
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymeric encapsulation of a ruthenium(II) polypyridyl complex: from synthesis to in vivo studies against high-grade epithelial ovarian cancer†
João P. M.
António
 ab,
Albert
Gandioso
a,
Fariba
Nemati
c,
Nancy
Soliman
ab,
Robin
Vinck
a,
Fan
Sun
d,
Carine
Robert
b,
Pierre
Burckel
e,
Didier
Decaudin
cf,
Christophe M.
Thomas
*b and
Gilles
Gasser
*a
ab,
Albert
Gandioso
a,
Fariba
Nemati
c,
Nancy
Soliman
ab,
Robin
Vinck
a,
Fan
Sun
d,
Carine
Robert
b,
Pierre
Burckel
e,
Didier
Decaudin
cf,
Christophe M.
Thomas
*b and
Gilles
Gasser
*a
aChimie ParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, Laboratory for Inorganic Chemical Biology, 75005 Paris, France. E-mail: gilles.gasser@chimieparistech.psl.eu; Web: https://www.gassergroup.com
bChimie ParisTech, PSL University, CNRS, Institut de Recherche de Chimie Paris, 75005 Paris, France. E-mail: christophe.thomas@chimieparistech.psl.eu; Web: https://www.ircp.cnrs.fr/la-recherche/equipe-cocp/
cTranslational Research Department, Laboratory of Preclinical Investigation, PSL University, Institut Curie, 26 rue d'Ulm, Paris 75248, France
dChimie ParisTech, PSL University, CNRS, Institut de Recherche de Chimie Paris, 75005 Paris, France
eUniversité de Paris, Institut de physique du globe de Paris, CNRS, F-75005 Paris, France
fDepartment of Medical Oncology, Institut Curie, 26 rue d'Ulm, Paris 75248, France
First published on 6th December 2022
Abstract
The in vitro to in vivo translation of metal-based cytotoxic drugs has proven to be a significant hurdle in their establishment as effective anti-cancer alternatives. Various nano-delivery systems, such as polymeric nanoparticles, have been explored to address the pharmacokinetic limitations associated with the use of these complexes. However, these systems often suffer from poor stability or involve complex synthetic procedures. To circumvent these problems, we report here a simple, one-pot procedure for the preparation of covalently-attached Ru–polylactide nanoparticles. This methodology relies on the ring-opening polymerization of lactide initiated by a calcium alkoxide derivative formed from calcium bis(trimethylsilyl amide) and a hydroxyl-bearing ruthenium complex. This procedure proceeds with high efficiency (near-quantitative incorporation of Ru in the polymer) and enables the preparation of polymers with varying molecular weights (2000–11000 Da) and high drug loadings (up to 68% w/w). These polymers were formulated as narrowly dispersed nanoparticles (110 nm) that exhibited a slow and predictable release of the ruthenium payload. Unlike standard encapsulation methods routinely used, the release kinetics of these nanoparticles is controlled and may be adjusted on demand, by tuning the size of the polymer chain. In terms of cytotoxicity, the nanoparticles were assessed in the ovarian cancer cell line A2780 and displayed potency comparable to cisplatin and the free drug, in the low micromolar range. Interestingly, the activity was maintained when tested in a cisplatin-resistant cell line, suggesting a possible orthogonal mechanism of action. Additionally, the internalization in tumour cells was found to be significantly higher than the free ruthenium complex (>200 times in some cases), clearly showcasing the added benefit in the drug's cellular permeation and accumulation of the drug. Finally, the in vivo performance was evaluated for the first time in mice. The experiments showed that the intravenously injected nanoparticles were well tolerated and were able to significantly improve the pharmacokinetics and biodistribution of the parent drug. Not only was the nanosystem able to promote an 18-fold increase in tumour accumulation, but it also allowed a considerable reduction of drug accumulation in vital organs, achieving, for example, reduction levels of 90% and 97% in the brain and lungs respectively. In summary, this simple and efficient one-pot procedure enables the generation of stable and predictable nanoparticles capable of improving the cellular penetration and systemic accumulation of the Ru drug in the tumour. Altogether, these results showcase the potential of covalently-loaded ruthenium polylactide nanoparticles and pave the way for its exploitation and application as a viable tool in the treatment of ovarian cancer.
Introduction
High-grade epithelial ovarian cancer is the deadliest gynaecologic cancer, ranking fifth overall in female cancer deaths, with a mortality-to-incidence ratio of 64% and 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 750 new cases in the USA in 2020.1 Most women have widespread intra-abdominal disease at the time of diagnosis and the 5 year survival rate for these women is only about 40% after receiving standard therapy (vs. 85% of breast cancer).2,3 Currently, the standard first-line treatment for ovarian cancer consists of surgical cytoreduction and platinum-based chemotherapy. Although this approach has proven to be the most effective treatment to date, many ovarian cancers exhibit primary platinum resistance, and most patients develop secondary platinum resistance throughout the treatment. In this setting, there is a paucity of approved therapeutic options and new effective therapies are required to improve patient survival rates of patients, especially in its advanced stages and platinum-resistant phenotypes.
750 new cases in the USA in 2020.1 Most women have widespread intra-abdominal disease at the time of diagnosis and the 5 year survival rate for these women is only about 40% after receiving standard therapy (vs. 85% of breast cancer).2,3 Currently, the standard first-line treatment for ovarian cancer consists of surgical cytoreduction and platinum-based chemotherapy. Although this approach has proven to be the most effective treatment to date, many ovarian cancers exhibit primary platinum resistance, and most patients develop secondary platinum resistance throughout the treatment. In this setting, there is a paucity of approved therapeutic options and new effective therapies are required to improve patient survival rates of patients, especially in its advanced stages and platinum-resistant phenotypes.
The clinical success of cisplatin, oxaliplatin and carboplatin has boosted the research directed at novel metal-based anticancer drugs. Among the potential metal-based candidates, ruthenium complexes have emerged as leading players by showing extremely promising results, with multiple Ru(III) candidates having entered clinical trials.4 Despite their raw potential, metal-based drugs are often plagued by various pharmacokinetic limitations, particularly their off-target toxicity, low solubility, fast systemic clearance and poor membrane permeability.5 One way to tackle these issues is through the development of macromolecular delivery systems. These versatile systems, such as polymeric nanoparticles, are able to convey an anticancer agent to a biological target, while protecting it from chemical and/or biological degradation, and release the encapsulated agent in a controlled fashion.6 In addition to the increased stability and membrane permeability, polymeric encapsulation of cytotoxic agents can also enable their targeted delivery to cancer cells through the enhanced permeability and retention (EPR) effect, although this is still under debate.7,8
Although extremely promising, the in vivo delivery of chemical entities to their molecular targets constitutes a significant challenge. The main hurdles encountered in the delivery of active payloads from biodegradable polymer matrices are low drug loadings, inconsistent encapsulation efficiencies and the fast, uncontrolled release from the matrix, particularly during the first 12 h (‘burst release’).6,9–11 These limitations are traditionally associated with physical encapsulation, the most common method of generating nanoparticles. Alternatively, novel encapsulation methodologies have been developed to overcome such limitations, where the payload is covalently attached to the polymer backbone.12–16 While covalent encapsulation approaches display numerous theoretical advantages, such as high stability (no burst release), loading efficiency and predictable drug release, its development is frequently hampered by low yields and complex synthetic schemes, often requiring multiple protection/deprotection steps.17
Recently, our group reported a simple and efficient procedure to generate covalent ruthenium–polylactide polymers without complicated purification steps.18,19 This method employed a ruthenium-bearing zinc complex as the initiator in a lactide ring-opening polymerization (ROP) process (Fig. 1). We could demonstrate that this system was able to improve the delivery of a Ru photosensitizer to cancer cells and displayed various advantages over traditional encapsulation methodologies. In this work, we further explore this technology by designing a biocompatible covalent drug delivery system that enables the safe and controlled delivery of a Ru(II) cytotoxic payload to ovarian cancer cells and, for the first time, evaluate its in vivo efficacy.
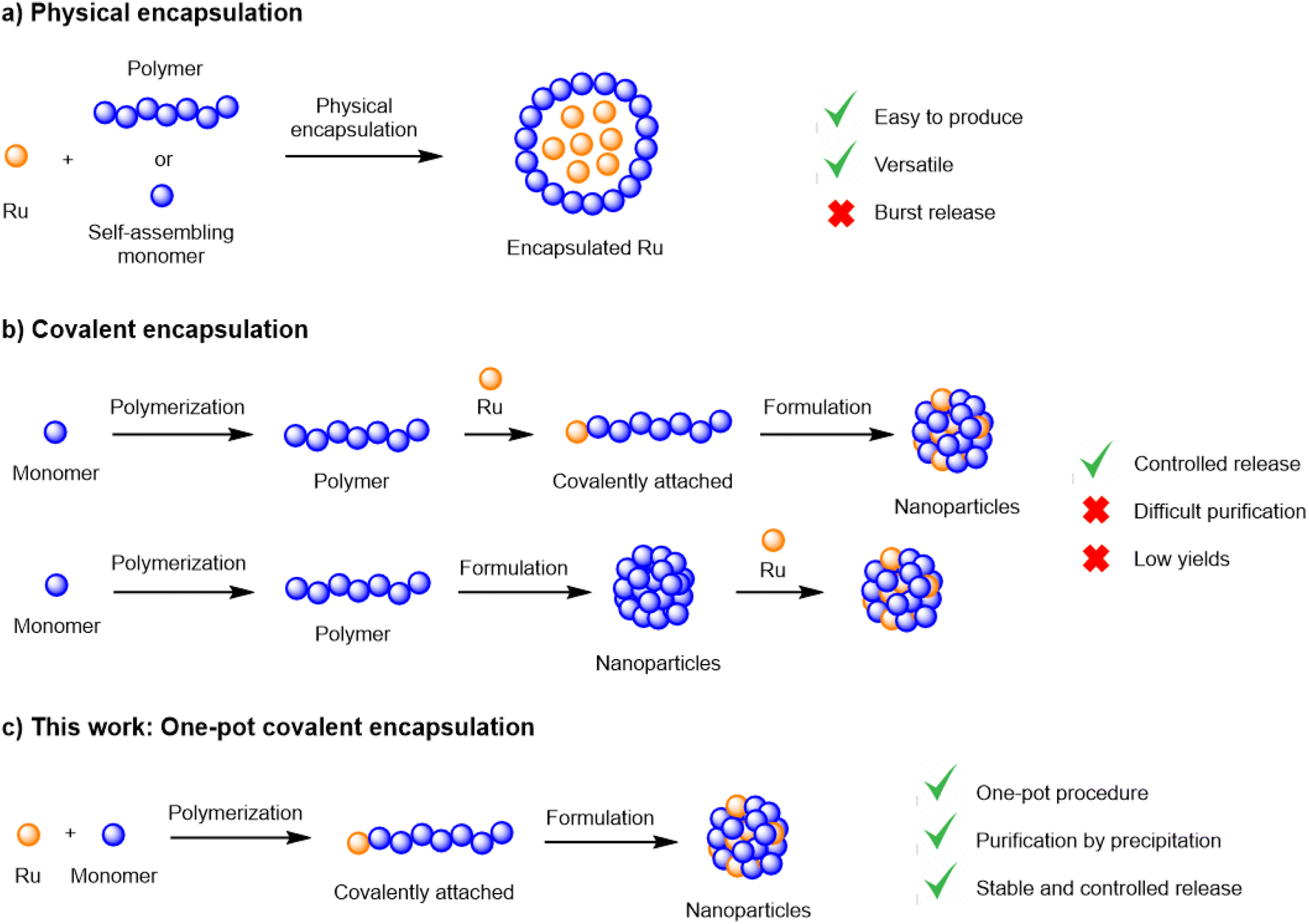 | ||
| Fig. 1 Encapsulation methodologies and their advantages and limitations; (a) physical encapsulation; (b) covalent encapsulation; (c) one-pot covalent encapsulation. | ||
Results and discussion
Synthesis of a cytotoxic ruthenium(II) complex
The first step was the selection of a suitable cytotoxic payload. Ruthenium complexes featuring phenyl-1H-imidazo-1,10-phenanthroline ligands are well known for their DNA binding properties and cytotoxic activity.20,21 Therefore, we envisaged the design of a similar ligand featuring a hydroxyl handle capable of initiating ROP. In our first attempt, we designed a ruthenium complex that contained a phenol group and explored its ability to promote the ROP of lactide (synthesis and characterization in the ESI†). Unfortunately, despite showing satisfactory polymerization competence, the resulting Ru–PLA polymer, which contained a phenolic ester, was shown to be unstable in aqueous solution and, therefore, unsuitable for biological applications. After this setback, we went back to the drawing board and devised a new ruthenium complex. To address the stability limitation, we elected to replace the phenol with a hydroxymethyl handle which, in theory, should generate a more stable ester. The synthesis of [Ru(2,2′-bipyridine)2(4-hydroxymethyl-phenyl-1H-imidazo-1,10-phenanthroline)] (PF6)2 (Ru) is illustrated in Scheme 1. Unlike other Ru–arene complexes, the bipyridine ligands are considerably stable, and Ru does not undergo aquation processes. This was confirmed by evaluating its stability in plasma, where it was shown to be stable for more than 10 days (Fig. S4†).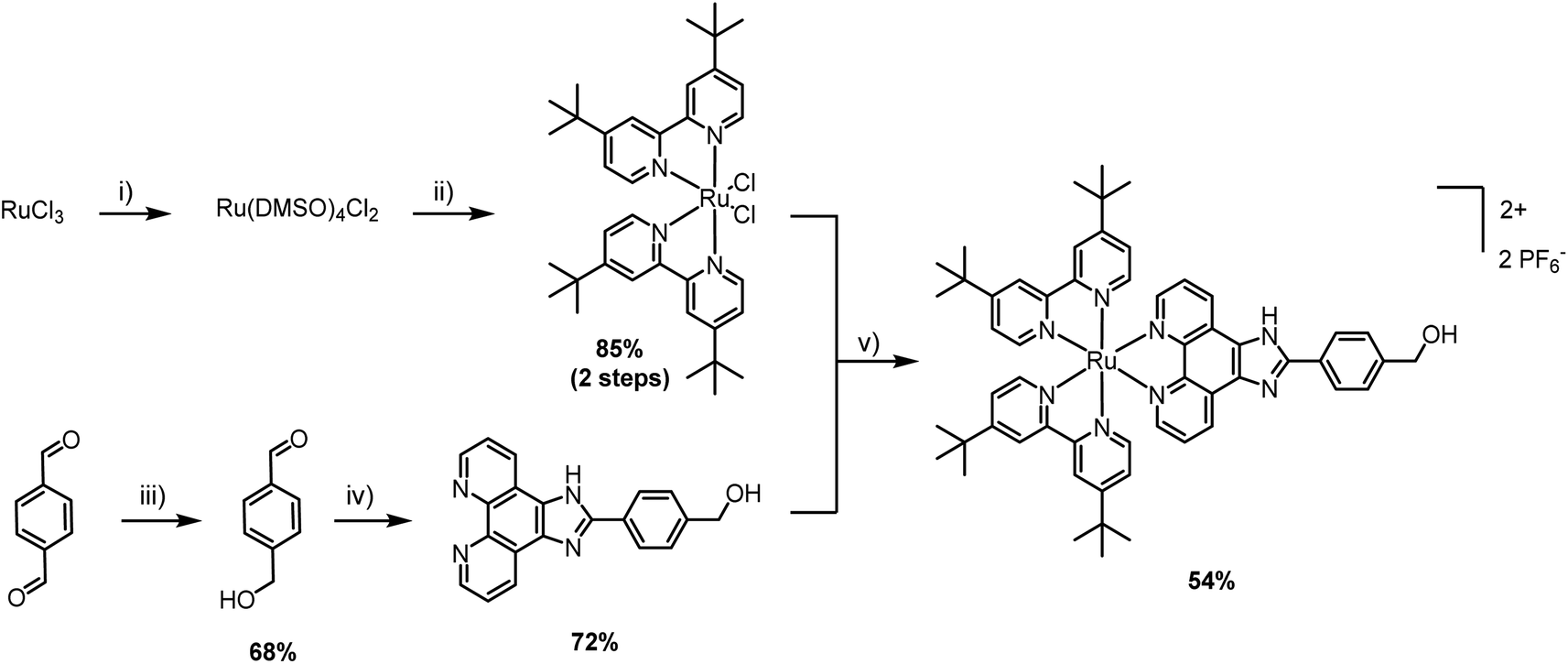 | ||
| Scheme 1 Synthetic route for the preparation of Ru: (i) EtOH, reflux, 3 h, then DMSO, reflux, 2 h; (ii) 4,4′-di-tert-butyl-2,2′-dipyridyl; LiCl; DMF; 120 °C, overnight; (iii) NaBH4, MeOH/THF, r.t., 6 h; (iv) 1,10-phenanthroline-5,6-dione, NH4OAc, AcOH, reflux, 2 h; (v) H2O/EtOH (1:1), 90 °C, overnight, then NH4PF6. | ||
The cytotoxicity of Ru after 48 h of incubation was confirmed in CT-26 colon cancer cell line (IC50 = 8.1 ± 0.4 μM) and A2780 ovarian carcinoma cell line (IC50 = 6.6 ± 1.0 μM). Cisplatin was used as a control and its cytotoxicity was comparable to Ru, in the lower micromolar range (IC50 = 7.1 ± 0.9 μM in A2780).
Ru–PLA preparation and characterization
Owing to its decent stability and reactivity, the commercially-available tin(II) bis(2-ethylhexanoate) Sn(Oct)2 is the traditional go-to catalyst for ROP. However, due to its substantial toxicity, the polymer manufacturing process must undergo multiple rounds of purification to ensure complete removal of the catalyst and compliance with FDA regulations. In addition, Sn(Oct)2 does not offer high activities or good control over the ROP parameters. To avoid those drawbacks, we decided to replace Sn(Oct)2 with a biocompatible calcium catalyst. Various metal trimethylsilyl amides, including Ca[N(SiMe3)2]2·2THF, have been shown to catalyse controlled polymerizations under mild conditions, producing polyesters with controlled molecular weight and a tailored macromolecular architecture.22,23In a preliminary polymerization attempt, two equivalents of Ru were dissolved in THF and added to one equivalent of Ca[N(SiMe3)2]2·2THF in THF. The solution was stirred for 5 minutes to allow the formation of the intermediary calcium bis-alkoxide, followed by the addition of lactide (LA). However, the in situ-generated alkoxide intermediate was insoluble in THF and precipitated. Despite being undesirable, the observed precipitation confirms the formation of the intermediate alkoxide, which is essential for the polymerization. To address this problem, we replaced THF with a 1:1 THF/CH2Cl2 system where the catalyst is dissolved in THF and Ru is dissolved in CH2Cl2. Pleasantly, the intermediary was soluble in this solvent system and the reaction proceeded with the addition of rac-lactide ([LA]final = 0.1 M). The reaction was stirred for 20 h at room temperature followed by precipitation in pentane/diethyl ether 1:1, to remove the unreacted monomer. This simple and efficient procedure, illustrated in Scheme 2, proceeds with high conversions and near quantitative incorporation of Ru in the polymer. Moreover, by adjusting the number of lactide equivalents, we were able to obtain four polymers, P1–P4, with different molecular weights: 2, 4, 7.5 and 11 kDa, respectively.
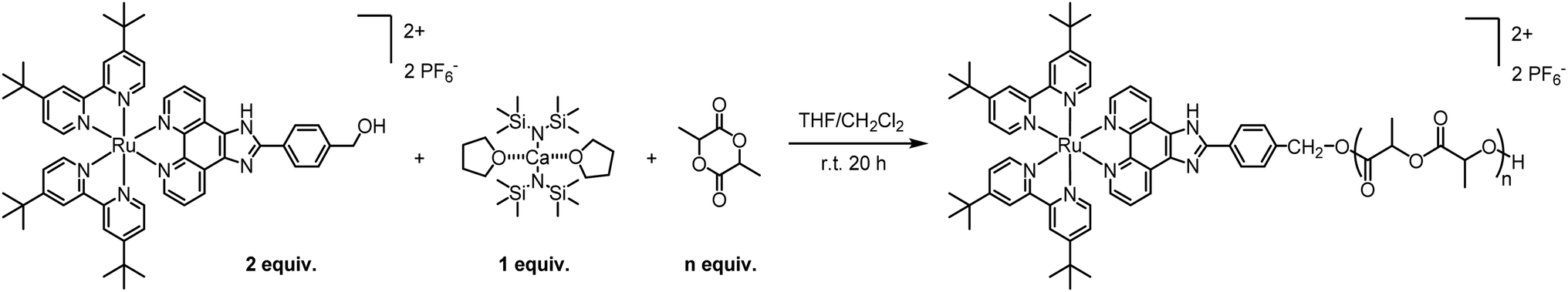 | ||
| Scheme 2 General procedure for the preparation of Ru–PLA polymers. | ||
The obtained amorphous orange solids were characterized by NMR spectroscopy, size-exclusion chromatography, and MALDI-TOF mass spectrometry. First, the conversion was determined by integration of the 1H NMR methine resonances of PLA (multiplet at 5.05–5.20 ppm) and the unreacted monomer (quadruplet at 4.99 ppm). In all four polymers, the conversion rates were >96%, indicating an excellent polymerization efficiency. The number-average molar mass (Mn), calculated through the ratio of LA/Ru peaks, was similar to the theoretical values, which confirms the controlled character of the polymerization. Furthermore, Ru incorporation in the polymer is confirmed by a downfield shift in the benzylic CH2 from 4.69 ppm (benzylic alcohol) to 5.27 ppm (benzylic ester) (Fig. S7†). These results are corroborated by MALDI-TOF analysis, which displays a peak distribution compatible with Ru–PLA and a peak interval of 72 (i.e., molecular weight of lactic acid) (Fig. 2 and S9†). This interval, instead of 144 (i.e., the molecular weight of lactide), suggests the existence of transesterification reactions. These side reactions occur when the growing chain reacts with itself or other polymeric chains instead of lactide, leading to polymer scrambling.24 These are relatively common in ROP initiated with homoleptic complexes, particularly in reactions with high conversions and longer reaction times. Finally, size-exclusion chromatography analysis of the obtained polymers revealed the presence of unimodal distributions with calculated Mn comparable to the theoretical ones with the dispersity (Đ) increasing proportionally to the polymer molecular weight (Table 1 and Fig. 3).
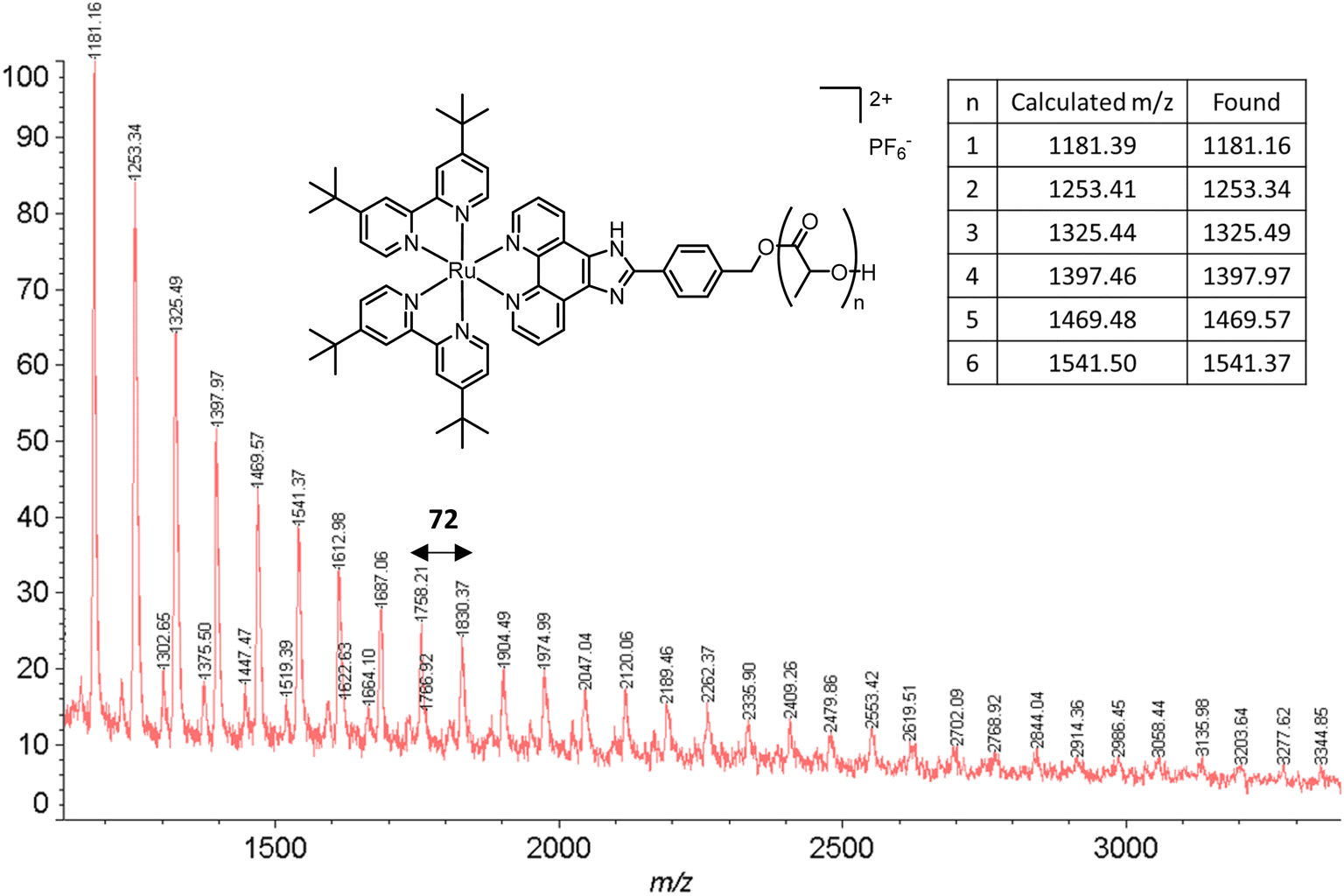 | ||
| Fig. 2 MALDI-TOF spectrum of P2. The experimental peaks found match the calculated series with a peak gap of 72 g mol−1. | ||
| LA/Ca | M n,theo (Da) | M n,SEC (Da) | Đ | M n,NMR (Da) | DPb | Conv.c (%) | % Rud | |
|---|---|---|---|---|---|---|---|---|
| a M n and Đ of polymer determined by SEC-RI in THF at RT using polystyrene standards and Mn corrected by the Mark–Houwink parameter (0.58). b Degree of polymerization (DP) and Mn,NMR were calculated by 1H-NMR spectroscopy in CD3CN. c Conversion was determined by the integration of 1H NMR methine resonances of lactide and PLA. d Theoretical ruthenium loading, calculated between the ratio of Ru molecular weight and polymer molecular weight. | ||||||||
| P1 | 10 | 2000 | 860 | 1.12 | 1840 | 4.1 | 96 | 68.0 |
| P2 | 40 | 4000 | 4360 | 1.32 | 3220 | 13.4 | 98 | 39.4 |
| P3 | 90 | 7500 | 6240 | 1.64 | 6820 | 33.5 | 98 | 20.6 |
| P4 | 140 | 11000 |
9750 | 1.76 | 11480 |
77 | 99 | 10.2 |
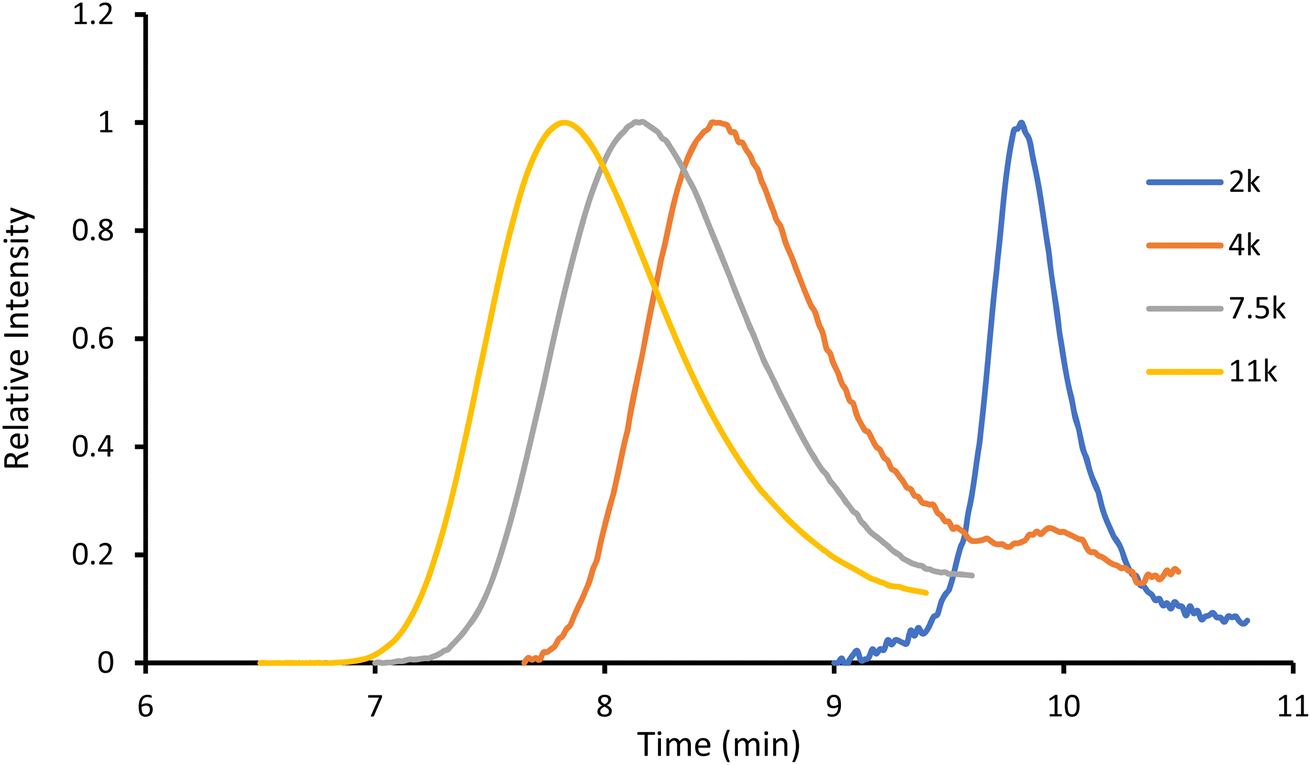 | ||
| Fig. 3 Gel permeation chromatography of P1–P4. | ||
Preparation and characterization of nanoparticles NP1–NP4
With the four polymers in hand, we proceeded to formulate the nanoparticles through a modified nanoprecipitation process.18P1–P4 were dissolved in 0.5 mL of acetone and added dropwise to 1 mL of an aqueous solution of Kolliphor P188 (1% w/v). Kolliphor P188 is a FDA-approved triblock copolymer routinely used as an adjuvant in the preparation of nanoparticle solutions.25 The mixture was stirred for 10 minutes, to stabilize the generated nanoparticles. Then, upon removal of the acetone under reduced pressure, a cloudy nanoparticle suspension was obtained. A final centrifugation step was performed to remove large aggregates and precipitated polymer, and the final clear suspension was obtained (Table 2). The size of the nanoparticles was assessed by dynamic light scattering (DLS) and showed a range of sizes between 104 and 134 nm (Table 2 and Fig. S11–S14†). This size is ideal for the targeted delivery of drugs, as they are small enough to leak from the blood vessels and accumulate in the tumour environment, but large enough to avoid toxicity and rapid elimination from circulation.26,27 Complementary transmission electronic microscopy (TEM) experiments confirmed the presence of spherical nanoparticles with a diameter ≈ 110 nm (Fig. 4, S20 and S21†). The zeta potential of NP3 was also evaluated and was found to be +80 mV. This highly positive value indicates a good electrostatic stability of the NPs. The concentration of Ru in the nanoparticles was determined by ultraviolet-visible (UV-vis) spectroscopy. After confirming the encapsulation did not change the absorption spectrum of Ru, a calibration curve was elaborated and used to calculate the concentration of Ru in the nanoparticles (Fig. S16–S18†).| Nanoparticle | Polymer | M n,theo | D z (nm) | PDIa | [Ru]b (μM) |
|---|---|---|---|---|---|
| a Average intensity diameter (Dz) and polydispersity index (PDI) were calculated by DLS. b Ru concentration was calculated by UV-vis. Absorbance at 472 nm was converted to concentration using a calibration curve. | |||||
| NP1 | P1 | 2000 | 134.5 ± 1.0 | 0.098 ± 0.027 | 649 |
| NP2 | P2 | 4000 | 104.0 ± 1.1 | 0.122 ± 0.007 | 1182 |
| NP3 | P3 | 7500 | 114.3 ± 0.2 | 0.078 ± 0.004 | 506 |
| NP4 | P4 | 11000 |
116.2 ± 0.2 | 0.066 ± 0.007 | 651 |
 | ||
| Fig. 4 TEM image of NP3 showing a spherical nanoparticle with a diameter of approximately 100 nm. | ||
NP1–NP4 release kinetics in PBS pH 7.4
Contrary to physical encapsulation where burst release is a major concern, covalently encapsulated drugs are commonly released in a controlled and predictable fashion. In the case of Ru–PLA, its release is dependent on the hydrolysis of the ester bond connecting the Ru and the polymer. Therefore, to mimic the stability of the synthesized nanoparticles in physiological conditions, we set out to evaluate their release kinetics in PBS pH 7.4 at 37 °C. The nanoparticles with the shortest polymer chain (NP1) displayed lower stability and achieved total payload release after 24 h (Fig. 5). It is also possible to observe an increase in stability proportional to the polymer chain length, with NP4 only reaching a release of 82% Ru after 48 h. After their preparation and characterization, we set out to evaluate the biological activity of NP1–NP4.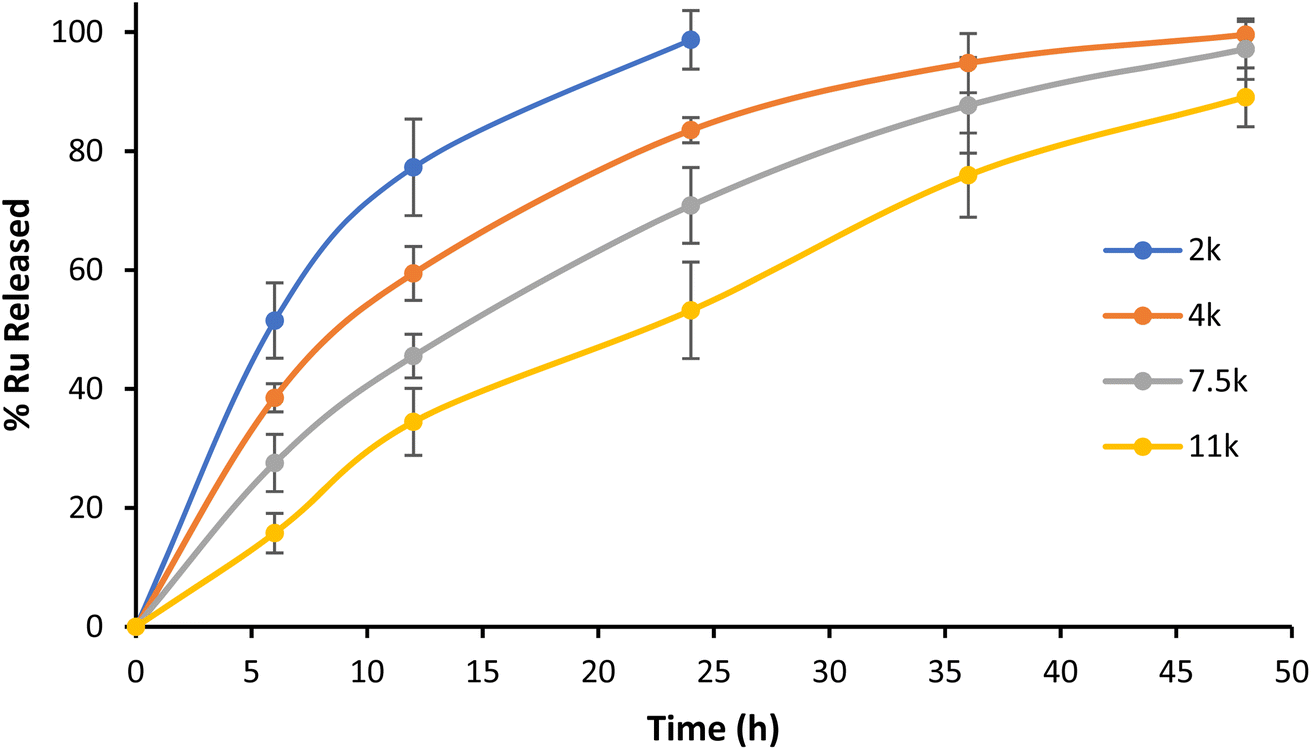 | ||
| Fig. 5 Kinetic profile of Ru release from NP1–NP4 in PBS pH 7.4 at 37 °C. Representative data from three independent experiments are shown. | ||
Cytotoxicity studies on 2D monolayer cells
The first step toward the biological investigation of the Ru complex and the corresponding nanoparticles (NP1–NP4) was the evaluation of their cytotoxicity in monolayer cultures of A2780 (human ovarian adenocarcinoma), A2780 cis (human ovarian adenocarcinoma cisplatin-resistant), and RPE-1 (human retinal pigment epithelial) cell lines using a fluorometric resazurin cell viability assay. Cisplatin was tested in the same cell lines as a positive control and the observed IC50 values are reported in Table 3.| A2780 | A2780 cis* | RPE-1 | ||
|---|---|---|---|---|
| 48 h | 72 h | 72 h | 72 h | |
| Cisplatin | 7.1 ± 0.9 | 5.3 ± 0.7 | 14.1 ± 0.2 | 38.5 ± 1.2 |
| Ru | 6.6 ± 1.0 | 7.4 ± 0.7 | 7.0 ± 0.5 | 21.2 ± 3.2 |
| NP1 | 24.8 ± 3.6 | 18.5 ± 0.6 | 10.1 ± 0.8 | >100 |
| NP2 | >100 | 20.4 ± 1.3 | 14.6 ± 0.7 | >100 |
| NP3 | >100 | 35.4 ± 2.9 | 26.2 ± 1.5 | >100 |
| NP4 | >100 | >100 | >100 | >100 |
Interestingly, while Ru and NP1 exert toxicity in the low micromolar range after 48 h (IC50 = 6.6 μM and 24.8 μM, respectively), NP2–NP4 did not show relevant toxicity (IC50 > 100 μM). While unexpected, these results can be rationalized through analysis of the nanoparticles' release kinetics. While NP1 liberates all Ru in under 24 h, higher molecular weight nanoparticles take longer to release the payload, with NP4 only reaching ≈80% release after 48 h. This slow release translates into a slower accumulation in the tumour and a delayed cell death, which may not be significant after only 48 h. Therefore, we repeated the experiment with an incubation time of 72 h and, pleasantly, NP2 and NP3 showed toxicity in the micromolar range (IC50 = 20.4 μM and 35.4 μM, respectively). NP4 on the other hand, displayed no relevant toxicity once again (IC50 > 100 μM).
Taking into account these results, subsequent cell viability assays were always performed with an incubation time of 72 h to ensure maximum release of cytotoxic Ru from the nanoparticles. Then, we evaluated our system in a cisplatin-resistant ovarian cancer cell line (A2780 cis). While cisplatin displayed a 3-fold decrease in activity, both Ru and NP1–NP3 showcased similar or even increased toxicity in this cell line. These results appear to indicate an orthogonal mechanism of action/intracellular trafficking compared to cisplatin. Finally, the compounds were also tested in the healthy RPE-1 cell line and showed considerably lower toxicity when compared to tumour cell lines.
Cellular uptake in cancer cells
To rationalize the results observed in the cell viability assays, we set out to evaluate the cellular uptake of the Ru(II) complex, both alone and connected to the nanoparticles, using inductively-coupled plasma mass spectrometry (ICP-MS). A2780 cells were incubated with 5 μM for 12 h, 24 h, 48 h, and 72 h and the results obtained are portrayed in Fig. 6.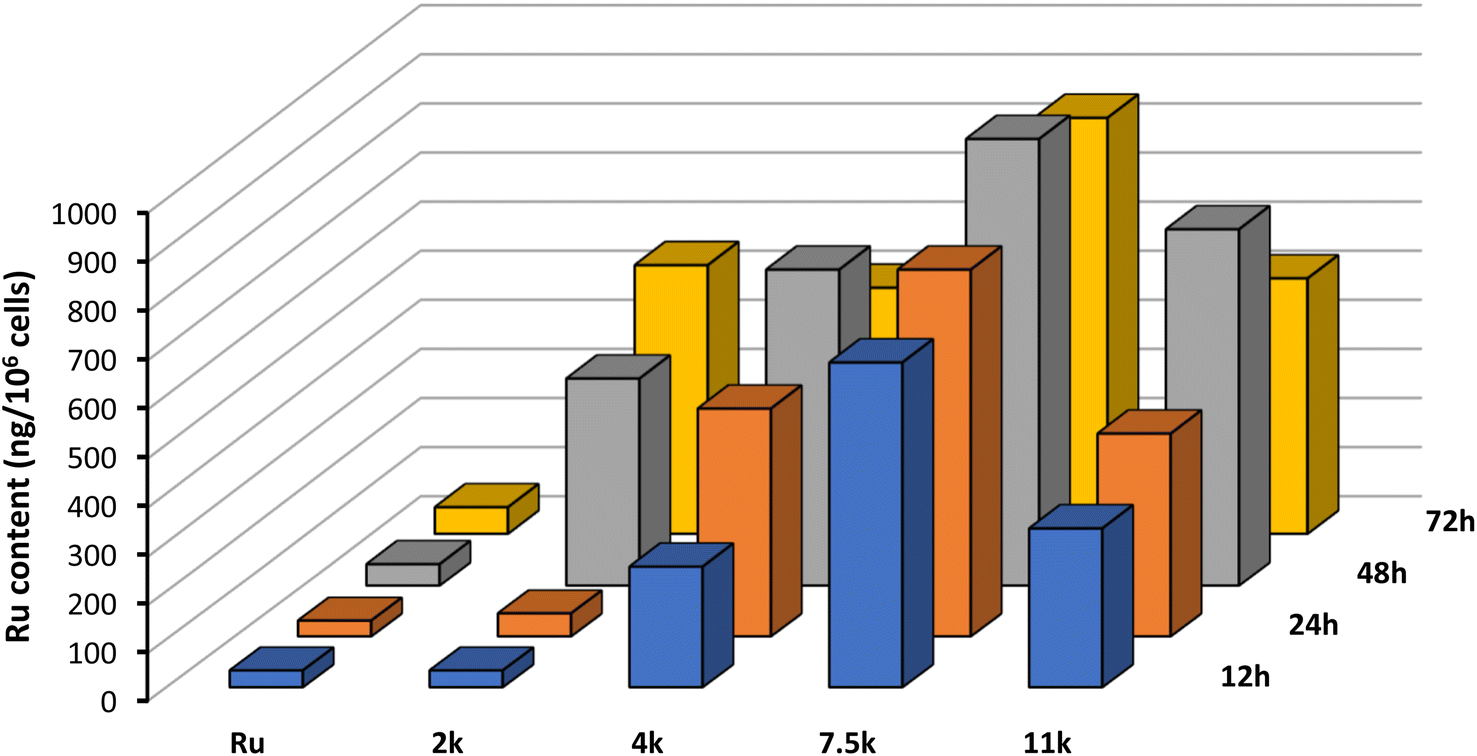 | ||
| Fig. 6 Cellular uptake of Ru and NP1–NP4 by ICP-MS of A2780 cells incubated at 5 μM for 12 h, 24 h, 48 h, and 72 h at 37 °C. Representative data from three independent experiments is shown. | ||
Interestingly, after just 12 h of incubation, the intracellular content of Ru is significantly higher for NP1–NP4 compared with the free complex. This result highlights the important role of this technology in increasing membrane permeation and cell accumulation. Moreover, the size of the polymer chain plays a critical role in the internalization, with longer polymers displaying increased accumulation. The additional lipophilicity of longer polymeric chains facilitates interaction with the hydrophobic cell membrane and promotes the internalization of the nanoparticles. However, 7500 Da appears to be the optimal polymer size, after which internalization becomes less efficient.
To confirm the ICP-MS observations, we decided to investigate the internalization using fluorescence microscopy in living cells. This was possible because Ru has an inherent luminescence, which enables its direct visualization in live cells. In these experiments, we aimed to assess the internalization efficiency and the subcellular localization of the nanoparticles vs. the free drug. Theoretically, we expected to observe differences in the internalization mechanism: the free Ru entering the cell by passive diffusion and localizing diffusely across the cytoplasm; nanoparticles entering through an endocytosis mechanism and localizing initially in the lysosome, and, as the polymer is hydrolysed, the Ru starts to diffuse in the cytoplasm. For this reason, instead of using NP3, which displayed higher cellular internalization, we elected to use NP2, which has a faster release of the payload and would accelerate the lysosome-cytoplasm translocation.
To evaluate this hypothesis, we incubated both compounds (10 μM) for 4 h, 8 h, 12 h, and 24 h. Unfortunately, it was not possible to identify any difference in cellular localization between the NPs and the free ruthenium, as both appear to be diffused evenly inside the cells. However, it is possible to observe a significant difference in luminescence intensity after 8 h, with the NPs displaying higher fluorescence than the free drug (Fig. S23†). These observations confirm previous ICP-MS results that suggested improved cellular accumulation of NPs compared with the free complex.
In vivo results
After validating their in vitro performance and confirming their ability to improve cellular internalization, we set out to evaluate the performance of our covalently-loaded nanoparticles in vivo. For these experiments, we elected to use NP3, as they embodied a satisfactory balance between internalization, activity, and controlled release. First, we evaluated the tolerability of Ru and NP3 in female Swiss nude mice, with vehicle and blank nanoparticles (NP) as controls. At a 6.6 mg kg−1 dose, both Ru and NP3 were well tolerated, as no impact was observed on the weight of the mice. This result is an auspicious indication of the safety and viability of our developed nanoparticles (Fig. 7A).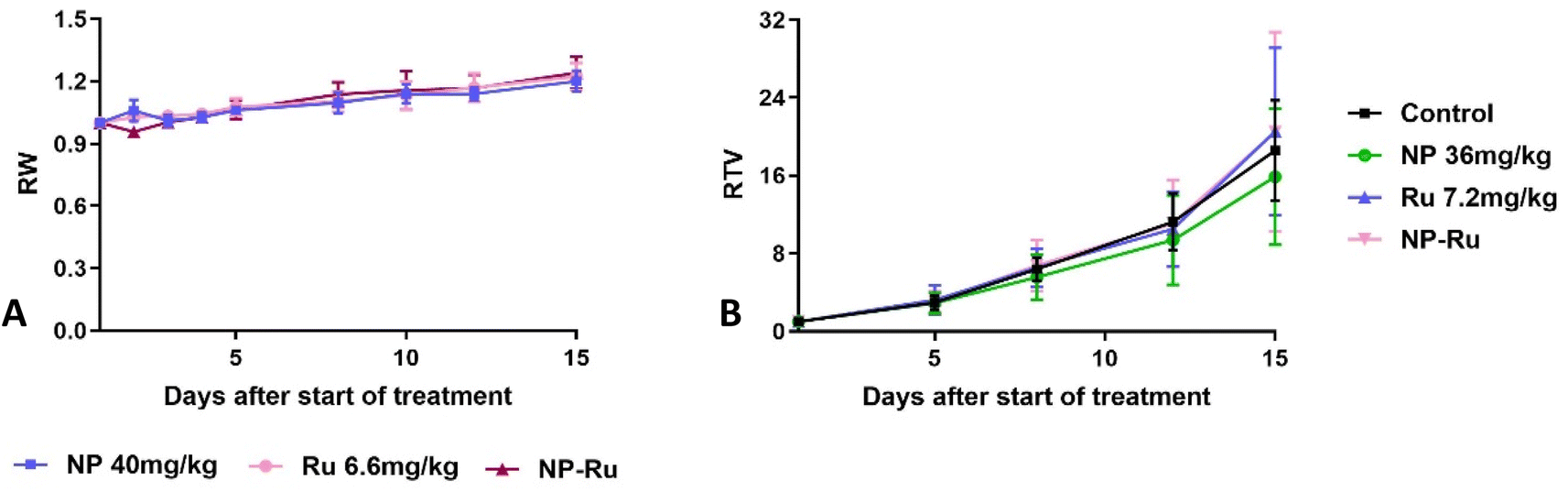 | ||
| Fig. 7 In vivo experiments: (A) evaluation of the toxicities of NP, Ru, and NP–Ru. (B) Relative tumour volume (RTV) after NP, Ru, and NP–Ru administration. Representative data from five independent experiments is shown. | ||
Then, we initiated experiments to determine the efficacy of Ru and NP3 in the treatment of ovarian carcinoma. Among the available in vivo models, patient-derived xenografts (PDXs) represent one of the most accurate, as it reduces (as far as possible) the gap between human tumours and preclinical models.28 In PDX models, tissue or cells of a patient's tumour are implanted into an immunodeficient or humanized mouse. The main characteristics of this approach are the stability of the gene expression profiles of PDXs in mice at first transplantation and during the in vivo maintenance of the model.29–31 Moreover, PDXs can represent highly-predictive models for therapeutic response in cancer patients, with a positive and a negative predictive value of 85% and 91%, respectively. All things accounted for, PDXs are a complex, but rewarding model to perform in vivo experiments.32 In this work, a PDX approach was selected where mice were xenografted with a 20–40 mm3 patient-derived tumour fragment of OV54 serous adenocarcinoma. Unfortunately, after 15 days of treatment, no significant differences in relative tumour volume (RTV) was observed between mice treated with Ru, NP3 and controls (Fig. 7B). While surprising, we theorized these disappointing results may be related to a lack of in vivo activity of the Ru complex and not necessarily associated with a subpar performance of our covalent nanoparticle technology. To confirm this hypothesis, we decided to perform a biodistribution experiment and assess the effect of the nanoparticles on improving the accumulation of the cytotoxic drug in the tumour.
Biodistribution experiments
The biodistribution of Ru and NP3 in this mouse model was studied by intravenous tail injection into nude mice. After 24 h, the mice were sacrificed and the major organs (i.e., tumour tissues, brain, lungs, liver and kidney) were separated, digested and the ruthenium content was determined by ICP-MS. The first important observation is that Ru and NP3 accumulate primarily in the liver, with a significant accumulation also visible in the kidneys and lungs. Being the two main centres for drug clearance and metabolism, drug accumulation of drugs in the liver and kidneys is well known and widely reported in the literature, both for both small molecules and nanoparticles.33 However, in the case of NP3, a noticeable decrease in ruthenium accumulation in peripheral tissues was observed, compared to Ru, with observed reductions ranging from 51% in the kidney, to 97% in the lungs (Fig. 8 and S27†). Importantly, with NP3 it was possible to achieve an 18-fold increase in ruthenium concentration in the tumour, when compared to Ru. This increased accumulation in the tumour, together with the diminished accumulation in peripheral tissues, validates our nanoparticle approach and confirms its ability to improve the pharmacokinetics and distribution of the payload.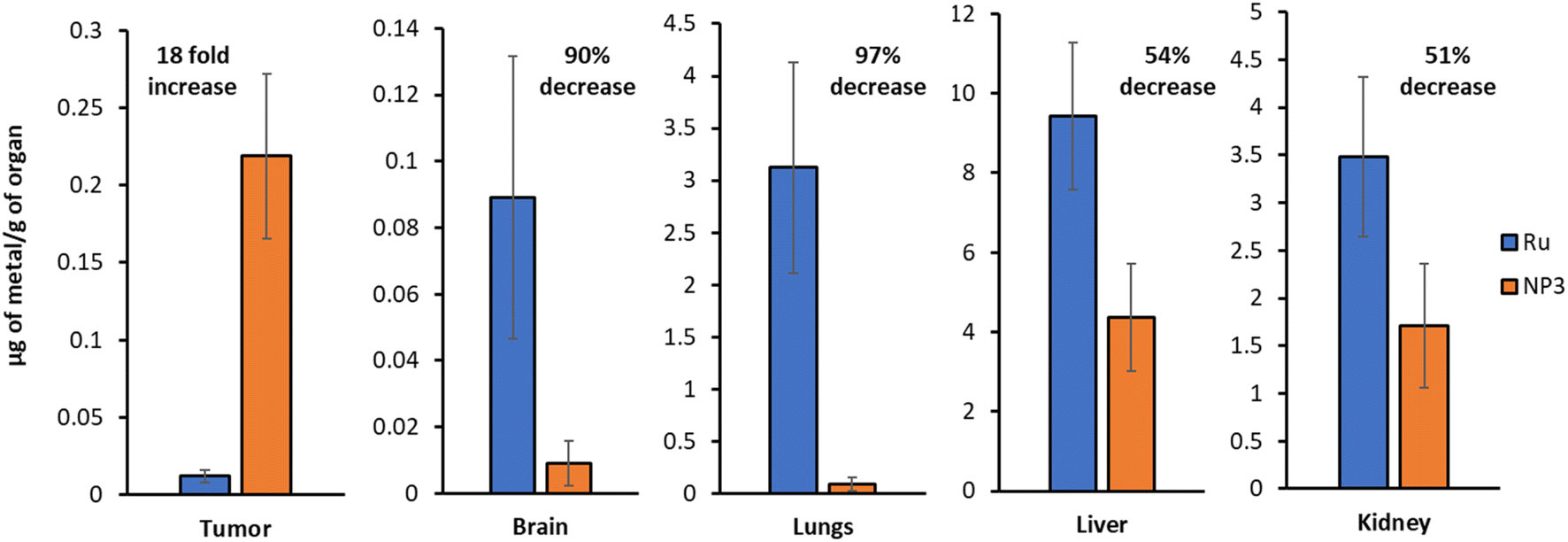 | ||
| Fig. 8 ICP-MS quantification of the ruthenium content in various organs (Ruvs.NP3). Representative data from five independent experiments is shown. | ||
Conclusions
In the present work, we reported the application of a ROP methodology to generate covalently attached cytotoxic ruthenium nanoparticles. This simple and straightforward one-pot protocol, catalysed by calcium bis(trimethylsilyl amide), proceeds with high conversions and quantitative incorporation of the ruthenium in the nanoparticles. Furthermore, unlike other methodologies, which require complicated purification stages, a single precipitation and washing step is performed to obtain the pure product. The reported procedure enabled the preparation of polymers with varying molecular weight (2000–11000 Da) and high drug loadings (up to 68% w/w). These polymers were formulated into narrowly dispersed nanoparticles (≈110 nm) that showed a slow and predictable release of the ruthenium payload. After confirming their cytotoxicity in A2780 cells, ICP-MS and fluorescence microscopy experiments demonstrated a positive effect of the nanoparticles in the cellular accumulation of the ruthenium payload.Unfortunately, the cytotoxic Ru that we designed displayed unsatisfactory cytotoxic potency in the animal experiments, which hindered the success of the nanoparticles. Nonetheless, we demonstrated in this work the advantages of covalently-loaded ruthenium nanoparticles in terms of stability, predictability and in vivo pharmacokinetic modulation. We believe that, when loaded with an adequate cytotoxic ruthenium complex, this technology will unlock its true potential and shine as a reliable alternative in the treatment of ovarian carcinomas. Thanks to its simplicity and efficiency, we have confidence that this ROP methodology may even be expanded beyond ruthenium, to design other metal-based covalent nanoparticles, and can be applied in the treatment of numerous different tumours. Hopefully, these are the first steps in the creation of a new class of therapeutics to aid us in the arduous fight against cancer.
Author contributions
João P. M. António and Nancy Soliman performed the synthesis and characterisation of the nanoparticles. Albert Gandioso and Robin Vinck performed the in vitro experiments. Fariba Nemati performed the in vivo experiments. Fan Sun performed the TEM experiments. Carine Robert performed the MALDI-Tof experiments. Pierre Burckel performed the ICP-MS experiments. Didier Decaudin, Christophe M. Thomas and Gilles Gasser supervised the project and corrected the manuscript. The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Chimie ParisTech – PSL and CNRS are thanked for financial support. Île-de-France Region is gratefully acknowledged for financial support of 500 MHz NMR spectrometer of Chimie ParisTech in the framework of the SESAME equipment project (no. 16016326). This work was financially supported by an ERC Consolidator Grant PhotoMedMet to G. G. (GA 681679) and has received support under the program “Investissements d'Avenir” launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G. G.) and ANR-l 7-CONV-0005 (bourse Qlife to J. P. M. A., G. G., C. M. T. and D. D.). C. M. T. is grateful to the Institut Universitaire de France. A. G. thanks the ARC Foundation for Cancer Research for a postdoctoral research fellowship. Part of the ICP-MS measurements was supported by IPGP multidisciplinary program PARI, and by Paris–IdF region SESAME Grant no. 12015908. We wish to thank the Animal Platform of the Institut Curie-Orsay and Paris, as well as Laura Sourd for her technical assistance. We would also like to thank Lisa Gourdon for performing some fluorescence spectroscopy experiments. All in vivo experimental procedures were specifically approved by the Ethics Committee of the Institut Curie CEEA-IC #118 (Authorization APAFiS# 25870-2020060410487032-v1 given by the National Authority) in accordance with the international guidelines.References
- R. L. Siegel, K. D. Miller, A. Jemal and C. A. Cancer, J. Clin., 2020, 70, 7–30 Search PubMed.
- J. S. Barnholtz-Sloan, A. G. Schwartz, F. Qureshi, S. Jacques, J. Malone and A. R. Munkarah, Am. J. Obstet. Gynecol., 2003, 189, 1120–1127 CrossRef PubMed.
- S. Lheureux, M. Braunstein, A. M. Oza and C. A. Cancer, J. Clin., 2019, caac.21559 Search PubMed.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- E. Villemin, Y. C. Ong, C. M. Thomas and G. Gasser, Nat. Rev. Chem., 2019, 3, 261–282 CrossRef CAS.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- A. E. Hansen, A. L. Petersen, J. R. Henriksen, B. Boerresen, P. Rasmussen, D. R. Elema, P. M. af Rosenschöld, A. T. Kristensen, A. Kjær and T. L. Andresen, ACS Nano, 2015, 9, 6985–6995 CrossRef CAS PubMed.
- S. Sindhwani, A. M. Syed, J. Ngai, B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, B. Ouyang, Z. Lin, L. Wang, M. Egeblad and W. C. W. Chan, Nat. Mater., 2020, 19, 566–575 CrossRef CAS PubMed.
- M. He, Z. Zhang, Z. Jiao, M. Yan, P. Miao, Z. Wei, X. Leng, Y. Li, J. Fan, W. Sun and X. Peng, Chin. Chem. Lett. DOI:10.1016/j.cclet.2022.05.088.
- S. Su and P. M. Kang, Nanomaterials, 2020, 10, 656 CrossRef CAS PubMed.
- B. K. Lee, Y. Yun and K. Park, Adv. Drug Delivery Rev., 2016, 107, 176–191 CrossRef CAS PubMed.
- Q. Yin, R. Tong, Y. Xu, K. Baek, L. W. Dobrucki, T. M. Fan and J. Cheng, Biomacromolecules, 2013, 14, 920–929 CrossRef CAS PubMed.
- J. Nicolas, Chem. Mater., 2016, 28, 1591–1606 CrossRef CAS PubMed.
- H. Phan, K. Kortsen, G. Englezou, B. Couturaud, A. J. Nedoma, A. K. Pearce and V. Taresco, J. Polym. Sci., 2020, 58, 1911–1923 CrossRef CAS.
- K. Cai, X. He, Z. Song, Q. Yin, Y. Zhang, F. M. Uckun, C. Jiang and J. Cheng, J. Am. Chem. Soc., 2015, 137, 3458–3461 CrossRef CAS PubMed.
- K. C. F. Dantas, J. dos S. Rosário and P. P. Silva-Caldeira, Pharmaceutics, 2022, 14, 1506 CrossRef CAS PubMed.
- N. Soliman, G. Gasser and C. M. Thomas, Adv. Mater., 2020, 32, 2003294 CrossRef CAS PubMed.
- N. Soliman, L. K. McKenzie, J. Karges, E. Bertrand, M. Tharaud, M. Jakubaszek, V. Guérineau, B. Goud, M. Hollenstein, G. Gasser and C. M. Thomas, Chem. Sci., 2020, 11, 2657–2663 RSC.
- K. Upitak and C. M. Thomas, Acc. Chem. Res., 2022, 55, 2168–2179 CrossRef CAS PubMed.
- R. K. Vuradi, K. Dandu, P. K. Yata, V. R. M., R. R. Mallepally, N. Chintakuntla, R. Ch, S. S. Thakur, C. M. Rao and S. S., New J. Chem., 2018, 42, 846–859 RSC.
- X. Hu, Y. Lu, C. Dong, W. Zhao, X. Wu, L. Zhou, L. Chen, T. Yao and S. Shi, Chem.–Eur. J., 2020, 26, 1668–1675 CrossRef CAS PubMed.
- Z. Zhong, P. J. Dijkstra, C. Birg, M. Westerhausen and J. Feijen, Macromolecules, 2001, 34, 3863–3868 CrossRef CAS.
- M. J.-L. Tschan, R. M. Gauvin and C. M. Thomas, Chem. Soc. Rev., 2021, 50, 13587–13608 RSC.
- C. Robert, T. E. Schmid, V. Richard, P. Haquette, S. K. Raman, M.-N. Rager, R. M. Gauvin, Y. Morin, X. Trivelli, V. Guérineau, I. del Rosal, L. Maron and C. M. Thomas, J. Am. Chem. Soc., 2017, 139, 6217–6225 CrossRef CAS PubMed.
- G. L. Allotey-Babington, H. Nettey, S. D'Sa, K. Braz Gomes and M. J. D'Souza, J. Drug Delivery Sci. Technol., 2018, 47, 181–192 CrossRef CAS.
- L. Tang, X. Yang, Q. Yin, K. Cai, H. Wang, I. Chaudhury, C. Yao, Q. Zhou, M. Kwon, J. A. Hartman, I. T. Dobrucki, L. W. Dobrucki, L. B. Borst, S. Lezmi, W. G. Helferich, A. L. Ferguson, T. M. Fan and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15344–15349 CrossRef CAS PubMed.
- S. Prabha, G. Arya, R. Chandra, B. Ahmed and S. Nimesh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 83–91 CrossRef CAS PubMed.
- A. T. Byrne, D. G. Alférez, F. Amant, D. Annibali, J. Arribas, A. V. Biankin, A. Bruna, E. Budinská, C. Caldas, D. K. Chang, R. B. Clarke, H. Clevers, G. Coukos, V. Dangles-Marie, S. G. Eckhardt, E. Gonzalez-Suarez, E. Hermans, M. Hidalgo, M. A. Jarzabek, S. de Jong, J. Jonkers, K. Kemper, L. Lanfrancone, G. M. Mælandsmo, E. Marangoni, J.-C. Marine, E. Medico, J. H. Norum, H. G. Palmer, D. S. Peeper, P. G. Pelicci, A. Piris-Gimenez, S. Roman-Roman, O. M. Rueda, J. Seoane, V. Serra, L. Soucek, D. Vanhecke, A. Villanueva, E. Vinolo, A. Bertotti and L. Trusolino, Nat. Rev. Cancer, 2017, 17, 254–268 CrossRef CAS PubMed.
- C. Laurent, D. Gentien, S. Piperno-Neumann, F. Némati, A. Nicolas, B. Tesson, L. Desjardins, P. Mariani, A. Rapinat, X. Sastre-Garau, J. Couturier, P. Hupé, L. de Koning, T. Dubois, S. Roman-Roman, M.-H. Stern, E. Barillot, J. W. Harbour, S. Saule and D. Decaudin, Mol. Oncol., 2013, 7, 625–636 CrossRef CAS PubMed.
- H. Bougherara, F. Némati, A. Nicolas, G. Massonnet, M. Pugnière, C. Ngô, M.-A. Le Frère-Belda, A. Leary, J. Alexandre, D. Meseure, J.-M. Barret, I. Navarro-Teulon, A. Pèlegrin, S. Roman-Roman, J.-F. Prost, E. Donnadieu and D. Decaudin, Oncotarget, 2017, 8, 99950–99965 CrossRef PubMed.
- F. Reyal, C. Guyader, C. Decraene, C. Lucchesi, N. Auger, F. Assayag, L. De Plater, D. Gentien, M.-F. Poupon, P. Cottu, P. De Cremoux, P. Gestraud, A. Vincent-Salomon, J.-J. Fontaine, S. Roman-Roman, O. Delattre, D. Decaudin and E. Marangoni, Breast Cancer Res., 2012, 14, R11 CrossRef CAS PubMed.
- E. Izumchenko, K. Paz, D. Ciznadija, I. Sloma, A. Katz, D. Vasquez-Dunddel, I. Ben-Zvi, J. Stebbing, W. McGuire, W. Harris, R. Maki, A. Gaya, A. Bedi, S. Zacharoulis, R. Ravi, L. H. Wexler, M. O. Hoque, C. Rodriguez-Galindo, H. Pass, N. Peled, A. Davies, R. Morris, M. Hidalgo and D. Sidransky, Ann. Oncol., 2017, 28, 2595–2605 CrossRef CAS PubMed.
- D. Van Haute and J. M. Berlin, Ther. Delivery, 2017, 8, 763–774 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05693c |
| This journal is © The Royal Society of Chemistry 2023 |