
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElastic organic semiconducting single crystals for durable all-flexible field-effect transistors: insights into the bending mechanism†
Ranita
Samanta
a,
Susobhan
Das
 a,
Saikat
Mondal
a,
Tamador
Alkhidir
b,
Sharmarke
Mohamed
*bd,
Satyaprasad P.
Senanayak
*c and
C. Malla
Reddy
*a
a,
Saikat
Mondal
a,
Tamador
Alkhidir
b,
Sharmarke
Mohamed
*bd,
Satyaprasad P.
Senanayak
*c and
C. Malla
Reddy
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur, Nadia, West Bengal 741246, India. E-mail: cmallareddy@gmail.com
bDepartment of Chemistry, Green Chemistry & Materials Modelling Laboratory, Khalifa University of Science and Technology, P.O. Box 127788, Abu Dhabi, United Arab Emirates. E-mail: sharmarke.mohamed@ku.ac.ae
cNanoelectronics and Device Physics Lab, School of Physical Sciences, National Institute of Science Education and Research, An OCC of HBNI, Jatni 752050, India. E-mail: satyaprasad@niser.ac.in
dAdvanced Materials Chemistry Center (AMCC), Khalifa University of Science and Technology, P.O. Box 127788, Abu Dhabi, United Arab Emirates
First published on 22nd December 2022
Abstract
Although many examples of mechanically flexible crystals are currently known, their utility in all-flexible devices is not yet adequately demonstrated, despite their immense potential for fabricating high performance flexible devices. Here, we report two alkylated diketopyrrolopyrrole (DPP) semiconducting single crystals, one of which displays impressive elastic mechanical flexibility whilst the other is brittle. Using the single crystal structures and density functional theory (DFT) calculations, we show that the methylated diketopyrrolopyrrole (DPP-diMe) crystals, with dominant π-stacking interactions and large contributions from dispersive interactions, are superior in terms of their stress tolerance and field-effect mobility (μFET) when compared to the brittle crystals of the ethylated diketopyrrolopyrrole derivative (DPP-diEt). Periodic dispersion-corrected DFT calculations revealed that upon the application of 3% uniaxial strain along the crystal growth (a)-axis, the elastically flexible DPP-diMe crystal displays a soft energy barrier of only 0.23 kJ mol−1 while the brittle DPP-diEt crystal displays a significantly larger energy barrier of 3.42 kJ mol−1, in both cases relative to the energy of the strain-free crystal. Such energy–structure–function correlations are currently lacking in the growing literature on mechanically compliant molecular crystals and have the potential to support a deeper understanding of the mechanism of mechanical bending. The field effect transistors (FETs) made of flexible substrates using elastic microcrystals of DPP-diMe retained μFET (from 0.019 cm2 V−1 s−1 to 0.014 cm2 V−1 s−1) more efficiently even after 40 bending cycles when compared to the brittle microcrystals of DPP-diEt which showed a significant drop in μFET just after 10 bending cycles. Our results not only provide valuable insights into the bending mechanism, but also demonstrate the untapped potential of mechanically flexible semiconducting crystals for designing all flexible durable field-effect transistor devices.
Introduction
Organic crystalline materials, which enable many applications in the fields of electronics,1–4 smart actuators,5 sensors,6,7etc., have recently gained tremendous attention in materials science due to their unique advantages like room temperature processability, tunability, low cost, etc.8–10 Although high crystallinity is critical for obtaining optimum device performance,10 this often comes with a counter effect, i.e., increased mechanical brittleness,11,12 severely hampering the durability of the devices. Contrary to the general perception that all organic crystalline materials are brittle, many recent studies have demonstrated that a small subset of organic crystals exhibit exceptional mechanical flexibility with practical relevance.12–15 Systematic studies have shown that the mechanical flexibility or stress tolerance ability of molecular crystals strongly depends on their packing and the nature of the intermolecular interactions among the molecules.12–27 Although significant progress has been made in understanding the structure–mechanical property correlations in the field of crystal engineering, controlling mechanical flexibility in crystalline materials still remains an extraordinary challenge. Recent reports on several organic single crystals with exceptional elastic flexibility have shown their attractive applications in wave-guides,18,19,28 ferroelectrics,20,29 and two terminal semiconducting devices with outstanding strain independent conductivity.11,21,30,31 In addition to designing flexible crystals, the development of strategies based on fabricating organic crystals with inherent twists has also been utilized to realize strain independent charge transport in the crystals.32Gong and co-workers recently found a unique way to grow bent single crystals of coronene and used different device geometries for demonstrating two terminal conductivity.31 Although they used rigid substrates in their devices, the findings revealed that the bent coronene crystals exhibited several orders of magnitude increase in their conductivity as compared to the pristine crystals. The authors used X-ray photoelectron spectroscopy (XPS) for the first time to demonstrate a change in the molecular orbital energy levels with variations in the radius of curvature of the bent crystals.
Utilizing the recent critical insights gained from the understanding of elastic molecular crystals,12,14,20,22,23,28,33,34 here we demonstrate the advantage of elasticity in semiconducting single crystals for fabricating mechanically flexible and durable electronic devices, as compared to a structurally close brittle crystalline analogue. Hence, our correlation study, connecting molecular and supramolecular level information to the mechanical flexibility in the studied semiconducting single crystals, has implications for the design of mechanically durable and efficient crystalline organic electronics for use in flexible field-effect transistors (FET) and other optoelectronic applications, via a bottom-up approach (molecule-to-crystal-to-device) using the principles of crystal engineering. There are reports of FETs with relatively high mobility values derived from non-flexible substrates, but we note that the mobility values of the FETs described in our work are comparable to that of other reported FETs fabricated using flexible substrates.35 Here, we showcase the advantages of flexible semiconducting crystals over their brittle counterparts by fabricating a durable all-flexible FET, demonstrating a proof-of-concept.
Results
We prepared two derivatives of a well-known p-type semiconductor molecule, diketopyrrolopyrrole (DPP), substituted with alkyl groups, namely dimethyldiketopyrrolopyrrole (DPP-diMe) and diethyldiketopyrrolopyrrole (DPP-diEt) (Fig. 1a and b). | ||
| Fig. 1 Molecular structure (inset) and optical images of single crystals of (a) DPP-diMe and (b) DPP-diEt. Micrographs showing the bending steps in single crystals of (c) DPP-diMe (elastic) and (d) DPP-diEt (brittle, during the qualitative three-point flexural mechanical tests). | ||
We introduced simple alkyl groups based on the design strategies reported for soft-crystals.11,12,36 Aromatic systems with such functional groups typically close pack via π-stacking and other dispersive interactions in crystal structures which tend to accommodate local molecular movements under stress, thus in general imparting mechanical softness to the crystals.11,36 Acicular single crystals of DPP-diMe were obtained from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dichloromethane and toluene solution, whereas single crystals of DPP-diEt were obtained from a toluene solution via the slow solvent evaporation method in 5–7 days. In both cases, the obtained single crystals were amenable for all further studies. The results from qualitative mechanical bending tests17 showed that crystals of DPP-diMe bend smoothly (Fig. 1c) into a loop. Upon release of external mechanical stress, the crystals recovered to their original shape without any sign of permanent deformation, confirming their elastic nature (see ESI Movies S1 and S2†). Upon testing the mechanical properties of the crystals of DPP-diEt with comparable morphologies under similar conditions, we observed no appreciable elastic flexibility. Instead, crystals of DPP-diEt fractured in a brittle manner (Fig. 1d, ESI Movies S1 and S2†), a property which is characteristic of most molecular crystals. The maximum strain (ε) that the elastic crystal (length: 2 mm; thickness: 0.01 mm) of DPP-diMe could withstand during mechanical bending was in the range of 3–6% (for details see ESI, Fig. S1†). On the other hand, the acicular crystals of DPP-diEt fractured without any noticeable bending deflection, confirming their poor strain tolerance when compared to the DPP-diMe crystals. To rationalize the difference in the macroscopic mechanical behaviour, we investigated the two crystals using single crystal X-ray diffraction (SCXRD), nanoindentation, energy frameworks (using CrystalExplorer 17.5) and density functional theory (DFT) methods (see the ESI† for details).
:1 dichloromethane and toluene solution, whereas single crystals of DPP-diEt were obtained from a toluene solution via the slow solvent evaporation method in 5–7 days. In both cases, the obtained single crystals were amenable for all further studies. The results from qualitative mechanical bending tests17 showed that crystals of DPP-diMe bend smoothly (Fig. 1c) into a loop. Upon release of external mechanical stress, the crystals recovered to their original shape without any sign of permanent deformation, confirming their elastic nature (see ESI Movies S1 and S2†). Upon testing the mechanical properties of the crystals of DPP-diEt with comparable morphologies under similar conditions, we observed no appreciable elastic flexibility. Instead, crystals of DPP-diEt fractured in a brittle manner (Fig. 1d, ESI Movies S1 and S2†), a property which is characteristic of most molecular crystals. The maximum strain (ε) that the elastic crystal (length: 2 mm; thickness: 0.01 mm) of DPP-diMe could withstand during mechanical bending was in the range of 3–6% (for details see ESI, Fig. S1†). On the other hand, the acicular crystals of DPP-diEt fractured without any noticeable bending deflection, confirming their poor strain tolerance when compared to the DPP-diMe crystals. To rationalize the difference in the macroscopic mechanical behaviour, we investigated the two crystals using single crystal X-ray diffraction (SCXRD), nanoindentation, energy frameworks (using CrystalExplorer 17.5) and density functional theory (DFT) methods (see the ESI† for details).
DPP-diMe crystallizes in the monoclinic space group P21/n with half a molecule in the asymmetric unit. Molecules with a nearly planar conformation are π-stacked along the a-axis (crystal growth axis) to form parallel columns in the structure (Fig. 2a–c). The interplanar distance of the stacks is 3.298 Å with a slipping angle of 45° (Fig. 2a). Molecules of adjacent stacked columns are mutually oriented such that the methyl groups of one column point to the thiophene ring of another column (Fig. 2d). The π-stacking is a dominant feature of the structure with only weak (sp2)C–H⋯O and other dispersive contacts in the structure. The energy framework calculations further confirm that the π-stacking (Etotal = −69 kJ mol−1, see ESI Fig. S2 and S3†) interactions dominate the packing (Fig. 2b). Contributions from other dispersive interactions are significantly smaller with Etotal = −17 kJ mol−1 and −15 kJ mol−1 (see ESI Fig. S2 and S3†). The orientation of columnar packing with respect to both major (011) and minor (01−1) faces is comparable (Fig. 2c and d). The nearly corrugated arrangement of molecules makes the crystal structure devoid of any flat slip planes despite the presence of dispersive interactions in orthogonal directions, i.e., strong π-stacking and relatively weak (sp2)C–H⋯O and other dispersive interactions.
 | ||
| Fig. 2 Crystal packing of DPP-diMe crystals. (a) π-Stacked columns, extended along the crystal growth direction, with a slip-stack angle of 45° and interplanar distance of 3.298 Å and (b) the corresponding pairwise total interaction energy, Etotal = −69 kJ mol−1 (calculated from the CIF obtained from SCXRD). (c and d) Packing patterns viewed along different directions. | ||
The presence of dispersive interactions with large energy contributions and planar conformation of molecules, and the absence of facile slip planes, seem to favour the appreciable elastic flexibility in crystals of DPP-diMe.11,12,14,34 Upon mechanical bending of the crystals on the major face (011), molecules are expected to experience local changes to accommodate an expansion of the outer arc and contraction of the inner arc (Fig. 5a).12,14,19,23,28,33,34 Upon the release of stress, the molecules regain their original positions, allowing the crystal to restore its original macroscopic shape. Generally, crystals with strong three-dimensional networks and those with rigid structures (specific interactions) deform in a brittle manner.11,16,36 Crystals dominated by large dispersive interactions, particularly the parallel columns with dominant π-stacking, have been shown to allow reversible local molecular movements, thus making them exceptionally elastic.14,19,23,28,33,34 Crystals with facile slip planes may show a small initial elastic response but eventually deform plastically, limiting their restoration ability.37,38 In the case of DPP-diMe, we did not observe any plastic deformation during bending experiments.
The DPP-diEt molecule also crystallizes in the monoclinic space group P21/n with half a molecule in the asymmetric unit. However, here the molecule is non-planar with a “Z-shaped” conformation as the alkyl substituents with an extra –(CH2)– group are oriented nearly perpendicular to the molecular plane (Fig. 3a). Similar to DPP-diMe, these crystals also stack along the a-axis via π-stacking with an interplanar distance of 3.373 Å and slipping angle of 40° (Fig. 3b). The (011) and (01−1) faces were found to be the major and minor faces, respectively. The π-stacking interactions are slightly weaker (Etotal = −61 kJ mol−1) here (Fig. 3c). There are also other dispersive interactions in the structure with Etotal = −18 kJ mol−1, −11 kJ mol−1 and −19 kJ mol−1. In Etotal = −19 kJ mol−1, the major contribution is (sp2)C–H⋯O having a maximum Eele component of −12.4 kJ mol−1 (see ESI Fig. S4 and S5†). In the structure, four DPP-diEt molecules form a tetramer through the moderately strong (sp2)C–H⋯O (d/Å; θ/°: 2.49 Å, 164.07°) hydrogen bonding interactions (Fig. 3d). The electrostatic potential surfaces show that the carbonyl oxygen of DPP-diEt is more polarizing than that in DPP-diMe, which is consistent with the observation of stronger (sp2)C–H⋯O hydrogen bonding interactions in the former (Fig. S6†). We have not observed a slip parallel to the needle axis in these crystals, which suggests that the activation energy for the sliding of layers is higher than the fracture energy.11,12,17 Our analysis reveals that the interactions perpendicular to the stacking direction are relatively stronger and more specific in DPP-diEt as compared to that in DPP-diMe. Moreover, the non-planarity of the DPP-diEt molecule also might be contributing to the rigidity of the stacks, preventing local movement (rotation or splaying of stacked molecules). Short range and long-range molecular movements form the basis for elastic (reversible) and plastic (irreversible) bending, respectively, in molecular crystals. Hence, the rigid structure of DPP-diEt can be attributed to the observed brittleness in its crystals upon stressing on (011)/(01−1).
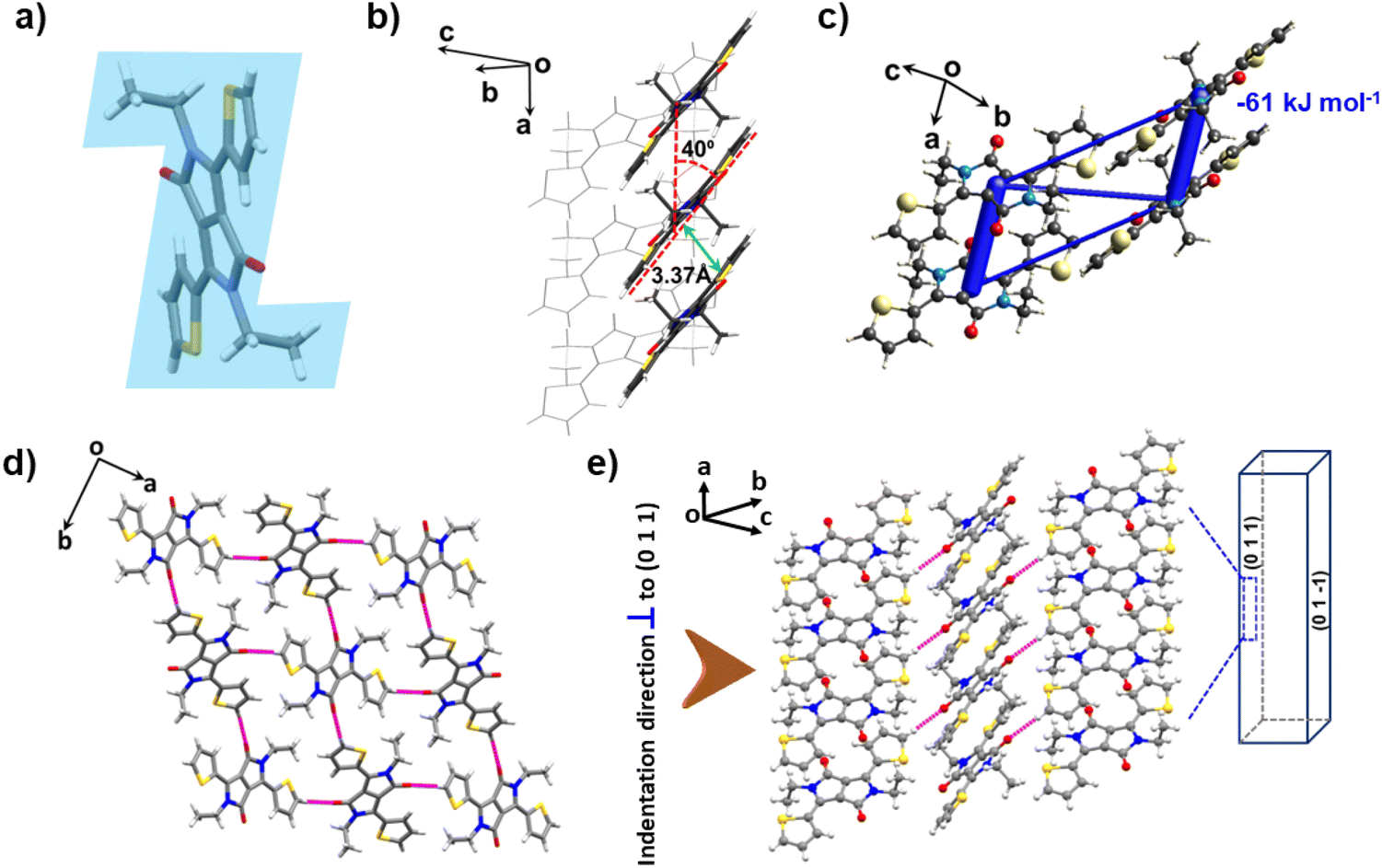 | ||
| Fig. 3 Crystal packing of DPP-diEt with the 3D topology of energy frameworks. (a) “Z-shaped” DPP-diEt molecule. (b) π-Stacked molecules with an interplanar distance of 3.373 Å and slip-stack angle of 40°. (c) Energy frameworks showing pairwise total intermolecular interaction energies (calculated from the CIF obtained from SCXRD). The thickness of the tube showcases the magnitude of the corresponding pairwise energy. (d) View of the hydrogen-bonded 2D sheet of DPP-diEt in the ab plane; (e) packing pattern showing the indentation direction. | ||
Furthermore, we quantified the mechanical properties39,40 of the two crystals by performing nanoindentation tests on the (011) major faces of DPP-diMe and DPP-diEt crystals at a peak load of 6 mN (ESI Fig. S7 and S8†). The typical load (P)–depth (h) curves obtained from the nanoindentation tests on the two crystals are given in Fig. 4a. The Young's modulus (E) and nanohardness (H) for DPP-diMe crystals are 6.47 ± 0.16 GPa and 0.24 ± 0.01 GPa, respectively (Fig. 4a). However, in the case of the DPP-diEt single crystal, the E and H are slightly higher, 8.64 ± 1.09 GPa and 0.27 ± 0.02 GPa, respectively (Fig. 4a). The representative two-dimensional (2D) profiles of the indent impressions of both the DPP-diMe and DPP-diEt single crystals along with their corresponding height profile diagrams are shown in Fig. 4b. The higher E (and marginally higher H) for the brittle DPP-diEt crystals is consistent with the higher share of contributions from specific C–H⋯O interactions perpendicular to the stacked columns, as compared to that of the elastic DPP-diMe crystals. The lower E and H in the case of DPP-diMe are consistent with its softer nature. Nevertheless, the nanoindentation results reveal that the mechanical properties of these crystals are comparable to other moderately soft molecular crystals.
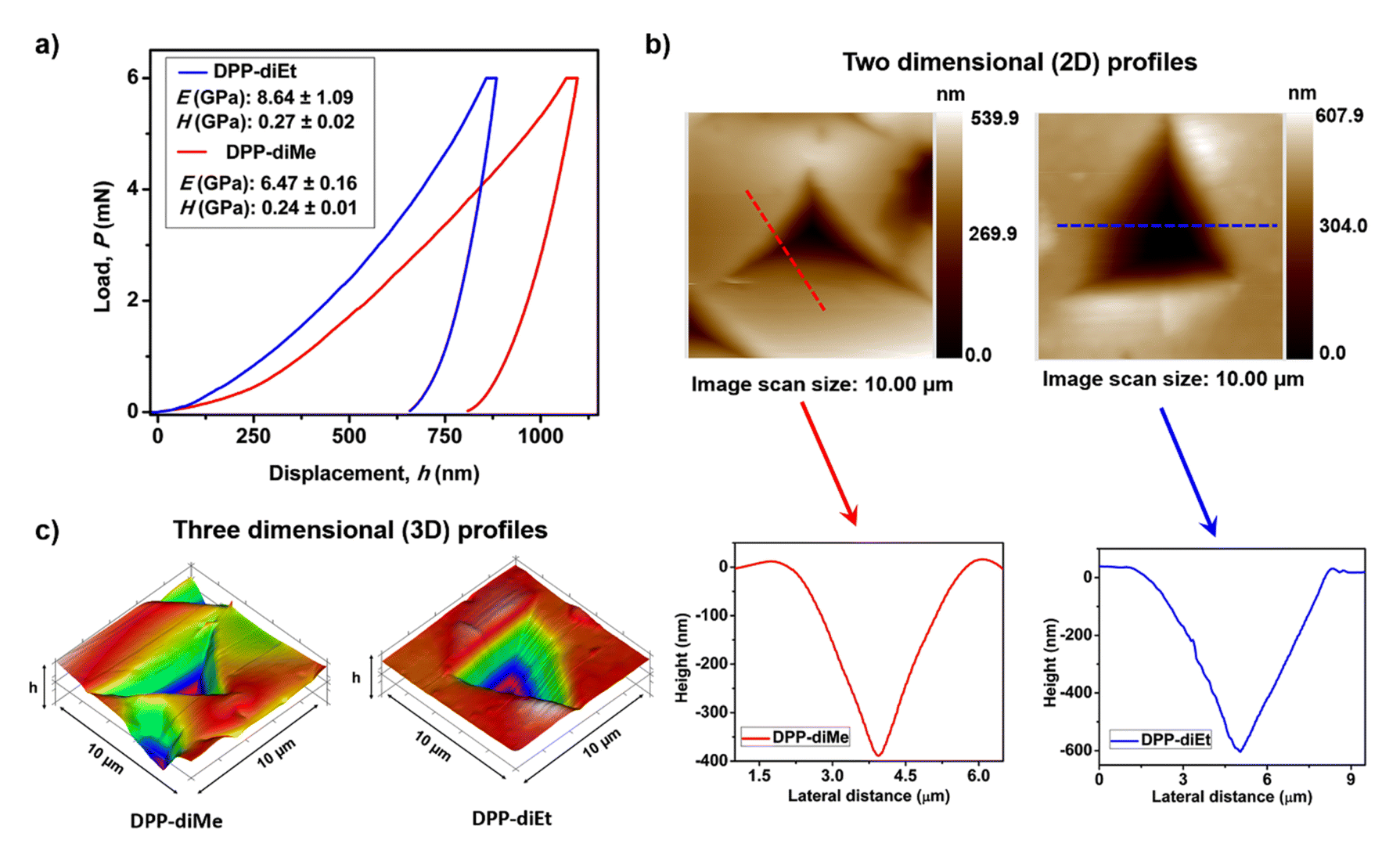 | ||
| Fig. 4 Quantification of the mechanical properties of DPP-diMe and DPP-diEt crystals via the nanoindentation technique. (a) The load (P) vs. depth (h) curves for DPP-diEt (blue) and DPP-diMe (red) crystals under 6 mN force with their corresponding average elastic modulus (E) and hardness (H) values (inset). (b) 2D scanning probe microscopy (SPM) images of impressions right after indentation on the major surfaces of DPP-diMe and DPP-diEt crystals with their corresponding height profile diagrams (lower panels). (c) 3D profiles of post indent impressions on the crystals. | ||
To obtain further insights into the distinct mechanical responses of DPP-diMe and DPP-diEt crystals, we investigated the two structures using periodic dispersion-corrected DFT methods (see ESI Section 4†). Upon application of flexural stress on the (011) face, crystals of DPP-diMe and DPP-diEt are expected to expand along the a-axis on the convex (outer arc) side of the needle but compress along the concave (inner arc) side (Fig. 5a). Uniaxial strain-energy calculations show that these changes are accommodated by an increase in the angle between molecular and (100) planes from the inner arc to outer arc (Fig. 5a). Our in silico results, which are obtained using widely available DFT codes, are in good agreement with the molecular level bending mechanisms observed in elastic crystals using synchrotron microfocus X-ray diffraction studies (which are not easily accessible).33
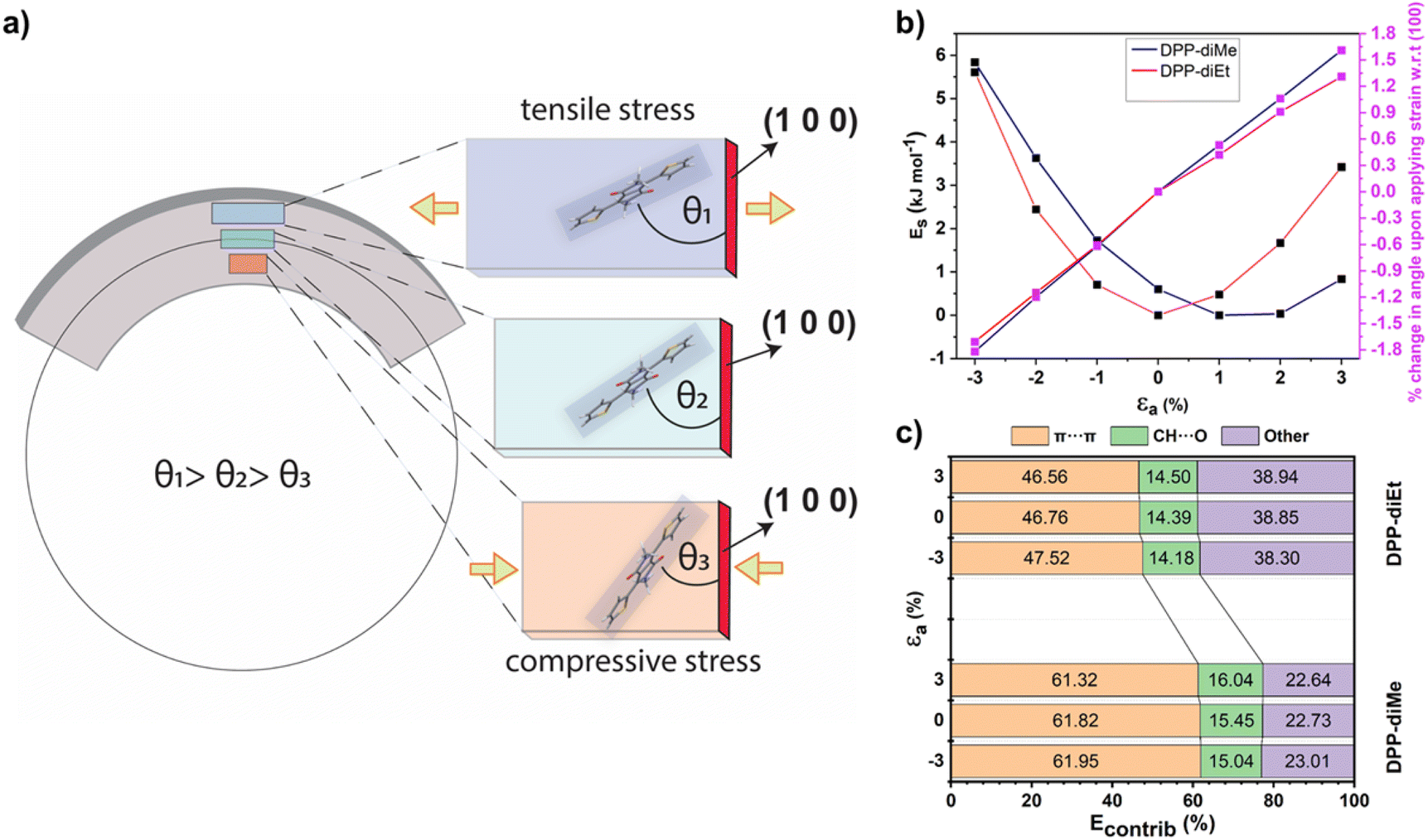 | ||
| Fig. 5 Suggested bending mechanism of flexible DPP-diMe crystals and the comparative strain tolerance penalties of DPP derivatives and DFT-energy profiles. (a) Bending mechanism showing expected expansion (outer arc) and compression (inner arc) along the crystal length in DPP-diMe, accommodated by a decrease in θ from the outer to the inner arc. (b) Strain-energy curves (black symbol) and the strain-percentage of change in angle between the molecular and (100) planes (pink symbols) for DPP-diMe (blue line) and DPP-diEt (pink line), where strain is applied along the needle or a-axis. (c) Energy framework contributions to the total crystal energy as a function of the intermolecular interactions observed with varying strain on the a-axis. | ||
Furthermore, we simulated the energy penalty (Es) for applying tensile and compressive strain on both crystals in order to obtain insights into their strain tolerance behaviour along the a-axis (εa). The resulting plots of the strain-energy and the strain-change in angle between the molecular and (100) planes (Fig. 5b) reveal that DPP-diMe is significantly more tolerant of tensile strain than DPP-diEt. The energy barrier for tensile strain along the a-axis is only 0.23 kJ mol−1 for DPP-diMe upon the application of a 3% uniaxial strain on the equilibrium strain-free crystal. The most stable structure of DPP-diMe in the strain–energy simulations is obtained upon applying a uniaxial tensile strain in the range of 1–2%. The fact that the most stable structure of DPP-diMe corresponds to a non-zero strain state, reflects the soft energy barrier for the elongation of the crystal along the crystal growth axis. The paucity of the reported DFT strain–energy data on elastically deformable molecular crystals limits our ability to compare these findings to the uniaxial strain behaviour of other elastically bendable molecular crystals. However, comparable periodic DFT calculations on rhombohedral crystalline GeTe (r-GeTe) suggests41 that the minimum in the strain-energy curve need not always be at zero strain, and in the case of r-GeTe it is as high as 2.47% uniaxial strain for its phase 2 structure. For DPP-diMe, the DFT results are in excellent agreement with the results of the experimental mechanical bending tests (see ESI, Fig. S1†), which showed that crystals of DPP-diMe can tolerate strains of up to 6% without fracturing. By contrast, the DFT-derived strain-energy curve for DPP-diEt showed that an energy barrier of 3.42 kJ mol−1 is required to apply a 3% tensile strain on the equilibrium strain-free crystal. This energy penalty is an order of magnitude higher than that observed for DPP-diMe. Hence, our results are in good agreement with the brittle nature of DPP-diEt crystals. For context, the energy barrier for applying a 3% tensile strain on crystals of DPP-diEt is of the order of the energy barrier observed in switching between crystal polymorphs.42 Whilst we do not have a sufficient number of published examples from DFT strain-energy calculations involving brittle and elastic molecular crystals, such a relatively large calculated energy barrier for applying a 3% strain in DPP-diEt suggests that it would not be energetically stable to withstand tensile strain during bending tests, in agreement with the results of the experiment. Both crystals display comparable energy barriers for tolerating compressive strains up to −3%. Energy framework calculations comparing the changes in the intermolecular interactions upon the application of crystal strain (Fig. 5c and see energy framework discussion in Section 2 & 4†), reveal that DPP-diMe is stabilized by π-stacking interactions to a greater extent than DPP-diEt. This remains true irrespective of the amount of uniaxial strain applied and indeed whether this strain is tensile or compressive (Fig. 5b). Upon the application of compressive strain, the π-stacking contribution to the total crystal energy becomes more stabilizing in both crystals, whilst upon the application of tensile strain, the opposite is the case (see Table S1†). Nevertheless, the dominance of π-stacking interactions in DPP-diMe, combined with the low strain energy barrier along the needle axis, explains the superior mechanical flexibility of DPP-diMe.
As a proof-of-concept for the mechanical durability of these single crystals under strain and to probe the effect of the same on the semiconducting performance, we fabricated field effect transistors (FETs) by carefully sticking the individual single crystals electrostatically to lithographically patterned Au S–D electrodes. However, the devices made using this method exhibited an extremely low channel current of ∼1 pA (Fig. S9†) indicating poor contact between the crystal and the electrode. We could overcome the metal–semiconductor contact issues by utilizing a drop casting technique which allowed the crystals to grow on top of a flexible substrate containing lithographically patterned Au S–D electrodes (Fig. S10†). PXRD measurements performed on these needle-like crystals (Fig. S11†) grown on top of the flexible substrates confirmed that they are identical to the single crystals used for qualitative and quantitative mechanical testing. The devices were then completed by spin coating the cytop layer and depositing a Au gate electrode (Fig. 6a and d; see the details in the ESI†). Devices fabricated with crystals from a 2 mg mL−1 (1:1) dichloromethane and toluene solution of DPP-diMe exhibited a p-type current modulation with a hole field-effect mobility (μFET) ∼10−3 cm2 V−1 s−1 (Fig. S12†). Upon increasing the concentration of the DPP-diMe solution to 10 mg mL−1, the density of the crystals in the channel increased beyond the percolation threshold (Fig. S12 and S13;† details of the calculation of the percolation threshold are provided in ESI Section 10†), resulting in an increase of channel current by two orders of magnitude, and correspondingly the maximum μFET value of 0.057 cm2 V−1 s−1 and an average mobility value of 0.034 ± 0.011 cm2 V−1 s−1 measured over 16 devices (Fig. 6c, S14a and S15†) were obtained.43,44 It was ensured that under this fabrication condition output characteristics exhibit a clean well-defined linear and saturation regime without any sign of contact limited transport (Fig. 6b and f and S14†). Furthermore, the value of contact resistance estimated from the transfer line method (Fig. S16†) was observed to be at least one or two orders of magnitude lower than the channel resistance indicating that the transport is not contact limited.
 | ||
| Fig. 6 Performance evaluation of flexible field effect transistors (FETs) fabricated from crystals of DPP-diMe and DPP-diEt. (a) Schematic presentation of the fabricated structure of the bottom contact top gate flexible FET device. (b) Output and (c) transfer characteristics measured on FETs (L = 20 μm, W = 1 mm) fabricated with DPP-diMe crystals. (d) Digital image of the flexible device fabricated with DPP-diMe crystals. (e) SEM images of single crystals of DPP-diMe (Left) and DPP-diEt (Right) over the fabricated device. (f) Output and (g) transfer characteristics measured on FETs (L = 20 μm, W = 1 mm) fabricated with DPP-diEt crystals. (h and i) Transfer characteristics measured on a typical bottom contact top gate flexible device (L = 20 μm, W = 1 mm) upon multiple cycles of bending with a radius of 5 mm for DPP-diMe FET and DPP-diEt FET, respectively. (j) Estimated μFET upon multiple cycles of straining at a bend radius of 5 mm for the DPP-diMe and DPP-diEt devices. (k) Statistical distribution depicting the change in mobility in FETs upon 10 cycles of bending with a radius of 5 mm for DPP-diMe and DPP-diEt FETs. Error bars are indicative of the mean deviation of measurement over three devices for figure (j). | ||
To ensure a proper comparison with the flexible FET of DPP-diMe, devices of DPP-diEt were also fabricated with the same concentration i.e., 10 mg mL−1 but with toluene solution of DPP-diEt (ensuring > 90% coverage of the channel area). These devices exhibited a maximum μFET value of 0.009 cm2 V−1 s−1 (Fig. 6g and S14b†) and an average mobility value of (0.007 ± 0.002) cm2 V−1 s−1 measured over 16 devices (Fig. S15†). The magnitude of mobility observed in these FETs is lower than that of DPP-diMe devices, but is comparable to many other well-known solution processable n-type and p-type semiconductors.30,43,45 This lower mobility of DPP-diEt as compared to DPP-diMe can possibly be attributed to its larger π-stacking distance (3.373 Å and 3.298 Å, respectively) between the molecular layers and smaller slipping angle (40° and 45° respectively).1 The variation in the π-stacking distance between the molecular layers and difference in their slipping angles could be associated with the difference in the bulkiness of the alkyl side chains.46–48
To analyse the mechanical durability of the crystals, these flexible FETs were subjected to a bending strain parallel to the channel. Typical transfer characteristics of the flexible FETs when strained by bending the devices with different radii are shown in Fig. S14a and b.† Devices fabricated from both DPP-diMe and DPP-diEt based flexible FETs exhibited a decrease in the channel current as the strain radius decreased from 20 mm (strain of 0.3%) to 5 mm (strain of 1.25%), respectively, and consequently the μFET also decreased (ESI Fig. S14c†). More importantly, the decrease in μFET is significantly higher for the DPP-diEt (∼30 times decrease) compared to DPP-diMe (∼5 times decrease). These observations are in excellent agreement with the inherent mechanical durability of the respective single crystals (ESI Fig. S14c†), studied by the experimental and computational mechanical assessment. Despite the demonstration of mobility values as low as 10−4 cm2 V−1 s−1 from the strained flexible devices fabricated from DPP-diEt, it is important to note that these measurements have been reliable over a number of devices (at least 3 devices) and such values of low mobility are not uncommon in solution processed organic semiconductors.41,43 Although it has been observed that the single crystals of DPP-diMe can withstand strains up to 3–6%, we still observed a decrease in μFET, for much lower strains in the device (strain range: 0.7–1.25%). This behaviour can be attributed to several factors such as the difference in strain between the layers involved in device fabrication, development of micro-cracks in the dielectric layer and the metal contacts with the crystal. All these factors play an important role in the performance of devices in real-life applications.49–51
We further evaluated the mechanical durability of the crystals in these flexible FETs by subjecting them to multiple cycles (10 to 40 cycles with a bend radius of 5 mm) of strain parallel to the channel (Fig. 6h and k). Interestingly, the devices fabricated with DPP-diEt exhibited at least a two orders of magnitude decrease in μFET after just 10 cycles of straining (Fig. 6j and k). However, under similar conditions, the flexible FETs fabricated with DPP-diMe devices exhibited only a marginal decrease in μFET (0.019 cm2 V−1 s−1 to 0.014 cm2 V−1 s−1) indicating the efficacy and practical utility of our molecular design in obtaining flexible electronic devices capable of reliable operation after multiple cycles of strain (Fig. 6j). To further elucidate the statistical reliability of the mechanical durability measurements we plot Δμ (=μinitial/μfinal, where μinitial is the mobility in the first cycle of strain with a bend radius of 5 mm and μfinal is the mobility after 10 cycles of strain with a bend radius of 5 mm) over 5 devices. As observed from Fig. 6k, devices fabricated from DPP-diEt exhibit Δμ > 102 whereas devices fabricated from DPP-diMe exhibit Δμ ∼ 1–2 indicating a relatively strain independent performance. Notably, the performance of the DPP-diMe based flexible FETs was retained even after further bending cycles up to 40 cycles and are projected to retain reasonable mobility even up to 275 cycles of strain, demonstrating our proof-of-concept (Fig. S17†). The poor durability of the DPP-diEt fabricated transistor is attributed to the brittle nature of DPP-diEt crystals. In comparison, elastic DPP-diMe crystals with higher stress tolerance can accommodate multiple cycles of strain. This behaviour is outstanding considering that our devices contain self-assembled single crystals and not the typical polycrystalline thin films (as evident from the SEM images and XRD measurements), hence demonstrating the promise of our study. Moreover, the extent of strain applied on the devices presented in this work is significantly higher than the typical strain that a flexible wearable electronic device is expected to withstand during operation.52
Conclusions
In conclusion, we demonstrate a fully flexible field effect transistor with two derivatives of diketopyrrolopyrrole (DPP). To the best of our knowledge, this is the first demonstration of an all-flexible FET using elastic molecular crystals. Moreover, we bring out a clear difference in the device behaviour when we fabricate devices from two different derivatives of the same conjugated core, one of which is flexible whilst the other is brittle. It was possible to demonstrate clean field effect performance for flexible FETs fabricated with both derivatives of DPP. More importantly, flexible FETs fabricated from flexible crystals retained the field effect mobility for up to 40 bending cycles without any drop in mobility and measurable mobility value is obtained till 275 cycles of strain with a narrow bend over a radius of 5 mm. Periodic dispersion-corrected DFT calculations show a soft energy barrier for crystal elongation along the growth axis in the case of DPP-diMe, with the most stable structure corresponding to a positive strain state. Due to its brittle nature, the same mechanical compliance to strain was not observed in experiments or in the DFT simulations for DPP-diEt. This was manifested in the significantly higher DFT energy penalty for crystal elongation in DPP-diEt. The DFT calculations also revealed the rotation of molecules for accommodating the contraction of the crystal growth axis from the outer to the inner arc in the bent region. Such energy–structure–function correlations using a combination of DFT and crystallographic analyses are currently lacking in the literature and have the potential to accelerate the discovery of new mechanically compliant molecular crystals. This work establishes connections from molecule to crystal to device performance, and paves the way for the fabrication of next-generation smart materials that feature mechanically compliant yet crystalline active materials.Data availability
The datasets supporting this article have been uploaded as part of the ESI material.†Author contributions
R. S. and C. M. R. designed the project. R. S., C. M. R., S. M. and S. P. S. prepared the manuscript. S. D., R. S., Sa. M. and S. P. S. conducted the experiments. S. M. and T. A performed the theoretical calculations. All authors contributed to the data analysis and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. S. thanks UGC-India for her fellowship and S. D. and Sa. M. thank CSIR-India for their fellowships [CSIR-SRF, File No. 09/921(0162)/2017-EMR-I and 09/921(0183)/2017-EMR-I, respectively]. C. M. R. is grateful to DST, India (SJF/CSA-02/2014–15) for a Swarnajayanti fellowship. We thank Dr Manjima Bhattacharya (IISER Kolkata) for her initial help with nanoindentation experiments. S. M. acknowledges financial support from Khalifa University under the CIRA program (CIRA-2018-068) and the Advanced Technology Research Council (ATRC) for an ASPIRE Award for Research Excellence 2020 (AARE-2020). The periodic DFT calculations were performed using the high-performance computing clusters of Khalifa University and the authors would like to acknowledge the support of the research computing department at Khalifa University. S. P. S. acknowledges funding support from the Royal Society through the Newton Alumni Fellowship, SERB-SRG/2020/001641, and SERB-IPA/2021/000096 and DAE funding through NISER and is thankful to Prof. Richard Friend, University of Cambridge for useful discussions.References
- T. He, M. Stolte and F. Würthner, Adv. Mater., 2013, 25, 6951–6955 CrossRef CAS PubMed.
- M. Mas-Torrent, P. Hadley, S. T. Bromley, X. Ribas, J. Tarrés, M. Mas, E. Mollins, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2004, 126, 8546–8553 CrossRef CAS PubMed.
- S. Guerin, A. Stapleton, D. Chovan, R. Mouras, M. Gleeson, C. McKeown, M. R. Noor, C. Silien, F. M. F. Rhen, A. L. Kholkin, N. Liu, T. Soulimane, S. A. M. Tofail and D. Thompson, Nat. Mater., 2018, 17, 180–186 CrossRef CAS PubMed.
- D. P. Karothu, G. Dushaq, E. Ahmed, L. Catalano, S. Polavaram, R. Ferreira, L. Li, S. Mohamed, M. Rasras and P. Naumov, Nat. Commun., 2021, 12, 1326 CrossRef CAS PubMed.
- S. C. Sahoo, S. B. Sinha, M. S. R. N. Kiran, U. Ramamurty, A. F. Dericioglu, C. M. Reddy and P. Naumov, J. Am. Chem. Soc., 2013, 135, 13843–13850 CrossRef CAS PubMed.
- H. Li, W. Shi, J. Song, H.-J. Jang, J. Dailey, J. Yu and H. E. Katz, Chem. Rev., 2019, 119, 3–35 CrossRef CAS PubMed.
- A. Ravi and K. M. Sureshan, Angew. Chem., Int. Ed., 2018, 57, 9362–9366 CrossRef CAS PubMed.
- L. Jiang, H. Dong and W. J. Hu, Mater. Chem., 2010, 20, 4994–5007 RSC.
- Z. Wang, P. K. Nayak, J. A. Caraveo-Frescas and H. N. Alshareef, Adv. Mater., 2016, 28, 3831–3892 CrossRef CAS PubMed.
- A. L. Briseno, R. J. Tseng, M.-M. Ling, E. H. L. Falcao, Y. Yang, F. Wudl and Z. Bao, Adv. Mater., 2006, 18, 2320–2324 CrossRef CAS.
- R. Devarapalli, S. B. Kadambi, C.-T. Chen, G. R. Krishna, B. R. Kammari, M. J. Buehler, U. Ramamurty and C. M. Reddy, Chem. Mater., 2019, 31, 1391–1402 CrossRef CAS.
- S. Saha, M. K. Mishra, C. M. Reddy and G. R. Desiraju, Acc. Chem. Res., 2018, 51, 2957–2967 CrossRef CAS PubMed.
- S. Takamizawa and Y. Miyamoto, Angew. Chem., Int. Ed., 2014, 53, 6970–6973 CrossRef CAS PubMed.
- S. Ghosh and C. M. Reddy, Angew. Chem., Int. Ed., 2012, 51, 10319–10323 CrossRef CAS PubMed.
- K. Wang, M. K. Mishra and C. C. Sun, Chem. Mater., 2019, 31, 1794–1799 CrossRef CAS.
- C. M. Reddy, K. A. Padmanabhan and G. R. Desiraju, Cryst. Growth Des., 2006, 6, 2720–2731 CrossRef CAS.
- C. M. Reddy, R. C. Gundakaram, S. Basavoju, M. T. Kirchner, K. A. Padmanabhan and G. R. Desiraju, Chem. Commun., 2005, 31, 3945–3947 RSC.
- M. Annadhasan, A. R. Agrawal, S. Bhunia, V. V. Pradeep, S. S. Zade, C. M. Reddy and R. Chandrasekar, Angew. Chem., Int. Ed., 2020, 59, 13852–13858 CrossRef CAS PubMed.
- S. Hayashi, S.-Y. Yamamoto, D. Takeuchi, Y. Ie and K. Takagi, Angew. Chem., Int. Ed., 2018, 57, 17002–17008 CrossRef CAS PubMed.
- M. Owczarek, K. A. Hujsak, D. P. Ferris, A. Prokofjevs, I. Majerz, P. Szklarz, H. Zhang, A. A. Sarjeant, C. L. Stern, R. Jakubas, S. Hong, V. P. Dravid and J. F. Stoddart, Nat. Commun., 2016, 7, 13108–13117 CrossRef CAS PubMed.
- H. Sun, S. K. Park, Y. Diao, E. P. Kvam and K. Zhao, Chem. Mater., 2021, 33, 1883–1892 CrossRef CAS.
- H. Liu, Z. Lu, Z. Zhang, Y. Wang and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8448–8452 CrossRef CAS PubMed.
- S. Ghosh, M. K. Mishra, S. B. Kadambi, U. Ramamurty and G. R. Desiraju, Angew. Chem., Int. Ed., 2015, 54, 2674–2678 CrossRef CAS PubMed.
- X. Liu, A. A. L. Michalchuk, B. Bhattacharya, N. Yasuda, F. Emmerling and C. R. Pulham, Nat. Commun., 2021, 12, 3871–3878 CrossRef CAS PubMed.
- B. B. Rath, G. Gallo, R. E. Dinnebier and J. J. Vittal, J. Am. Chem. Soc., 2021, 143, 2088–2096 CrossRef CAS PubMed.
- B. B. Rath, M. Gupta and J. J. Vittal, Chem. Mater., 2022, 34, 178–185 CrossRef CAS.
- H. Liu, Z. Lu, B. Tang, C. Qu, Z. Zhang and H. Zhang, Angew. Chem., Int. Ed., 2020, 59, 12944–12950 CrossRef CAS PubMed.
- V. Gude, P. S. Choubey, S. Das, B. N. Shivakiran Bhaktha, C. M. Reddy and K. Biradha, J. Mater. Chem. C, 2021, 9, 9465–9472 RSC.
- J. Harada, T. Shimojo, H. Oyamaguchi, H. Hasegawa, Y. Takahashi, K. Satomi, Y. Suzuki, J. Kawamata and T. Inabe, Nat. Chem., 2016, 8, 946–952 CrossRef CAS PubMed.
- D. P. Karothu, G. Dushaq, E. Ahmed, L. Catalano, M. Rasras and P. Naumov, Angew. Chem., Int. Ed., 2021, 60, 26151–26157 CrossRef CAS PubMed.
- Y. Chen, Z. Chang, J. Zhang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 22424–22431 CrossRef CAS PubMed.
- Y. Yang, Y. Zhang, C. T. Hu, M. Sun, S. Jeong, S. S. Lee, A. G. Shtukenberg and B. Kahr, Chem. Mater., 2022, 34, 1778–1788 CrossRef CAS.
- A. Worthy, A. Grosjean, M. C. Pfrunder, Y. Xu, C. Yan, G. Edwards, J. K. Clegg and J. C. McMurtrie, Nat. Chem., 2018, 10, 65–69 CrossRef CAS PubMed.
- S. Hayashi, F. Ishiwari, T. Fukushima, S. Mikage, Y. Imamura, M. Tashiro and M. Katouda, Angew. Chem., Int. Ed., 2020, 59, 16195–16201 CrossRef CAS PubMed.
- T. Yamamoto and K. Takimiya, J. Am. Chem. Soc., 2007, 129, 2224–2225 CrossRef CAS PubMed.
- G. R. Krishna, R. Devarapalli, G. Lal and C. M. Reddy, J. Am. Chem. Soc., 2016, 138, 13561–13567 CrossRef CAS PubMed.
- S. Das, G. Vreeman, U. Ramamurty, C. C. Sun and C. M. Reddy, ChemRxiv, 2021, Cambridge Open Engage, Cambridge, preprint, DOI:10.26434/chemrxiv-2021-7jh31.
- S. Das, S. Saha, M. Sahu, A. Mondal and C. M. Reddy, Angew. Chem., Int. Ed., 2022, 61, e202115359 CAS.
- S. Varughese, M. S. R. N. Kiran, U. Ramamurty and G. R. Desiraju, Angew. Chem., Int. Ed., 2013, 52, 2701–2712 CrossRef CAS PubMed.
- M. S. R. N. Kiran, S. Varughese, C. M. Reddy, U. Ramamurty and G. R. Desiraju, Cryst. Growth Des., 2010, 10, 4650–4655 CrossRef CAS.
- W. J. Yang, H. Park, D. S. Kim, T. Ha, S. J. Park, M. Ahn, J. H. Kim, Y.-K. Kwon and M.-H. Cho, Sci. Rep., 2019, 9, 12816 CrossRef PubMed.
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC.
- B. Narayan, S. P. Senanayak, V. Jain, K. S. Narayan and S. J. George, Adv. Funct. Mater., 2013, 23, 3053–3060 CrossRef CAS.
- M.-Z. Li, S.-T. Han and Y. Zhou, Adv. Intell. Syst. Comput., 2020, 2, 2000113 CrossRef.
- P. Jonkheijm, N. Stutzmann, Z. Chen, D. M. de Leeuw, E. W. Meijer, A. P. H. J. Schenning and F. Würthner, J. Am. Chem. Soc., 2006, 128, 9535–9540 CrossRef CAS PubMed.
- Z. Bao and A. J. Lovinger, Chem. Mater., 1999, 11, 2607–2612 CrossRef CAS.
- Z. Ma, H. Geng, D. Wang and Z. Shuai, J. Mater. Chem. C, 2016, 4, 4546–4555 RSC.
- Y. Guo, G. Han and Y. Yi, J. Energy Chem., 2019, 35, 138–143 CrossRef.
- S. Yuvaraja, A. Nawaz, Q. Liu, D. Dubal, S. G. Surya, K. N. Salama and P. Sonar, Chem. Soc. Rev., 2020, 49, 3423–3460 RSC.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- T. Sekitani, U. Zschieschang, H. Klauk and T. Someya, Nat. Mater., 2010, 9, 1015–1022 CrossRef CAS PubMed.
- L. Gao, Small, 2017, 13, 1603994 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2102226. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05217b |
| This journal is © The Royal Society of Chemistry 2023 |