
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAzapeptide activity-based probes for the SARS-CoV-2 main protease enable visualization of inhibition in infected cells†
Roeland
Vanhoutte
 a,
Marta
Barniol-Xicota
a,
Winston
Chiu
b,
Laura
Vangeel
b,
Dirk
Jochmans
b,
Steven
De Jonghe
b,
Hadeer
Zidane
c,
Haim M.
Barr
c,
Nir
London
d,
Johan
Neyts
b and
Steven H. L.
Verhelst
*ae
a,
Marta
Barniol-Xicota
a,
Winston
Chiu
b,
Laura
Vangeel
b,
Dirk
Jochmans
b,
Steven
De Jonghe
b,
Hadeer
Zidane
c,
Haim M.
Barr
c,
Nir
London
d,
Johan
Neyts
b and
Steven H. L.
Verhelst
*ae
aDepartment of Cellular and Molecular Medicine, Laboratory of Chemical Biology, KU Leuven, Herestraat 49 box 802, 3000 Leuven, Belgium. E-mail: steven.verhelst@kuleuven.be
bLaboratory of Virology and Chemotherapy, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, KU Leuven, Herestraat 49, Box 1043, 3000, Leuven, Belgium
cMaurice and Vivienne Wohl Institute for Drug Discovery, The Nancy and Stephen Grand Israel National Center for Personalized Medicine, The Weizmann Institute of Science, Rehovot 7610001, Israel
dDepartment of Chemical and Structural Biology, Weizmann Institute of Science, Rehovot 7610001, Israel
eAG Chemical Proteomics, Leibniz Institute for Analytical Sciences ISAS, Otto-Hahn-Str. 6b, 44227 Dortmund, Germany
First published on 6th January 2023
Abstract
The COVID-19 pandemic has revealed the vulnerability of the modern, global society. With expected waves of future infections by SARS-CoV-2, treatment options for infected individuals will be crucial in order to decrease mortality and hospitalizations. The SARS-CoV-2 main protease is a validated drug target, for which the first inhibitor has been approved for use in patients. To facilitate future work on this drug target, we designed a solid-phase synthesis route towards azapeptide activity-based probes that are capped with a cysteine-reactive electrophile for covalent modification of the active site of Mpro. This design led to the most potent ABP for Mpro and one of the most potent inhibitors reported thus far. We demonstrate that this ABP can be used to visualize Mpro activity and target engagement by drugs in infected cells.
Introduction
The SARS-CoV-2 virus is the causative agent of COVID-19 and has resulted in the largest worldwide viral pandemic in a century. The virus belongs to the family of betacoronaviruses, which have been known to infect humans since 19661,2 and have caused two previous pandemics: the SARS pandemic in 2002![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 and the MERS pandemic in 2012.4 Although the development of vaccines has lowered the incidence of hospitalizations and fatalities due to COVID-19, new mutants of the virus have been emerging for which the vaccines are less effective.5 Despite the lower disease severity of the new mutants,6 the infectivity has increased. As a result, future waves of infection with associated hospitalizations, hygienic measures and general societal burden are expected. Therefore, the development of antiviral drugs remains necessary in the battle against SARS-CoV-2.
3 and the MERS pandemic in 2012.4 Although the development of vaccines has lowered the incidence of hospitalizations and fatalities due to COVID-19, new mutants of the virus have been emerging for which the vaccines are less effective.5 Despite the lower disease severity of the new mutants,6 the infectivity has increased. As a result, future waves of infection with associated hospitalizations, hygienic measures and general societal burden are expected. Therefore, the development of antiviral drugs remains necessary in the battle against SARS-CoV-2.
The main protease (Mpro) of SARS-CoV-2 has gained a lot of attention as a drug target, as it is responsible for the proteolytic release of twelve of the sixteen non-structural proteins (nsps) from the two polyproteins expressed by the virus.7 A wide variety of studies has therefore been conducted to generate inhibitors for Mpro, many of which employ compounds that bear cysteine-reactive warheads.8,9 Various reversible covalent inhibitors have been reported. For example, Zhang et al. found that a previously discovered substrate-based inhibitor with an α-ketoamide warhead for alpha- and betacoronavirus Mpro and enterovirus 3C proteases was active against SARS-CoV-2 Mpro.10 Similar substrate-based peptide aldehydes were reported around the same time as potent SARS-CoV-2 Mpro inhibitors.11 GC376, a bisulfite adduct of a peptide aldehyde with a similar peptide sequence, which had already been developed as a prodrug against the feline coronavirus, also turned out to be potent against SARS-CoV-2 Mpro.12,13 In December 2021, the FDA approved Pfizer's reversible covalent peptide nitrile nirmatrelvir14 as a combination therapy with ritonavir, for the treatment of COVID-19.
Irreversible covalent inhibitors for Mpro have been reported based on various cysteine-reactive warheads, including acyloxy methyl ketones (AOMKs),15 azanitriles and pyridyl esters,16 and Michael acceptors such as peptide vinyl esters17 and vinyl sulfones.18 Additionally, some covalent drugs or drug candidates, such as carmofur and ebselen, were reported as SARS-CoV-2 Mpro inhibitors.17
Several irreversible covalent warheads have been used to create activity-based probes (ABPs) for Mpro. Rut et al. used a vinyl sulfone warhead attached to a tetrapeptide containing natural and unnatural amino acids optimized for the P4–P1 preference of Mpro, with a fluorophore or biotin detection tag.18 The group of Böttcher created Mpro-reactive ABPs in a library synthesis by coupling an amine-containing ligand with a chloroacetamide-containing activated ester.19 Although the above studies provided useful probes, both used solution-phase chemistry for the construction of these molecules. Solid-phase chemistry would allow creation of a larger variety of structures in a shorter time. Van de Plassche et al. implemented solid-phase peptide synthesis (SPPS) to create ABPs with an AOMK warhead.20 However, this solid-phase approach is restricted to this particular warhead. Therefore, we aimed at designing a solid-phase strategy that would allow variation of the peptidic element as well as the reactive warhead. In this work, we report the on-resin synthesis of azapeptide-derived ABPs and their application in visualizing Mpro activity and inhibition in infected cells.
Results & discussion
For our synthetic design, we aimed at using a SPPS-based method that does not only allow rapid synthesis, but also enables the introduction of the reactive electrophilic warhead in a last step for easy diversification of this essential element of the ABP. Of note, cysteine-reactive electrophiles span a wide range of reactivities,21–23 and finding the right reactivity may be crucial to obtain an optimal ABP. In addition, not all electrophiles may be compatible with peptide elongation conditions. Coupling the electrophile in a final step is therefore highly desirable. Conveniently, the primary specificity element of SARS-CoV-2 Mpro comprises a Gln residue in the P1 position (Fig. 1A), which opens the possibility to anchor the molecule to the solid support at this position. As the warhead is placed at the C-terminal end of the P3–P1 peptide recognition element, we opted for the use of an azapeptide, introducing a secondary nitrogen in the peptide backbone to which a warhead can be coupled (Fig. 1B). Covalent azapeptidic inhibitors were previously developed for various cysteine proteases, including caspases24 and Mpro of the SARS-CoV-1 virus, which emerged in 2002.25 Recently, a covalent inhibitor of the SARS-CoV-2 papain-like protease based on an azaglycine-type scaffold has also been reported.26 In addition, we have previously provided proof of principle that solid-phase synthesis is possible for caspase-targeting inhibitors with a P1 aza-Asp residue.27,28 | ||
| Fig. 1 (A) Substrates of Mpro contain a Gln residue in the P1 position, directly N-terminal to the scissile bond (in red). (B) Design of azapeptide ABPs. The Gln side chain serves as a solid-phase anchor. It is introduced as an aza-amino acid, in which the α-carbon is replaced by nitrogen (blue). The aza-amino acid can then be capped with a cysteine-reactive warhead. The detection tag at the N-terminus allows detection of the covalent probe–Mpro complex. | ||
Initially, we aimed at constructing the azapeptide completely on a solid support by coupling bromopropionate to Rink resin, followed by reaction with Fmoc-hydrazine (Scheme 1A). The substitution unfortunately failed, likely due to the low nucleophilicity of Fmoc-hydrazine and the substantially lower electrophilicity of the bromopropionate compared with bromoacetate, for which substitution can be readily achieved.27 Performance of this reaction in solution-phase synthesis also failed, supporting this explanation (Scheme 1B). To enable SPPS of the desired azapeptides, we therefore constructed key building block 4 in solution, starting from Boc-protected hydrazine and tert-butyl 3-bromopropanoate (Scheme 1C), which was reported to proceed in better yields than for Fmoc-hydrazine.29 After the substitution, the N1 was protected using Alloc chloride to give intermediate 3, from which both the Boc and tert-butyl ester were removed using HCl. Finally, the N2 was reprotected with Fmoc chloride to form P1 building block 4.
 | ||
| Scheme 1 Synthesis of the azapeptide building block. (A) An attempt to synthesize a P1 azaGln fragment on a Rink amide resin did not work. (a) 3-Bromo propionyl bromide, DIEA, and DCM. (b) Fmoc-hydrazine, DIEA, DMF, 40 °C. (B) In solution, substitution by Fmoc-hydrazine did not function. (c) Fmoc-hydrazine, DIEA, DMF, 40 °C. (C) Synthesis of central orthogonally protected building block 4, (d) Boc-hydrazine, DIEA, DMF, 40 °C. (e) Alloc-Cl, DIEA, DMF, rt, 26% over two steps. (f) TFA, DCM, rt. (g) Fmoc-Cl, DIEA, DCM, rt, 69% over two steps. | ||
Compound 4 represents the cornerstone for the on-resin synthesis of the ABPs, as it serves a threefold purpose: the anchor point to the resin, the site for attachment of the peptide recognition element and the position for connection of the warhead. Building block 4 was first coupled to Rink amide resin via its carboxylic acid function, yielding the desired Gln side chain attached to the solid support. Next, we used Fmoc-based SPPS to couple an L-Leu and L-Tle residue to the N2 of the P1 azapeptide element. After capping the peptide with a hexynoic acid alkyne tag, the Alloc on the N1 was removed and capped with a cysteine-reactive warhead. We installed five different reactive electrophiles via an amide or sulfonamide bond: an epoxysuccinate (7a), a vinylsulfonamide (7b), a methylfumarate (7c), a chloroacetamide (7d) and a 2-chloro-5-nitrobenzamide (7e). These warheads react with cysteines in four different ways, namely epoxide ring opening (7a), Michael addition (7b and 7c), SN2 substitution (7d) and SNAr substitution (7e). After coupling of the warheads, the probes were cleaved from the resin using a mixture of TFA, TIS and water, which was compatible with all warheads installed on the probes (Scheme 2).
 | ||
| Scheme 2 On-resin synthesis of ABPs 7a–e. (a) (1) Deprotection with DMF/piperidine 4/1; (2) 4, HBTU, DIEA, DMF, rt, overnight. (b) Elongation by repeated cycles of: (1) DMF/piperidine 4/1, 5 minutes; (2) Fmoc-Leu-OH (overnight), Fmoc-Tle-OH or hexynoic acid (3 h), HBTU, DIEA, DMF, rt. (c) Pd(PPh3)4, PhSiH3, DCM, 30 minutes. (d) Coupling of either: (1) mono-ethyl epoxysuccinate, HBTU, DIEA, DMF, rt, overnight; (2) 2-chloroethane sulfonylchloride, DIEA, DCM, rt, overnight; (3) mono-methyl fumarate, HBTU, DIEA, DMF, rt, overnight; (4) chloroacetyl chloride, DIEA, DCM, rt, overnight; or (5) 2-chloro-5-nitrobenzoic acid, HBTU, DIEA, DMF, rt, overnight. (e) TFA/TIS/water 95/2.5/2.5, 30 minutes. | ||
A first assessment of the probes was done by competitive activity-based protein profiling (ABPP). Herein, Mpro was pretreated with 7a–e and residually active enzyme was labeled with the previously reported20 TAMRA-Tle-Leu-Gln-AOMK probe. At 10 μM concentration, probes 7a, 7c and 7d showed strong inhibition of Mpro, whereas probes 7b and 7e showed none or little (Fig. S1†). Next, apparent IC50 values (ICApp50) were determined with a kinetic assay using a quenched fluorescent substrate. This revealed a large difference in activity between the probes. While probes 7a, 7c and 7d display low-to-mid nanomolar ICApp50, 7b and 7e are much less active with ICApp50 values exceeding 45 μM (Table 1). This difference in activity is striking, as the vinyl sulfone warhead of 7b is the strongest Cys-reactive warhead used in this study.21 A potential explanation lies in the distance between the site of attack of the nucleophilic Cys residue on the warhead and the α-position of the P1 residue. For probes 7a, 7c and 7d, this distance is two bonds, whereas for probes 7b and 7e, this is three bonds (Fig. 2A). It would thus appear that the two-bond distance is a strict requirement for this type of Mpro inhibitor to engage in a productive reaction with the active site cysteine, although a larger number of warheads should be tested to confirm this hypothesis. To gain some more insight into the difference in reactivity, the warhead, P1 and P2 residues of the most active compound 7d and the inactive vinylsulfonamide 7b were docked into the active site of Mpro. The top docking poses that displayed good overlap with a known peptidic Mpro inhibitor (Fig. 2B and C), showed that the electrophilic carbon of the chloroacetamide is in close distance of the active site C145, whereas the vinyl moiety of 7b is localized much further and points towards the opposite direction (Fig. 2D and E).
| Compound | ICApp50a | EC50b | CC50c | CC50d |
|---|---|---|---|---|
| a Measured on purified SARS-CoV-2 Mpro. b Measured on VeroE6 cells infected with SARS-CoV-2. c Measured on Huh7 cells. d Measured on VeroE6 cells. | ||||
| 7a | 206 nM | >10 μM | 36.7 μM | N.D. |
| 7b | Inactive | N.D. | N.D. | N.D. |
| 7c | 44.0 nM | >10 μM | 62.5 μM | N.D. |
| 7d | 20.8 nM | 3.25 μM | 50.0 μM | >10 μM |
| 7e | >45 μM | N.D. | N.D. | N.D. |
 | ||
| Fig. 2 Engagement of the warhead by the active site cysteine. (A) Comparison of the position of attack on a substrate versus the here-synthesized inhibitors with chloroacetamide (7d) and vinylsulfonamide (7b). (B) Overlay of the crystal structure of SARS-CoV-2 Mpro with an aldehyde inhibitor (PDB code: 6LZE) with a docked structure of a vinylsulfonamide. Mpro is depicted as a semitransparent surface with a protein backbone in a cartoon format. Active site cysteine C145 is colored in magenta, the original inhibitor in cyan and the docked structure in green. (C) Overlay of the same crystal structure with a docked structure of a chloroacetamide. (D) Position of the vinylsulfonamide relative to the active site cysteine. Note that the electrophilic carbon points away from the nucleophile. (E) Position of the chloroacetamide relative to the active site cysteine. Pictures were rendered in PyMol.30 | ||
Next, the selectivity of probes 7a, 7c, 7d and 7e for Mpro over human proteins was assessed in a whole proteome by ABPP. To this end, HEK293 lysates were spiked with Mpro and treated with a 1 μM probe concentration. Covalently modified probe targets were then visualized via CuAAC (copper(I)-catalyzed azide alkyne cycloaddition) with TAMRA-azide. At pH 7.5, which is around the optimal pH for Mpro, we detected only one gel band for probes 7a, 7c and 7d. Compound 7e did not yield any signal, further confirming its inactivity. The fluorescent gel band was identified as Mpro, as it corresponds to the right molecular mass of the protein and the same band was detected when using the known Mpro-reactive ABP TAMRA-Tle-Leu-Gln-AOMK on HEK-lysates spiked with wild type Mpro, but not when the lysate was spiked with a catalytically dead C145A Mpro mutant. Because various reported covalent Mpro inhibitors show cross reactivity with cysteine cathepsins,15,16,31 the same experiment was also performed at pH 5.5, which is the optimal pH for cysteine cathepsin proteases. The occurrence of cathepsins in HEK-lysates was confirmed by using fluorescently labelled DCG-04, a pan-reactive cathepsin ABP.32 None of the probes (7a, 7c, 7d and 7e) showed any protein labelling at this pH (other than Mpro), illustrating the lack of off-targets. Striking was the clear labeling of Mpro by 7c and 7d at this acidic pH, showcasing the strong reactivity of these probes, even under sub-optimal conditions.
We next assessed the sensitivity of the two most active probes, 7c and 7d for labelling Mpro. To this end, 20 nM Mpro was incubated with decreasing concentrations of ABP and visualized via CuAAC with TAMRA-azide. Probe 7c gave a detectable Mpro signal down to 42 nM concentration and probe 7d detected Mpro even at a substoichiometric probe concentration of 14 nM (Fig. S2A†). The detection limit of probe 7d was also determined in a whole proteome by spiking HEK293T lysates (1 mg mL−1 total protein) with decreasing amounts of Mpro. With 1 μM 7d, it was possible to detect active Mpro down to 0.05% of the total protein (Fig. S2B†). This suggests that probe 7d may be used to detect active Mpro in infected cells.
With these highly active and selective probes at hand, we now set out to detect Mpro activity and its inhibition in infected cells. First, we determined the capacity of probes 7a, 7c and 7d to inhibit SARS-CoV-2 replication in VeroE6 cells. Compounds 7a and 7c showed low potency (EC50 > 10 μM), while compound 7d displayed single digit micromolar activity. The cellular toxicity (CC50) of all compounds was in the mid-micromolar range (Table 1). To illustrate that probe 7d can detect endogenous levels of Mpro, SARS-CoV-2-infected cells were lysed and treated with 7d. As controls, lysates of non-infected cells with or without spiked exogenous recombinant Mpro were treated under the same conditions (Fig. 3C). A fluorescently-labeled band was only visible in the sample from infected cells as well as in the spiked sample, whereas not a single fluorescent band was observed in the non-infected sample (Fig. 3D), confirming the high probe selectivity seen in Fig. 3A and B. The presence of Mpro was confirmed by western blot, which revealed distinct bands that overlay with the fluorescently-labeled bands of probe detection (Fig. 3D).
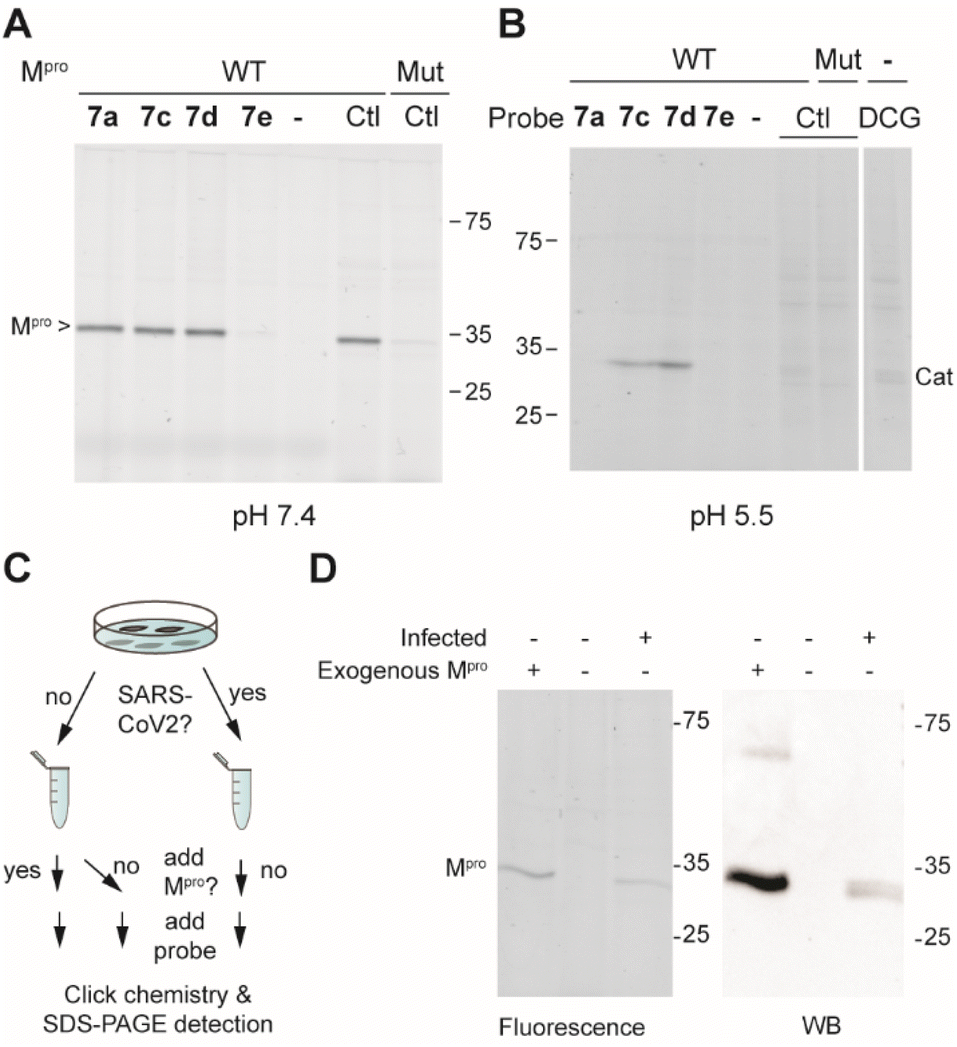 | ||
| Fig. 3 (A) Labeling of Mpro-spiked HEK lysates (1 mg mL; 0.1% Mpro) at pH 7.5 with probes 7a, 7c, 7d and 7e (1 μM for 1 h) with visualization via CuAAC with TAMRA-N3. Mut = C145A active site Mpro mutant and Ctl = control Mpro probe TAMRA-Tle-Leu-Gln-AOMK, which labels the active Mpro but not the catalytically dead mutant. (B) Same as in panel (A), but at pH 5.5. DCG = TAMRA-conjugated DCG-04 pan cathepsin probe. (C) Schematic workflow of Mpro detection in samples of infected and uninfected cells. (D) Detection of Mpro that is fluorescently labeled by probe 7d (left panel) and of total Mpro by western blot (right panel). | ||
With the ability to label endogenous Mpro, the most effective azapeptide ABP 7d was now used for detection of Mpro activity in virally infected cells by fluorescence microscopy. Hence, VeroE6 cells were incubated with SARS-CoV-2 for 24 h, followed by a 2 h treatment with an Mpro inhibitor or a DMSO vehicle. Next, ABP 7b (10 μM) was added to detect residually active Mpro. Labeled Mpro was then visualized after cell fixation by CuAAC with TAMRA-azide, whereas presence of the virus was detected with an antibody against the SARS-CoV-2 nucleocapsid protein. Samples were subjected to automated fluorescence microscopy with a ThermoFisher ArrayScan XTI system (Fig. 4A). Non-infected cells showed a low probe-derived TAMRA signal and low Alexafluor 647-derived signal of the viral nucleocapsid protein, indicating high selectivity of the in situ click reaction and the antibody, respectively (Fig. 4B). Infected cells not treated with probe 7b also displayed low TAMRA fluorescence, further confirming the in situ click selectivity (Fig. 4C). In contrast, virus-infected cells were well visualized by using probe 7d as demonstrated by the red fluorescence, which overlays with the false-green colored antibody fluorescence (Fig. 4D). Importantly, the probe signal disappeared almost completely in cells treated with nirmatrelvir (Fig. 4E and S3–S4†) as well as with carmofur (Fig. S3†), an antineoplastic drug that also inhibits SARS-CoV-2 Mpro. Although the fluorescence was relatively weak compared with the background, quantification of the signal shows a reduction in the fluorescence of nirmatrelvir-treated cells to levels close to the background (Fig. S5†). This confirms that the fluorescence signal indeed stems from the reaction of active Mpro with probe 7d and demonstrates the utility in detection of this viral drug target and its inhibition in live cells.
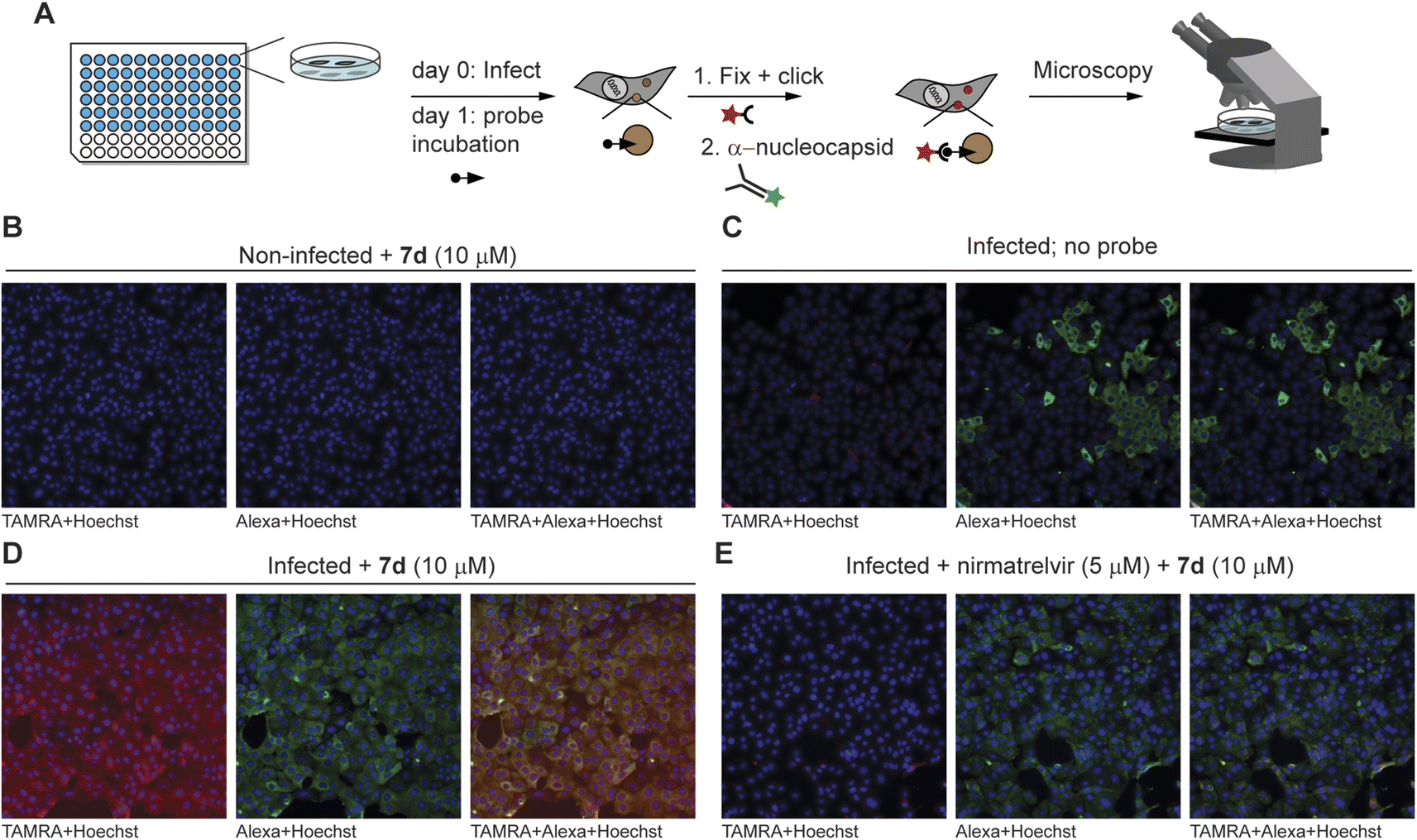 | ||
| Fig. 4 Fluorescence microscopy detection of Mpro inhibition in SARS-CoV-2-infected cells. (A) Summary of workflow. One day after infection of VeroE6 cells, probes were added to cells – with or without inhibitor pretreatment. Click chemistry with TAMRA-N3 as well as antibody staining against the nucleocapsid protein of the virus were performed after fixation, followed by fluorescence microscopy. The minimum intensity for visualization was set above the background intensity of no probe control samples. (B) Non-infected control cells do not show TAMRA staining derived from the probe (in red) or Alexa647 staining of the SARS-CoV-2 anti-nucleocapsid protein (in green). (C) Infected cells without probe treatment do not show red fluorescence in the cells positive for the SARS-CoV-2 virus (green). (D) Treatment with probe 7d results in red staining of infected cells, which can be seen by yellow co-staining in the overlay panel. The staining can be attributed to Mpro activity, as uninfected cells (panel (B)) and cells not treated with the probe (panel (C)) do not show this. (E) Mpro inhibitor nirmatrelvir reduces the signal by probe 7d, further indicating that 7d reacts with active Mpro. See Fig. S4† for overlay channels and bright light images of cells. | ||
Conclusions
Mpro is an essential enzyme for SARS-CoV-2 replication and therefore represents an attractive drug target. To enable the detection of active Mpro within infected cells, here we developed highly potent and selective ABPs. A multifunctional design of azapeptide building block 4 allowed solid-phase synthesis and rapid variation of the reactive electrophile, which is a crucial element of ABPs. We found that azapeptide chloroacetamide 7d was highly active against SARS CoV-2 Mpro in a biochemical assay. This type of reactive warhead had a large influence on the potency of inhibition. Interestingly, the vinyl sulfonamide derivative, an electrophile with the highest reported reactivity, was inactive, likely because of the unfavorable placement in relation to the nucleophilic cysteine in the active site. Although compound 7d has high potency in vitro, the activity in cells is substantially lower, which is likely due to poor cell permeability. This can be attributed to the primary amide that mimics the glutamine side chain, as a related azapeptide inhibitor with a pyrrolidone as a cyclic glutamine side chain analog, reported during the course of this work, displays nanomolar potency.33 We therefore expect that the cellular potency of the here-reported probes, and thereby the applicability in cells, can be further fine-tuned by optimization of the peptide element as well as the reactive group. In this regard, the recent discovery of di- and trihaloacetamides as reactive electrophiles for SARS-CoV-2 Mpro may also be useful for future design of novel azapeptide Mpro inhibitors and probes.34 The solid-phase method described here will enable rapid synthesis of libraries of such compounds. Additionally, variation of the P1 position and the reactive warhead may also facilitate the development of ABPs for other viral proteases. The here-reported probe 7d permits the visualization of drug target engagement in a cellular context. We expect that this may aid future drug discovery efforts directed towards SARS-CoV-2 Mpro.Data availability
The coordinates of the docked compounds are available from the Research Data Repository of KU Leuven at: https://www.rdr.kuleuven.be.Author contributions
RV and SV conceived the project, RV performed chemical synthesis and biochemical evaluation, MBX performed protein expression and biochemical evaluation, WC performed experiments with the live virus, HZ and HMB performed IC50 measurements, SV performed in silico docking and mutagenesis, SV, NL, LV, DJ, SDJ, and JN supervised the experiments, and RV and SV performed data analysis and wrote the paper. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank Rita Derua for help in recording HRMS, Luc Baudemprez for NMR, Joost Schepers, Tina Van Buyten and Thibault Francken for assistance with SARS-CoV-2 assays, and the COVID Moonshot consortium for contributing reagents. We acknowledge financial support from KU Leuven (PDM grant PDMT2/21/074 to RV), Research Foundation Flanders FWO (project no. G0E3617N and post-doc fellowship 12A2723N), the Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfalen, the Regierende Bürgermeister von Berlin, and the Bundesministerium für Bildung und Forschung. Part of this research work was performed using the ‘Caps-It’ research infrastructure (project ZW13-02) that was financially supported by the Hercules Foundation, Belgium and Rega Foundation, KU Leuven.Notes and references
- D. A. Tyrrell and M. L. Bynoe, Lancet, 1966, 1, 76–77 CrossRef CAS PubMed.
- D. Hamre and J. J. Procknow, Proc. Soc. Exp. Biol. Med., 1966, 121, 190–193 CrossRef CAS PubMed.
- T. Kuiken, R. A. M. Fouchier, M. Schutten, G. F. Rimmelzwaan, G. Van Amerongen, D. Van Riel, J. D. Laman, T. De Jong, G. Van Doornum, W. Lim, A. E. Ling, P. K. S. Chan, J. S. Tam, M. C. Zambon, R. Gopal, C. Drosten, S. Van Der Werf, N. Escriou, J. C. Manuguerra, K. Stöhr, J. S. M. Peiris and A. D. M. E. Osterhaus, Lancet, 2003, 362, 263–270 CrossRef CAS PubMed.
- A. M. Zaki, S. van Boheemen, T. M. Bestebroer, A. D. M. E. Osterhaus and R. A. M. Fouchier, N. Engl. J. Med., 2012, 367, 1814–1820 CrossRef CAS PubMed.
- H. F. Tseng, B. K. Ackerson, Y. Luo, L. S. Sy, C. A. Talarico, Y. Tian, K. J. Bruxvoort, J. E. Tubert, A. Florea, J. H. Ku, G. S. Lee, S. K. Choi, H. S. Takhar, M. Aragones and L. Qian, Nat. Med., 2022, 28, 1063–1071 CrossRef CAS PubMed.
- A. Sigal, R. Milo and W. Jassat, Nat. Rev. Immunol., 2022, 22, 267–269 CrossRef CAS PubMed.
- S. Ullrich and C. Nitsche, Bioorg. Med. Chem. Lett., 2020, 30, 127377 CrossRef CAS PubMed.
- H. Yang and J. Yang, RSC Med. Chem., 2021, 12, 1026–1036 RSC.
- C. S. B. Chia, W. Xu and P. Shuyi Ng, ChemMedChem, 2022, 17, e202100576 CAS.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- W. Dai, B. Zhang, X. M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L. K. Zhang, Y. Xu, H. Yang and H. Liu, Science, 2020, 368, 1331–1335 CrossRef CAS.
- W. Vuong, M. B. Khan, C. Fischer, E. Arutyunova, T. Lamer, J. Shields, H. A. Saffran, R. T. McKay, M. J. van Belkum, M. A. Joyce, H. S. Young, D. L. Tyrrell, J. C. Vederas and M. J. Lemieux, Nat. Commun., 2020, 11, 5409 CrossRef CAS.
- C. Ma, M. D. Sacco, B. Hurst, J. A. Townsend, Y. Hu, T. Szeto, X. Zhang, B. Tarbet, M. T. Marty, Y. Chen and J. Wang, Cell Res., 2020, 30, 678–692 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. A. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586–1593 CrossRef CAS.
- B. Bai, A. Belovodskiy, M. Hena, A. S. Kandadai, M. A. Joyce, H. A. Saffran, J. A. Shields, M. B. Khan, E. Arutyunova, J. Lu, S. K. Bajwa, D. Hockman, C. Fischer, T. Lamer, W. Vuong, M. J. Van Belkum, Z. Gu, F. Lin, Y. Du, J. Xu, M. Rahim, H. S. Young, J. C. Vederas, D. L. Tyrrell, M. J. Lemieux and J. A. Nieman, J. Med. Chem., 2022, 65, 2905–2925 CrossRef CAS PubMed.
- J. Breidenbach, C. Lemke, T. Pillaiyar, L. Schäkel, G. Al Hamwi, M. Diett, R. Gedschold, N. Geiger, V. Lopez, S. Mirza, V. Namasivayam, A. C. Schiedel, K. Sylvester, D. Thimm, C. Vielmuth, L. Phuong Vu, M. Zyulina, J. Bodem, M. Gütschow and C. E. Müller, Angew. Chem., Int. Ed., 2021, 60, 10423–10429 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- W. Rut, K. Groborz, L. Zhang, X. Sun, M. Zmudzinski, B. Pawlik, X. Wang, D. Jochmans, J. Neyts, W. Młynarski, R. Hilgenfeld and M. Drag, Nat. Chem. Biol., 2021, 17, 222–228 CrossRef CAS PubMed.
- L. Peñalver, P. Schmid, D. Szamosvári, S. Schildknecht, C. Globisch, K. Sawade, C. Peter and T. Böttcher, Angew. Chem., Int. Ed., 2021, 60, 6799–6806 CrossRef PubMed.
- M. A. T. van de Plassche, M. Barniol-Xicota and S. H. L. Verhelst, ChemBioChem, 2020, 21, 3383–3388 CrossRef CAS.
- J. S. Martin, C. J. MacKenzie, D. Fletcher and I. H. Gilbert, Bioorg. Med. Chem., 2019, 27, 2066–2074 CrossRef CAS.
- E. Resnick, A. Bradley, J. Gan, A. Douangamath, T. Krojer, R. Sethi, P. P. Geurink, A. Aimon, G. Amitai, D. Bellini, J. Bennett, M. Fairhead, O. Fedorov, R. Gabizon, J. Gan, J. Guo, A. Plotnikov, N. Reznik, G. F. Ruda, L. Díaz-Sáez, V. M. Straub, T. Szommer, S. Velupillai, D. Zaidman, Y. Zhang, A. R. Coker, C. G. Dowson, H. M. Barr, C. Wang, K. V. M. Huber, P. E. Brennan, H. Ovaa, F. Von Delft and N. London, J. Am. Chem. Soc., 2019, 141, 8951–8968 CrossRef CAS PubMed.
- L. Petri, P. Ábrányi-Balogh, P. R. Varga, T. Imre and G. M. Keserű, Bioorg. Med. Chem., 2020, 28, 115357 CrossRef CAS PubMed.
- J. L. Asgian, K. E. James, Z. Z. Li, W. Carter, A. J. Barrett, J. Mikolajczyk, G. S. Salvesen and J. C. Powers, J. Med. Chem., 2002, 45, 4958–4960 CrossRef CAS PubMed.
- T. W. Lee, M. M. Cherney, C. Huitema, J. Liu, K. E. James, J. C. Powers, L. D. Eltis and M. N. G. James, J. Mol. Biol., 2005, 353, 1137–1151 CrossRef CAS PubMed.
- B. Sanders, S. Pohkrel, A. Labbe, I. Mathews, C. Cooper, R. Davidson, G. Phillips, K. Weiss, Q. Zhang, H. O’Neill, M. Kaur, L. Ferrins, J. Schmidt, W. Reichard, S. Surendranathan, D. Kumaran, B. Andi, G. Babnigg, N. Moriarty, P. Adams, A. Joachimiak, C. Jonsson, S. Wakatsuki, S. Galanie, M. Head and J. Parks, Res. Sq., 2022, rs.3.rs-1840200 Search PubMed.
- D. Kato, S. H. L. Verhelst, K. B. Sexton and M. Bogyo, Org. Lett., 2005, 7, 5649–5652 CrossRef CAS PubMed.
- K. B. Sexton, D. Kato, A. B. Berger, M. Fonovic, S. H. L. Verhelst and M. Bogyo, Cell Death Differ., 2007, 14, 727–732 CrossRef CAS PubMed.
- O. Busnel and M. Baudy-Floc'h, Tetrahedron Lett., 2007, 48, 5767–5770 CrossRef CAS.
- W. L. Delano, The PyMol Molecular Graphics System, https://www.pymol.org Search PubMed.
- K. Steuten, H. Kim, J. C. Widen, B. M. Babin, O. Onguka, S. Lovell, O. Bolgi, B. Cerikan, C. J. Neufeldt, M. Cortese, R. K. Muir, J. M. Bennett, R. Geiss-Friedlander, C. Peters, R. Bartenschlager and M. Bogyo, ACS Infect. Dis., 2021, 7, 1457–1468 CrossRef CAS PubMed.
- D. Greenbaum, A. Baruch, L. Hayrapetian, Z. Darula, A. Burlingame, K. F. Medzihradszky and M. Bogyo, Mol. Cell. Proteomics, 2002, 1, 60–68 CrossRef CAS PubMed.
- Y. Hirose, N. Shindo, M. Mori, S. Onitsuka, H. Isogai, R. Hamada, T. Hiramoto, J. Ochi, D. Takahashi, T. Ueda, J. M. M. Caaveiro, Y. Yoshida, S. Ohdo, N. Matsunaga, S. Toba, M. Sasaki, Y. Orba, H. Sawa, A. Sato, E. Kawanishi and A. Ojida, J. Med. Chem., 2022, 65, 13852–13865 CrossRef CAS PubMed.
- C. Ma, Z. Xia, M. D. Sacco, Y. Hu, J. A. Townsend, X. Meng, J. Choza, H. Tan, J. Jang, M. V. Gongora, X. Zhang, F. Zhang, Y. Xiang, M. T. Marty, Y. Chen and J. Wang, J. Am. Chem. Soc., 2021, 143, 20697–20709 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04147b |
| This journal is © The Royal Society of Chemistry 2023 |