
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgrammable synthesis of well-defined, glycosylated iron(II) supramolecular assemblies with multivalent protein-binding capabilities†
Jake H.
Schwab
 ,
Jake B.
Bailey
,
Milan
Gembicky
and
Julia M.
Stauber
*
,
Jake B.
Bailey
,
Milan
Gembicky
and
Julia M.
Stauber
*
Department of Chemistry and Biochemistry, University of California, 9500 Gilman Dr, La Jolla, San Diego, CA, USA. E-mail: jstauber@ucsd.edu
First published on 20th December 2022
Abstract
Multivalency plays a key role in achieving strong, yet reversible interactions in nature, and provides critical chemical organization in biological recognition processes. Chemists have taken an interest in designing multivalent synthetic assemblies to both better understand the underlying principles governing these interactions, and to build chemical tools that either enhance or prevent such recognition events from occurring in biology. Rationally tailoring synthetic strategies to achieve the high level of chemical control and tunability required to mimic these interactions, however, is challenging. Here, we introduce a systematic and modular synthetic approach to the design of well-defined molecular multivalent protein-binding constructs that allows for control over size, morphology, and valency. A series of supramolecular mono-, bi-, and tetrametallic Fe(II) complexes featuring a precise display of peripheral saccharides was prepared through coordination-driven self-assembly from simple building blocks. The molecular assemblies are fully characterized, and we present the structural determination of one complex in the series. The mannose and maltose-appended assemblies display strong multivalent binding to model lectin, Concanavalin A (Kd values in μM), where the strength of the binding is a direct consequence of the number of saccharide units decorating the molecular periphery. This versatile synthetic strategy provides chemical control while offering an easily accessible approach to examine important design principles governing structure–function relationships germane to biological recognition and binding properties.
Introduction
Saccharide–protein interactions feature prominently in biological systems and are responsible for a diverse array of cellular functions including cell-signaling, communication, and regulation processes.1,2 Such interactions take place via the recognition of cluster, or oligomeric-type carbohydrate biomolecules that engage with protein targets through multiple, simultaneous interactions, which is known as the multivalency effect.3–5 These interactions result in enhanced binding across several orders of magnitude when compared with the binding of the single-saccharide, monovalent analogues.6,7 Such behaviour has driven efforts to design multivalent glycoassemblies with controlled spatial and topology parameters to guide the development of effective therapeutics and to better understand and address underlying principles governing protein-glycan recognition.8,9 A range of synthetic platforms including glycopeptides,10,11 glycodendrimers,12,13 glycopolymers,14–16 glyconanoparticles,17 among others,3 have been employed to probe saccharide-based multivalent interactions (Fig. 1). However, it remains challenging to systematically vary and control parameters such as valency, spacing, and molecular precision, which are critical design components of any system meant to interface with the complex compositions at protein binding sites. Well-defined, multivalent saccharide-appended constructs that enable control over composition and size have the potential to provide critical insight into important structure–function relationships germane to their efficacy; however, such structurally precise frameworks are rare.18–21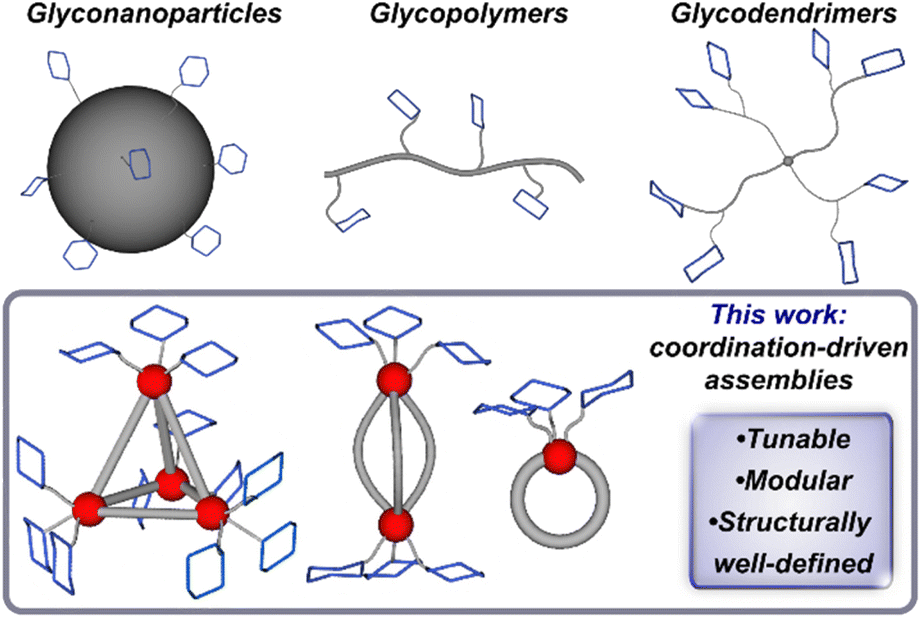 | ||
| Fig. 1 Multivalent glycoassemblies including nanoparticles, polymers and dendrimers, and this work, which introduces a new class of glycosylated supramolecular multivalent complexes. | ||
The use of coordination-based supramolecular systems offers a platform to explore new classes of structurally complex, yet well-defined multivalent assemblies that are amenable to architectural tuning. Coordination-driven self-assembly has provided a foundation for the development of nano-scale supramolecular systems with wide ranging applications22 including but not limited to catalysis,23 molecular sensing,24 nanomedicine,25,26 and fundamental investigations.27,28 Work pioneered by Fujita,29,30 Stang,31,32 Raymond,33 and others34,35 has led to the development of a variety of three-dimensional scaffolds by providing a rational and versatile approach to the design of discrete frameworks with predictable directionality and structural morphology.36 Through judicious choice of organic subcomponents and metal ions, chemists have and continue to develop synthetic strategies that enable the preparation of complex supramolecular assemblies while maintaining precise control over their spatial, topological, stability, and solubility properties.
Here, we introduce a new approach to the synthesis of structurally well-defined multivalent glycosylated architectures through coordination-driven self-assembly. Employing this versatile and modular strategy, we have prepared a series of programmable and robust saccharide-grafted supramolecular Fe(II) complexes from simple organic building blocks. Mono, di-, and tetrametallic Fe(II) assemblies (Fig. 1) that feature distinct spatial arrangements of three, six and twelve appended carbohydrates, respectively, have been synthesized and spectroscopically characterized. We also report the single-crystal X-ray structure of one complex, which enabled us to gain a key understanding of the molecular structure on the atomic level. We investigated the binding of these hybrid assemblies to plant-based protein, Concanavalin A, and showcase how this strategy offers chemical tools for systematic and rational control over molecular recognition properties. This series has the potential to provide insight into how modular tuning of the valency and morphology of hierarchical constructs affects their binding behaviour, while offering a rational approach to building systems of high affinity and specificity.
Results and discussion
Subcomponent design and synthesis
Here, we present a bottom-up approach to the synthesis of glycosylated supramolecular coordination-based structures based on a subcomponent self-assembly strategy first introduced by Nitschke et. al.27 In this straightforward process, simple building blocks including formylpyridines and di-, tri-, or tetratopic amines spontaneously self-assemble around metal ion templates through the formation of covalent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and dative N→metal bonds.37 This method has successfully been used to prepare a variety of metal–organic architectures including helicates, catenanes, rotaxanes, cages, and grids.38,39 Employing this approach, we first prepared a series of saccharide-substituted formyl pyridine building blocks. These subcomponents were synthesized in two steps starting from the acetate-protected carbohydrate precursors, which were converted to the corresponding 2-bromoethyl derivatives via substitution at the anomeric position with bromoethanol. Each derivative was subsequently treated with 5-hydroxypicolinaldehyde in the presence of K2CO3 in DMF, which resulted in clean O-glycosylation to generate the 5-substituted α-mannose, β-glucose, β-galactose, and β-maltose-appended picolinaldehydes (A–D). The presence of the acetate protecting groups rendered these derivatives soluble in polar organic solvents and enabled their straightforward purification by column chromatography.
N and dative N→metal bonds.37 This method has successfully been used to prepare a variety of metal–organic architectures including helicates, catenanes, rotaxanes, cages, and grids.38,39 Employing this approach, we first prepared a series of saccharide-substituted formyl pyridine building blocks. These subcomponents were synthesized in two steps starting from the acetate-protected carbohydrate precursors, which were converted to the corresponding 2-bromoethyl derivatives via substitution at the anomeric position with bromoethanol. Each derivative was subsequently treated with 5-hydroxypicolinaldehyde in the presence of K2CO3 in DMF, which resulted in clean O-glycosylation to generate the 5-substituted α-mannose, β-glucose, β-galactose, and β-maltose-appended picolinaldehydes (A–D). The presence of the acetate protecting groups rendered these derivatives soluble in polar organic solvents and enabled their straightforward purification by column chromatography.
Synthesis and characterization of glucose-functionalized Fe4 tetrahedron
With the goal of building a new class of carbohydrate-functionalized systems capable of binding to protein targets, aqueous compatibility and solubility considerations were imperative. Incorporation of water-soluble building blocks has been shown to enable the subcomponent self-assembly of metal-anchored cages that are both soluble and stable in aqueous media.27,40,41 Specifically, the use of organic building blocks bearing polar42 or charged27,43 functional groups combined with the use of water-soluble metal salts and counterions44,45 has permitted the successful synthesis of water-stable cages. Metal-ion complexation has also been shown to stabilize the typically water reactive imine bond against hydrolysis.46With these considerations in mind, we initially selected benzidinedisulfonate as a polar, charged, ditopic amine subcomponent that has been shown to form tetrahedral assemblies in aqueous solution.47–49 Accordingly, treatment of glucose-substituted subcomponent A with commercial benzidinedisulfonic acid and Fe(BF4)2·6H2O in the presence of [NMe4][OH] in water (Scheme 1) resulted in the formation of one major species as assayed by 1H NMR spectroscopy. Notably, these reaction conditions were optimized to include an excess of hydroxide ion, which was used to both deprotonate benzidinedisulfonic acid, and to achieve de-O-acetylation of acetate-protected subcomponent Ain situ. The one-pot self-assembly and ester hydrolysis of A conveniently and cleanly enabled rapid access to the desired deprotected product and simplified the synthetic procedure by eliminating an independent de-O-acetylation step. After precipitation from the crude reaction mixture, the product was easily purified away from the low molecular weight by-products by size-exclusion chromatography.
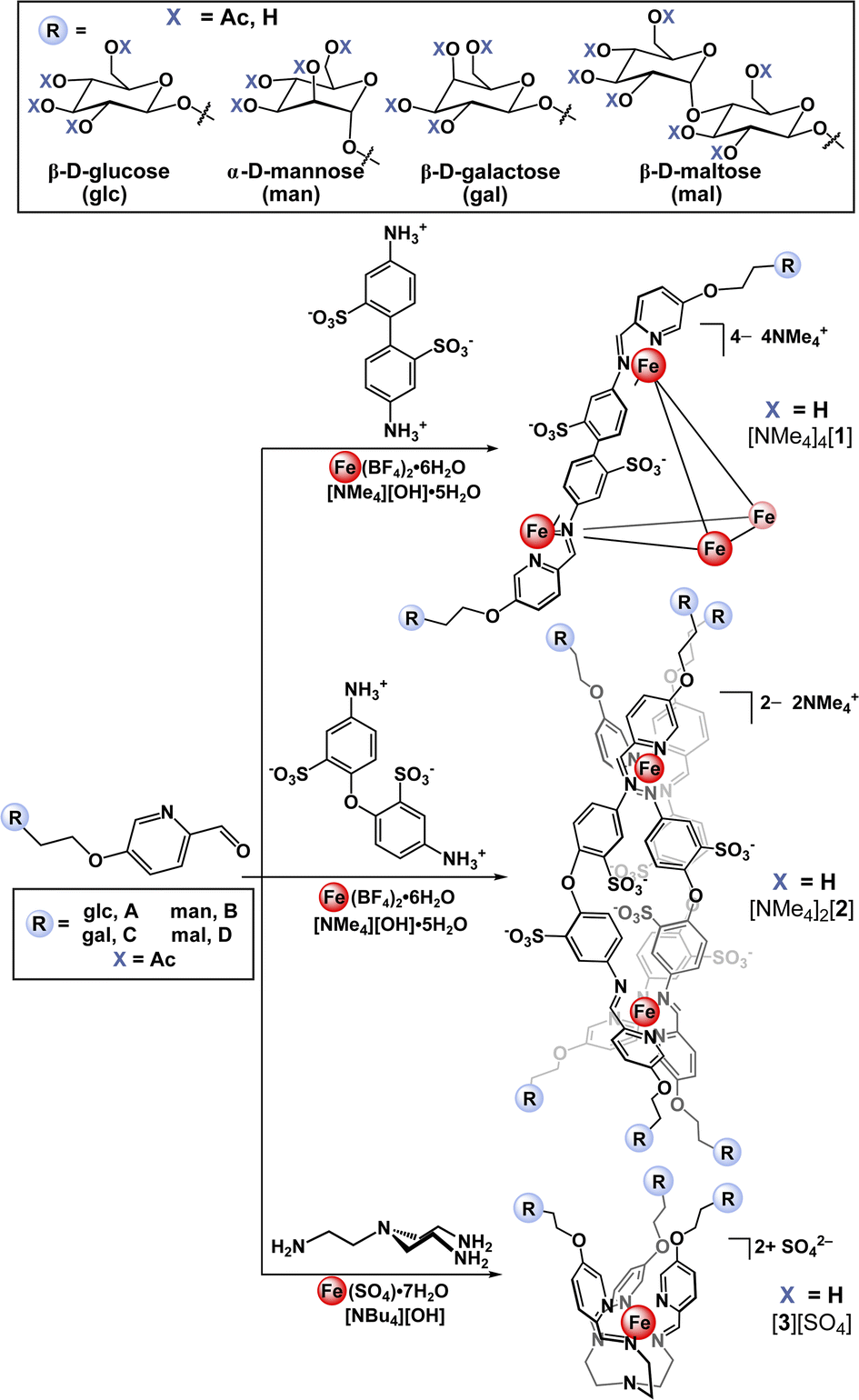 | ||
| Scheme 1 Synthetic scheme for the preparation of tetrahedral ([NMe4]4[1]), helical ([NMe4]2[2]), and mononuclear ([3][SO4]) glycosylated self-assemblies containing β-D-glucose (glc), α-D-mannose (man), β-D-galactose (gal), and β-D-maltose (mal) appendages. | ||
The [NMe4]4[1-glc] product contains Fe(II) in the low-spin, S = 0, state as indicated by resonances exclusively located in the diamagnetic region of the 1H NMR spectrum (Fig. 2a). The intense magenta colour (λ = 505 nm [ε 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1], 545 nm [ε 24000 M−1 cm−1], Fig. 2b) of [NMe4]4[1-glc] is also ascribed to strong metal-to-ligand charge transfer excitations characteristic of low-spin Fe(II) in an iminopyridine coordination environment.47,50,51 Only one set of aromatic signals were observed by 1H and 13C NMR spectroscopy, which is in good agreement with the isolation of a single, highly symmetric species in solution. The 1H NMR aromatic resonances of [NMe4]4[1-glc] are slightly broadened (Δν = 7–18 Hz) when compared with the signals of the A precursor, which is attributed to slow molecular tumbling of the rigid cage core in solution, and typical of large molecular-weight molecules (MW of [1-glc]4– = 6009 g mol−1).18,52 Imine bond formation during self-assembly was supported by the disappearance of the aldehyde 1H NMR signal of subcomponent A (Fig. 2a, δ 9.95 ppm, CDCl3), and the appearance of a new up-field shifted signal attributed to the imine proton located at δ 9.09 ppm in the 1H NMR spectrum of the product (Fig. 2a, D2O). Infrared spectroscopy also confirmed the formation of an imine bond and coordination of the nitrogen donor atom to the Fe centre as evidenced by the absence of an aldehyde vibration in the IR spectrum of [NMe4]4[1-glc], and the appearance of a strong vibration ascribed to the imine CN stretch (νCN 1558 cm−1, ESI Fig. S33†).53,54 Formation of the twelve-fold glucose-substituted assembly was unequivocally confirmed by high-resolution electrospray mass spectrometry (HR-ESI-MS(–)) by the presence of the tetraanionic molecular ion peak (1502.2307 m/z, Fig. 2c), which established both the charge and the intact Fe4L6 metal–ligand stoichiometry of [1-glc]4–.
000 M−1 cm−1], 545 nm [ε 24000 M−1 cm−1], Fig. 2b) of [NMe4]4[1-glc] is also ascribed to strong metal-to-ligand charge transfer excitations characteristic of low-spin Fe(II) in an iminopyridine coordination environment.47,50,51 Only one set of aromatic signals were observed by 1H and 13C NMR spectroscopy, which is in good agreement with the isolation of a single, highly symmetric species in solution. The 1H NMR aromatic resonances of [NMe4]4[1-glc] are slightly broadened (Δν = 7–18 Hz) when compared with the signals of the A precursor, which is attributed to slow molecular tumbling of the rigid cage core in solution, and typical of large molecular-weight molecules (MW of [1-glc]4– = 6009 g mol−1).18,52 Imine bond formation during self-assembly was supported by the disappearance of the aldehyde 1H NMR signal of subcomponent A (Fig. 2a, δ 9.95 ppm, CDCl3), and the appearance of a new up-field shifted signal attributed to the imine proton located at δ 9.09 ppm in the 1H NMR spectrum of the product (Fig. 2a, D2O). Infrared spectroscopy also confirmed the formation of an imine bond and coordination of the nitrogen donor atom to the Fe centre as evidenced by the absence of an aldehyde vibration in the IR spectrum of [NMe4]4[1-glc], and the appearance of a strong vibration ascribed to the imine CN stretch (νCN 1558 cm−1, ESI Fig. S33†).53,54 Formation of the twelve-fold glucose-substituted assembly was unequivocally confirmed by high-resolution electrospray mass spectrometry (HR-ESI-MS(–)) by the presence of the tetraanionic molecular ion peak (1502.2307 m/z, Fig. 2c), which established both the charge and the intact Fe4L6 metal–ligand stoichiometry of [1-glc]4–.
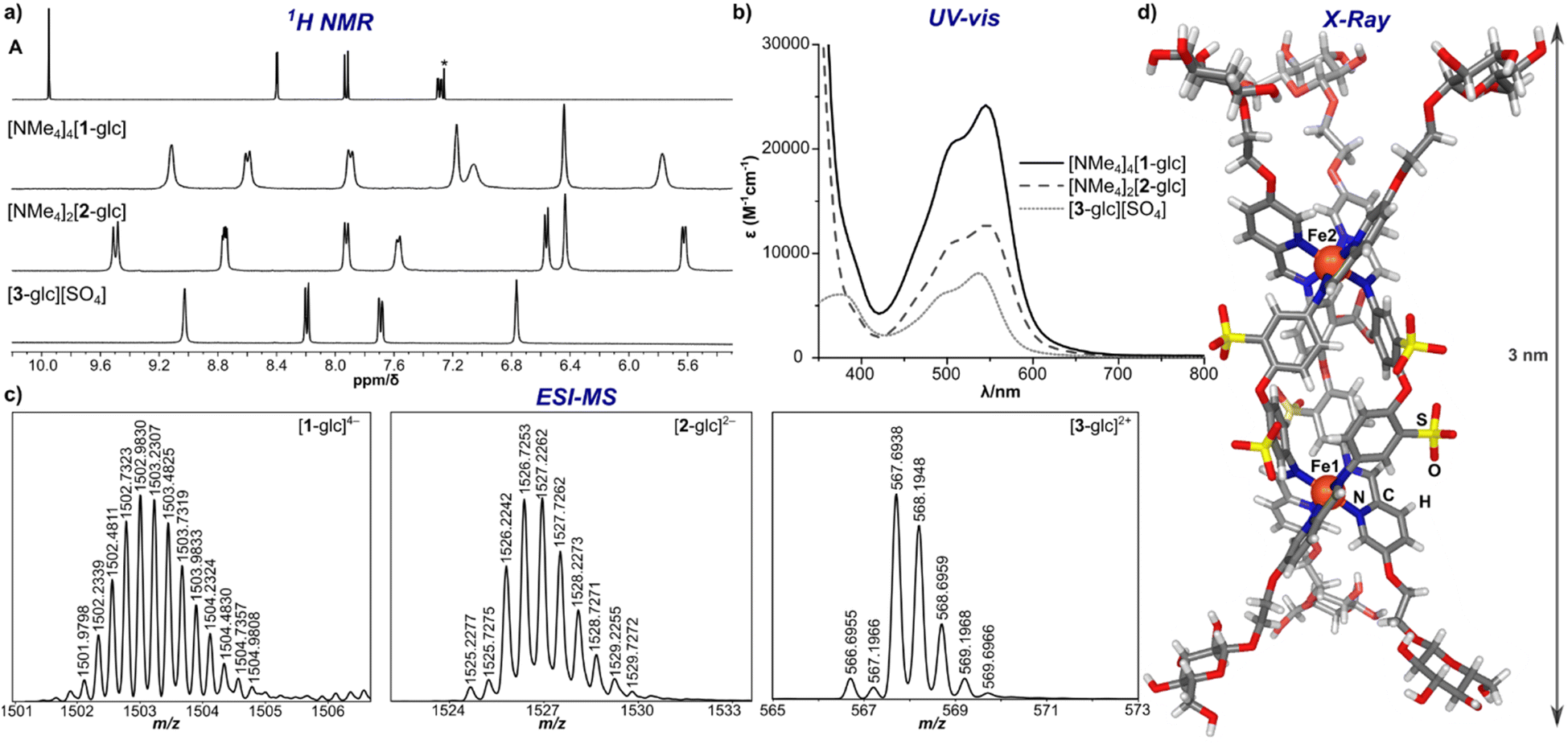 | ||
| Fig. 2 (a) 1H NMR spectra of subcomponent A (CDCl3 (*), 25 °C, 400 MHz), [NMe4]4[1-glc], [NMe4]2[2-glc], and [3-glc][SO4] (D2O, 25 °C, 400 MHz); (b) UV-vis spectra of [NMe4]4[1-glc], [NMe4]2[2-glc], and [3-glc][SO4] (50 μM, H2O); (c) HR-ESI-MS of [1-glc]4−, [2-glc]2−, and [3-glc]2+; (d) solid-state molecular structure of [2-glc]2− (Fe, orange; N, blue; O, red; S, yellow; C, grey; H, white). | ||
With a protocol for the clean synthesis of [NMe4]4[1-glc] in place, we next conducted an analogous self-assembly procedure employing either NaOH or KOH as the hydroxide source to prepare the sodium and potassium salts of [1-glc]4–. The facile preparation of Na4[1-glc] and K4[1-glc] (ESI Section S2.3.2 and S2.3.3†) showcases the versatility of this methodology and the ease of which salts of various countercations can be prepared. Ready access to alkali metal salts of [1-glc]4– also provides an excellent opportunity to study this system and others with higher potential biological compatibility when compared with the [NMe4]+ analogues.
Scope of saccharide-functionalized assemblies
Applying the modularity of this synthetic approach, we next sought out other simple amine building blocks that would offer the ability to easily program the size, shape, and valency of resulting assemblies. Tetrahedral cage [1-glc]4− bears twelve peripheral glucose groups as a consequence of coordination at each of the four Fe(II) centres by three glycosylated iminopyridine fragments. Therefore, the preparation of analogously-substituted mono- and bimetallic Fe(II) complexes would offer structures tethered with three and six glucose groups, respectively, thereby providing rational control over saccharide substitution. We selected 4,4′-diaminodiphenyl ether-2,2′-disulfonic acid and tris(2-aminoethyl)amine (tren) as di- and tritopic amine subcomponents to pair with subcomponent A on account of the reported ability of these building blocks to direct assemblies into di- and mononuclear structures, respectively.55,56Previous reports have demonstrated that the reaction between 2-formylpyridine and 4,4′-diaminodiphenyl ether-based subcomponents in the presence of divalent transition metal ions results in the formation of bimetallic triple-stranded helicates as opposed to tetrahedral assemblies on account of the enhanced flexibility in the ligand backbone.56,57 With this precedent, we employed a protocol analogous to the one described for the synthesis of [NMe4]4[1-glc] to prepare a bimetallic glucose-functionalized helicate. Treatment of 4,4′-diaminodiphenyl ether-2,2′-disulfonic acid with A in the presence of Fe(BF4)2·6H2O and [NMe4][OH]·5H2O resulted in clean conversion to the triple-stranded helix, [NMe4]2[2-glc], as judged by 1H NMR spectroscopic characterization of the crude product. Complex [NMe4]2[2-glc] was purified via size-exclusion chromatography, and the purified product was characterized by 1H (Fig. 2a) and 13C NMR spectroscopy (ESI, Fig. S37†). NMR characterization revealed two sets of resonances that are likely due to the presence of the right-handed, ΔΔ, and left-handed, ΛΛ, configurations (one set of resonances),58 and the presence of the ΛΔ isomer (second set of resonances), which contains two different configurations at the metal centres (Fig. 2a). Indeed, mixtures of ΔΔ, ΛΛ, and ΛΔ configurations are reported for other dinuclear M2L3 helicates in the literature.59,60 The presence of both ΔΔ and ΛΛ configurations in this product was confirmed by an X-ray diffraction study conducted on a single crystal of the salt (vide infra and ESI Section S7†). Complex [NMe4]2[2-glc] was also characterized by UV-vis (Fig. 2b) and infrared (ESI Fig. S41†) spectroscopy, with spectroscopic signatures consistent with those observed for the tetrametallic [NMe4]4[1-glc] analogue. The identity of [2-glc]2– was confirmed as the sixfold-glucose substituted bimetallic complex by HR-ESI-MS(−) (Fig. 2c).
To establish the structures of this new class of glycosylated molecules, we focused efforts on growing single crystals of the isolated assemblies for X-ray diffraction analysis. Crystals of [NMe4]2[2-glc] of sufficient quality for structural determination were grown by vapor diffusion of acetone into a saturated aqueous solution of the complex at 25 °C over the course of 48 h. An X-ray diffraction study of these crystals permitted structural determination of the [2-glc]2− anion, and the solid-state structure is displayed in Fig. 2d. As shown in the molecular structure, the framework of [2-glc]2− spans a 3 nm distance from end-to-end, in which each end of the helix is capped by three glucose fragments. The solid-state structure also displays all six sulfonate groups directed towards the exterior of the helicate, which enforces the C3 symmetry of the molecular core that is reflected in the high symmetry observed by NMR spectroscopy. Such crystallographic characterization is critical to this work by enabling a more complete understanding of the molecular structure of this class of assemblies.
We next expanded the series of glycosylated assemblies to include a monometallic Fe(II) complex that would enforce a trivalent saccharide framework. Treatment of tris(2-aminoethyl)amine with subcomponent A in the presence of stoichiometric amounts of [NBu4][OH] and FeSO4 in H2O at 70 °C resulted in the formation of mononuclear, [3-glc][SO4] (Scheme 1), as evidenced by 1H NMR spectroscopic analysis of the product. This self-assembly reaction is straightforward and proceeds cleanly to generate C3-symmetric [3-glc]2+ as supported by the high symmetry observed in the 1H and 13C NMR spectra of the product. Monometallic [3-glc][SO4] was also characterized by UV-vis and infrared spectroscopy, and the identity of this product as the triply-substituted glucose complex was confirmed by HR-ESI-MS(+) (Fig. 2c).
With a successful self-assembly procedure established, we extended this synthetic approach to the preparation of analogous glycosylated complexes employing subcomponents B (α-D-mannose), C (β-D-galactose), and D (β-D-maltose) as shown in Scheme 1. The mannose-, galactose- and maltose-substituted tetra-, bi-, and monometallic analogues were prepared following similar synthetic procedures described for the glucose-derivatives. This versatile synthetic strategy allowed us to easily and rapidly establish a molecular library of well-defined saccharide-appended assemblies that includes twelve unique examples. All complexes were characterized by NMR spectroscopy (1H and 13C), IR and UV-vis spectroscopy, and the identities of all products were confirmed by HR-ESI-MS (ESI Section 2.3†).
Stability studies
Employing [NMe4]4[1-glc] as a model complex, the stability of the present glycosylated assemblies in biologically-relevant media was evaluated. Complex [NMe4]4[1-glc] was exposed to fetal bovine serum cell-culture media, solutions of various buffers and pH environments (4–10), as well as the common biological nucleophile, glutathione,61 for extended periods of time (72 h) with minimal observable signs of degradation as assayed by UV-vis spectroscopy (ESI Section S4.2†). The excellent stability profile of [NMe4]4[1-glc] under these conditions is a direct consequence of the robust framework of the tetrahedral cage and the metal–ligand bonding strength. The high water solubility of [NMe4]4[1-glc] (302 g L−1, ESI Section S3†) also plays a critical role in the overall biological compatibility of the complex and its ability to remain intact in solution.46 The [NMe4]4[1-glc] complex also has an excellent shelf-life and can be stored on the benchtop as a solid under open-atmosphere conditions and in aqueous solution for extended periods of time. No decomposition of [NMe4]4[1-glc] in D2O (5 mM, 25 °C) was detected even after nine months as monitored by 1H NMR spectroscopic analysis (ESI Section S4.1†).Binding studies
Given the high compatibility of [NMe4]4[1-glc] with biological conditions, we were further motivated to probe the use of the present assemblies as agents to interrogate multivalent binding interactions with protein targets. The ability of the current saccharide-appended systems to bind lectins was assessed employing model protein, Concanavalin A (Con A), which has been widely used to study physiologically relevant protein–carbohydrate interactions.62,63 Con A is derived from jack bean (Canavalia ensiformis) seeds,64 and is an aggregate composed of 26 kDa monomeric units that exists as a dimer in the 5.0–5.6 pH range, while the tetramer is the predominant form at physiological pH.65 Each monomeric unit contains one saccharide-binding site that selectively recognizes α-D-mannoside and α-D-glucoside resides.3,66Con A and multivalent binders have been shown to form noncovalent, cross-linked aggregates that can precipitate out of solution depending on the strength of the binding interaction.15,18,66,67 Since Con A binds both terminal mannose and maltose groups, we evaluated the binding between the series of mannose and maltose-tethered systems with Con A through assessing the lectin–ligand aggregation by turbidimetric analysis. When dodeca- ([NMe4]4[1-man], [NMe4]4[1-mal]) and hexa-substituted ([NMe4]2[2-man], [NMe4]2[2-mal]) complexes were mixed with Con A at pH 7.0, the solutions became opaque within seconds of mixing, indicating the formation of colloidal lectin–ligand aggregates and direct evidence of protein binding. The turbidity of the mixtures was monitored by measuring the increase in absorbance at λ 545 nm over the course of 180 min. In the case of dodeca-, and hexa-substituted complexes, the absorbance reached a plateau after ca. 20 min, and remained constant until the end of the measurement (Fig. 3), suggesting these assemblies were able to cross-link and precipitate nearly all of the Con A in solution. In contrast, negligible changes in absorption were observed upon addition of trivalent [3-man][SO4] and [3-mal][SO4] to solutions of Con A. The lack of aggregate formation for the trivalent derivatives is attributed to lower binding affinity of [3-man]2+ and [3-mal]2+ with Con A, and indicative that tri-substituted complexes do not have suitable valency or peripheral substitution to crosslink lectin binding sites. This result is consistent with the report from Toone et al.,65 in which agglutination assays demonstrated no multivalency effect for bi- and trivalent dendrimers with Con A, whereas glycomolecules of higher valency showed a considerably enhanced cluster glycoside effect.3,68
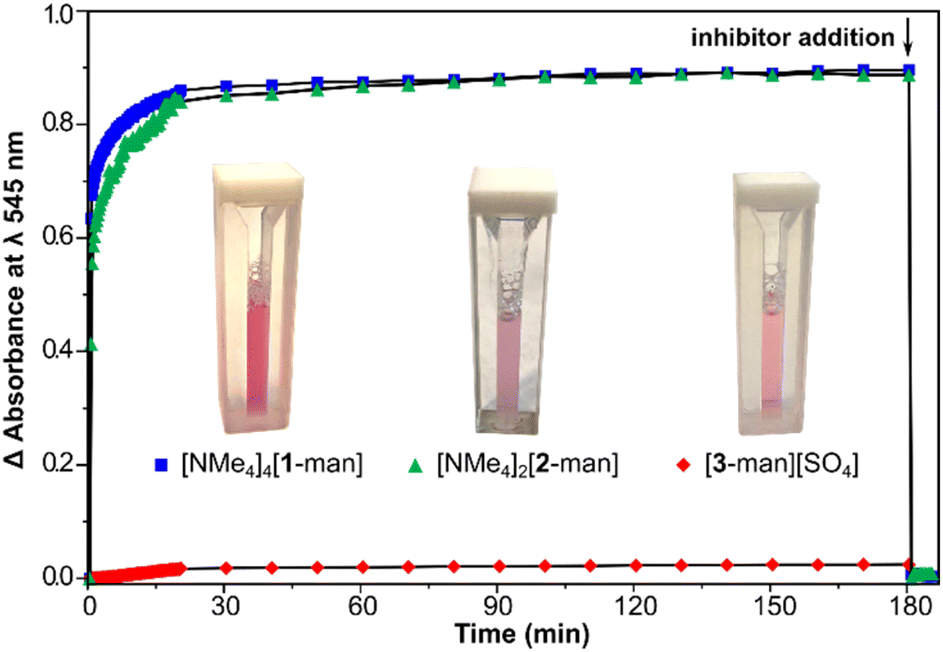 | ||
Fig. 3 Turbidity changes over the course of 200 min as monitored by the increase in absorbance at λ 545 nm upon mixing [NMe4]4[1-man] ( ), [NMe4]2[2-man] ( ), [NMe4]2[2-man] ( ), and [3-man][SO4] ( ), and [3-man][SO4] ( ) with Con A (pH 7.0, 10 mM HEPES buffer, 1 mM MnCl2, 1 mM CaCl2, 100 mM NaCl). The arrow indicates the addition of competitive inhibitor, methyl-α-D-mannose. Images of the mixtures are shown after 180 min immediately before inhibitor addition. ) with Con A (pH 7.0, 10 mM HEPES buffer, 1 mM MnCl2, 1 mM CaCl2, 100 mM NaCl). The arrow indicates the addition of competitive inhibitor, methyl-α-D-mannose. Images of the mixtures are shown after 180 min immediately before inhibitor addition. | ||
After 180 min, an excess (1000 equiv.) of methyl-α-D-mannose was added to each mixture (shown in Fig. 3 with arrow), which resulted in immediate deaggregation (deagglutination) of the protein clusters. This result indicates that methyl-α-D-mannose can serve as a competitive inhibitor and that the multivalent crosslinking is reversible. Aggregate formation was not observed in the case of experiments conducted with complexes bearing β-glucose and β-galactose groups, which is consistent with the lack of binding between these assemblies and Con A (vide infra), and the known selectivity of the protein for α-mannose and α-glucose.3
Thermodynamic parameters associated with the binding of maltose and mannose-grafted complexes to Con A were next determined via isothermal titration calorimetry (ITC). It is well-known that the precipitation of cross-linked aggregates during ITC experiments has a significant negative effect on the reliability of thermodynamic data.64,69 Therefore, in order to minimize the potential of precipitation during measurements, all experiments in this study were performed at low protein concentration, low salt concentrations (NaCl, CaCl2, MnCl2), and in acidic conditions (pH 4.8) with Con A in its predominantly dimeric form.64
Calorimetric data for the titration of dimeric Con A with [NMe4]4[1-man] are shown in Fig. 4a, which display a monotonic decrease in the exothermic heat of binding with each addition of analyte. The titration was conducted a total of three times, and consistent results were obtained throughout the measurements (ESI Section S6.1†). The dissociation constant, Kd, for the interaction between [NMe4]4[1-man] and Con A was determined to be 2.4 μM, which corresponds to a 350-fold increase in affinity when compared with the Kd value determined for the monovalent binding epitope, D-mannose (Kd = 0.83 × 10−3 M, ESI Section S6.7†).70 The significant difference in Kd values reveals that the multivalent presentation of mannose groups on [1-man]4– results in a considerably stronger binding profile per saccharide equivalent. These findings are consistent with data reported for other systems that exhibit affinity enhancements of up to three to four orders of magnitude relative to the binding of monovalent D-mannose due to multivalency effects.15,71–74
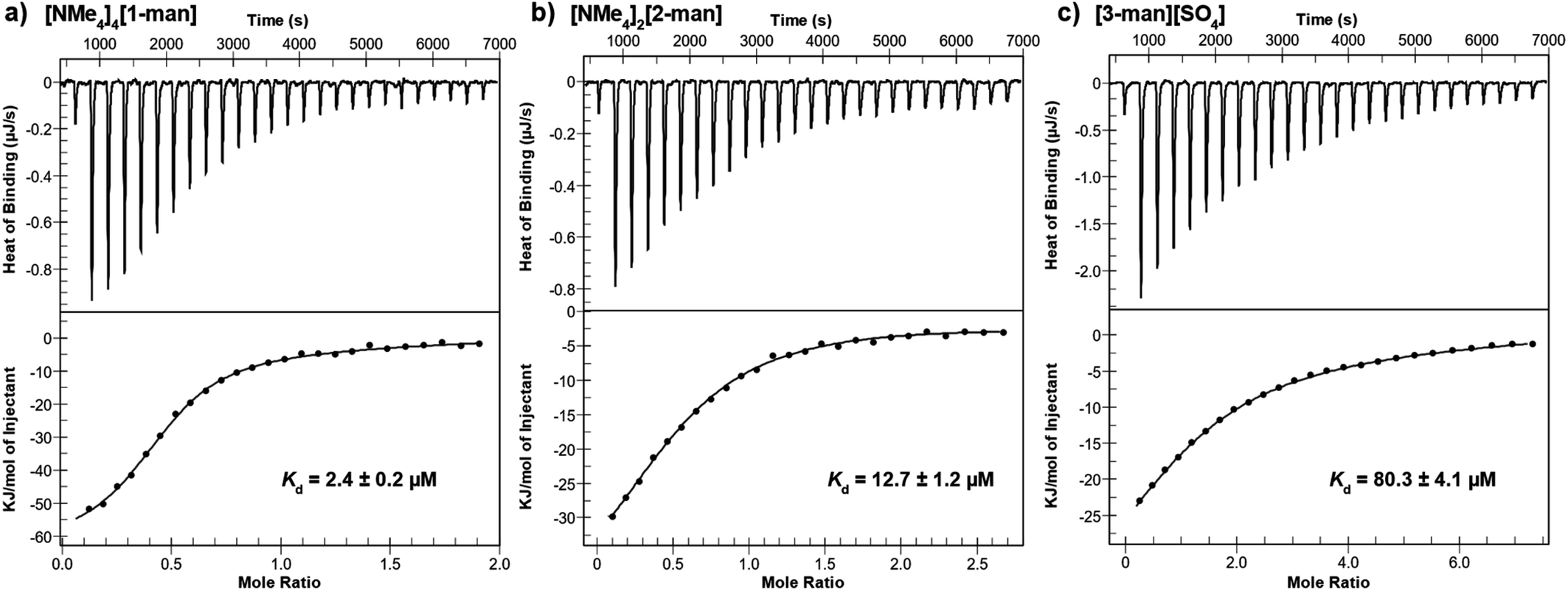 | ||
| Fig. 4 Isothermal titration calorimetry (ITC) thermograms (top) and fitted binding isotherms (bottom) for the binding of (a) [NMe4]4[1-man], (b) [NMe4]2[2-man], and (c) [3-man][SO4] to dimeric Con A and the Kd values determined from each measurement. | ||
We next investigated the impact of saccharide density on binding strength by determining the dissociation constants for the hexavalent, [2-man]2–, and trivalent, [3-man]2+, analogues. A Kd value of 12.7 μM was determined for the binding of [2-man]2– with Con A (Fig. 4b) under similar experimental conditions used for measurements with [1-man]4–. These data display that dodecavalent [1-man]4– exhibits an over five-fold enhancement in binding to Con A when compared with the affinity of [2-man]2–, which bears half the number of mannose units. We next evaluated the binding of the trivalent derivative, [3-man]2+, with Con A. Measurements indicate an over 30-fold decrease in binding strength of [3-man]2+ (Kd = 80.3 μM, Fig. 4c) when compared with the binding of [1-man]4–. The Kd values determined from ITC experiments demonstrate that the multivalent binding event strength increases exponentially as the valency associated with the molecular architecture increases, which is a result consistent with the cluster glycoside effect.3,5,68 These experiments establish the markedly enhanced binding avidity, which is the overall cooperative binding force,75 that results from the multivalent presentation of saccharides, and highlight the relationship between molecular architecture and affinity. The observed trend in Kd values of [1-man]4– < [2-man]2– < [3-man]2+ is governed by rational, systematic, and programmable molecular design and provides a valuable opportunity to expand these concepts to higher multivalent binding events through the development of even more effective constructs.
We note that the overall charge of each complex in this series varies from 4− to 2+, and that there is the potential for charge to play a role in the binding strength of each system with Con A. A benefit of the current synthetic approach is that the interchangeable nature of organic subcomponents provides an excellent opportunity to systematically modify only the charge of a molecule without significantly affecting the size, shape, or valency by using neutral ditopic amines in the case of 1, and 2, and a charged tritopic amine for 3. The influence of charge on binding strength is a question that is currently under investigation using the presented synthetic approach.
The ITC measurements also allowed us to calculate the stoichiometry of binding, n, which is defined as the number of analyte molecules per protein binding site. For example, the n value for the binding of methyl-α-D-mannose and trimannose to Con A are both 1.0,64 indicating one mannose epitope binds to a single Con A binding site.76 However, n values less than 1.0 are typical for the binding of multivalent carbohydrates to Con A.63,72 ITC-derived values of n that are less than 1.0 suggest crosslinking between multivalent proteins and multivalent analytes in solution. In agreement with this known behaviour, the n values for [1-man]4–, [2-man]2–, and [3-man]2+ binding to each Con A monomer were determined to be 0.22, 0.30, and 0.73, respectively, which are all considerably lower than 1.0.
It is important to note that the functional valence, which is defined as the exact number of epitopes participating in binding (N = 1/n), is lower than the structural valence, or the actual number of epitopes available for binding for the present systems.69 Although the structural valence of [1-man]4– is twelve, its functional valence is four, which indicates that one-third of the available mannose units of [1-man]4– participate in binding to each Con A monomer, and that [1-man]4– is involved in tetravalent cross-linking interactions with the lectin. A functional valence of four agrees well with the structure of [1-man]4– and suggests the three mannose groups at each of the four Fe-vertices of the tetrahedron engage in monovalent binding to Con A. This result is expected given the distance between each monomeric binding site of a Con A is ca. 6.5 nm,12 which is significantly greater than the distance between the three mannose units at each Fe vertex of [1-man]4– (ca. 2–3 nm). In comparison, the functional valence of [2-man]2– and [3-man]2+ are lower than that of [1-man]4–, which is in line with the lower structural valence and weaker binding of these complexes.63,64,72,77,78
The functional valence values for [1-man]4–, [2-man]2–, and [3-man]2+ of 4.55, 3.33, and 1.37, respectively, are slightly higher than the expected values of 4, 2, and 1, which suggests that each mannose epitope of these systems can interact with more than one Con A binding site at a given time by crosslinking proteins. In fact, it has been reported that short mannoside chains can crosslink two Con A binding sites such that the proteins can approach without making substantial contact.65,79
We further probed the binding capabilities of the current system by determining the Kd values of the maltose-decorated complexes ([1-mal]4–, [2-mal]2–, [3-mal]2+) with Con A by ITC.80 Direct comparison between the dissociation constants determined for the mannose and maltose derivatives indicate the maltose systems bind Con A with approximately three times lower affinity (ESI Section S6.2† and Fig. 5). These results compare well with the known selectivity exhibited by Con A, which binds D-mannose six times stronger than it binds D-maltose (Kd(mal)/Kd(man) 6:1).81,82
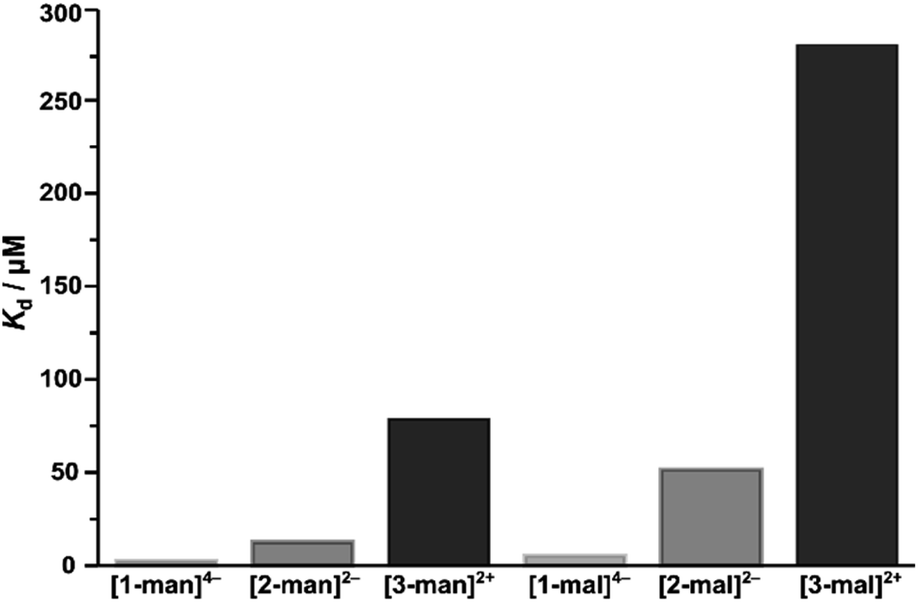 | ||
| Fig. 5 Dissociation constants (Kd) for the binding of mannose- and maltose-functionalized complexes with Con A, displaying the trend in binding strength associated with the structural forms of [1]4–, [2]2– and [3]2+. | ||
The extent of nonspecific binding was probed by investigating the interaction between the β-glucose ([1-glc]4–) and β-galactose ([1-gal]4–) substituted Fe4 tetrahedra with Con A. Even at high concentration, [1-glc]4– and [1-gal]4– fail to engage in binding with Con A as evidenced by negligible heat release upon titration. Similarly, minimal heat release was observed when a solution of the unfunctionalized parent Fe4 cage, [NMe4]4[Fe4L6] (L = bis[2-sulfonato-4(2-pyridylmethyleneamino)benzene]),27,47 was analysed at high injection concentration as a negative control. These findings highlight the high affinity and selectivity of the current system.
Conclusions
With this work, we have introduced a systematic and facile method for the preparation of a series of twelve glycosylated Fe(II)-anchored systems by coordination-driven self-assembly. The mannose- and maltose-decorated complexes were evaluated as multivalent binders to Con A, and the results obtained verify that the multivalent saccharide interactions are proportional to the saccharide density, which in the case of the present work, are governed by rational and systematic molecular design. The ease of synthesis, versatility, and modular nature of this methodology provides a new element of control and predictability to the synthesis of molecular multivalent constructs,8,18,20,21 and introduces a versatile class of supramolecular complexes primed for use in biological applications. Importantly, this approach of building hierarchical architectures provides a platform to investigate and better understand the principles governing structure–activity relationships critical to multivalent biological recognition events. Ultimately, this work serves as a valuable demonstration that supramolecular coordination complexes can be employed for biologically-relevant applications,18,19,26,83–85 and provides a framework for designing well-defined molecules that can potentially be programmed to compliment complex protein binding sites. Current work is focused on expanding the library of hierarchical systems and leveraging the host-guest behavior86–88 of coordination-cage cavities to explore this class of multivalent molecules in targeted drug delivery applications.Data availability
All experimental data are available in the ESI† of this article.Author contributions
J. M. S. devised the study and designed and performed all synthetic work and characterized all compounds. J. H. S. conducted ITC experiments, and J. B. B. and M. G. contributed crystallographic characterization. J. M. S. wrote the manuscript and all authors have given final approval.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Department of Chemistry and Biochemistry at UCSD. We thank Professor Jerry Yang at UCSD for generously allowing us to use his ITC instrument and laboratory space, and Aashish Shivkumar is thanked for assistance with ITC experiments. Professor Fleur Ferguson (UCSD) is thanked for helpful discussions and Professor Arnold Rheingold (UCSD) is thanked for assistance with crystallographic characterization.Notes and references
- Z. Zhang and E. Fan, Methods Enzymol., 2003, 362, 209–218 CAS.
- Y. Watanabe, J. D. Allen, D. Wrapp, J. S. McLellan and M. Crispin, Science, 2020, 369, 330–333 CrossRef CAS.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef PubMed.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Angew. Chem., Int. Ed., 2006, 45, 2348–2368 CrossRef PubMed.
- C. Müller, G. Despras and T. K. Lindhorst, Chem. Soc. Rev., 2016, 45, 3275–3302 RSC.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2454–2794 CrossRef.
- A. Bernardi, J. Jiménez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K. E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penadés, F. Peri, R. J. Pieters, O. Renaudet, J. L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schäffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Curr. Opin. Chem. Biol., 2000, 4, 696–703 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2012, 134, 4112–4115 CrossRef CAS PubMed.
- S. André, O. Renaudet, I. Bossu, P. Dumy and H. J. Gabius, J. Pept. Sci., 2011, 17, 427–437 CrossRef PubMed.
- M. L. Wolfenden and M. J. Cloninger, J. Am. Chem. Soc., 2005, 127, 12168–12169 CrossRef.
- V. Percec, P. Leowanawat, H. J. Sun, O. Kulikov, C. D. Nusbaum, T. M. Tran, A. Bertin, D. A. Wilson, M. Peterca, S. Zhang, N. P. Kamat, K. Vargo, D. Moock, E. D. Johnston, D. A. Hammer, D. J. Pochan, Y. Chen, Y. M. Chabre, T. C. Shiao, M. Bergeron-Brlek, S. André, R. Roy, H. J. Gabius and P. A. Heiney, J. Am. Chem. Soc., 2013, 135, 9055–9077 CrossRef.
- M. D. Disney, J. Zheng, T. M. Swager and P. H. Seeberger, J. Am. Chem. Soc., 2004, 126, 13343–13346 CrossRef PubMed.
- C. W. Cairo, J. E. Gestwicki, M. Kanai and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 1615–1619 CrossRef PubMed.
- K. Godula and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 9963–9965 CrossRef PubMed.
- M. Marradi, F. Chiodo, I. García and S. Penadés, Chem. Soc. Rev., 2013, 42, 4728–4745 RSC.
- N. Kamiya, M. Tominaga, S. Sato and M. Fujita, J. Am. Chem. Soc., 2007, 129, 3816–3817 CrossRef PubMed.
- F. Zhou, S. Li, T. R. Cook, Z. He and P. J. Stang, Organometallics, 2014, 33, 7019–7022 CrossRef.
- F. J. Feher, K. D. Wyndham and D. J. Knauer, Chem. Commun., 1998, 2393–2394 RSC.
- E. A. Qian, A. I. Wixtrom, J. C. Axtell, A. Saebi, D. Jung, P. Rehak, Y. Han, E. H. Moully, D. Mosallaei, S. Chow, M. S. Messina, J. Y. Wang, A. T. Royappa, A. L. Rheingold, H. D. Maynard, P. Král and A. M. Spokoyny, Nat. Chem., 2017, 9, 333–340 CrossRef PubMed.
- H. Vardhan, M. Yusubov and F. Verpoort, Coord. Chem. Rev., 2016, 306, 171–194 CrossRef.
- D. H. Leung, D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2004, 43, 963–966 CrossRef PubMed.
- J. Zhao, Z. Zhou, G. Li, P. J. Stang and X. Yan, Natl. Sci. Rev., 2021, 8, nwab045 CrossRef PubMed.
- C. He, D. Liu and W. Lin, Chem. Rev., 2015, 115, 11079–11108 CrossRef PubMed.
- A. Casini, B. Woods and M. Wenzel, Inorg. Chem., 2017, 56, 14715–14729 CrossRef.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1700 CrossRef PubMed.
- J. Roukala, J. Zhu, C. Giri, K. Rissanen, P. Lantto and V. V. Telkki, J. Am. Chem. Soc., 2015, 137, 2464–2467 CrossRef PubMed.
- D. Fujita, K. Suzuki, S. Sato, M. Yagi-Utsumi, Y. Yamaguchi, N. Mizuno, T. Kumasaka, M. Takata, M. Noda, S. Uchiyama, K. Kato and M. Fujita, Nat. Commun., 2012, 3, 1093–1097 CrossRef.
- M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchlt and K. Ogura, Nature, 1995, 378, 469–471 CrossRef.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef PubMed.
- P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502–518 CrossRef.
- D. L. Caulder and K. N. Raymond, J. Chem. Soc., Dalton Trans., 1999, 1185–1200 RSC.
- B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef.
- S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052–3064 CrossRef.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef.
- M. He and J. M. Lehn, Chem.–Eur. J., 2021, 27, 7516–7524 CrossRef PubMed.
- C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC.
- T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476–2490 RSC.
- T. Jiao, G. Wu, Y. Zhang, L. Shen, Y. Lei, C. Wang, A. C. Fahrenbach and H. Li, Angew. Chem., Int. Ed., 2020, 59, 18350–18367 CrossRef PubMed.
- E. G. Percástegui, T. K. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480–13544 CrossRef PubMed.
- M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Chem. Sci., 2013, 4, 2744–2751 RSC.
- D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez and J. R. Nitschke, Angew. Chem., Int. Ed., 2018, 57, 3717–3721 CrossRef CAS PubMed.
- B. Roy, E. Zangrando and P. S. Mukherjee, Chem. Commun., 2016, 52, 4489–4492 RSC.
- M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645–5647 CrossRef CAS.
- E. G. Percástegui, J. Mosquera, T. K. Ronson, A. J. Plajer, M. Kieffer and J. R. Nitschke, Chem. Sci., 2019, 10, 2006–2018 RSC.
- P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297–8301 CrossRef CAS PubMed.
- J. A. Foster, R. M. Parker, A. M. Belenguer, N. Kishi, S. Sutton, C. Abell and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 9722–9729 CrossRef CAS PubMed.
- S. Xi, L. Bao, Z. Xu, Y. Wang, Z. Ding and Z. Gu, Eur. J. Inorg. Chem., 2017, 3533–3541 CrossRef CAS.
- T. F. Miller, L. R. Holloway, P. P. Nye, Y. Lyon, G. J. O. Beran, W. H. Harman, R. R. Julian and R. J. Hooley, Inorg. Chem., 2018, 57, 13386–13396 CrossRef CAS.
- A. M. McDaniel, A. K. Rappé and M. P. Shores, Inorg. Chem., 2012, 51, 12493–12502 CrossRef CAS.
- M. P. Foster, C. A. McElroy and C. D. Amero, Biochemistry, 2007, 46, 331–340 CrossRef CAS PubMed.
- T. J. Ozumerzifon, R. F. Higgins, J. P. Joyce, J. L. Kolanowski, A. K. Rappé and M. P. Shores, Inorg. Chem., 2019, 58, 7785–7793 CrossRef PubMed.
- P. Shejwalkar, N. P. Rath and E. B. Bauer, Synth, 2014, 46, 57–66 Search PubMed.
- C. Giri, P. K. Sahoo, R. Puttreddy, K. Rissanen and P. Mal, Chem.–Eur. J., 2015, 21, 6390–6393 CrossRef CAS PubMed.
- P. Mal and J. R. Nitschke, Chem. Commun., 2010, 46, 2417–2419 RSC.
- H. Cheng, D. Chun-ying, F. Chen-jie and M. Qing-jin, J. Chem. Soc., Dalton Trans., 2000, 2419–2424 RSC.
- L. Fabbrizzi, J. Org. Chem., 2020, 85, 12212–12226 CrossRef PubMed.
- F. Cui, S. Li, C. Jia, J. S. Mathieson, L. Cronin, X. J. Yang and B. Wu, Inorg. Chem., 2012, 51, 179–187 CrossRef.
- D. Yang, L. K. S. von Krbek, L. Yu, T. K. Ronson, J. D. Thoburn, J. P. Carpenter, J. L. Greenfield, D. J. Howe, B. Wu and J. R. Nitschke, Angew. Chem., Int. Ed., 2021, 60, 4485–4490 CrossRef PubMed.
- H. J. Forman, H. Zhang and A. Rinna, Mol. Aspect. Med., 2009, 30, 1–12 CrossRef PubMed.
- Z. Derewenda, J. Yariv, J. R. Helliwell, A. J. Kalb, E. J. Dodson, M. Z. Papiz, T. Wan and J. Campbell, EMBO J., 1989, 8, 2189–2193 CrossRef PubMed.
- S. L. Mangold and M. J. Cloninger, Org. Biomol. Chem., 2006, 4, 2458 RSC.
- T. K. Dam, R. Roy, S. K. Das, S. Oscarson and C. F. Brewer, J. Biol. Chem., 2000, 275, 14223–14230 CrossRef CAS PubMed.
- S. M. Dimick, S. C. Powell, S. A. McMahon, D. N. Moothoo, J. H. Naismith and E. J. Toone, J. Am. Chem. Soc., 1999, 121, 10286–10296 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS PubMed.
- S. Ordanini, W. Celentano, A. Bernardi and F. Cellesi, Beilstein J. Nanotechnol., 2019, 10, 2192–2206 CrossRef CAS PubMed.
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321–327 CrossRef CAS.
- T. K. Dam, M. L. Talaga, N. Fan and C. F. Brewer, in Methods in Enzymology, Academic Press Inc., 2016, vol. 567, pp. 71–95 Search PubMed.
- Q. Sha, J. Fei, C. Tu, B. F. Liu, Z. Hu and X. Liu, Microchim. Acta, 2022, 189, 1–11 CrossRef PubMed.
- E. M. Munoz, J. Correa, R. Riguera and E. Fernandez-Megia, J. Am. Chem. Soc., 2013, 135, 5966–5969 CrossRef CAS PubMed.
- R. S. Loka, M. S. McConnell and H. M. Nguyen, Biomacromolecules, 2015, 16, 4013–4021 CrossRef CAS PubMed.
- T. K. Dam, R. Roy, D. Pagé and C. F. Brewer, Biochemistry, 2002, 41, 1359–1363 CrossRef CAS PubMed.
- B. M. Illescas, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2017, 139, 6018–6025 CrossRef CAS PubMed.
- P. I. Kitov and D. R. Bundle, J. Am. Chem. Soc., 2003, 125, 16271–16284 CrossRef CAS PubMed.
- J. H. Naismith, C. Emmerich, J. Habash, S. J. Harrop, J. R. Helliwell, W. N. Hunter, J. Raftery, A. J. Kalb (Gilboa) and J. Yariv, Acta Crystallogr. Sect. D Biol. Crystallogr., 1994, 50, 847–858 CrossRef CAS PubMed.
- T. K. Dam, R. Roy, D. Pagé and C. F. Brewer, Biochemistry, 2002, 41, 1351–1358 CrossRef CAS PubMed.
- M. Nagao, M. Kichize, Y. Hoshino and Y. Miura, Biomacromolecules, 2021, 22, 3119–3127 CrossRef CAS PubMed.
- M. François-Heude, A. Méndez- Ardoy, V. Cendret, P. Lafite, R. Daniellou, C. O. Mellet, J. M. García Fernández, V. Moreau and F. Djedaïni-Pilard, Chem.–Eur. J., 2015, 21, 1978–1991 CrossRef PubMed.
- S. S. Banerjee and D. H. Chen, Chem. Mater., 2007, 19, 3667–3672 CrossRef CAS.
- F. P. Schwarz, K. D. Puri, R. G. Bhat and A. Surolia, J. Biol. Chem., 1993, 268, 7668–7677 CrossRef CAS PubMed.
- D. K. Mandal, N. Kishore and C. F. Brewer, Biochemistry, 1994, 33, 1149–1156 CrossRef CAS PubMed.
- S. Datta, M. L. Saha, N. Lahiri, G. Yu, J. Louie and P. J. Stang, Org. Lett., 2018, 20, 7020–7023 CrossRef CAS PubMed.
- J. Zhu, C. J. E. Haynes, M. Kieffer, J. L. Greenfield, R. D. Greenhalgh, J. R. Nitschke and U. F. Keyser, J. Am. Chem. Soc., 2019, 141, 11358–11362 CrossRef CAS.
- J. Han, A. F. B. Räder, F. Reichart, B. Aikman, M. N. Wenzel, B. Woods, M. Weinmüller, B. S. Ludwig, S. Stürup, G. M. M. Groothuis, H. P. Permentier, R. Bischoff, H. Kessler, P. Horvatovich and A. Casini, Bioconjugate Chem., 2018, 29, 3856–3865 CrossRef CAS PubMed.
- S. Mecozzi and J. Rebek, Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef.
- O. Yanshyna, M. J. Białek, O. V. Chashchikhin and R. Klajn, Commun. Chem., 2022, 5, 1–12 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2190699. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05689e |
| This journal is © The Royal Society of Chemistry 2023 |