
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of thread-like monomer structure on the synthesis of poly[n]catenanes from metallosupramolecular polymers†
Marissa M.
Tranquilli
a,
Benjamin W.
Rawe
 b,
Guancen
Liu
a and
Stuart J.
Rowan
*abc
b,
Guancen
Liu
a and
Stuart J.
Rowan
*abc
aDepartment of Chemistry, University of Chicago, Chicago, IL, USA. E-mail: stuartrowan@uchicago.edu
bPritzker School of Molecular Engineering, University of Chicago, Chicago, IL, USA
cChemical and Engineering Sciences, Argonne National Laboratory, Lemont, IL, USA
First published on 15th February 2023
Abstract
The main-chain poly[n]catenane consists of a series of interlocked rings that resemble a macroscopic chain-link structure. Recently, the synthesis of such intriguing polymers was reported via a metallosupramolecular polymer (MSP) template that consists of alternating units of macrocyclic and linear thread-like monomers. Ring closure of the thread components has been shown to yield a mixture of cyclic, linear, and branched poly[n]catenanes. Reported herein are studies aimed at accessing new poly[n]catenanes via this approach and exploring the effect the thread-like monomer structure has on the poly[n]catenane synthesis. Specifically, the effect of the size of the aromatic linker and alkenyl chains of the thread-like monomer is investigated. Three new poly[n]catenanes (with different ring sizes) were prepared using the MSP approach and the results show that tailoring the structure of the thread-like monomer can allow the selective synthesis of branched poly[n]catenanes.
Introduction
The field of mechanically interlocked molecules (MIMs)1–3 is characterized by a distinctive mechanical bond, in which components are spatially associated (permanently entangled) rather than chemically bound. One of the most easily identifiable members of the MIM family is the catenane: a molecule composed of two or more interlocked rings (or macrocycles) that are distinct from each other but cannot be separated without breaking a covalent bond. The simplest example of this structure is a [2]catenane, with the “2” referring to the number of rings (Fig. 1). As a result of the mechanical bond, the catenanes' macrocycles are capable of a range of large amplitude motions, such as rotation, elongation, and twisting of the rings,4–6 that are difficult or cannot be achieved in non-interlocked molecules.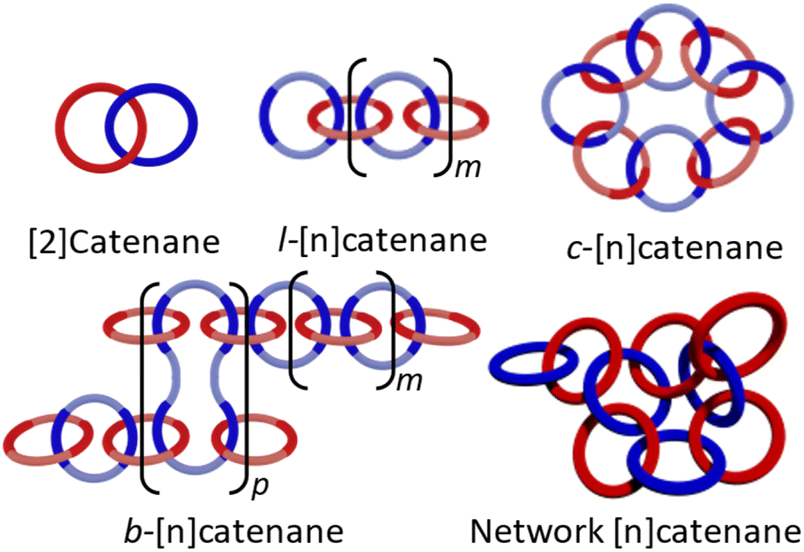 | ||
| Fig. 1 Schematic representation of a molecular [2]catenane and poly[n]catenane architectures: linear (l-), cyclic (c-), branched (b-), and network. | ||
Since the inception of synthetic interlocked molecules like the catenane,7–10 scientists within the field have looked to exploit their unique mobility to access new materials.11,12 An ideal way to do this is through the synthesis of mechanically interlocked polymers (MIPs),13–15 where the motion of the interlocked components can directly impact their material properties. However, to study and fully utilize these materials, synthetic protocols to access specific MIP architectures need to be developed. For some MIP architectures, such as polycatenanes,16–18 the synthetic obstacles are particularly pronounced: the yields of ring closing reactions (required in the synthesis of a catenane) toward a specific macrocycle are generally not high yielding.19 Furthermore, ring formation is favoured at low monomer concentrations, while higher reaction concentrations are generally required to access high molar mass polymers. None-the-less, a number of synthetic approaches have been explored, that have resulted in the synthesis of higher order catenanes20–22 and polycatenane architectures, including side-chain23 and main-chain poly[2]catenanes,23–28 poly[2]catenane cross-linked networks,29,30 polymeric [2]catenanes,31,32 and radial polycatenanes.33–35
The synthesis of polycatenanes in which the polymer backbone consists only of interlocked rings, such as main-chain poly[n]catenanes (the molecular equivalent of a macroscopic chain) or Olympic gels (interlocked ring networks), have proven to be more challenging.36 Entirely catenated polymers can be found in several different architectural varieties (Fig. 1): the cyclic (c-), the linear (l-), the branched (b-), or the network poly[n]catenane. There have been several examples of oligomeric [n]catenanes,37–43 most notably the l-[5]catenane (or Olympiadane)37 and a branched b-[7] catenane35 synthesized by Stoddart and co-workers. More recently a linear [6]catenane was prepared by Barnes and coworkers.43 While they are all major synthetic achievements, these compounds were synthesized in a step-wise manner, which is less conducive to accessing high molar mass MIPs. In a different approach, Meijer and Di Stefano used ring-opening metathesis of a [2]catenane to yield oligomeric [n]catenated molecules.44 Poly[n]catenane networks (Olympic gels) have been proposed as by-products in 1,2-dithiane polymerizations45,46 and more recently polydimethylsiloxane-based catenated networks have been accessed during the polymerization of telechelic polydimethylsiloxane (PDMS) macromonomers.47 Catenated materials have also been found to occur naturally in kinetoplast DNA48 and DNA polymeric catenated structures not found in nature can also be accessed.49,50 None-the-less examples of fully catenated, synthetic polymeric materials are rare.
Recently, a route to main-chain poly[n]catenanes, which utilized a self-assembled metallosupramolecular polymer (MSP) template, has been reported.51,52 The resulting poly[n]catenane, which is composed of macrocycle 1 (Fig. 2a) and a macrocycle formed from the linear thread-like monomer 2(xan-6) (Fig. 2b), was synthesized via a ring-closing metathesis reaction of the pre-assembled MSP template. Both monomers (1 and 2(xan-6)) contain two 2,6-bisbenzimidazolylpyridine (Bip) ligands53,54 which allows them to self-assemble into the MSP template upon addition of metal ions, such as Zn(II). The thread component consists of three distinct regions: the Bip ligand (dark blue, Fig. 2b), a rigid aromatic linker (3,6-disubstituted 9,9-dimethylxanthene) (xan) and a reactive alkenyl tail (R = 4-hexenyl). As this monomer is the reactive component in the critical ring closing metathesis reaction, it was designed to encourage the formation of the ring closed macrocycle (3(xan-6)),55 by approximately matching the size of the rigid aromatic linker and the alkoxy chain formed upon reaction of the two alkenyl tails.
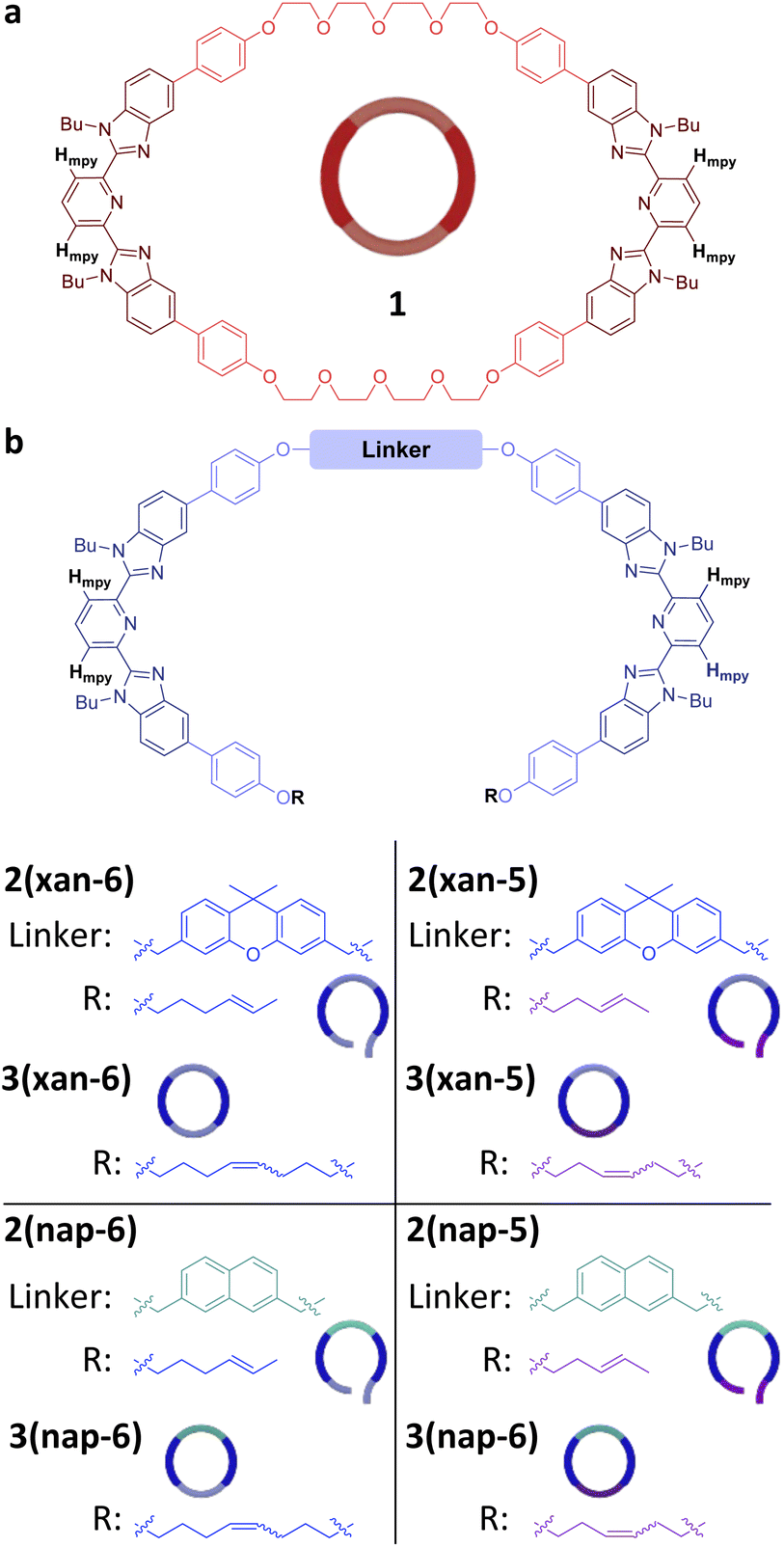 | ||
| Fig. 2 Chemical structures of the components used in the poly[n]catenane synthesis. (a) Macrocycle (1) with Bip ligand emphasized in dark red. (b) Structures of the open thread molecules (2) and their ring-closed counterparts (3) with the ligand (Bip) emphasized in dark blue, the linker indicated as such, and the alkenyl tail given as R. The diagnostic Hmpy proton is identified in the structures. | ||
The poly[n]catenane synthesis started with the self-assembly of macrocycle 1, thread 2(xan-6) and two equivalents Zn(II) ions to yield both the linear (l-1·2(xan-6)·Zn(II)2) and cyclic MSP (c-1·2(xan-6)·Zn(II)2) (Fig. 3).51 A ring-closing metathesis reaction using the Hoveyda–Grubbs generation II catalyst was then carried out allowing access to the metalated poly[n]catenates. After demetallation, the compounds were characterized by a combination of nuclear magnetic resonance spectroscopy (NMR) and size exclusion chromatography-multiangle light scattering (SEC-MALS) to characterize the size and architecture of the products. Those studies showed that the synthesis resulted in a mixture of linear, cyclic, and branched main-chain poly[n]catenanes (l-4(xan-6), c-4(xan-6), b-4(xan-6), respectively) as well as a series of non-interlocked side-products (1, 2(xan-6), 3(xan-6), and 5(xan-6)) (Fig. 3). The presence of branched poly[n]catenanes as well as the non-interlocked structures suggest that the alkenes in the thread-like component of the MSP do more than undergo the intra-thread reactions that yield the l- and c-4(xan-6); for example, they can also react with alkenes in other thread components (inter-thread reactions).
 | ||
Fig. 3 The poly[n]catenane synthesis starts with the self-assembly of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of macrocycle 1 and thread 2(xan-6) with 2 equivalents of zinc di[bis(trifluoromethylsulfonyl)imide] (Zn(NTf2)2) to yield both the linear and cyclic metallosupramolecular polymer (l-1·2(xan-6)·Zn(II)2 and c-1·2(xan-6)·Zn(II)2, respectively) (depending on concentration). Reaction of MSP with Hoveyda–Grubbs G2 catalyst results in a series of metalated polycatenates. Once demetallated, the reaction mixture contains linear, branched, and cyclic poly[n]catenanes (4(xan-6)) as well as several non-interlocked byproducts (1, 2(xan-6), 3(xan-6), 5(xan-6)). :1 ratio of macrocycle 1 and thread 2(xan-6) with 2 equivalents of zinc di[bis(trifluoromethylsulfonyl)imide] (Zn(NTf2)2) to yield both the linear and cyclic metallosupramolecular polymer (l-1·2(xan-6)·Zn(II)2 and c-1·2(xan-6)·Zn(II)2, respectively) (depending on concentration). Reaction of MSP with Hoveyda–Grubbs G2 catalyst results in a series of metalated polycatenates. Once demetallated, the reaction mixture contains linear, branched, and cyclic poly[n]catenanes (4(xan-6)) as well as several non-interlocked byproducts (1, 2(xan-6), 3(xan-6), 5(xan-6)). | ||
To date, only the combination of 1 and 2(xan-6) has been used to access poly[n]catenanes using the MSP-template approach. As such it is important to explore the capabilities of this synthetic methodology to access poly[n]catenanes beyond the original system. For example, as computational modeling has shown that ring size impacts the properties of the poly[n]catenane in both the solution56 and melt state,57,58 it is important to explore if this route allows access to poly[n]catenanes with different ring sizes? Such studies require the use of new monomers raising the question of how the monomer structure impacts the yield or architecture of the formed poly[n]catenanes.
To start to address such questions the focus of this work was to access new poly[n]catenanes with different ring sizes and investigate how changing the length of the aromatic linker (a xanthene vs. a naphthalene moiety) and the alkenyl tails (R = 4-hexenyl vs. 3-pentenyl) of the linear-thread impacts the formation of both the MSPs and poly[n]catenanes.
Results and discussion
In order to explore the synthesis of new poly[n]catenanes with different ring sizes, three new threads were targeted (Fig. 2b), 2(xan-5), 2(nap-5) and 2(nap-6). 2(xan-5) was chosen to explore how the shorter 3-pentenyl tails (relative to the 4-hexenyl tails in 2(xan-6)) impacts the poly[n]catenane synthesis. As prior work has shown that the nature of the aromatic linker group is important to access [3]catenanes in high yield,55 it was decided to explore the smaller, but geometrically similar, 2,7-disubstituted naphthalene (nap) aromatic moiety in place of the 3,6-disubstituted 9,9-dimethylxanthene (xan) linker. Therefore, two new threads were prepared using this shorter naphthalene linker, one with the 3-pentenyl alkyl units (2(nap-5)) and one with the 4-hexenyl alkyl tails (2(nap-6)).Threads 2(xan-5), 2(nap-5) and 2(nap-6) were synthesized in a similar fashion to thread 2(xan-6)51 (for experimental details and characterization see ESI page S4–S8 and Fig. S1–S3†). The three new MSPs were then prepared in a similar manner to that reported previously.51,52 Specifically, a 1:1 ratio of macrocycle (1) and the appropriate thread (2(xan-5), 2(nap-5), or 2(nap-6)) was titrated with zinc di[bis(trifluoromethylsulfonyl)imide] (Zn(NTf2)2) (monitored by the integration of the Hmpy protons) until no unbound Hmpy peaks were observed, corresponding to an MSP with a ratio of 1:1:2 (1:2:Zn(II)2). Based on the principle of maximal site occupancy,59 the resulting assemblies should be the MSPs 1·2(xan-5)·Zn(II)2, 1·2(nap-5)·Zn(II)2, and 1·2(nap-6)·Zn(II)2, respectively, in which monomers 1 and 2 are alternating along the assembled backbone.
Characterization of the original MSP 1·2(xan-6)·Zn(II)2 using both 1H NMR spectroscopy and DOSY (diffusion ordered spectroscopy) NMR,51 allowed identification of two distinct MSP populations (linear, l-MSP, and cyclic, c-MSP). Following this approach, the three new MSPs were characterized at a series of concentrations (0.25, 2.5, 10.0 mM w.r.t. to 1, Fig. S4†). Fig. 4a shows that, at 2.5 mM (w.r.t. 1), each of the new MSPs have peak populations within the regions that have previously been attributed to l- and c-MSP in 1·2(xan-6)·Zn(II)2.51 As such, the Hmpy peaks shifted upfield (δH = 8.62–8.72 ppm) are assigned as corresponding to the c-MSP. If such peaks do correspond to c-MSPs, then the intensities of these peaks should increase at lower concentrations, where the formation of c-MSP would be favoured on account of ring-chain equilibria.52,60,61 Indeed 1H NMR of all four MSPs at lower concentrations (e.g., 0.25 mM, Fig. 4b) show a significant increase in the intensity of the upfield peaks. Each MSP was further characterized by DOSY NMR at a concentration of 2.5 mM to determine diffusion coefficients of the two distinct regions (Fig. 4, S5–S7, Table S1†). For all four MSPs, the protons in the region 8.72–8.80 ppm have diffusion coefficients between 1.9–2.3 × 10−10 m2 s−1. The upfield peaks (8.62–8.72 ppm) exhibit higher diffusion coefficients, with values between 3.8–5.8 × 10−10 m2 s−1, suggesting lower molar mass assemblies and is consistent with these peaks corresponding to c-MSPs.
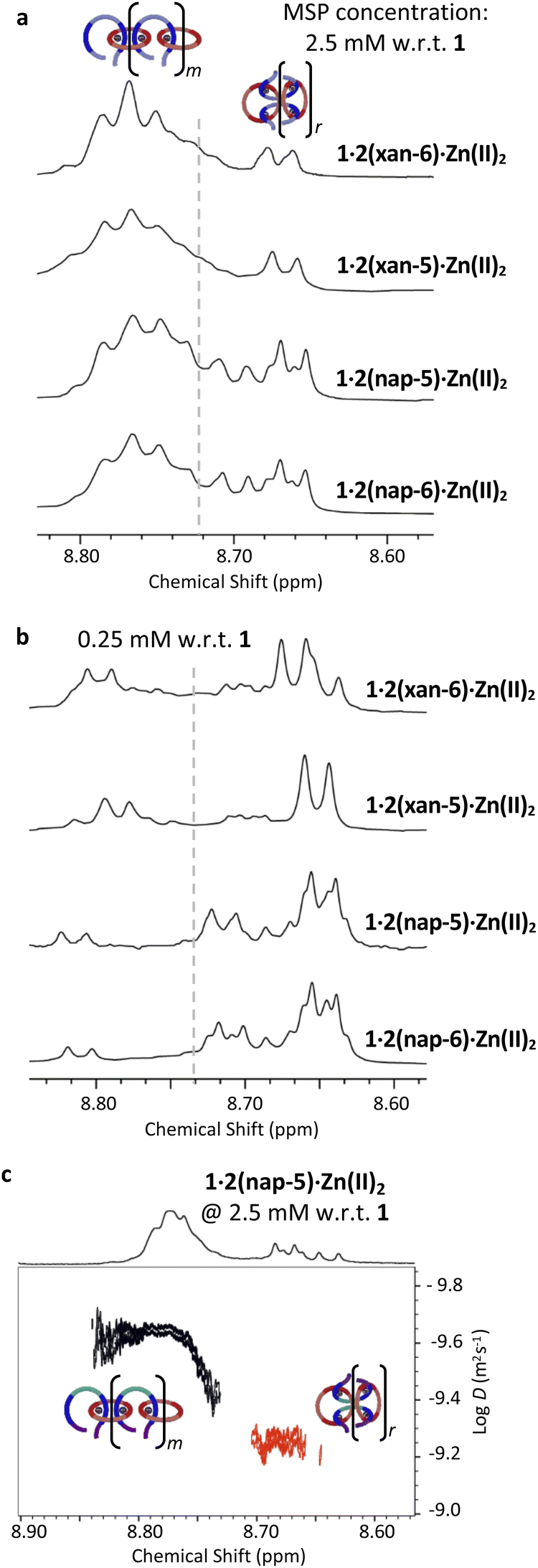 | ||
| Fig. 4 Region of the 1H NMR spectra (500 MHz, 1:5 CD3CN:CDCl3, 298 K) corresponding to the Hmpy protons for each MSP at (a) 2.5 mM and (b) 0.25 mM. (c) Analysis of DOSY NMR (500 MHz, CD2Cl2, 298 K) data of the MSP 1·2(nap-5)·Zn(II)2 at a concentration of 2.5 mM. Two distinctive regions of linear (black, diffusion coefficient of 2.3 × 10−10 m2 s−1) and cyclic (red, 5.8 × 10−10 m2 s−1) MSP can be seen. | ||
At 2.5 mM (w.r.t. 1) the MSPs based on the xanthene containing threads (1·2(xan-6)·Zn(II)2 and 1·2(xan-5)·Zn(II)2) show similar peak shapes (Fig. 4a) and both contain ca. 20% of their total MSP in the upfield (“c-MSP”) region. In contrast, the MSPs formed from the threads with the naphthalene linker (1·2(nap-5)·Zn(II)2 and 1·2(nap-6)·Zn(II)2) show a significant increase in c-MSP population, with ca. 34% of their MSP peak population in the upfield region. At lower concentrations, such as 0.25 mM, where cyclic MSP is more favoured, the naphthalene containing MSPs show over 80% of their peak population in the 8.62–8.72 ppm region while the xanthene containing threads are ca. 60% (Fig. 4b). Thus, the nature of the aromatic linker (xan vs. nap) impacts the MSP structure, with the shorter naphthalene linker resulting in an increase in the amount of lower molar mass c-MSP. The alkenyl tail, on the other hand, appears to have little-to-no effect on the MSP.
With this information in hand, each MSP 1·2·Zn(II)2 was subjected to a ring-closing metathesis reaction, following the previously published procedure,51 at a concentration of 2.5 mM w.r.t. 1. After deactivating the catalyst and removing the Zn(II) from the resulting product (the mass recovered for these reactions is generally equivalent to the mass of the reactants added), each of the crude reaction mixtures were characterized by 1H NMR (Fig. S8†). Fig. 5a shows the region of the 1H NMR that has been assigned to the Hmpy protons in 4(xan-6),51 in which the peaks above 8.27 correspond to the Hmpy protons of the non-interlocked products (i.e., macrocycle 1, thread 2 and/or the Acyclic Diene Metathesis, ADMET, product 5) and peaks between 8.10–8.27 ppm correspond to the Hmpy protons in the catenated material.51 All three of the crude reaction mixtures using the new thread-like monomers show peaks in both these regions. Samples of pure ADMET (5(xan-5), 5(nap-5), 5(nap-6)) and ring closed thread (3(xan-5), 3(nap-5), 3(nap-6)) were synthesized for each new thread to confirm the 1H NMR shifts of these compounds (Fig. S9–S14†). Using this data, it was possible to confirm the assignment of the peaks ca. 8.35 ppm to monomer 2 and/or its corresponding ADMET polymer (5) and the peaks around 8.30 ppm to macrocycle 1 (Fig. 5a). To explore the nature of the products whose Hmpy protons appear between 8.10–8.27 ppm, it was necessary to purify the crude reaction mixtures. Preparatory size exclusion chromatography (preparative SEC, 1:3 DMF:THF) was used to remove the residual macrocycle (1). Additional purification could be achieved by either partial metalation followed by precipitation of the metalated catenated material (4(xan-5) and 4(nap-6)),51 or separating out the catenated material using a preparatory-silica plate (4(nap-5)) (ESI page S16†). These purified poly[n]catenanes were used only in the following NMR studies to help confirm their interlocked nature. Correlated Spectroscopy (COSY) experiments were carried out to fully assign the 1H NMR peaks (Fig. S15–S17†). It is important to note that the COSY data confirms that the peaks above 8.10 ppm do correspond to the Hmpy protons in the three new polymers. The interlocked nature of the new poly[n]catenanes was confirmed via low temperature Nuclear Overhauser Effect Spectroscopy (NOESY) (Fig. S18–S23†). Fig. 5b shows the partial NOESY spectrum of 4(nap-5), highlighting the main intercomponent cross-peaks. These cross-peaks were not observed in the mixture of 1 and 3(nap-5) at the same concentration (Fig. S21†), affirming the interlocked nature of 4(nap-5). Similar results are seen in both 4(xan-5) and 4(nap-6) (Fig. S19 and S23,† respectively). As such, the NOESY data strongly supports the synthesis of three new poly[n]catenanes.
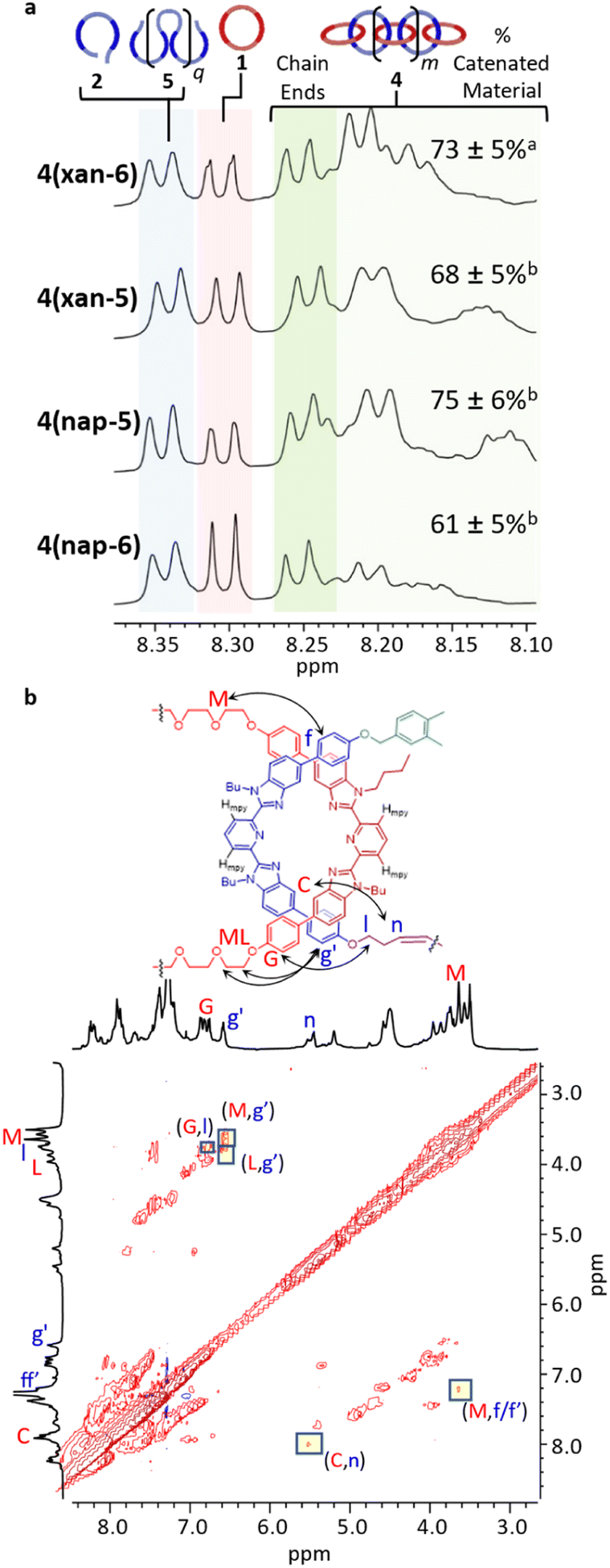 | ||
| Fig. 5 (a) Region of the 1H NMR spectra (500 MHz, CDCl3, 298 K) corresponding to the Hmpy protons for the demetallated crude reaction mixtures for poly[n]catenane 4(xan-6)27,28 (top) and the new poly[n]catenanes, 4(xan-5), 4(nap-5) and 4(nap-6). (b) NOE spectroscopy for 4(nap-5) highlighting the crosspeaks between the interlocked components. aError for data set taken at 95% confidence interval (n = 5). bError for data set taken at 95% confidence interval (n = 3). | ||
The 1H NMR can also be used to provide information on the polymer chain ends, which when combined with molar mass data obtained from SEC-MALS can be used to determine the poly[n]catenane architecture51 (see ESI† for more details). As was previously shown for 4(xan-6),51 the higher mobility of the singly-threaded chain-ends should result in a larger T1 value for the chain end peaks than for peaks that correspond to the doubly-threaded interior rings. NMR relaxation experiments on all three new polymers (4(xan-5), 4(nap-5) and 4(nap-6)) show that the peaks in the region 8.24–8.27 ppm show higher T1 values than the more upfield shifted protons (Fig. S24, Table S2†), consistent with those peaks corresponding to the chain ends of these poly[n]catenanes.
With the Hmpy chemical shifts of the non-interlocked and catenated materials assigned using these purified materials, it was possible to obtain the reaction yield of the three poly[n]catenanes by comparing the integration of the non-interlocked peaks (8.28–8.37 ppm) and interlocked peaks (8.09–8.27 ppm) in the 1H NMRs of the crude reaction mixtures (Fig. 5a). The poly[n]catenane yield is similar for all four linkers (Table 1), with perhaps only the 4(nap-6) showing a drop in yield (ca. 61%). There does appear to be some benefit of size matching the aromatic linker and alkoxy units, as the use of the smaller 3-pentenyl tails with the naphthalene linker in the thread results in a slight improvement in the yield of poly[n]catenane 4(nap-5) (ca. 75%) relative to 4(nap-6). However, in all cases the key unproductive side reaction in the poly[n]catenane synthesis is the formation of the ADMET side product 5. Interestingly, simply shortening the alkenyl tails does not appear to significantly alter the amount of 5 obtained in reaction (Table 1). What appears to have more of an effect on the amount of 5 formed is the use of a mismatched longer tail with the shorter aromatic linker.
| Sample | Average% product distribution in crude reaction mixturea |
|
|
|
Poly[n]catenane architecture (% based on catenated products)g | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 1 | 2 + 5 | [<5]Cath | Cyclic | Linear | Branched | ||||
| a Conversion of MSP to catenane calculated by integrating Hmpy peaks corresponding to non-interlocked products 1 and 5 (8.36–8.28 ppm) against Hmpy peaks corresponding to 4 (8.28–8.10 ppm). b Error for data set taken at 95% confidence interval (n = 5). c Error for data set taken at 95% confidence interval (n = 3). Exact yields given in ESI. d Number average molecular weight in g mol−1 of the crude catenane sample (n = 2) determined by MALS coupled with an RI detector using dn/dc values that were determined from the crude reaction mixture (see text and ESI for more details). e Number average degree of polymerization, which also corresponds to the average number of rings, calculated via eqn (S1). f Average number of chain ends calculated via eqn (S2). g Architecture breakdown reported as % of total interlocked products as determined via deconvolution of SEC RI trace data. h Value includes the total amount of [2] and [3]catenanes identified. i Value based on the total area of Peak C in the deconvolution of the 4(xan-5) SEC trace although data suggests that this peak corresponds to both cyclic and oligomeric linear catenane. j While data suggests that no l-poly[n]catenane (DP > 5) is formed when the pentenyl-based thread-like monomer is used, low molar mass linear l-[n]catenanes (DP < 5) are formed and are included [<5]Cat total. | ||||||||||
| 4(xan-6) | 73 ± 5b | 12 ± 3b | 15 ± 2b | 17700 |
11 | 2.9 | 8 | 12 | 51 | 29 |
| 4(xan-5) | 68 ± 5c | 15 ± 3c | 17 ± 3c | 14500 |
9 | 4.4 | 29i | <8i | 0j | 63j |
| 4(nap-5) | 75 ± 6c | 11 ± 4c | 14 ± 2c | 18300 |
12 | 5.4 | 22 | 0 | 0j | 78j |
| 4(nap-6) | 61 ± 5c | 17 ± 3c | 22 ± 2c | 13800 |
9 | 3.0 | 5 | 23 | 45 | 27 |
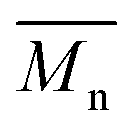
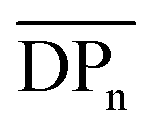

Given that the structure of the thread has a relatively small impact on interlocked polymer yield, it is of interest to determine if the different thread-like monomers play a role in the distribution of the poly[n]catenane architecture, i.e., cyclic, linear or branched.51,52 To this end, size exclusion chromatography-multi-angle light scattering (SEC-MALS) experiments of the crude reaction mixtures were undertaken and analyzed in conjunction with the 1H NMR data.
The SEC-MALS traces of the three poly[n]catenane crude reaction mixtures (Fig. 6) clearly demonstrate that polymeric materials are formed in all three reactions. The dn/dc values (Instruments and methods, S3†) of the crude mixtures were obtained to allow determination of the average molar mass for each sample (Fig. 6). It is important to note that while the dn/dc values are specific to a given polymer and independent of molar mass at higher values, the measured dn/dc value from the crude reaction mixture can only be used to obtain approximate molar masses of these compounds, especially for the lower molar mass compounds62 (i.e., [2]–[4]catenane or small cyclic catenanes) or non-interlocked side products. As such, the molar masses obtained from the MALS of these species are used as an approximation in the following analysis and are combined with appropriate 1H NMR data and starting material reference standards.
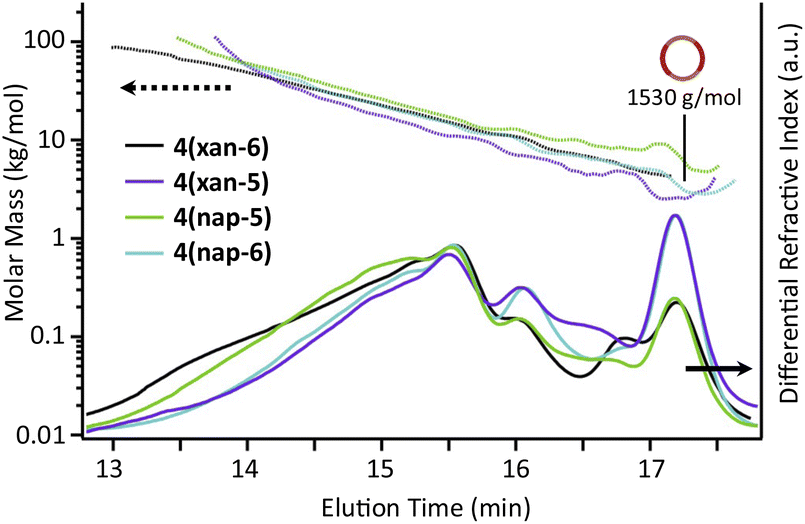 | ||
| Fig. 6 SEC (mobile phase – 25% DMF in THF) refractive index traces for 4(xan-6), 4(xan-5), 4(nap-5), and 4(nap-6) (bottom) and absolute molar mass data as determined by MALS (top). | ||
As would be expected for a step growth polymerization, the data shows that the poly[n]catenane obtained in the lowest yield (4(nap-6)) has the lowest observable maximum molar mass. The SEC traces of all the crude reaction mixtures clearly show that multiple different products with a range of molar masses obtained (Fig. 6); however, differences in the general peak shapes suggest that the different thread-like components do result in different product distributions.
To understand the product distribution trends in more detail, the SEC traces of the crude reaction mixtures were analyzed via Gaussian deconvolution using a procedure similar to the prior work.52Fig. 7a–d shows the Gaussian deconvolution of the crude reaction mixture for 4(xan-6), 4(xan-5), 4(nap-5), and 4(nap-6), respectively. Fig. 7a shows the assigned peaks of the 4(xan-6) product distribution based on the prior work.51,52 Using a similar protocol the three new poly[n]catenane mixtures were analyzed. To do this the refractive index trace (RI) is deconvoluted into the minimum number of peaks required to resolve the trace. It is important to point out that there are more than one set of peaks that can resolve such a complex chromatogram. The peaks were chosen based on standards (1, the threads (2) and their ring closed macrocycles (3), Fig. S25†), the SECs of fractionated samples (see below) and to be consistent with the NMR data. For example, the SEC curve of the crude reaction mixture of 4(xan-5) could be deconvoluted into 7 different peaks A-G (the sum of these peaks matched the SEC trace almost perfectly, Fig. 7b). Peaks A and B can be assigned as 1 and 2(xan-5), based on standards (Fig. S25†). To determine the rest of the deconvoluted peaks, preparatory SEC (1:3 DMF:THF) was employed to isolate fractions of the crude 4(xan-5) mixture. Seven individual fractions were collected (based on elution time) and analyzed via1H NMR (Fig. 8a) and SEC-MALS (Fig. 8b): the highest Mn fraction is the first to elute (fraction 1) and the lowest Mn fraction being the last to elute (fraction 7). The molar masses obtained from SEC-MALS of these materials (Fig. 8b) shows relatively good correlation between the higher Mn fractions, consistent with the measured dn/dc obtained from the crude reaction mixture (0.2042) corresponding to the higher molar mass poly[n]catenanes of 4(xan-5). Differences in the calculated molar masses (from MALS) are observed in the later fractions, especially for those that contain the lower Mn compounds. It is worthwhile pointing out that the molar mass of 1 is 1530 g mol−1 (shown as the dotted line in Fig. 8b) and the Mn obtained for 1 using the dn/dc value measured on the crude sample is ca. 2000 g mol−1. As such, while the measured mass does not exactly match these lower molar mass species, it is possible to get an approximation of their Mn using this approach. Given that it was not possible to isolate all the low molar mass products to determine their individual dn/dc values, the number average degree of polymerization (DPn) assigned to these low molar mass species is <5 (Fig. 8b) and other techniques, such as NMR, were used to further support the (in some cases tentative) assignment of what these peaks correspond to.
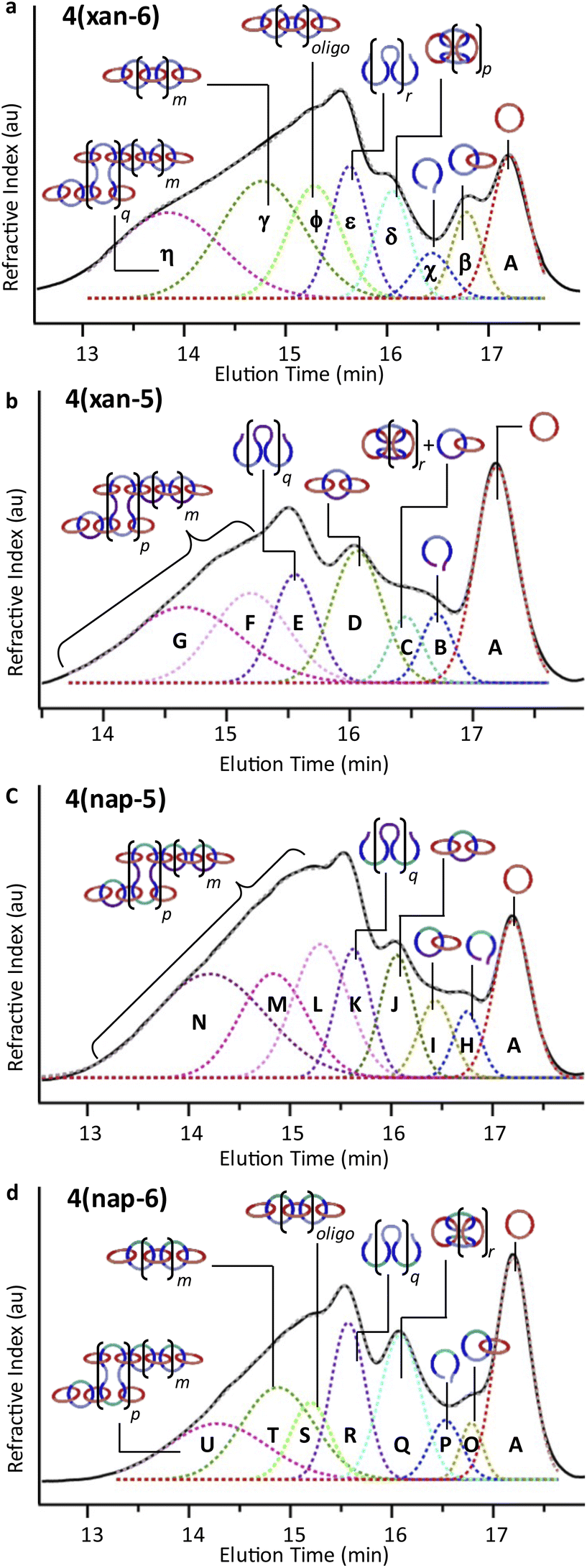 | ||
| Fig. 7 SEC (mobile phase – 25% DMF in THF) refractive index traces for (a) 4(xan-6), (b) 4(xan-5) (c) 4(nap-5), and (d) 4(nap-6). Deconvolution performed using a Gaussian fit, peak assignments made based on SEC analysis shown in Fig. 8, S26 and S28.† Sum of the deconvoluted peaks fits for (a)–(d) designated by the grey dashed line. | ||
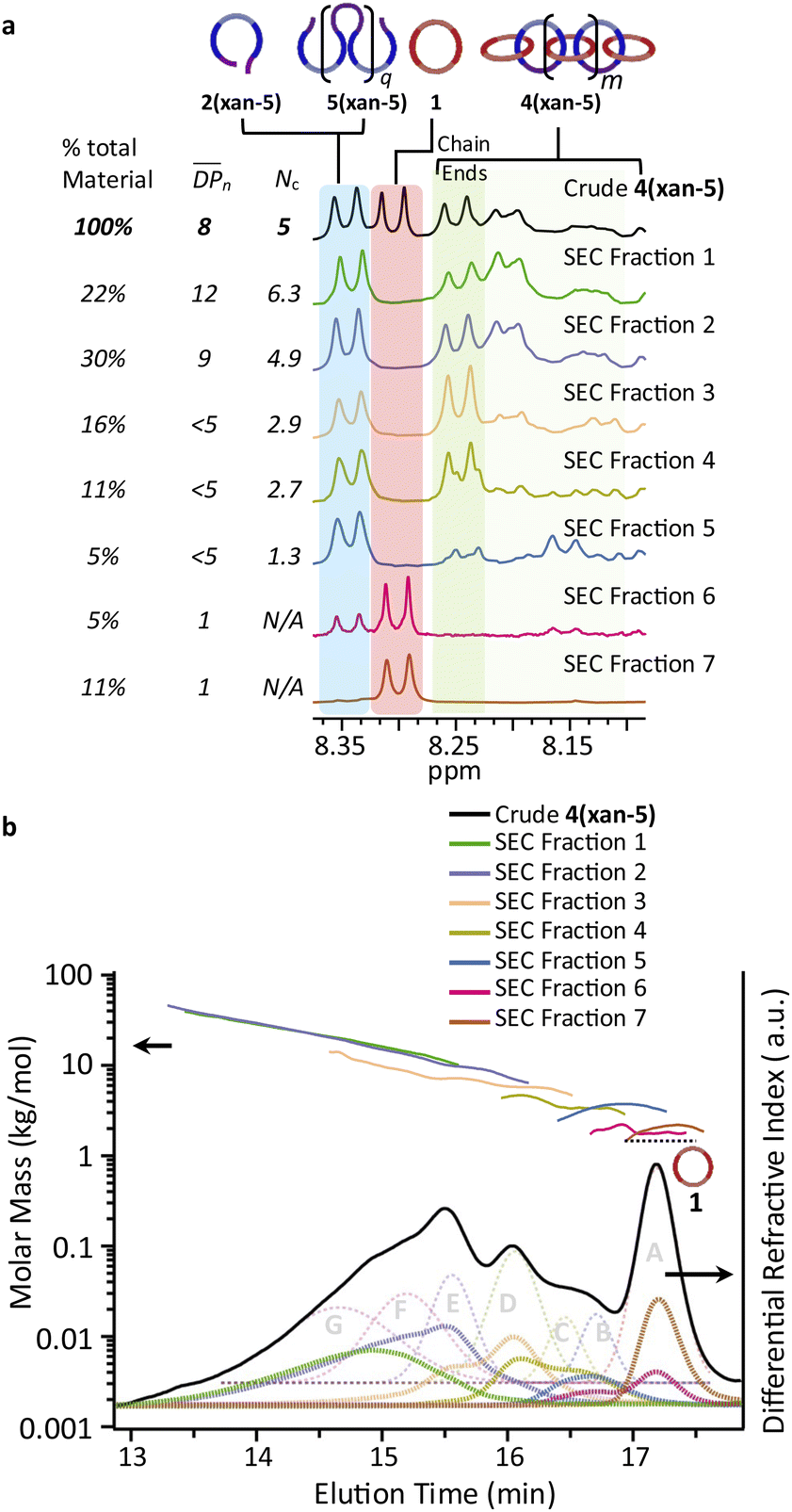 | ||
Fig. 8 (a) 1H NMR traces for all fractionated 4(xan-5) samples along with relative weight percent, the calculated number average degree of polymerization (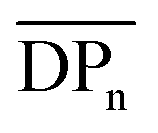 ) and number of chain ends (Nc) of each fraction using MALS analysis ( ) and number of chain ends (Nc) of each fraction using MALS analysis (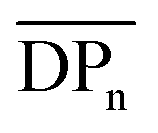 from eqn (S1)) (Nc from eqn (S2)). (b) The SEC RI traces (bottom) for each fractionated sample and the absolute molar mass (from MALS) using the dn/dc value (0.2042) obtained for 4(xan-5). The black dotted line shows the molar mass of 1 which can be compared to the measured molar mass (using the above dn/dc) of fraction 7 (brown) that contains predominantly 1. from eqn (S1)) (Nc from eqn (S2)). (b) The SEC RI traces (bottom) for each fractionated sample and the absolute molar mass (from MALS) using the dn/dc value (0.2042) obtained for 4(xan-5). The black dotted line shows the molar mass of 1 which can be compared to the measured molar mass (using the above dn/dc) of fraction 7 (brown) that contains predominantly 1. | ||
1H NMR of fractions 6 and 7 confirmed the assignments of Peaks A and B as starting materials. To help with the assignment of the subsequent deconvoluted peaks the average number of chain ends (Nc) in each fraction was determined (using a combination 1H NMR and DPn from MALS and Eqn (S1) and (S2)).52 A key difference between cyclic (c-), linear (l-) or branched (b-) poly[n]catenanes is their number of chain ends, Nc, (0, 2 and >3, respectively) and thus average Nc gives some insight into the polymer architectures in a given fraction. It is worthy of note that only fraction 5 has an average Nc < 2, suggesting that very little c-4(xan-5) is formed overall in this reaction. The MALS data of fraction 5, which primarily corresponds to a mixture of peak B (2(xan-5)) and C, suggests that this fraction has relatively low molar mass species (DPn < 5). Its calculated average Nc of 1.3 suggests the presence of cyclic poly[n]catenane. Thus, it is possible that Peak C corresponds to c-4(xan-5). However, in prior work it was shown that the chain ends for the majority of poly[n]catenanes are derived from macrocycle 1 (only one doublet observed between 8.21–8.27 ppm).51 This makes sense, as any incomplete reaction of the linear blue thread component would result in a red macrocycle chain end after demetallation. As the NMR of fraction 5 shows at least two different doublets in the region 8.21–8.27, consistent with the presence of low Mn catenanes with chain ends derived from both 1 and 3(xan-5), the data suggests that Peak C corresponds to both c-4(xan-5) and other low molar mass linear species (e.g. [2]catenane). Based on this analysis it appears that the amount of c-4(xan-5) formed is <8% (Table 1) but it is hard to get more specific with the current data set. However, even if it is assumed that peak C only corresponds to c-4(xan-5), the yield of c-poly[n]catenane is at least half of that observed in the 4(xan-6) synthesis, suggesting shortening the alkenyl chain inhibits its formation.
Both fractions 3 and 4 have a DPn < 5 and Nc < 3. As these fractions primarily correspond to Peak D, it suggests that this peak most likely corresponds to low Mn linear catenanes (Nc = 2), probably l-[3–5]catenane. Peak E is assigned to the ADMET polymer 5(xan-5) based on a combination of its presence in the 1H NMR of fractions 1–4, the amount 5(xan-5) determined in the 1H NMR of the crude reaction mixture (Fig. 5a, Table 1), and the comparative assignment of the peaks in the 4(xan-6) reaction (Fig. 7a). Finally, fractions 1 and 2, which correspond predominantly to Peaks F and G, contain the highest Mn materials and both fractions have average Nc's > 4, suggesting that these materials are b-4(xan-5). It is worthy of note that, unlike with 4(xan-6), no peaks could be assigned to corresponding to a polymeric l-4(xan-5) (only linear species observed correspond to low molar mass species such [2–5]catenane). Thus, while it is not possible to exclude the formation of higher molecular weight l-4(xan-5), it appears that shortening the alkenyl chain reduces the efficiency of the intramolecular cyclization of 2(xan-5) in the MSP that is required to yield both the linear and cyclic poly[n]catenanes. The consequence of which is the shorter alkenyl chain on the thread results in the formation of more branched poly[n]catenane, b-4(xan-5) and an increase in the amount of low molar mass [n]catenanes (Table 1).
If the shorter alkenyl chain is the reason for the enhanced formation the b-poly[n]catenane, then it would be expected that the 1·2(nap-5)·Zn(II)2 MSP would yield predominantly b-4(nap-5). Thus, a similar peak deconvolution analysis was performed on the crude 4(nap-5) reaction mixture (see Fig. S26† for more details). The data summarized in Fig. 7c confirms that the product distribution closely follows what is observed in 4(xan-5), with the branched b-4(nap-5) being the dominant poly[n]catenane architecture and little-to-no c-4(nap-5) or l-4(nap-5) being produced. Of course, with the current data it is not possible to entirely rule out their formation altogether, however, it is clear that the amount of c- or l-poly[n]catenanes formed in these reactions is significantly lower than for the original poly[n]catenane that used the longer alkenyl chain. It is worth noting that it is possible to use the NMR and SEC of the crude reaction mixture to obtain an average Nc of the poly[n]catenanes synthesized (Table 1). Doing this shows that, irrespective the aromatic linker in the thread, the shorter 3-pentenyl tail results in a much higher average Nc (4.4 or 5.4) than using the threads with the longer 4-hexenyl tail (Nc = 2.9 or 3). This further supports the idea that the shorter alkenyl tails favours the formation of b-poly[n]catenanes, presumably by disfavouring the intra-thread ring closing reaction that is required to make both the c- and l-poly[n]catenanes.
The fact that no evidence was found for the formation of a significant amount of c-4(nap-5) might be surprising as the NMR of the MSP precursor 1·2(nap-5)·Zn(II)2 showed a higher percentage of c-MSP (Fig. 4a) than in the xan-derivatives. However, it is consistent with the idea that the shorter alkenyl tail inhibits the ring closing of the thread in the MSP (irrespective of the MSP being linear or cyclic). If this occurs, then the major products should be b-4(nap-5) and/or low molecular weight oligomers, consistent with the distribution analysis data. To explore this more prior work51 had shown that it was possible to access c-poly/oligo[n]catenanes with 1 and 2(xan-6) using lower reaction concentrations and Fe(II) as the templating metal ion. Thus, the synthesis of c-4(nap-5) was attempted following the same procedures (Fig. S27†). In this experiment, no c-4(nap-5) could be isolated, supporting the idea that the shorter alkenyl chains do indeed disfavour the desired intramolecular ring closing reaction. It is worthwhile mentioning that the difficulty to ring-close these threads is also supported by the yields obtained for the synthesis of their corresponding macrocycles (3). The crude yield (from 1H NMR) of 3(xan-5) and 3(nap-5) obtained from the ring closing of 2(xan-5) and 2(nap-5) were 35% and 27%, respectively. In contrast the conversion of 2(nap-6) to 3(nap-6) under similar reaction conditions occurred in ca. 47% crude yield.
If the shorter alkenyl chain hinders c- and l- poly[n]catenane formation, then it made sense to explore the formation of 4(nap-6), which has the longer hexenyl chain and the shorter aromatic (nap) linker. After using the same deconvolution method as before (Fig. S28† for more details) the data suggests that, even though there is a drop in yield of the poly[n]catenane (ca. 61%), the distribution of poly[n]catenanes obtained (Fig. 7c) appear similar to those observed in 4(xan-6),51,52 with a few notable differences. The mismatch of the shorter linker and the longer tail does yield a larger amount of the non-interlocked ADMET product (5(nap-6)) than is observed in the 4(xan-6) synthesis, 22% vs. 15% respectively based on NMR (Table 1). As might be expected, a higher percentage of non-interlocked product corresponds to a drop in the overall degree of polymerization of 4(nap-6) (Table 1). However, as was the case for 4(xan-6), the data obtained from the fractioned samples of 4(nap-6) shows that the use of the longer alkenyl tails allow access to the formation of 1-4(nap-6), 4nap-64(nap-6) and b-4(nap-6) polymers. In fact, the overall product distribution of three 4(nap-6) architectures is similar to that of 4(xan-6), with perhaps a slight tendency to form more c-4(nap-6), which is consistent with the higher percentage of c-MSP that is formed in 1·2(nap-6)·Zn(II)2 relative to 1·2(xan-6)·Zn(II)2 (Fig. 4a).
Conclusions
This study has shown for the first time that the MSP-templated approach to poly[n]catenanes is possible with some structural variance in the thread-like monomer, allowing access to three new poly[n]catenanes with different ring sizes. It also shows that careful design of the thread component can be employed to preferentially access different poly[n]catenane architectures by this approach. The clearest example of this is the impact the length of the reactive alkenyl tails have on the product distribution. The use of the shorter 3-pentenyl tails hinders the ring closing reaction of the thread in the MSP and therefore inhibits the formation of the l- and c-poly[n]catenanes. Consequently, the use of such threads results in the predominant formation of the b-poly[n]catenanes, suggesting that 2(nap-5) or 2(xan-5) would be threads of choice for targeting branched polycatenanes while 2(xan-6) is currently the better option for the accessing linear poly[n]catenanes. While the size of the aromatic linker plays a role in the nature of the MSP, with the shorter naphthalene-based linker favouring the formation of the c-MSP, its role in determining the distribution of the poly[n]catenane architectures is not as significant as the size of the alkenyl tails in these systems.Overall, this study shows that is important to develop an understanding of the monomer structure/product distribution relationships for the synthesis of this class of complex interlocked polymers. This type of information can be important when specifically targeting new classes of catenated materials, e.g., network polycatenanes, where an increased formation of b-poly[n]catenanes will allow for easier network formation and open the door to the investigation of more complex catenane-containing materials.
Data availability
The authors declare that all data supporting the findings of this study are available within the article and ESI,† and raw data files are available from the corresponding author upon reasonable request.Author contributions
SJR proposed the study. BWR conducted preliminary studies. GL and MMT conducted further synthesis and analysis of poly[n]catenanes and comparative molecules. SJR supervised the work. MMT and SJR wrote the manuscript. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by National Science Foundation (NSF) grant number CHE-1903603. This work made use of the shared facilities at the University of Chicago Materials Research Science and Engineering Center (MRSEC), supported by National Science Foundation under award number DMR-2011854. Parts of this work were carried out at the Soft Matter Characterization Facility of the University of Chicago. We would like to thank the director of this facility, Dr Phillip Griffin, for assistance with SEC characterization. We would also like to thank the University of Chicago Chemistry NMR Facility and the facility manger Dr Josh Kurutz.References
- B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS PubMed.
- J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- P.-G. de Gennes, Scaling concepts in polymer physics, Cornell University Press, Ithaca, 1979 Search PubMed.
- E. Raphaël, C. Gay and P.-G. de Gennes, J. Stat. Phys., 1997, 89, 111–118 CrossRef.
- P.-G. de Gennes, Phys. A, 1999, 271, 231–237 CrossRef CAS.
- E. Wasserman, J. Am. Chem. Soc., 1960, 82, 4433–4434 CrossRef CAS.
- G. Schill and H. Zollenkopf, Justus Liebigs Ann. Chem., 1969, 721, 53–74 CrossRef CAS.
- C. O. Dietrich-Buchecker, J. P. Sauvage and J. M. Kern, J. Am. Chem. Soc., 1984, 106, 3043–3045 CrossRef CAS.
- P. R. Ashton, C. L. Brown, E. J. T. Chrystal, T. T. Goodnow, A. E. Kaifer, K. P. Parry, D. Philp, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 634 RSC.
- S. F. M. Van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2014, 43, 99–122 RSC.
- V. Balzani, M. Gómez-López and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405–414 CrossRef CAS.
- L. F. Hart, J. E. Hertzog, P. M. Rauscher, B. W. Rawe, M. M. Tranquilli and S. J. Rowan, Nat. Rev. Mater., 2021, 6, 508–530 CrossRef CAS.
- T. Takata, N. Kihara and Y. Furusho, Adv. Polym. Sci., 2004, 171, 1–75 CrossRef CAS.
- L. Fang, M. A. Olson, D. Benítez, E. Tkatchouk, W. A. Goddard III and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17–29 RSC.
- Z. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024–6046 CrossRef CAS PubMed.
- J.-P. Sauvage and C. Dietrich-Buchecke, Molecular catenanes, rotaxanes and knots: a journey through the world of molecular topology, John Wiley & Sons, 2008 Search PubMed.
- G. Liu, P. M. Rauscher, B. W. Rawe, M. M. Tranquilli and S. J. Rowan, Chem. Soc. Rev., 2022, 51, 4928–4948 RSC.
- F. Diederich, P. J. Stang and R. R. Tykwinski, Modern Supramolecular Chemistry, Wiley, 2008 Search PubMed.
- N. Hoyas Pérez and J. E. M. Lewis, Org. Biomol. Chem., 2020, 18, 6757–6780 RSC.
- H. Y. Au-Yeung, C.-C. Yee, A. W. Hung Ng and K. Hu, Inorg. Chem., 2018, 57, 3475–3485 CrossRef CAS PubMed.
- G. Gil-Ramirez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef CAS PubMed.
- M. A. Olson, A. Coskun, L. Fang, A. N. Basuray and J. Fraser Stoddart, Angew. Chem., Int. Ed., 2010, 49, 3151–3156 CrossRef CAS PubMed.
- C.-A. Fustin, C. Bailly, G. J. Clarkson, T. H. Galow and D. A. Leigh, Macromolecules, 2004, 37, 66–70 CrossRef CAS.
- C.-A. Fustin, G. J. Clarkson, D. A. Leigh, F. Van Hoof, A. M. Jonas and C. Bailly, Macromolecules, 2004, 37, 7884–7892 CrossRef CAS.
- J.-L. Weidmann, J.-M. Kern, J.-P. Sauvage, Y. Geerts, D. Muscat and K. Müllen, Chem. Commun., 1996, 1243–1244 RSC.
- D. Muscat, W. Köhler, H. J. Räder, K. Martin, S. Mullins, B. Müller, K. Müllen and Y. Geerts, Macromolecules, 1999, 32, 1737–1745 CrossRef CAS.
- J.-L. Weidmann, D. Muscat, S. Mullins, C. Rosenauer, K. Martin, J.-M. Kern, J.-P. Sauvage, Y. Geerts, H. J. Räder and W. Köhler, Chem.–Eur. J., 2002, 5, 1841–1851 CrossRef.
- G. Li, L. Wang, L. Wu, Z. Guo, J. Zhao, Y. Liu, R. Bai and X. Yan, J. Am. Chem. Soc., 2020, 142, 14343–14349 CrossRef CAS PubMed.
- M. A. Nosiglia, N. D. Colley, M. K. Danielson, M. S. Palmquist, A. O. Delawder, S. L. Tran, G. H. Harlan and J. C. Barnes, J. Am. Chem. Soc., 2022, 144, 9990–9996 CrossRef CAS PubMed.
- A. Bunha, P.-F. Cao, J. Mangadlao, F.-M. Shi, E. Foster, K. Pangilinan and R. Advincula, Chem. Commun., 2015, 51, 7528–7531 RSC.
- P.-F. Cao, J. D. Mangadlao, A. de Leon, Z. Su and R. C. Advincula, Macromolecules, 2015, 48, 3825–3833 CrossRef CAS.
- K. Morita, K. Motoyama, A. Kuramoto, R. Onodera and T. Higashi, J. Inclusion Phenom. Macrocyclic Chem., 2021, 100, 169–175 CrossRef CAS.
- Z. Zhang, J. Zhao, Z. Guo, H. Zhang, H. Pan, Q. Wu, W. You, W. Yu and X. Yan, Nat. Commun., 2022, 13, 1393 CrossRef CAS PubMed.
- T. Higashi, K. Morita, X. Song, J. Zhu, A. Tamura, N. Yui, K. Motoyama, H. Arima and J. Li, Commun. Chem., 2019, 2, 78 CrossRef.
- N. Watanabe, Y. Ikari, N. Kihara and T. Takata, Macromolecules, 2004, 37, 6663–6666 CrossRef CAS.
- D. B. Amabilino, P. R. Ashton, A. S. Reder, N. Spencer and J. F. Stoddart, Angew. Chem., Int. Ed., 1994, 33, 1286–1290 CrossRef.
- D. B. Amabilino, P. R. Ashton, S. E. Boyd, J. Y. Lee, S. Menzer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed., 1997, 36, 2070–2072 CrossRef CAS.
- H. Iwamoto, S. Tafuku, Y. Sato, W. Takizawa, W. Katagiri, E. Tayama, E. Hasegawa, Y. Fukazawa and T. Haino, Chem. Commun., 2016, 52, 319–322 RSC.
- K. Wang, C.-C. Yee and H. Y. Au-Yeung, Chem. Sci., 2016, 7, 2787–2792 RSC.
- A. W. H. Ng, C. Yee and H. Y. Au-Yeung, Angew. Chem., Int. Ed., 2019, 58, 17375–17382 CrossRef CAS PubMed.
- A. W. H. Ng, S. K. Lai, C. Yee and H. Y. Au-Yeung, Angew. Chem., Int. Ed., 2022, 61, e202110200 CrossRef CAS PubMed.
- N. D. Colley, M. A. Nosiglia, S. L. Tran, G. H. Harlan, C. Chang, R. Li, A. O. Delawder, Y. Zhang and J. C. Barnes, ACS Cent. Sci., 2022, 8, 1672–1682 CrossRef CAS PubMed.
- J. A. Berrocal, L. M. Pitet, M. M. L. Nieuwenhuizen, L. Mandolini, E. W. Meijer and S. Di Stefano, Macromolecules, 2015, 48, 1358–1363 CrossRef CAS.
- K. Endo, T. Shiroi, N. Murata, G. Kojima and T. Yamanaka, Macromolecules, 2004, 37, 3143–3150 CrossRef CAS.
- K. Endo, T. Shiroi and N. Murata, Polym. J., 2005, 37, 512–516 CrossRef CAS.
- P. Hu, J. Madsen, Q. Huang and A. L. Skov, ACS Macro Lett., 2020, 9, 1458–1463 CrossRef CAS PubMed.
- J. C. Wang, Annu. Rev. Biochem., 1985, 54, 665–697 CrossRef CAS PubMed.
- M. A. Krasnow and N. R. Cozzarelli, J. Biol. Chem., 1982, 257, 2687–2693 CrossRef CAS PubMed.
- Y. S. Kim, B. Kundukad, A. Allahverdi, L. Nordensköld, P. S. Doyle and J. R. C. van der Maarel, Soft Matter, 2013, 9, 1656–1663 RSC.
- Q. Wu, P. M. Rauscher, L. Xiaolong, R. J. Wojtecki, J. J. de Pablo, M. J. Hore and S. J. Rowan, Science, 2017, 358, 1434–1439 CrossRef CAS PubMed.
- M. M. Tranquilli, Q. Wu and S. J. Rowan, Chem. Sci., 2021, 12, 8722–8730 RSC.
- B. M. McKenzie, A. K. Miller, R. J. Wojtecki, J. C. Johnson, K. A. Burke, K. A. Tzeng, P. T. Mather and S. J. Rowan, Tetrahedron, 2008, 64, 8488–8495 CrossRef CAS.
- J. E. Hertzog, V. J. Maddi, L. F. Hart, B. W. Rawe, P. M. Rauscher, K. M. Herbert, E. P. Bruckner, J. J. de Pablo and S. J. Rowan, Chem. Sci., 2022, 13, 5333–5344 RSC.
- R. J. Wojtecki, Q. Wu, J. C. Johnson, D. G. Ray, L. T. J. Korley and S. J. Rowan, Chem. Sci., 2013, 4, 4440–4448 RSC.
- P. M. Rauscher, S. J. Rowan and J. J. de Pablo, ACS Macro Lett., 2018, 7, 938–943 CrossRef CAS PubMed.
- P. M. Rauscher, K. S. Schweizer, S. J. Rowan and J. J. De Pablo, Macromolecules, 2020, 53, 3390–3408 CrossRef CAS.
- P. M. Rauscher, K. S. Schweizer, S. J. Rowan and J. J. de Pablo, J. Chem. Phys., 2020, 152, 214901 CrossRef CAS PubMed.
- R. Kramer, J. M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci., 1993, 90, 5394–5398 CrossRef CAS PubMed.
- J. A. Semlyen, Mechanisms of Polyreactions-Polymer Characterization, Springer Berlin Heidelberg, Berlin, Heidelberg, pp. , pp. 41–75 Search PubMed.
- J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS.
- P. C. Hiemenz and T. P. Lodge, Polymer Chemistry, CRC press, Boca Raton, 2nd, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H NMR spectra, DOSY NMR analysis, NMR analysis calculations, SEC MALS deconvolution and MALS analysis, chain end analysis calculations. See DOI: https://doi.org/10.1039/d2sc05542b |
| This journal is © The Royal Society of Chemistry 2023 |