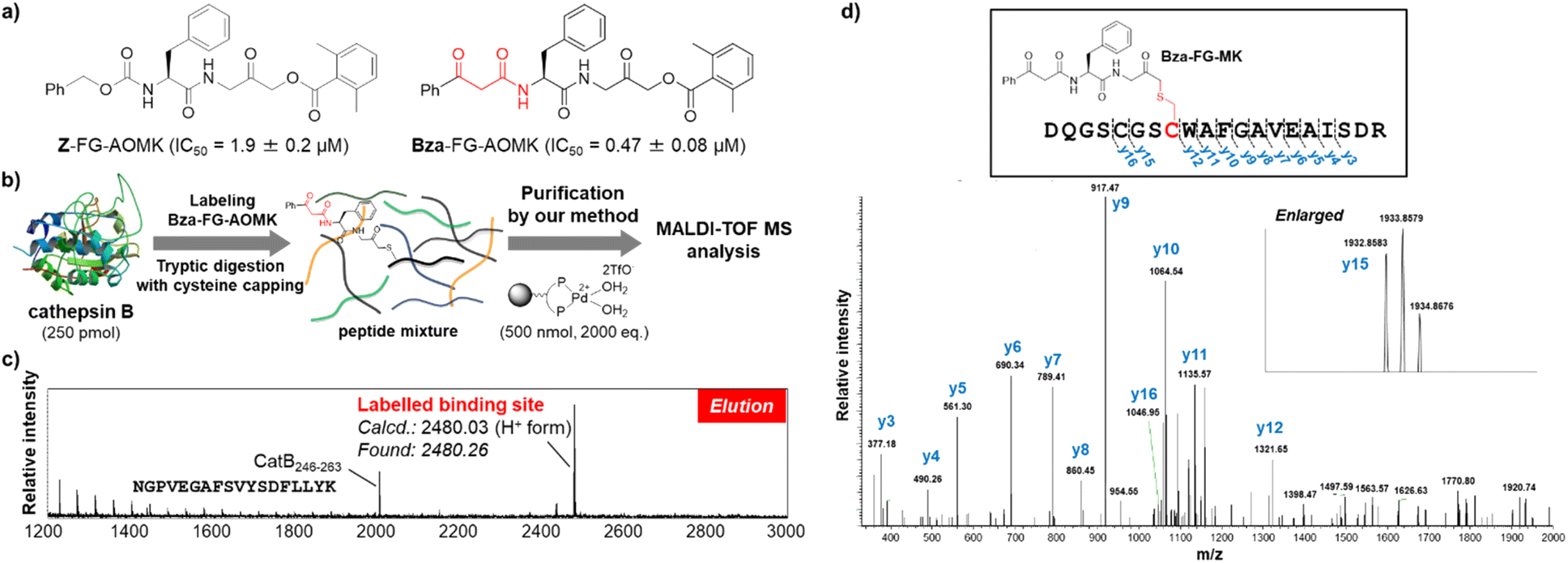 | ||
| Fig. 6 Identification of the FG-AOMK probe binding site. (a) Structures and CatB-inhibitory activities of Z-FG-AOMK and Bza-FG-AOMK. (b) Purification of CatB peptide labelled with Bza-FG-AOMK using TentaGel-supported Pd complex. (c) MALDI-TOF MS analysis of the peptides purified by the TentaGel-supported Pd complex. The amino acid sequence of CatB246–263 is shown. (d) LC-MS/MS analysis of labelled peptide purified by TentaGel-supported Pd complex. The N-terminal fifth cysteine residue was carbamidomethyl modified. | ||
Conclusions
The successful purification and identification of the CatB site labelled with the FG-AOMK probe demonstrated that the affinity and selectivity of the reaction between the palladium complex and β-ketoamide tag were sufficient for effective purification of labelled peptides. Since the β-ketoamide moiety is small and easy to introduce into target compounds,42 our method should be suitable for the identification of binding proteins and binding sites of bioactive compounds. Moreover, β-ketoacids are common metabolites, and this method can contribute to the purification of new natural products containing β-ketoamide functionality.43 In this study, we used the Pd aqua complex, which was originally developed to catalyse asymmetric reactions, for affinity purification. Although metal complexes have previously been used for the purification of biomolecules,44 our strategy is the first to use the unique interaction of a metal catalyst, which functions as a pseudo-enzyme, with its substrate for selective purification. This concept may also be applicable to other metal catalysts with high substrate specificity.Data availability
Additional experimental procedures, supplementary data, and spectral data are available in ESI.†Author contributions
K. D. and M. S. envisioned the project and designed the experiments. K. H. and K. K. synthesized the compounds and performed labelling experiments and purification of labelled peptides. K. H., K. K., and H. E. prepared and analysed Tentagel-supported Pd complex. K. H., K. K., and K. D. performed enzymatic experiments. K. H., K. K., and M. A. performed MS analysis. K. H., K. D., and M. S. drafted the manuscript with inputs from all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partly supported by RIKEN, Japan Science and Technology Agency, Japan Society for the Promotion of Science KAKENHI (grant numbers 23651244 (K. D. and H. E.), 26560439 (K. D.), and 20K20591 (M. S. and K. D.)), AMED-CREST, AMED (grant number JP18gm0710004 (M. S.)), Extramural Collaborative Research Grant of Cancer Research Institute, Kanazawa University (K. D.), and the Eisai Award in Synthetic Organic Chemistry, Japan (K. D.). K. H. was supported by the Junior Research Associate Program in RIKEN. We thank the Support Unit for biomaterial analysis and RIKEN CBS Research Resources Division for technical assistance with peptide synthesis and LC-MS/MS analysis.Notes and references
- J. Shonberg, R. C. Kling, P. Gmeiner and S. Löber, Bioorg. Med. Chem., 2015, 23, 3880–3906 CrossRef CAS PubMed.
- I. M. S. de Vera, P. K. Giri, P. Munoz-Tello, R. Brust, J. Fuhrmann, E. Matta-Camacho, J. Shang, S. Campbell, H. D. Wilson, J. Granados, W. J. Gardner, T. P. Creamer, L. A. Solt and D. J. Kojetin, ACS Chem. Biol., 2016, 11, 1795–1799 CrossRef CAS PubMed.
- L. Q. Al-Mawsawi, V. Fikkert, R. Dayam, M. Witvrouw, T. R. Burke, C. H. Borchers and N. Neamati, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10080–10085 CrossRef CAS PubMed.
- Y. Hatanaka, Chem. Pharm. Bull., 2015, 63, 1–12 CrossRef CAS PubMed.
- M. Chen, M. Asanuma, M. Takahashi, Y. Shichino, M. Mito, K. Fujiwara, H. Saito, S. N. Floor, N. T. Ingolia, M. Sodeoka, K. Dodo, T. Ito and S. Iwasaki, Cell Chem. Biol., 2021, 28, 475–486 CrossRef CAS PubMed.
- N. R. Burton, P. Kim and K. M. Backus, Org. Biomol. Chem., 2021, 19, 7792–7809 RSC.
- F. Kotzyba-Hibert, I. Kapfer and M. Goeldner, Angew. Chem., Int. Ed. Engl., 1995, 34, 1296–1312 CrossRef CAS.
- D. W. Goldman, J. S. Pober, J. White and H. Bayley, Nature, 1979, 280, 841–843 CrossRef CAS PubMed.
- R. E. Galardy, L. C. Craig, J. D. Jamieson and M. P. Printz, J. Biol. Chem., 1974, 249, 3510–3518 CrossRef CAS PubMed.
- M. S. Cohen, C. Zhang, K. M. Shokat and J. Taunton, Science, 2005, 308, 1318–1321 CrossRef CAS PubMed.
- G. Chen, A. Heim, D. Riether, D. Yee, Y. Milgrom, M. A. Gawinowicz and D. Sames, J. Am. Chem. Soc., 2003, 125, 8130–8133 CrossRef CAS PubMed.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Nature, 2016, 534, 570–574 CrossRef CAS PubMed.
- S. M. Hacker, K. M. Backus, M. R. Lazear, S. Forli, B. E. Correia and B. F. Cravatt, Nat. Chem., 2017, 9, 1181–1190 CrossRef CAS PubMed.
- M. E. Abbasov, M. E. Kavanagh, T.-A. Ichu, M. R. Lazear, Y. Tao, V. M. Crowley, C. W. Am Ende, S. M. Hacker, J. Ho, M. M. Dix, R. Suciu, M. M. Hayward, L. L. Kiessling and B. F. Cravatt, Nat. Chem., 2021, 13, 1081–1092 CrossRef CAS PubMed.
- T. Miki, S. H. Fujishima, K. Komatsu, K. Kuwata, S. Kiyonaka and I. Hamachi, Chem. Biol., 2014, 21, 1013–1022 CrossRef CAS PubMed.
- T. Yamaguchi, M. Asanuma, S. Nakanishi, Y. Saito, M. Okazaki, K. Dodo and M. Sodeoka, Chem. Sci., 2014, 5, 1021–1029 RSC.
- S. Wakayama, S. Kiyonaka, I. Arai, W. Kakegawa, S. Matsuda, K. Ibata, Y. L. Nemoto, A. Kusumi, M. Yuzaki and I. Hamachi, Nat. Commun., 2017, 8, 14850 CrossRef CAS PubMed.
- S. H. L. Verhelst, M. Fonović and M. Bogyo, Angew. Chem., Int. Ed. Engl., 2007, 46, 1284–1286 CrossRef CAS PubMed.
- M. Fonović, S. H. L. Verhelst, M. T. Sorum and M. Bogyo, Mol. Cell. Proteomics, 2007, 6, 1761–1770 CrossRef PubMed.
- A. E. Speers and B. F. Cravatt, J. Am. Chem. Soc., 2005, 127, 10018–10019 CrossRef CAS PubMed.
- K. D. Park, R. Liu and H. Kohn, Chem. Biol., 2009, 16, 763–772 CrossRef CAS PubMed.
- Y. Yang and S. H. L. Verhelst, Chem. Commun., 2013, 49, 5366–5368 RSC.
- J. D. Hirsch, L. Eslamizar, B. J. Filanoski, N. Malekzadeh, R. P. Haugland, J. M. Beechem and R. P. Haugland, Anal. Biochem., 2002, 308, 343–357 CrossRef CAS PubMed.
- A. S. Pina, Í. L. Batalha and A. C. A. Roque, Methods Mol. Biol., 2014, 1129, 147–168 CrossRef CAS PubMed.
- A. Tsuchiya, M. Asanuma, G. Hirai, K. Oonuma, M. Muddassar, E. Nishizawa, Y. Koyama, Y. Otani, K. Y. J. Zhang and M. Sodeoka, Mol. BioSyst., 2013, 9, 1026–1034 RSC.
- J. Ando, M. Asanuma, K. Dodo, H. Yamakoshi, S. Kawata, K. Fujita and M. Sodeoka, J. Am. Chem. Soc., 2016, 138, 13901–13910 CrossRef CAS PubMed.
- H. Egami, S. Kamisuki, K. Dodo, M. Asanuma, Y. Hamashima and M. Sodeoka, Org. Biomol. Chem., 2011, 9, 7667–7670 RSC.
- A. Miyazaki, M. Asanuma, K. Dodo, H. Egami and M. Sodeoka, Chem. – Eur. J., 2014, 20, 8116–8128 CrossRef CAS PubMed.
- E. Hagiwara, A. Fujii and M. Sodeoka, J. Am. Chem. Soc., 1998, 120, 2474–2475 CrossRef CAS.
- A. Fujii, E. Hagiwara and M. Sodeoka, J. Am. Chem. Soc., 1999, 121, 5450–5458 CrossRef CAS.
- Y. Hamashima, D. Hotta and M. Sodeoka, J. Am. Chem. Soc., 2002, 124, 11240–11241 CrossRef CAS PubMed.
- Y. Hamashima, K. Yagi, H. Takano, L. Tamás and M. Sodeoka, J. Am. Chem. Soc., 2002, 124, 14530–14531 CrossRef CAS PubMed.
- Y. Hamashima and M. Sodeoka, Chem. Rec., 2004, 4, 231–242 CrossRef CAS PubMed.
- Y. Hamashima, N. Sasamoto, N. Umebayashi and M. Sodeoka, Chem.–Asian J., 2008, 3, 1443–1455 CrossRef CAS PubMed.
- N. Umebayashi, Y. Hamashima, D. Hashizume and M. Sodeoka, Angew. Chem., Int. Ed. Engl., 2008, 47, 4196–4199 CrossRef CAS PubMed.
- K. Hayamizu, N. Terayama, D. Hashizume, K. Dodo and M. Sodeoka, Tetrahedron, 2015, 71, 6594–6601 CrossRef CAS.
- J. Rayo, N. Amara, P. Krief and M. M. Meijler, J. Am. Chem. Soc., 2011, 133, 7469–7475 CrossRef CAS PubMed.
- A. Fujii and M. Sodeoka, Tetrahedron Lett., 1999, 40, 8011–8014 CrossRef CAS.
- Y. Otomaru, T. Senda and T. Hayashi, Org. Lett., 2004, 6, 3357–3359 CrossRef CAS PubMed.
- D. Kato, K. M. Boatright, A. B. Berger, T. Nazif, G. Blum, C. Ryan, K. A. H. Chehade, G. S. Salvesen and M. Bogyo, Nat. Chem. Biol., 2005, 1, 33–38 CrossRef CAS PubMed.
- H. Kawagoe, J. Ando, M. Asanuma, K. Dodo, T. Miyano, H. Ueda, M. Sodeoka and K. Fujita, Sci. Rep., 2021, 11, 15742 CrossRef CAS PubMed.
- W. Li, Y. Zheng, E. Qu, J. Bai and Q. Deng, Eur. J. Org. Chem., 2021, 2021, 5151–5192 CrossRef CAS.
- T. Nogawa, A. Terai, K. Amagai, J. Hashimoto, Y. Futamura, A. Okano, M. Fujie, N. Satoh, H. Ikeda, K. Shin-Ya, H. Osada and S. Takahashi, J. Nat. Prod., 2020, 83, 3598–3605 CrossRef CAS PubMed.
- R. C. F. Cheung, J. H. Wong and T. B. Ng, Appl. Microbiol. Biotechnol., 2012, 96, 1411–1420 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplemental data, and spectral data. See DOI: https://doi.org/10.1039/d2sc03112d |
| This journal is © The Royal Society of Chemistry 2023 |