
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical reactions of a diamidocarbene: cyclopropanation of bromonaphthalene, addition to pyridine, and activation of sp3 C–H bonds†
Tharushi A.
Perera‡
a,
William V.
Taylor
a,
M. Brenton
Gildner
 a,
Eric W.
Reinheimer
b,
Sho
Ito
c,
Anna
Nelson
a,
Shane R.
Yost
a and
Todd W.
Hudnall
*a
a,
Eric W.
Reinheimer
b,
Sho
Ito
c,
Anna
Nelson
a,
Shane R.
Yost
a and
Todd W.
Hudnall
*a
aDepartment of Chemistry and Biochemistry, Texas State University, 601 University Dr, San Marcos, TX 78666, USA. E-mail: hudnall@txstate.edu
bRigaku Americas Corporation, 9009 New Trails Dr, The Woodlands, TX 77381, USA
cRigaku Corporation, 3-9-12, Matsubara, Akishima, Tokyo 196-8666, Japan
First published on 10th May 2023
Abstract
We report unprecedented photochemistry for the diamidocarbene 1. Described within are the double cyclopropanation of 1-bromonaphthalene, the double addition to pyridine, and remarkably, the insertion into the unactivated sp3 C–H bonds of cyclohexane, tetramethylsilane, and n-pentane to give compounds 2–6, respectively. All compounds have been fully characterized, and the solid state structure of 4 was obtained using single crystal electron diffraction.
1 Introduction
The use of light as a renewable, green reagent to alter chemical reactivity or to activate or catalyse a chemical reaction remains ever important with dwindling global resources. Indeed, recent advances in photochemistry have made a profound impact on chemical synthesis; leading to a renaissance of contemporary photochemical studies in areas ranging from catalysis, switchable reactivity, materials science, medicinal chemistry, sensors, and fine chemical synthesis.1–6For example, photoredox catalysts derived from Ru/Ir polypyridyl complexes, dihydrophenazines, or simple dyes such as Eosin Y and Rose Bengal have been extensively investigated in recent years to mediate the single electron oxidation or reduction of organic molecules in exquisite organic synthesis.7–9 Similarly, photocatalytic pyrylium salts can initiate ring-opening metathesis polymerization of strained olefins,10 and photoswitchable carbenes have been developed where light can modulate the electronic properties of the carbene center.11
Photocatalytic C–H activation has been shown to occur in heterogeneous systems, but rarely with molecular photocatalysts.3,12–14 Indeed, there have been many reports of C–H functionalization using organophotoredox catalysis, but the challenge of C–H activation, especially among unactivated C–H bonds, has remained.8,15–17 Thus, the use of precious metal catalysts, high temperatures, and directing group manipulations are typically required.17 Additionally, photochemically generated carbenes have been reported previously, performing group transfer of aryl groups, but not C–H activation.18
We recently demonstrated that the singlet ground state of the N,N′-diamidocarbene (DAC) 1 could be photochemically switched to a triplet excited state through intersystem crossing from an excited singlet state.19 The new excited state was determined to originate from a triplet spin state carbene through a combination of experimental and computational approaches (Fig. 1). Interestingly, we found that the triplet excited state of carbene 1 undergoes Büchner ring expansion reactions in the presence of various arenes to give thermally reversible products (Fig. 1). While triplet carbenes had been previously reported to undergo Büchner ring expansion reactions, the thermal reversibility observed with the triplet DAC was unprecedented and allowed for the photochemical interconversion between Büchner ring expansion products.
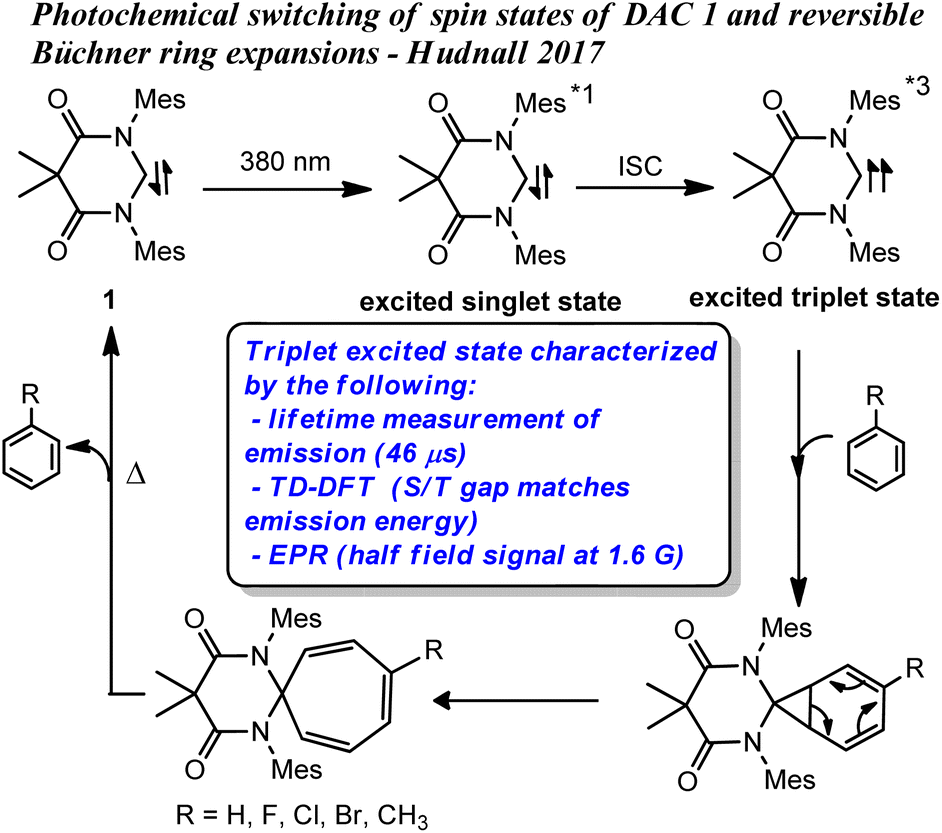 | ||
| Fig. 1 Previously reported photochemical Büchner reactions with DAC 1, *1 and *3 denotes excited singlet and triplet states, respectively. | ||
Similar to our study, Aldridge and co-workers found that the aluminyl complex, I, could undergo reversible Büchner ring expansion with benzene under thermal conditions (Scheme 1a).20 They also found that compound I could thermally insert into the C–H bonds of naphthalene and other arenes including benzene, toluene, n-butylbenzene, and xylenes (Scheme 1b).20,21 In contrast to C–H activation, Kira and co-workers observed that silylene II could undergo single, and double cyclopropanation reactions when irradiated in the presence of naphthalene, among other ring insertion reactions (Scheme 1c).22 Additionally, Ollevier and co-workers have performed cyclopropenation reactions from photolytically generated carbenes derived from diazirines,23 and Nguyen and Koenigs demonstrated that norcaradienes (i.e. cyclopropanated benzene derivatives) could be obtained from blue light irradiation of diazoacetates.24 In 2011, Braunschweig was able to trap the parent borylene (BH) through a formal [2 + 1] cycloaddition reaction in the presence of naphthalene that is analogous to the aforementioned cyclopropanation reactions (Scheme 1d).25
 | ||
| Scheme 1 Thermal insertion and C–H activation reactions with I (a and b), photochemical cyclopropanation of naphthalene with II (c), and trapping of parent borylene with naphthalene (d). | ||
Inspired by these results, we herein report novel photochemical reactivity of DAC 1, including the cyclopropanation of arenes, addition to heterocyclic arenes, and insertion into unactivated sp3 C–H bonds.
2 Results and discussion
After discovering that DAC 1 could undergo reversible Büchner ring expansion reactions upon irradiation, we became interested in exploring the photochemistry of this carbene further. We first decided to explore the photochemical reactions of 1 with naphthalene, taking inspiration from Kira and Aldridge, as described above.Following the reported procedure from Kira, we irradiated a hexanes solution of DAC 1 and 10 molar equivalents of naphthalene at 380 nm for two hours. Unfortunately, the 1H NMR spectrum of the crude reaction product revealed an intractable mixture of several different compounds. Importantly, we noted that the majority of these compounds appeared to be products from the insertion of 1 into C–H bonds of the solvent. Due to the constitutional isomeric mixture of hexanes, and the multiple different C–H bonds, there are numerous potential C–H insertion products. While this reaction was murky, it did expose an unexpected reactivity profile: unactivated sp3 C–H bond insertion.26 Given the inherent complication of attempting additional photolysis reactions in organic solvents, we began exploring reactions with liquid substrates to reduce potential unwanted side-reactions. To this end, DAC 1 was irradiated at 380 nm in neat 1-bromonaphthalene, pyridine, cyclohexane, and tetramethylsilane (Scheme 2).
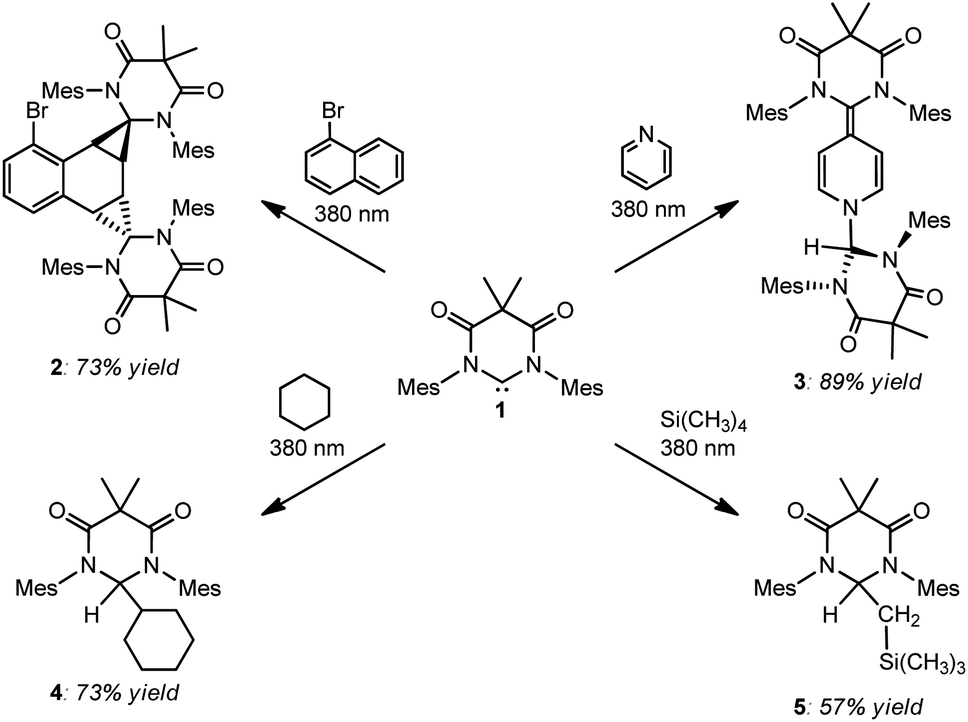 | ||
| Scheme 2 Photochemical reactions of DAC 1 with 1-bromonaphthalene (upper left), pyridine (upper right), cyclohexane (lower left), and tetramethylsilane (lower right). | ||
2.1 Photochemical reaction of DAC 1 with 1-bromonaphthalene
As can be seen in Scheme 2, irradiation of 1 at 380 nm in neat 1-bromonaphthalene, led to clean conversion to the doubly cyclopropanated naphthalene compound (2) in good yield (73%). While Kira observed both single and double cyclopropanation products of naphthalene by silylene II, irradiation of 1 with 1-bromonaphthalene gave exclusively compound 2.27 This is in stark contrast to the thermally reversible cyclopropanation reported by Kira with silylene II and naphthalene.22a While DAC 1 has been shown to undergo thermal [2 + 1] cycloaddition reactions with olefins, aldehydes, and nitriles,28 our study represents the first example where 1 was shown to cyclopropanate an aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.
C bond.
Compound 2 was fully characterized by 1H and 13C NMR spectroscopy, and the identity of the compound was further verified by single crystal X-ray diffraction (Fig. 2). Salient spectroscopic features that were observed in the 1H NMR spectrum (CDCl3) of compound 2 include four inequivalent cyclopropyl protons which were all observed as doublets at 1.15 ppm (J = 12 Hz), 1.36 ppm (J = 12 Hz), 2.02 ppm (J = 8 Hz) and 2.11 ppm (J = 8 Hz). Additionally, two sets of signals for the inequivalent carbenes were observed, along with a doublet at 6.94 ppm (J = 7.7 Hz), a triplet at 7.03 ppm (J = 7.7 Hz), and a doublet at 7.44 (J = 7.8 Hz) which corresponded to the bromonaphthalene protons. A 1H–1H NMR correlation spectroscopy (COSY) experiment was also performed on compound 2 in CDCl3 to properly assign the cyclopropyl protons (see ESI† for full discussion). Fig. 3, which shows an expanded region of the COSY spectrum of compound 2 from 0.6 to 3.0 ppm, clearly shows that the proton labelled H2a is strongly correlated to H3a, and H5a is likewise correlated to H6a as expected for vicinal (J3) coupling. Importantly, no correlation between H3a and H5a was observed, consistent with the Karplus equation, which states that nuclear spin–spin coupling is maximized as the dihedral angle between neighbouring nuclei approaches either 0° or 180° and minimized at 90°.29 For H3a and H5a, the dihedral angle as measured by single crystal X-ray diffraction was 83.0(4)°, consistent with the COSY data.
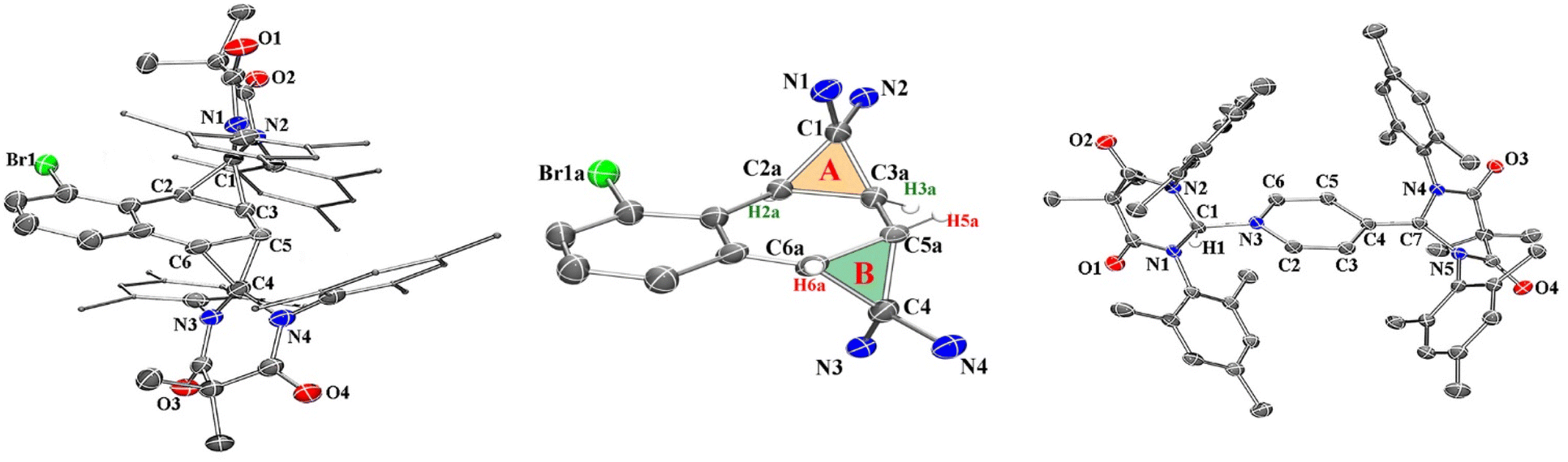 | ||
| Fig. 2 Crystal structures of 2 (far left and middle) and 3 (far right), thermal ellipsoids rendered at 50% probability. Hydrogen atoms have been removed for clarity with the exception of H2a, H3a, H5a, and H6a in compound 2 as well as H1 in compound 3. Pertinent metrical parameters are provided in the text. | ||
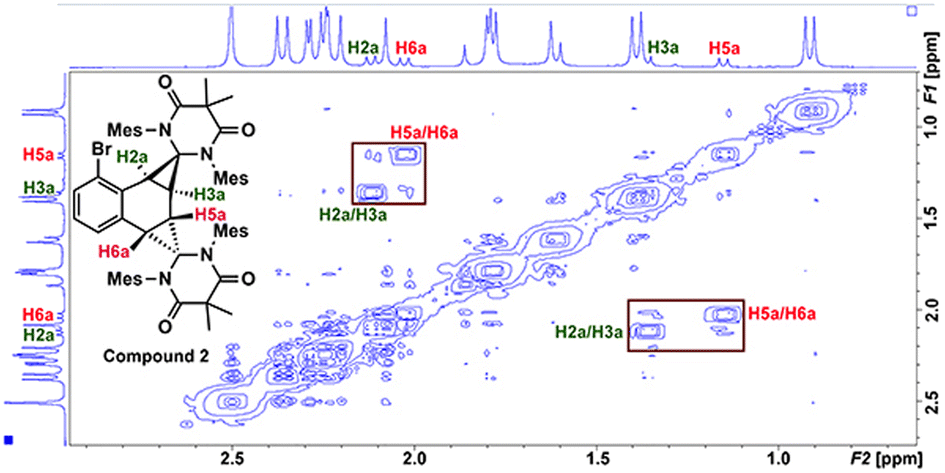 | ||
| Fig. 3 1H–1H correlation spectrum (COSY) of compound 2 from 0.6–3.0 ppm. | ||
In the solid state, the two independent cyclopropyl moieties exhibit the expected bond angles: C1–C2a–C3a = 60.0(18)°, C2a–C1–C3a = 57.9(14)°, C1–C3a–C2a = 62.1(16)° (ring A), and C4–C5a–C6a = 60.7(15)°, C5a–C4–C6a = 61.1(13)°, C4–C6a–C5a = 58.2(19)° (ring B), which give 180° for the sum of the internal angles for each ring, consistent with the formation of cyclopropanes. As mentioned in the previous paragraph, the dihedral angle Θ = 83.0(4)° between H3a and H5a is consistent with the apparent lack of coupling between these two vicinal protons in the 1H NMR and attendant COSY spectra.
2.2 Photochemical reaction of DAC 1 with pyridine
We next investigated the photochemical reaction of DAC 1 with pyridine as we wanted to explore the effect of a heteroatom on the regiochemistry of the Büchner ring expansion reaction described in Fig. 1. To this end, a solution of 1 in pyridine was irradiated at 380 nm for 30 minutes (Scheme 2) to give exclusive formation of compound 3 in excellent yield (89%).When the 1H NMR (C6D6) of 3 was first examined, we had initially thought that the expected Büchner ring expansion had occurred due to the appearance of olefinic protons spanning 4–6 ppm (Fig. 4). However, closer inspection into the integration of the olefinic signals compared to signals arising from the former carbene revealed that the product obtained had incorporated two carbene moieties into a single pyridine molecule. Additionally, a singlet at 5.06 ppm was observed which is consistent with addition of a hydrogen atom to the carbene centre of 1,30 indicating that the carbene had inserted into the C–H bond of the pyridine. The identity of compound 3 was further confirmed by single crystal X-ray diffraction (Fig. 2).
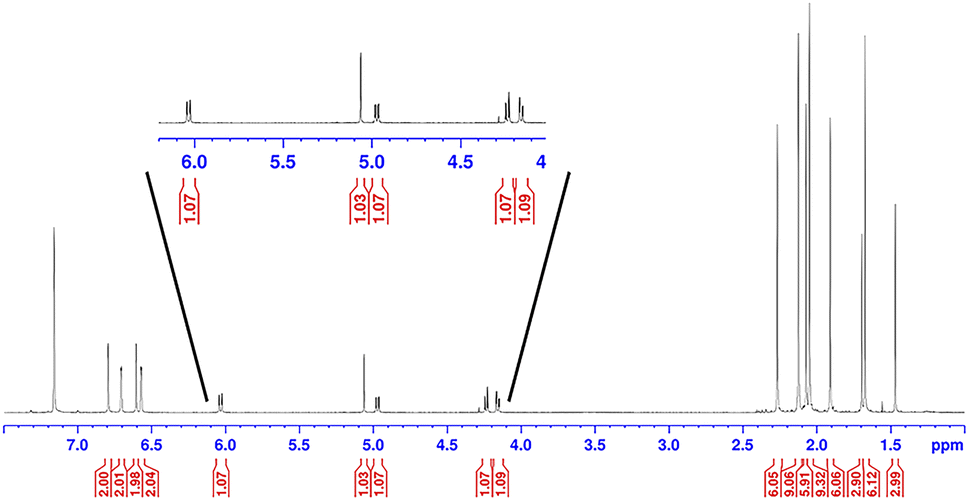 | ||
| Fig. 4 1H NMR of compound 3 taken in C6D6. | ||
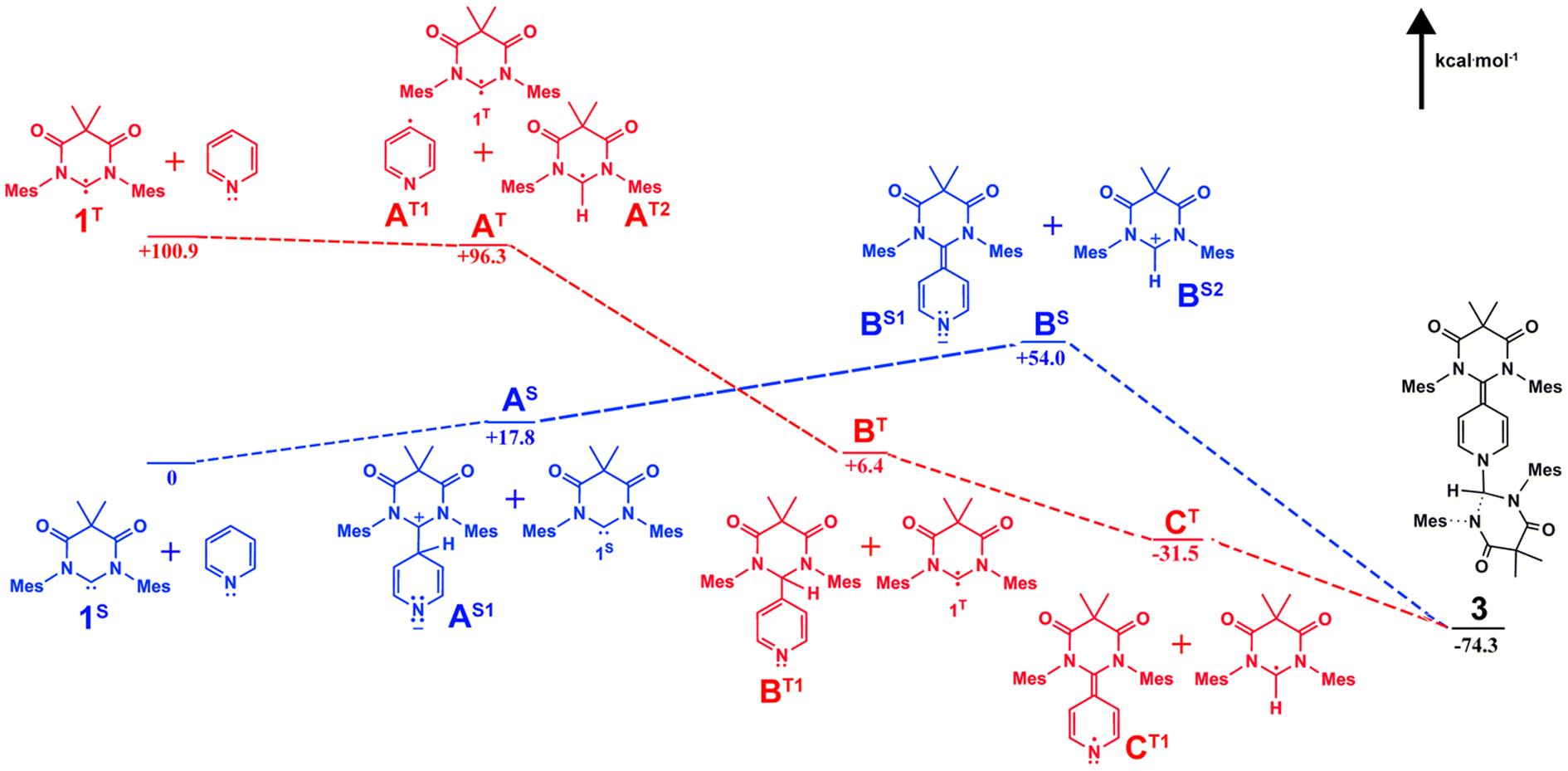
In the solid state structure of 3, the quinoidal geometry of the central – former pyridyl – ring was easily observed with short C7–C4, C5–C6, and C3–C2 distances of 1.373(2), 1.344(2), and 1.340(2) Å, respectively and longer C3–C4 and C4–C5 distances of 1.459(2) and 1.460(2) Å, respectively. Moreover, it was evident that the C7 carbon centre of a former carbene is trigonal planar and sp2-hybridized with the ΣC7 angles = 360.0. However, the C1 centre – the carbon atom of the second carbene molecule, adopts a distorted tetrahedral geometry, ΣC1 angles = 335.32°, consistent with the presence of the attached H1 atom. Based on this data, we hypothesize that one molecule of the triplet excited state DAC 1 inserted into the para C–H bond of pyridine followed by deprotonation or H-atom abstraction by a second equivalent of 1. Finally, combination of the two fragments resulted in formation of 3 (Fig. 5 below).
 | ||
| Fig. 5 Calculated singlet (blue) and triplet (red) pathways for the formation of compound 3 with probable intermediates. | ||
It has recently been shown by Bertrand and Stephan that carbenes including 1 can thermally insert into the C–F bonds of pentafluoropyridine31 and tris(pentafluorophenyl)borane,32 respectively; and more recently Hansmann33 showed that 1 could insert into the para C–H bond of pyridinium cations (Scheme 3). However, the electron poor substrates, such as those described in Hansmann's work are well known to undergo nucleophilic aromatic substitution in the presence of Lewis bases.31,32,34 In very stark contrast to these other thermal reactions, the formation of compound 3 was only observed when 1 was irradiated in the presence of pyridine, heating solutions of 1 only gave the known intramolecular C–H activation product.30c
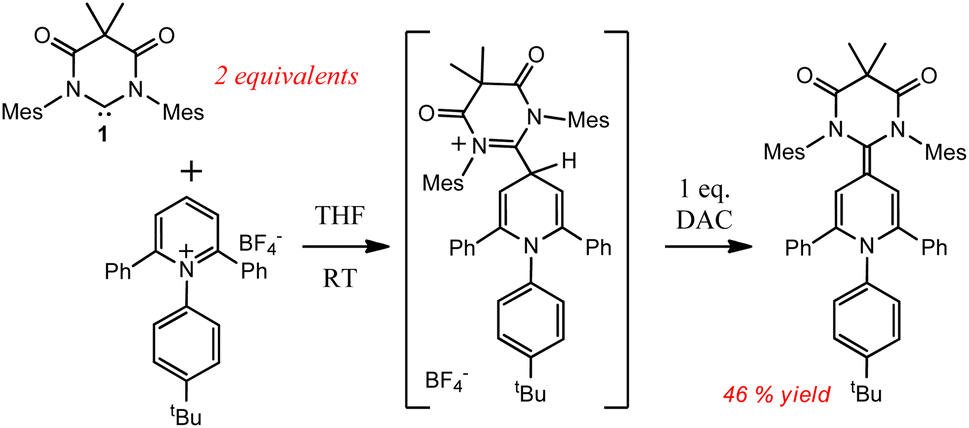 | ||
| Scheme 3 Known thermal insertion reactions of DAC 1 with pyridinium cations. | ||
To better understand this reactivity, we calculated probable thermal (singlet) and photochemical (triplet) intermediates for the formation of compound 3 starting from pyridine and singlet DAC 1 (1S) which have been defined as the zero-point energy (Fig. 5) using Q-Chem 5-package35 with the wB97x-D functional and 6-31G*/cc-pvtz basis sets for the optimization/energy calculations, respectively.36,37 As described, the singlet pathway (shown as blue structures) requires an activation energy of at least 54.0 kcal mol−1 to obtain the highest energy intermediates, BS1 and BS2 for the thermal reaction to proceed to the product 3. For the thermal pathway, addition of one molecule of DAC to the para carbon of pyridine gives the adduct AS1 which is 17.8 kcal mol−1 above 1S. Next, deprotonation by another DAC molecule results in formation of the highest energy intermediates BS. Coupling of these BS intermediates affords 3 (−74.3 kcal mol−1 relative to the zero-point energy).
On the other hand, the photochemical pathway (red structures) begins with a higher energy (+100.9 kcal mol−1), yet photochemically accessible triplet state (1T). From there, the remainder of the triplet pathway is downhill, first proceeding through H atom abstraction from pyridine by 1T to give intermediates AT at approximately 4 kcal mol−1 lower energy. Next, another 90 kcal mol−1 of energy is released upon coupling of pyridyl and 1T-derived radicals affords intermediate BT. Hydrogen atom abstraction from BT by an additional equivalent of 1T releases an additional 37 kcal mol−1 giving radical intermediates CT which undergo radical coupling to afford the final product 3.
2.3 Photochemical reactions of DAC 1 with cyclohexane and tetramethylsilane
Earlier, it was described that photochemical reactions of 1 in hydrocarbon solvents such as hexanes were complicated due to what appeared to be insertion into the multiple different C–H bonds in the solvent. To verify if photochemical C–H activation was possible using DAC, we next focused on the photolysis of 1 in liquid hydrocarbons featuring only a single type of C–H bond. First, we investigated the photochemical reaction of 1 with cyclohexane.Gratifyingly, irradiation of 1 in cyclohexane at 380 nm for 2 hours resulted in the formation of the C–H activation product, 4, in 73% yield. The identity of 4 was first confirmed by 1H NMR (C6D6) which revealed two diagnostic signals. First, a doublet (3J = 1.4 Hz) centred at 5.32 ppm, was observed which was consistent with a hydrogen atom (H1) bound to the former carbene C1 carbon atom (vide supra), and second, a multiplet centred at 1.62 was observed which corresponded to the remaining cyclohexyl proton (H2) attached to the C2 atom. A COSY NMR of compound 4 was performed to verify that H1 was indeed coupled to H2 (Fig. 6). As expected a correlation between the protons labelled H1 and H2 in the COSY spectrum was observed as indicated by the red boxes.
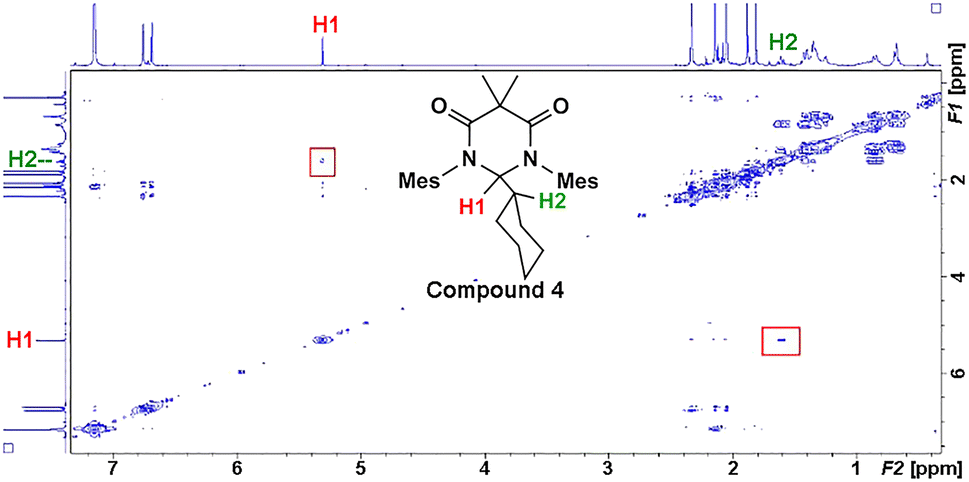 | ||
| Fig. 6 1H–1H correlation (COSY) spectrum of compound 4. | ||
To obtain further confirmation that C–H insertion into a cyclohexane molecule had been achieved, we next attempted to grow single crystals suitable for an X-ray diffraction analysis. Despite several solvent(s)/solvent combinations and various temperatures, we were only able to obtain microcrystalline powder. We confirmed that this powder was crystalline material out past 30° 2Θ using powder XRD (see ESI†). With this microcrystalline sample in hand, we were able to obtain publishable electron diffraction (ED) data which unequivocally confirmed the identity of compound 4 (Fig. 7).
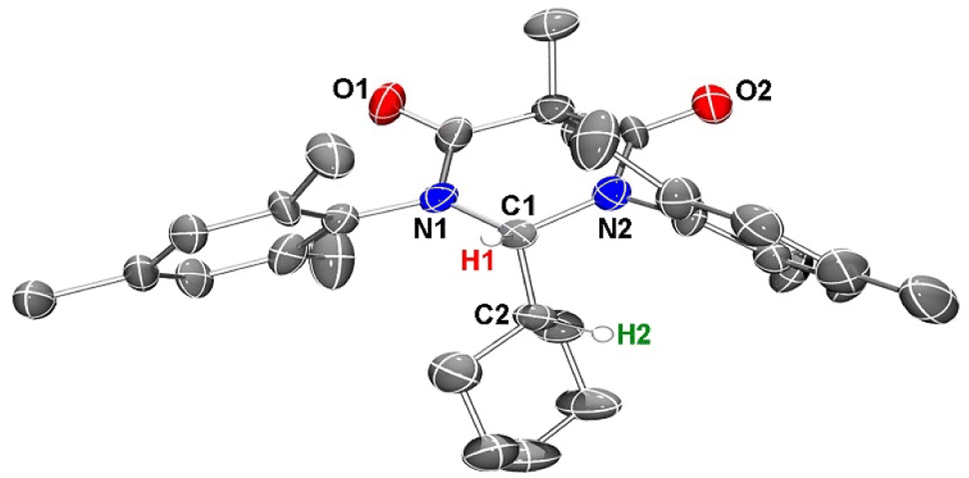 | ||
| Fig. 7 Single crystal electron diffraction structure of compound 4, thermal ellipsoids rendered at 30% probability. Hydrogen atoms except for H1 and H2 were removed for clarity. Pertinent metrical parameters are provided in the text. | ||
In the crystal structure of 4, the pyramidalization of the former carbene carbon atom, C1, was apparent with a ΣC1 angles = 340.18° and a 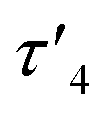 parameter38 of 0.92. It was also clear that the DAC inserted into the H1–C2 bond of the cyclohexyl substituent. Moreover, the H1–H2 dihedral angle of 72.6° was consistent with the weak coupling observed in the 1H NMR and COSY spectra obtained for compound 4. The C1–C2 distance of 1.605(17) Å was also slightly elongated when compared to the average C–C distance within the cyclohexyl moiety (1.541 Å), and most likely arises from steric strain. Unremarkably, the cyclohexyl group adopts the lowest energy chair conformation as would be expected.
parameter38 of 0.92. It was also clear that the DAC inserted into the H1–C2 bond of the cyclohexyl substituent. Moreover, the H1–H2 dihedral angle of 72.6° was consistent with the weak coupling observed in the 1H NMR and COSY spectra obtained for compound 4. The C1–C2 distance of 1.605(17) Å was also slightly elongated when compared to the average C–C distance within the cyclohexyl moiety (1.541 Å), and most likely arises from steric strain. Unremarkably, the cyclohexyl group adopts the lowest energy chair conformation as would be expected.
In an effort to interrogate if C–H or C–Si insertion reaction(s) could take place we explored the photochemical reaction of DAC 1 with tetramethylsilane (TMS). Although this reaction proved to be sluggish, taking up to 4 days with poor to moderate conversion, we observed insertion in the C–H bond of a methyl group on the TMS to afford compound 5 in 57% yield (based on purity of 1H NMR, see ESI† for detailed discussion). The most salient spectroscopic feature in the 1H NMR (C6D6) that allowed us to identify the structure of 5 was the characteristic Ccarbene–H resonance centred at 5.32 ppm. Unlike the NMR for compound 4, this resonance was a triplet (3JH–H = 4.0 Hz) that was coupled to a doublet centred at 1.32 (3JH–H = 4.0 Hz) which integrated to two protons corresponding to the methylene CH2–Si(CH3)3 protons. Additionally, a high-field singlet which integrated to 9 protons was observed at −0.64 ppm corresponding to the Si(CH3)3 group. Similar to compound 4, the coupling of the triplet (proton Ha) at 5.32 ppm and the doublet (proton Hb) at 1.32 ppm was observed in the COSY spectrum of compound 5 (Fig. 8). Indeed, there is a correlation between the protons labelled Ha and Hb in the COSY spectrum of compound 5 as indicated in the red boxes.
 | ||
| Fig. 8 1H–1H correlation spectrum (COSY) of compound 5 from 0–5.6 ppm. | ||
2.4 Photochemical reactions of DAC 1 with n-pentane
Finally, we sought to preliminarily investigate if DAC 1 has a preference for inserting into various C–H bonds, i.e. primary, secondary, or tertiary. It must be stated that to fully establish accurate reactivity trends, it would require the screening of numerous substrates of varying degrees of sterics and/or electronic features–which is beyond the scope of this paper. To further investigate the reactivity of this system, we have performed an additional study using n-pentane as a substrate.Irradiation of a suspension 1 in n-pentane at 380 nm for 12 hours resulted in the dissolution of the majority of the solid material. The yellow supernatant solution was separated from the residual solid by filtration followed by the evaporation of all volatile materials to afford compounds 6a–c as a mixture of isomers in 87.6% yield which, to date, we have been unable to separate (Scheme 4). Characterization of the residual solid from the reaction revealed minor amounts of unreacted DAC 1 and some DAC-ether which forms from trace water in the reaction. Although we have been unable to separate the isomeric mixture, we have confirmed the identity of these compounds using HRMS, 1H NMR spectroscopy, and computational methods.
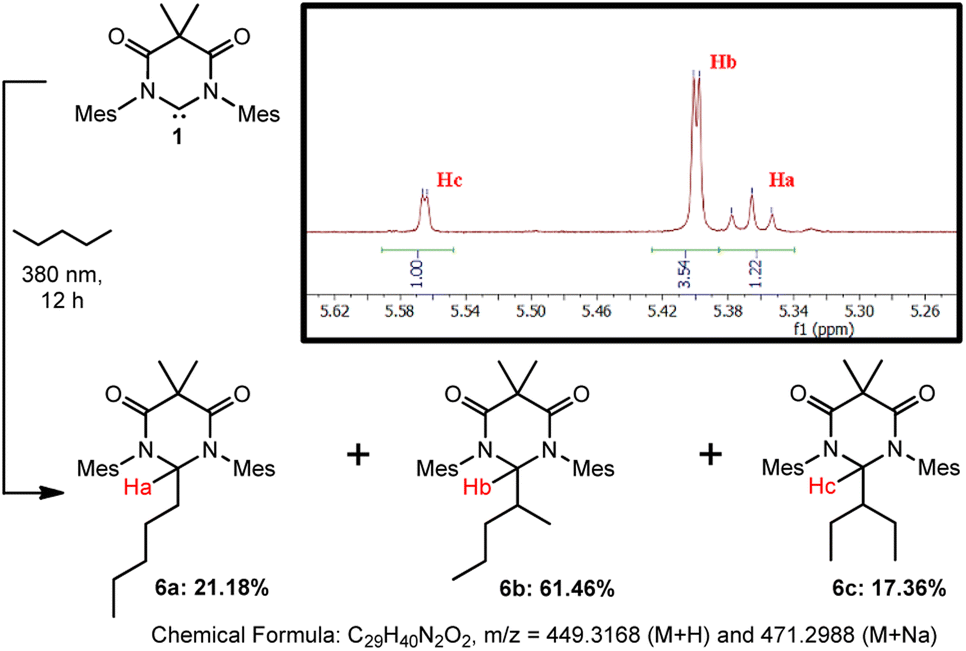 | ||
| Scheme 4 Photochemical reaction of DAC 1 with n-pentane to afford isomeric mixture of 6a–c. Inset shows 1H NMR (C6D6) of product mixture from 5.24–5.64 ppm. | ||
First, we performed HRMS (ESI†) to confirm the identity of the mixture of compounds and observed signals corresponding to the M + H (m/z = 449.3166) and M + Na (m/z = 471.2987) species which were nearly identical with the predicted values shown in Scheme 4. Second, we were able to identify the proton Ha corresponding to compound 6a, which appeared as a triplet at 5.366 ppm (C6D6, 3J = 6.2 Hz) due to coupling to the adjacent methylene group, where the DAC had inserted into the C–H bond of either the C1 or C5 carbon atoms. To identify Hb and Hc corresponding to 6b and 6c, respectively, we utilized DFT methods. The structures of compounds 6a–c were optimized using the B3LYP functional and 6-31G+d basis set and GAIO-SCF (benzene) NMR calculations were performed to correlate the observed experimental NMR signals to the appropriate products (see ESI† for full discussion).
The experimental and calculated NMR chemical shifts are provided in Table 1. The calculated chemical shifts were collectively upfield relative to the experimental values by an average of 0.098 ppm, but were all in good agreement. To differentiate between compounds 6b and 6c, the dihedral angles between Hb or Hc and their corresponding vicinal hydrogen atoms obtained from the optimized structures were inputted into the Karplus equation29 and the 3JH–H coupling constants were calculated. Although there is a larger deviation between the calculated and experimental coupling constants, the magnitude of the coupling allowed us to assign the NMR signal at 5.399 ppm to 6b and at 5.565 ppm to 6c. Statistically, this is what one would expect as there are four available secondary C–H bonds on carbons C2/C4 of pentane versus only two available C–H bonds on carbon C3, resulting in 6b forming preferentially over 6c. Moreover, the fact that 6b and 6c were not formed in the statistical 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, indicates that additional factors, such as the more sterically encumbered C–H bonds on C3 to give 6c, may play a role in the observed selectivity.
:1 ratio, indicates that additional factors, such as the more sterically encumbered C–H bonds on C3 to give 6c, may play a role in the observed selectivity.
| Proton | Exp. chemical shift (ppm) | Calc. chemical shift (ppm) | Delta of exp. vs. calc. chemical shift (ppm) |
|---|---|---|---|
| Ha | 5.366 | 5.276 | 0.090 |
| Hb | 5.399 | 5.301 | 0.098 |
| Hc | 5.565 | 5.459 | 0.106 |
| Proton | Exp. 3JH–H (Hz) | Calc. 3JH–H (Hz) | Delta of exp. vs. calc. 3JH–H (Hz) | Calc. dihedral angle (°) |
|---|---|---|---|---|
| Ha | 6.2 | 6.14 | 0.06 | 166/79 |
| Hb | 1.65 | 1.22 | 0.43 | 78.29 |
| Hc | 1.45 | 1.16 | 0.29 | 80.02 |
Admittedly, these preliminary results do not provide a full structure–reactivity relationship as they do not address steric or electronic factors, nor do they evaluate tertiary C–H bonds. However, these results clearly demonstrate that the excited state triplet DAC preferentially reacts with secondary over primary C–H bonds. This is evidenced by the distribution of 78.82% of the products (6b and 6c) being derived from secondary C–H bonds whereas only 21.18% (6a) comes from insertion into a primary C–H bond. Importantly, statistics do not play a role in this observed preference as there are exactly six secondary and six primary C–H bonds available for insertion.
3 Conclusions
The photochemical reactions of diamidocarbene 1 with 1-bromonaphthalene, pyridine, cyclohexane, and tetra-methylsilane have been explored. These reactions demonstrated that the photochemically accessible triplet excited state of 1 can: (i) cyclopropanate polyaromatic compounds, (ii) insert into unactivated aromatic C–H bonds, and (iii) can insert into unactivated sp3 C–H bonds.Compounds 2, 3, and 4 were fully characterized through 1D and 2D NMR spectroscopic experiments and their single crystal X-ray (compounds 2 and 3) and electron diffraction (compound 4) structures were determined to unequivocally verify their identity. The formation of compound 2 revealed that norcaradiene intermediates in our previously reported Büchner ring expansion reactions with DAC 1 could be obtained when appropriate arene substrates are employed. Interestingly, the photolysis of 1 in pyridine did not afford the anticipated Büchner ring expansion product, but rather a double addition product, 3. While the thermal insertion of carbenes, including 1, into pyridyl and pyridinium derivatives have been previously demonstrated, DFT computations revealed that the formation of 3 is only possible through a triplet pathway.
Finally, the insertion into unactivated sp3 C–H bonds marks the first example of such reactivity with a singlet carbene. While the photochemistry of DAC 1 is still an ongoing area of research in our group, we envision that this novel C–H activation chemistry, coupled with our previously reported reversible Büchner ring expansion reactions, could pave the way for new chemistry relevant to the functionalization of hydrocarbons.
Data availability
The experimental procedures, analytical data and computational details supporting the findings of this study are available within the manuscript and its ESI file.†Author contributions
T. W. H., T. A. P, W. V. T., and M. B. G. co-wrote the paper. T. W. H. conceived of, and designed the study. T. A. P., W. V. T., and M. B. G. contributed equally to the scientific experimentation. E. W. R. assisted with crystallographic measurements and M. B. G. and S. R. Y. performed the computational analyses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TWH gratefully acknowledges the Robert A. Welch Foundation (AI-1993-20190330) and the National Science Foundation (CHE-1955396) for their generous support. The authors thank Dr David Schilter for assistance with HRMS samples.Notes and references
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and Z. A. Von, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- J. P. Holland, M. Gut, S. Klingler, R. Fay and A. Guillou, Chem.–Eur. J., 2020, 26, 33–48 CrossRef CAS PubMed.
- S. Verma, R. B. Nasir Baig, M. N. Nadagouda and R. S. Varma, Catal. Today, 2018, 309, 248–252 CrossRef CAS PubMed.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- Ü. Tastan, F. Guba and D. Ziegenbalg, Chem. Eng. Technol., 2017, 40, 1418–1424 CrossRef.
- J. Ma, S. Chen, P. Bellotti, R. Guo, F. Schäfer and A. Heusler, et al. , Science, 2021, 371, 1338–1345 CrossRef CAS PubMed.
- M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543–9547 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- K. A. Ogawa, A. E. Goetz and A. J. Boydston, J. Am. Chem. Soc., 2015, 137, 1400–1403 CrossRef CAS PubMed.
- A. J. Teator, Y. Tian, M. Chen, J. K. Lee and C. W. Bielawski, Angew. Chem., Int. Ed., 2015, 54, 11559–11563 CrossRef CAS PubMed.
- B. Chen, L.-Z. Wu and C.-H. Tung, Acc. Chem. Res., 2018, 51, 2512–2523 CrossRef CAS PubMed.
- L. Wang, Z. Huang, S. Xie, Q. Zhang, H. Wang and Y. Wang, Catal. Commun., 2021, 153, 106300 CrossRef CAS.
- X. Cao, T. Han, Q. Peng, C. Chen and Y. Li, Chem. Commun., 2020, 56, 13918–13932 RSC.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- B. Sahoo, J.-L. Li and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 11577–11580 CrossRef CAS PubMed.
- T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed.
- S. Jana, C. Pei, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2021, 60, 13271–13279 CrossRef CAS PubMed.
- T. A. Perera, E. W. Reinheimer and T. W. Hudnall, J. Am. Chem. Soc., 2017, 139, 14807–14814 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, A. Heilmann, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 20376–20380 CrossRef CAS PubMed.
- (a) M. Kira, S. Ishida, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 2002, 124, 3830–3831 CrossRef CAS PubMed; (b) K. Uchiyama, S. Nagendran, S. Ishida, T. Iwamoto and M. Kira, J. Am. Chem. Soc., 2007, 129, 10638–10639 CrossRef CAS PubMed.
- N. Tanbouza, V. Carreras and T. Ollevier, Org. Lett., 2021, 23, 5420–5424 CrossRef CAS PubMed.
- Y. Guo, T. V. Nguyen and R. M. Koenigs, Org. Lett., 2019, 21, 8814–8818 CrossRef CAS PubMed.
- P. Bissinger, H. Braunschweig, K. Kraft and T. Kupfer, Angew. Chem., Int. Ed., 2011, 50, 4704–4707 CrossRef CAS PubMed.
- C. M. Weinstein, G. P. Junor, D. R. Tolentino, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 9255–9260 CrossRef CAS PubMed.
- (a) R. Huisgen and G. Juppe, Chem. Ber., 1961, 94, 2332 CrossRef CAS; (b) G. M. Badger, B. J. Christie, H. J. Rodda and J. M. Pryke, J. Chem. Soc., 1958, 1179–11184 RSC; (c) A. Oku, K. Harada, T. Yagi and Y. Shirahase, J. Am. Chem. Soc., 1983, 105, 4400 CrossRef CAS; (d) A. Oku, H. Tsuji, M. Yoshida and N. Yoshiura, J. Am. Chem. Soc., 1981, 103, 1244 CrossRef CAS.
- (a) J. P. Moerdyk and C. W. Bielawski, Nat. Chem., 2012, 4, 275–280 CrossRef CAS PubMed; (b) J. P. Moerdyk and C. W. Bielawski, J. Am. Chem. Soc., 2012, 134, 6116–6119 CrossRef CAS PubMed; (c) J. P. Moerdyk, D. Schilter and C. W. Bielawski, Acc. Chem. Res., 2016, 49, 1458–1468 CrossRef CAS PubMed.
- (a) M. Karplus, J. Chem. Phys., 1959, 30, 11–15 CrossRef CAS; (b) M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870–2871 CrossRef CAS.
- (a) T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039–16041 CrossRef CAS PubMed; (b) T. W. Hudnall, J. P. Moerdyk and C. W. Bielawski, Chem. Commun., 2010, 46, 4288–4290 RSC; (c) J. P. Moerdyk and C. W. Bielawski, Chem.–Eur. J., 2013, 19, 14773–14776 CrossRef CAS PubMed; (d) V. César, N. Lugan and G. Lavigne, Eur. J. Inorg. Chem., 2010, 361–365 CrossRef.
- S. Styra, M. Melaimi, C. E. Moore, A. L. Rheingold, T. Augenstein, F. Breher and G. Bertrand, Chem.–Eur. J., 2015, 21, 8441–8446 CrossRef CAS PubMed.
- R. J. Andrews and D. W. Stephan, Chem.–Eur. J., 2020, 26, 7194–7198 CrossRef CAS PubMed.
- P. W. Antoni, T. Bruckhoff and M. M. Hansmann, J. Am. Chem. Soc., 2019, 141, 9701–9711 CrossRef CAS PubMed.
- (a) G. C. Welch, J. R. R. San, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed; (b) K. C. Mondal, P. P. Samuel, H. W. Roesky, B. Niepoetter, R. Herbst-Irmer and D. Stalke, et al. , Chem. - Eur. J., 2014, 20, 9240–9245 CrossRef CAS PubMed.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao and N. Mardirossian, et al. , J. Chem. Phys., 2021, 155, 084801 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2201797–2201799. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05122b |
| ‡ Current address: Department of Natural Sciences, School of Health and Natural Sciences, Mercy College, 555 Broadway, Dobbs Ferry, NY, 10522, USA, E-mail: E-mail: nperera@mercy.edu |
| This journal is © The Royal Society of Chemistry 2023 |