
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlacing gold on a π+-surface: ligand design and impact on reactivity†‡
Wei-Chun
Liu
 and
François P.
Gabbaï
*
and
François P.
Gabbaï
*
Department of Chemistry, Texas A&M University, College Station, TX 77843, USA. E-mail: francois@tamu.edu
First published on 22nd November 2022
Abstract
We describe a novel gold chloride complex supported by an ambiphilic phosphine/xanthylium ligand in which the AuCl moiety interacts with the π+ surface of the xanthylium unit as indicated by structural studies. Energy decomposition analyses carried out on a model system indicates the prevalence of non-covalent interactions in which the electrostatic and dispersion terms cumulatively dominate. The presence of these AuCl–π+ interactions correlates with the high catalytic activity of this complex in the cyclisation of 2-(phenylethynyl)phenylboronic acid, N-propargyl-t-butylamide, and 2-allyl-2-(2-propynyl)malonate. Comparison with the significantly less active acridinium and the 9-oxa-10-boraanthracene analogues reinforces this conclusion.
Introduction
Sporadic results from the past two decades have shown that electron-rich late transition metal complexes may interact with π-acidic systems to form stacked supramolecular aggregates. Early examples of such complexes were obtained with trinuclear gold(I) complexes and fluoroaromatics (A, Fig. 1) or nitrated fluorenone as acceptors.1 More recently, this approach has been extended to the case of Pd(II) and Pt(II) complexes (B and C, Fig. 1).2 Theoretical investigations carried out on some of these systems, including C suggest that the cohesion of these supramolecular structures is largely of electrostatic origin, with donor–acceptor bonding playing a minor role. These calculations also indicate that the stabilisation energy of the stacking motif may be substantial, reaching values in the 20–35 kcal mol−1 range for compounds involving [Pt(ppy)acac] and fluorinated aromatics.2c These past investigations have also shown that the formation of these supramolecular complexes provides a handle over the luminescent properties of the late transition metal complex.1a–d,1f,2c,d However, to our knowledge, exploiting these interactions to adjust the reactivity of the transition metal centre has not been previously considered, even if electron-poor π surfaces have been shown to affect the reactivity of organic molecules. For example, as demonstrated by Matile, π-acidic surfaces such as that presented by naphthalene diimides can readily acidify organic functionalities forced in their proximity.3 The same effects formed the basis of the anion π-catalysis concept pioneered by the same group.4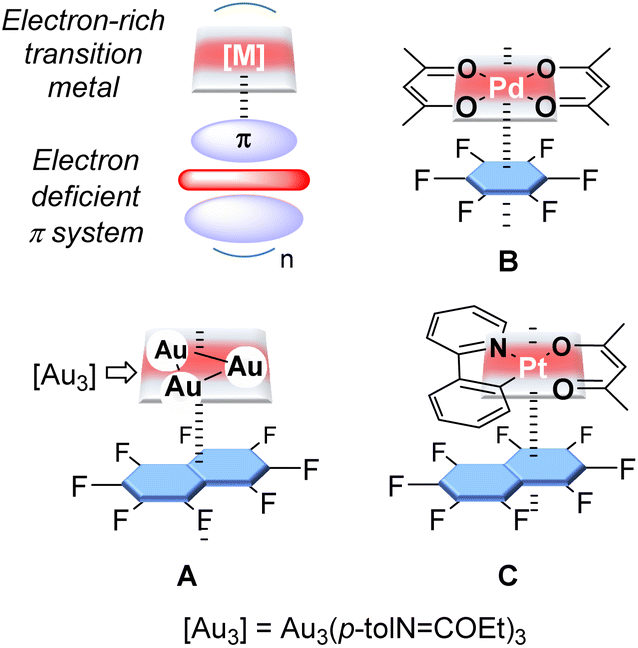 | ||
| Fig. 1 Supramolecular constructs involving late transition metal complexes and π-acidic systems. | ||
To test whether electron-poor surfaces could also affect the reactivity of transition metals, we have now decided to target complexes in which a gold(I) centre is positioned directly above a π-acidic system. Recognising the role that charges exert over the strength of anion π-interactions, we have chosen to consider a cationic π-acidic system.5 In this article, we illustrate this approach using complexes featuring an acridinium or a xanthylium unit as the π-acidic system, with a phosphine-ligated gold chloride moiety held in close proximity by a peri-substituted acenaphthene backbone.
Results and discussion
To begin, 5-diphenylphosphino-6-lithioacenaphthene was treated with xanthone at −78 °C to afford carbinol 1xant. The same approach using N-methylacridone afforded 1acr (Fig. 2). Dehydroxylation of 1xant and 1acr with HBF4 proceeded swiftly to afford the corresponding phosphonium derivatives [2xant]+ and [2acr]+, respectively, rather than the phosphine carbenium derivatives thus illustrating the tendency of phosphines to coordinate to carbenium ions.6 The tetrafluoroborate salts of both phosphonium cations are colourless air-stable solids. Their 13C NMR spectra reveal diagnostic resonance at 64.2 ppm (JC–P = 48.1 Hz) for [2xant][BF4] and 70.1 ppm (JC–P = 41.6 Hz) for [2acr][BF4] corresponding to the phosphine-neutralised Ccarb atom. 31P NMR spectra display a single downfield resonance at 52.0 ppm for [2xant][BF4] and 50.0 ppm for [2acr][BF4], as expected for phosphonium species. Their crystal structures, which confirm the coordination of the phosphorus atom to the adjacent carbenium, indicate that the P–Ccarb bond is slightly shorter in the case of the [2xant][BF4] (1.940(2) Å vs. 1.962(2) Å in [2acr][BF4]). A comparison of the pKR+ of 9-phenyl-N-methyl-acridinium (11.0) and 9-phenyl-xanthylium (1.8) suggests that this shortening is caused by the greater acidity of the xanthylium unit in [2xant][BF4].7 It follows that the newly formed P–Ccarb bond should be more stable in [2xant][BF4] than in [2acr][BF4]. | ||
| Fig. 2 (a) Synthesis of [2xant][BF4], [2acr][BF4], [3xant][OTf], [3acr][BF4] and [3acr][OTf]. (b) Crystal structures of [2xant][BF4] and [2acr][BF4]. (c and d) Crystal structures of [3xant][OTf] and [3acr][BF4] and ESP maps of [3xant]+ and [3acr]+. All ESP maps are drawn with an isosurface value of 0.001. For all crystal structure shown, the thermal ellipsoids in the structures are drawn at the 50% probability level. Hydrogen atoms, counter anions and interstitial solvent molecules omitted for clarity. | ||
In agreement with this conclusion, we observed that [2xant][BF4] does not react with (tht)AuCl in CH2Cl2. By contrast, [2acr][BF4] underwent a smooth P–Ccarb heterolytic bond activation reaction to afford the corresponding gold chloride complex [3acr]BF4 as an orange powder. Salt [3acr]BF4 could also be obtained by auration of the carbinol 1acr followed by in situ dehydroxylation with HBF4 (Fig. 2). A similar auration/dehydroxylation approach was considered for the xanthylium derivative [3xant]BF4. However, when the aurated carbinol 1xant-AuCl was treated with HBF4, the only identifiable product was the phosphonium cation [2xant]+ as indicated by NMR spectroscopy. Gratifyingly, a more selective reaction was observed when 1xant-AuCl was treated with TMSOTf, leading to the formation of [3xant]OTf as a deep purple solid in 92% yield (Fig. 2). The same procedure can be used to generate the triflate salt of [3acr]+. These salts have been analysed by 13C NMR spectroscopy which shows a signal at 176.3 ppm for [3xant]OTf and 161.2 ppm for [3acr]OTf corresponding to the resonance of the carbenium Ccarb centre of the xanthylium and acridinium unit, respectively.8 The phosphorus chemical shifts of both complexes (30.2 ppm for [3xant]OTf and 30.7 ppm for [3acr]OTf) are consistent with the presence of a triarylphosphine–AuCl motif.9
The structures of [3xant]OTf and [3acr]BF4 have been determined using X-ray diffraction. The steric constraints imposed by the rigid backbone position the AuCl motif of both complexes close to the xanthylium or acridinium unit, respectively. The proximity of these units can be measured by the shortest distance separating the mean plane (mpln) containing the xanthylium or acridinium unit and the gold or chlorine atom, as shown in Fig. 2. These distances, of 3.26 Å and 3.11 Å for [3xant]+ and [3acr]+, respectively, show that the electron-rich gold chloride moiety is positioned over the positively charged π+-surface of the aromatic cationic unit, as illustrated by the electrostatic potential (ESP) maps shown in Fig. 2. Such metal halide π+-interactions, which have been occasionally observed,10 can be regarded as the charge-reverse analogues of classical cation–π interactions involving electron-rich aromatic systems.11 In the case of [3xant]OTf, it is worth pointing out a rather short separation of 3.26 Å between the carbenium centre (Ccarb) and the gold atom. However, in agreement with prior studies on a related gold-xanthylium complex,12 NBO studies indicate negligible donor–acceptor bonding, suggesting that the interaction is mostly of electrostatic and dispersive origin.13 In the case of the acridinium derivative [3acr]BF4, the gold chloride moiety resides over the centre of the central pyridyl ring, leading to an even larger separation (3.49 Å) between the gold atom and the carbenium centre. Thus, both structures are characterised by the absence of significant Au → Ccarb charge–transfer interactions, despite the enforced proximity imposed by the rigid acenaphthene backbone. In agreement with the similarity seen in these two structures, we found that the computed steric and electronic parameters of the phosphine/carbenium present in [3xant]+ and [3acr]+ are almost identical (see ESI‡). This situation is reminiscent of that occurring in previously reported gold(I) complexes such as [4xant]+ and [4acr]+, featuring an intramolecularly installed xanthylium or acridinium unit (Scheme 1).12–14 In the case of [3acr]+, the gold-bound chloride anion of this complex forms a short contact with a hydrogen atom of the N-methyl group, indicating a hydrogen bonding interaction.
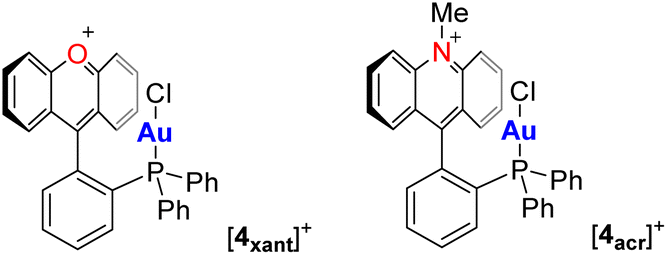 | ||
| Scheme 1 Examples of gold(I) complexes with an intramolecularly installed xanthylium or acridinium unit. | ||
To compare the structure and properties of these cations to those of a neutral derivative, we endeavoured to synthesise the isolelectronic boron analogue of [3xant]+. With this in mind, we first synthesised the phosphinoborane 5 as described in Fig. 3. Although the 11B (−3.3 ppm) and 31P NMR (7.0 ppm) resonances of 5 indicate P → B bond formation,15 we found that 5 readily reacted with (tht)AuCl to afford 6. Complex 6 displays a 11B NMR resonance at 49.6 ppm and an Au–B distance of 2.955(2) Å. This distance is comparable to that found in the AuCl complex of (o-(Ph2P)C6H4)BCy2 which also possesses a weak Au–B interaction.16 According to NBO calculations, the separation measured in 6 corresponds to an Au → B donor–acceptor interaction associated with a 2nd order perturbation energy of 5.2 kcal mol−1. The ESP map of this derivative shown in Fig. 3 shows that the electrostatic interaction between the AuCl and the neutral π-surface of the oxaborine unit may be unfavourable. Finally, buried volume calculations and frequency analysis of the corresponding putative nickel tricarbonyl complex shows that the steric and electronic parameters of the phosphine/borane present in 6 are very similar to those of [3xant]+ and [3acr]+ (see ESI‡).
 | ||
| Fig. 3 (a): Synthesis of the phosphinoborane derivative 6; (b) crystal structure and optimised structure, with ESP map, of 6. | ||
Aiming to better understand the forces arising from the confinement of the AuCl moiety over the π+ face of the cationic units in [3xant]+ and [3acr]+, we considered [H3PAuCl-Xant]+ and [H3PAuCl-Acr]+ (Xant = xanthylium and Acr = N-methyl acridinium) as simplified models. The structure of these models was optimised around a P–C9 distance arbitrarily set at 3.4 Å, a separation close that found in the crystal structure of [3xant]OTf (P–C9 = 3.41 Å) and [3acr]BF4 (P–C9 = 3.37 Å). The AuCl moiety of the resulting optimised structures is projected over the surface of the cationic heterocycle, leading to Au–mpln distances of 3.35 Å for [H3PAuCl-Xant]+ and 3.44 Å for [H3PAuCl-Acr]+ approaching those experimentally observed in the crystal structures. These model systems were next subjected to an energy decomposition analysis (EDA) as implemented in the ADF program.17 This analysis reveals that the [H3PAuCl-Acr]+ and [H3PAuCl-Xant]+ are stabilised by a interaction energy (ΔEInt) of −10.14 kcal mol−1 and −7.92 kcal mol−1 with respect to the individual components (Fig. 4). Similar forces have been invoked to rationalise the formation of stacks involving electron-rich trinuclear gold complexes and π-acidic aromatic derivatives.1g It is interesting to note that [H3PAuCl-Acr]+ is more stabilised than [H3PAuCl-Xant]+, an effect that we correlate to the involvement of hydrogen bonding interactions between the chloride anion and the nitrogen-bound methyl group of [H3PAuCl-Acr]+. The interactions energies determined for [H3PAuCl-Xant]+ and [H3PAuCl-Acr]+ can be further decomposed into their individual components. This decomposition shows that for both model complexes, the sum of the electrostatic (ΔEEl) and dispersion terms (ΔEDis) is significantly more negative than the orbital term (ΔEOrb), indicating that non-covalent forces dominate this interaction. It is important to note that the [Xant]+ cation is more π-acidic than the [Acr]+ cation. The greater π-acidity of the [Xant]+ cation is confirmed by its lower LUMO energy (−7.55 eV vs. −6.90 eV for [Acr]+) and its higher out-of-plane maximum electrostatic surface potential (VS,max) value which indicates the existence of a deeper π hole (Fig. 4).18 Thus, even if the interaction energy ΔEInt is larger in the case of the acridinium model complex because of involvement of the N-bound methyl group in hydrogen bonding interaction with the chloride, we contend that the AuCl–π+ interactions will be more intense in the case of the xanthylium derivative. Finally, efforts to optimise the structure of the boron isoelectronic analogue of [H3PAuCl-Xant]+ led to a structure in which the gold chloride moiety is oriented away from the oxaborine unit. The divergence observed during the optimisation of this model complex (H3PAuCl-oxaborine) speaks to the importance of the cationic charge in [H3PAuCl-Xant]+ and its influence on the stability of the model complexes.
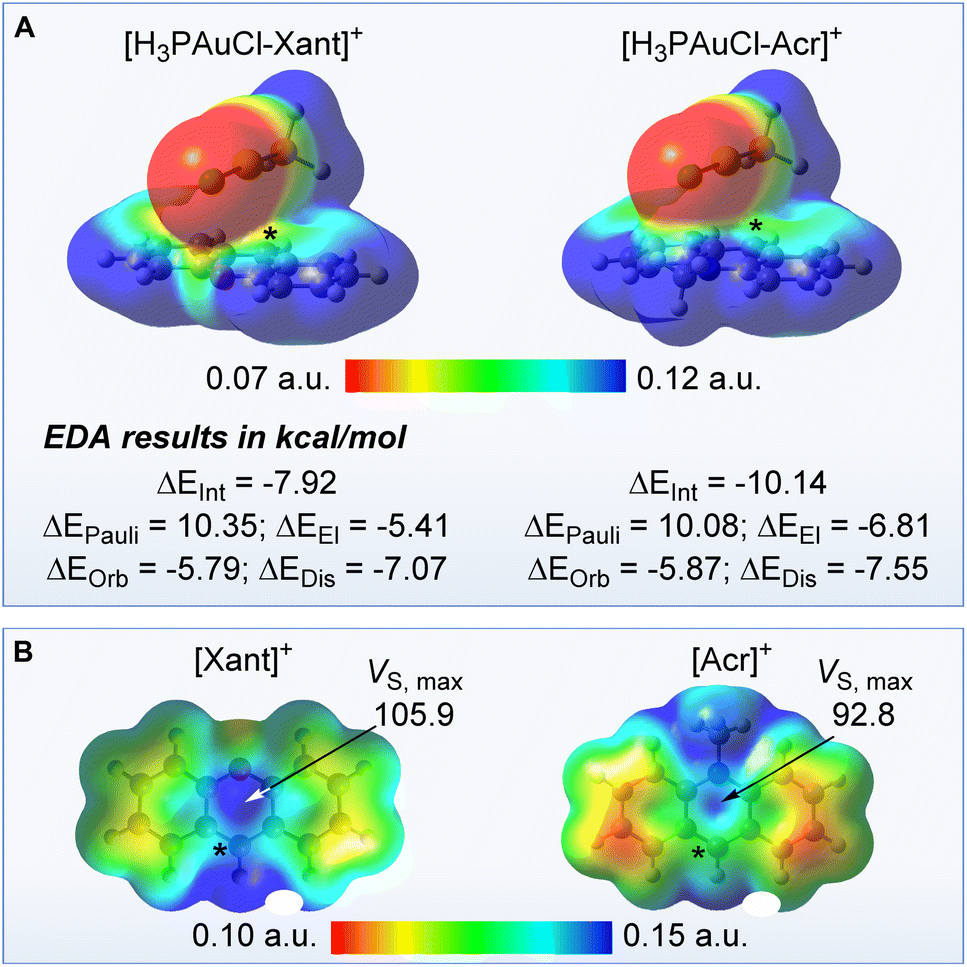 | ||
| Fig. 4 (a) Optimised structures, with ESP maps, of the model complexes [H3PAuCl-Xant]+ and [H3PAuCl-Acr]+, along with energy decomposition analysis results; (b) optimised structure and ESP maps of the [Xant]+ and [Acr]+ cations. The asterisk indicates the position of the C9 atom. The VS,max values, given in kcal mol−1, were obtained using the Multiwfn software. | ||
To test whether the AuCl–π+ interactions described above influence the reactivity of the metal centre, we first investigated the reactivity of the complexes toward chloride anions in CDCl3.12 While no reaction was observed for [3acr]OTf and 6, [3xant]OTf proved to be much more reactive and immediately decomposed, leading to the formation of the phosphonium [2xant]+ as the sole phosphorus-containing species. Surprised by this outcome which illustrates the elevated reactivity of [3xant]+, we decided to investigate the behaviour of this cationic complex in the presence of an alkynyl substrate prone to an isomerisation reaction. To start, we selected the alkynyl boronic acid a, a substrate that can be cyclised into b (reaction 1, Fig. 5) but only in the presence of carbophilic gold species such as Ph3PAuNTf2 which benefits from the presence of a weakly coordinating triflimide anion.19 Surprisingly, and despite the presence of a gold chloride moiety in [3xant]+, we observed that this acenaphthene/xanthylium-based, cationic gold complex was also active. Indeed, when present in a 2 mol% ratio in d3-MeCN at 50 °C, [3xant]+ promoted almost quantitative conversion (>95%) of a into b in 2 h. By contrast, no conversion was observed with Ph3PAuCl pointing to the favourable influence of the xanthylium unit. The ability of the xanthylium-containing complex [3xant]+ to cyclise the substrate appears to be unique as its acridinium counterpart [3acr]+ only afforded a 28% conversion of a into b at the same time point, under the same conditions. We also tested the previously reported6e,12 xanthylium phosphine gold chloride derivative [4xant]+ as a triflate salt and only observed a conversion of 11% (Fig. 5). A similar observation was made for the neutral borane derivative 6 which only led to 8% conversion after 2 h. These results underscore the unique carbophilic properties of [3xant]+. At first sight, it appears that a parallel can be established between the reactivity of this gold chloride complex and the π-acidity of the flanking cationic group. Indeed, based on the ESP map shown in Fig. 4b, the xanthylium cation features the highest VS,max, making it the most π-acidic unit considered in this study. These considerations, however, do not allow to readily explain the lower reactivity of the ortho-phenylene system [4xant]+.
 | ||
| Fig. 5 Left: benchmark reactions used to evaluate the catalytic properties of [3xant][OTf], [3acr][OTf], 6 and [4xant][OTf]. Top right: computed relative stability of the complexes formed by the gold chloride complexes and the reaction substrate a. Bottom right: figure showing the positioning of the gold atom with respect to xanthylium π hole in [3xant]+ and [4xant]+. | ||
To further elucidate the factors that govern this chemistry, we resorted to a simple computational modelling study. Assuming that the efficiency of these catalysts is correlated to their ability to activate the alkynyl functionality of a, we calculated the structure of the putative complex ([3xant-a]+) formed by the catalyst and the substrate. A survey of different possible conformers led to the identification of the structure shown in Fig. 5 as the lowest energy structure, with a binding energy of 25.8 kcal mol−1. This structure is characterised by the presence of a phosphine-ligated chloroauracyclopropene unit that is co-facially oriented with respect to the π+ surface of the xanthylium unit. A similar computational survey carried out with the other three catalysts considered in this study led to similar structures with a chloroauracyclopropene unit positioned over the surface of the adjacent heterocycle. These structures are, however, much less stable than that of [3xant-a]+ as indicated in Fig. 5. The lesser stability of [3acr-a]+ and 6-a can be correlated to the lower π-acidity of the acridinium and oxaborine units of [3acr]+ and 6, respectively. More surprising is the difference that exists between the acenaphthene-based system [3xant-a]+ and the ortho-phenylene-based system [4xant-a]+, with the latter being significantly less stable, even if both possess a xanthylium cation as a π-acidic unit. We propose that this difference arises, at least in part, from the positioning of the gold atom with respect to the xanthylium unit. In the case of [3xant]+, the gold atom is situated almost directly above the centroid of the xanthylium unit which also corresponds to the π hole as indicated by the ESP map in Fig. 4b. In the case of [4xant]+, the gold resides above the Ccarb atom, at the edge of the xanthylium ring offset with respect to the position of the π hole.18 This comparison leads us to hypothesise that, by siting over the area where the surface potential is maximum, the gold atom of [3xant]+ will see its electrophilic character enhanced via Au–π hole interactions. Another factor may be of entropic origin. Binding the alkyne to the less geometrically congested [4xant]+ will come at a higher entropic cost, elevating the free energy of the intermediate [4xant-a]+. Conversly, we contend that the congested and thus rigid structure of [3xant]+ will lower the entropic penalty involved in substrate binding.
To solidify the superior properties of [3xant]+, it became important to test the properties of these gold complexes in additional reactions that are typically not promoted by simple phosphine gold chloride complexes such as Ph3PAuCl. With this in mind, we selected the cycloisomerisation of the propargyl amide c (reaction 2) and enyne e (reaction 3) as two additional benchmarks of reactivity (Fig. 5). For reaction 2, we observed full conversion of c into d in 20 min when [3xant]+ was used in a 2 mol% loading. By contrast, [3acr]+ and [4xant]+ showed only 16% and 21% conversion, respectively, under the same conditions. Complex 6 only afforded traces of the product d, a result comparable to that observed with Ph3PAuCl. Similar results were obtained with reaction 3 which was carried out with a gold complex loading of 2 mol% and a temperature of 50 °C. Indeed, we found [3xant]+ to be the most reactive complex, as indicated by a conversion of e into f of 69% after 1 h. Under the same conditions, 6 showed no measurable product formation while the acridinium catalyst [3acr]+ and xanthylium catalyst [4xant]+ showed low activity, with conversions of only 11% and 2%, respectively. These additional results allow us to generalise the superior catalytic properties of [3xant]+. Its activity in the cyclisation of enyne e is particularly noteworthy as this substrate does not possess a protic functionality, thus ruling out activation of the catalyst by involvement of the Au–Cl bond in hydrogen-bonding interactions.20
Conclusions
The results reported herein allow us to introduce a novel strategy for enhancing the reactivity of late transition metal centres via non-covalent interactions. Indeed, our results show that positioning an AuCl moiety over the π-surface of a charged heterocycle elevates the carbophilic reactivity of the gold centre. A correlation is also established between the acidity of the π-system employed and the catalytic activity, with the xanthylium group outcompeting the less π-acidic acridinium group or its neutral isoelectronic oxaborine analogue. The difference in the properties of the acenaphthene-based system [3xant]+ and the ortho-phenylene-based system [4xant]+shows that the nature of the π-system employed is not the only factor since both contain a xanthylium unit. Instead, the difference seen in the behaviour of these two complexes forces us to consider the positioning of the gold atom over the π+ surface as another very important determinant. By analogy with the use of π-acidic systems for enhancing the protic character of an approaching substrates,4 we propose that the positive potential of the xanthylium unit elevates the Lewis acidity or electrophilicity of the gold centre, leading to the observed carbophilic reactivity enhancement. We also postulate that the more rigidly preorganised structure [3xant]+ attenuates entropy loss during the substrate binding step.Data availability
The data supporting this article have been uploaded as part of the ESI.‡ CCDC 2191151 (1xant), 2191152 ([2xant][BF4]), 2191153 ([2acr][BF4]), 2191154 ([3xant][OTf]-0.5(CH2Cl2)-0.5(H2O)), 2191155 ([3acr][BF4]-(CH2Cl2)), and 2191156 ([4xant][OTf]-(MeCN)), and 2191157 (6) contain the supplementary crystallographic data for this paper.Author contributions
WCL: investigation, conceptualization, formal analysis, writing – original draft preparation. FPG: conceptualization, formal analysis, funding acquisition, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the National Science Foundation (CHE-2154972), the Donors of the American Chemical Society Petroleum Research Fund (61541-ND3) and the Welch Foundation (A-1423), All calculations were conducted with the advanced computing resources provided by Texas A & M High Performance Research Computing.Notes and references
- (a) M. A. Rawashdeh-Omary, M. A. Omary, J. P. Fackler, R. Galassi, B. R. Pietroni and A. Burini, J. Am. Chem. Soc., 2001, 123, 9689–9691 CrossRef CAS PubMed; (b) M. M. Olmstead, F. Jiang, S. Attar and A. L. Balch, J. Am. Chem. Soc., 2001, 123, 3260–3267 CrossRef CAS PubMed; (c) A. A. Mohamed, M. A. Rawashdeh-Omary, M. A. Omary and J. J. P. Fackler, Dalton Trans., 2005, 2597–2602 RSC; (d) M. A. Omary, A. A. Mohamed, M. A. Rawashdeh-Omary and J. P. Fackler, Coord. Chem. Rev., 2005, 249, 1372–1381 CrossRef CAS; (e) E. R. T. Tiekink and J. Zukerman-Schpector, CrystEngComm, 2009, 11, 1176–1186 RSC; (f) O. Elbjeirami, M. D. Rashdan, V. Nesterov and M. A. Rawashdeh-Omary, Dalton Trans., 2010, 39, 9465–9468 RSC; (g) R. Hahn, F. Bohle, W. Fang, A. Walther, S. Grimme and B. Esser, J. Am. Chem. Soc., 2018, 140, 17932–17944 CrossRef CAS PubMed.
- (a) C. Browning, J. M. Hudson, E. W. Reinheimer, F.-L. Kuo, R. N. McDougald, H. Rabaâ, H. Pan, J. Bacsa, X. Wang, K. R. Dunbar, N. D. Shepherd and M. A. Omary, J. Am. Chem. Soc., 2014, 136, 16185–16200 CrossRef CAS PubMed; (b) A. V. Rozhkov, M. A. Krykova, D. M. Ivanov, A. S. Novikov, A. A. Sinelshchikova, M. V. Volostnykh, M. A. Konovalov, M. S. Grigoriev, Y. G. Gorbunova and V. Y. Kukushkin, Angew. Chem., Int. Ed., 2019, 58, 4164–4168 CrossRef CAS PubMed; (c) A. V. Rozhkov, I. V. Ananyev, R. M. Gomila, A. Frontera and V. Y. Kukushkin, Inorg. Chem., 2020, 59, 9308–9314 CrossRef CAS PubMed; (d) S. A. Katkova, K. V. Luzyanin, A. S. Novikov and M. A. Kinzhalov, New J. Chem., 2021, 45, 2948–2952 RSC; (e) Y. V. Torubaev, I. V. Skabitsky, A. V. Rozhkov, B. Galmés, A. Frontera and V. Y. Kukushkin, Inorg. Chem. Front., 2021, 8, 4965–4975 RSC; (f) L. E. Zelenkov, A. A. Eliseeva, S. V. Baykov, D. M. Ivanov, A. I. Sumina, R. M. Gomila, A. Frontera, V. Y. Kukushkin and N. A. Bokach, Inorg. Chem. Front., 2022, 9, 2869–2879 RSC.
- Y. Zhao, N. Sakai and S. Matile, Nat. Commun., 2014, 5, 3911 CrossRef CAS PubMed.
- (a) Y. Zhao, Y. Cotelle, L. Liu, J. Lopez-Andarias, A.-B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255–2263 CrossRef CAS PubMed; (b) A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef CAS PubMed.
- (a) D. Quiñonero, A. Frontera, D. Escudero, P. Ballester, A. Costa and P. M. Deyà, ChemPhysChem, 2007, 8, 1182–1187 CrossRef PubMed; (b) C. Estarellas, A. Frontera, D. Quinonero and P. M. Deya, J. Chem. Theory Comput., 2008, 4, 1981–1989 CrossRef CAS PubMed; (c) A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564–9583 CrossRef CAS; (d) P. Manna, S. K. Seth, M. Mitra, S. R. Choudhury, A. Bauzá, A. Frontera and S. Mukhopadhyay, Cryst. Growth Des., 2014, 14, 5812–5821 CrossRef CAS; (e) R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2015, 17, 14437–14453 RSC.
- (a) K. Chansaenpak, M. Yang and F. P. Gabbaï, Philos. Trans. R. Soc., A, 2017, 375, 20170007 CrossRef; (b) E. Follet, P. Mayer and G. Berionni, Chem.–Eur. J., 2017, 23, 623–630 CrossRef CAS PubMed; (c) A. C. Shaikh, J. M. Veleta, J. Moutet and T. L. Gianetti, Chem. Sci., 2021, 12, 4841–4849 RSC; (d) J. Zhou, L. L. Cao, L. Liu and D. W. Stephan, Dalton Trans., 2017, 46, 9334–9338 RSC; (e) E. D. Litle, L. C. Wilkins and F. P. Gabbaï, Chem. Sci., 2021, 12, 3929–3936 RSC.
- (a) J. W. Bunting, V. S. F. Chew, S. B. Abhyankar and Y. Goda, Can. J. Chem., 1984, 62, 351–354 CrossRef CAS; (b) C. D. Ritchie, Can. J. Chem., 1986, 64, 2239–2250 CrossRef CAS.
- D. T. Hogan and T. C. Sutherland, J. Phys. Chem. Lett., 2018, 9, 2825–2829 CrossRef CAS.
- N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133–4136 CrossRef PubMed.
- L. Mei, J. M. Veleta, J. Bloch, H. J. Goodman, D. Pierce-Navarro, A. Villalobos and T. L. Gianetti, Dalton Trans., 2020, 49, 16095–16105 RSC.
- S. Handa and L. M. Slaughter, Angew. Chem., Int. Ed., 2012, 51, 2912–2915 CrossRef CAS PubMed.
- L. C. Wilkins, Y. Kim, E. D. Litle and F. P. Gabbaï, Angew. Chem., Int. Ed., 2019, 58, 18266–18270 CrossRef CAS PubMed.
- W.-C. Liu, Y. Kim and F. P. Gabbaï, Chem.–Eur. J., 2021, 27, 6701–6705 CrossRef CAS PubMed.
- (a) J. Zhou, E. D. Litle and F. P. Gabbaï, Chem. Commun., 2021, 57, 10154–10157 RSC; (b) G. Park, M. Karimi, W.-C. Liu and F. P. Gabbaï, Angew. Chem., Int. Ed., 2022, 61, e202206265 CAS.
- (a) S. Bontemps, M. Devillard, S. Mallet-Ladeira, G. Bouhadir, K. Miqueu and D. Bourissou, Inorg. Chem., 2013, 52, 4714–4720 CrossRef CAS PubMed; (b) Y.-F. Li, Y. Kang, S.-B. Ko, Y. Rao, F. Sauriol and S. Wang, Organometallics, 2013, 32, 3063–3068 CrossRef CAS; (c) J. Beckmann, E. Hupf, E. Lork and S. Mebs, Inorg. Chem., 2013, 52, 11881–11888 CrossRef CAS PubMed; (d) O. Sadek, G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2021, 50, 5777–5805 RSC.
- S. Bontemps, G. Bouhadir, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 12056–12057 CrossRef CAS PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. v. Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- (a) A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496–2517 CrossRef; (b) J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541–548 CrossRef CAS.
- (a) C. Körner, P. Starkov and T. D. Sheppard, J. Am. Chem. Soc., 2010, 132, 5968–5969 CrossRef PubMed; (b) T. Kaehler, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2019, 58, 11379–11384 CrossRef CAS PubMed.
- (a) N. V. Tzouras, A. Gobbo, N. B. Pozsoni, S. G. Chalkidis, S. Bhandary, K. Van Hecke, G. C. Vougioukalakis and S. P. Nolan, Chem. Commun., 2022, 58, 8516–8519 RSC; (b) S. Sen and F. P. Gabbaï, Chem. Commun., 2017, 53, 13356–13358 RSC; (c) O. Seppänen, S. Aikonen, M. Muuronen, C. Alamillo-Ferrer, J. Burés and J. Helaja, Chem. Commun., 2020, 56, 14697–14700 RSC; (d) A. Franchino, À. Martí, S. Nejrotti and A. M. Echavarren, Chem.–Eur. J., 2021, 27, 11989–11996 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to Warren Piers on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Additional experimental and computational details and crystallographic data in cif format. CCDC 2191151–2191157. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05574k |
| This journal is © The Royal Society of Chemistry 2023 |