
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rate and equilibrium constants for the addition of triazolium salt derived N-heterocyclic carbenes to heteroaromatic aldehydes†
Zhuan
Duan
 a,
Claire M.
Young
a,
Jiayun
Zhu
b,
Alexandra M. Z.
Slawin
a,
AnnMarie C.
O'Donoghue
*b and
Andrew D.
Smith
*a
a,
Claire M.
Young
a,
Jiayun
Zhu
b,
Alexandra M. Z.
Slawin
a,
AnnMarie C.
O'Donoghue
*b and
Andrew D.
Smith
*a
aEaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, Fife KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
bDepartment of Chemistry, Durham University, South Road, Durham DH1 3LE, UK. E-mail: annmarie.odonoghue@durham.ac.uk
First published on 14th November 2022
Abstract
Heteroaromatic aldehydes are often used preferentially or exclusively in a range of NHC-catalysed processes that proceed through the generation of a reactive diaminoenol or Breslow Intermediate (BI), with the reason for their unique reactivity currently underexplored. This manuscript reports measurement of rate and equilibrium constants for the reaction between N-aryl triazolium NHCs and heteroaromatic aldehydes, providing insight into the effect of the NHC and heteroaromatic aldehyde structure up to formation of the BI. Variation in NHC catalyst and heteroaromatic aldehyde structure markedly affect the observed kinetic parameters of adduct formation, decay to starting materials and onward reaction to BI. In particular, large effects are observed with both 3-halogen (Br, F) and 3-methyl substituted pyridine-2-carboxaldehyde derivatives which substantially favour formation of the tetrahedral intermediate relative to benzaldehyde derivatives. Key observations indicate that increased steric hindrance leads to a reduction in both k2 and k−1 for large (2,6-disubstituted)-N-Ar groups within the triazolium scaffold, and sterically demanding aldehyde substituents in the 3-position, but not in the 6-position of the pyridine-2-carboxaldehyde derivatives. As part of this study, the isolation and characterisation of twenty tetrahedral adducts formed upon addition of N-aryl triazolium derived NHCs into heteroaromatic aldehydes are described. These adducts are key intermediates in NHC-catalysed umpolung addition of heteroaromatic aldehydes and are BI precursors.
Introduction
The umpolung activation of aldehydes using N-heterocyclic carbene (NHC) catalysts is a widely applicable reaction mode and is generally regarded as a cornerstone of organocatalysis. These reactions harness the ability of NHCs to generate a key catalytic intermediate diaminoenol, commonly referred to as the Breslow Intermediate (BI, Scheme 1), in a remarkable range of synthetic processes.1 In recent reports, a significant trend has emerged in the preferred or sometimes exclusive use of heteroaromatic aldehydes as substrates, with furyl-, pyridyl- and quinolyl-derivatives commonplace despite reasons for their preferential reactivity remaining relatively unexplored.2 As well as providing unique reactivity, the incorporation of these heteroaromatic motifs in NHC-catalysed reactions is of widespread interest due to their importance in target bioactive compounds and natural products.3 As representative examples, interception of the BI (derived from heteroaromatic aldehydes and the NHC catalyst and acting as an acyl anion equivalent) with alternative electrophiles has been harnessed in NHC-catalysed transformations (Scheme 1). In a rare example of cross-benzoin reactions with heteroaromatic aldehydes, Gravel demonstrated the diastereoselective benzoin reaction of heteroaromatic aldehydes (predominantly using furfural) with enantiopure alkyl amino aldehydes (Scheme 1a).4 In related reports, the observed anti-diastereoselectivity was rationalised to be dependent upon the N–H substituent, with use of tertiary amines leading to syn-diastereoselectivity.5 In 2013 Gaunt reported the formation of ketones from a range of heteroaromatic aldehydes via arylation of the corresponding Breslow intermediate using iodonium salts to give a wide range of heteroaromatic ketones in good to excellent yields (Scheme 1b).6 Furthermore, in 2009, Rovis demonstrated the enantioselective intermolecular Stetter-type addition of heteroaromatic aldehydes (predominantly 2-pyridine carboxaldehyde) to nitroolefins giving the addition products in excellent yield and er (Scheme 1c).7a Based upon invariance in reactivity and selectivity with electronic variation within the heteroarene, it was suggested that the pyridine nitrogen lone pair was not acting as a base in this transformation but may bias reactivity due to steric effects when compared with benzaldehydes.7b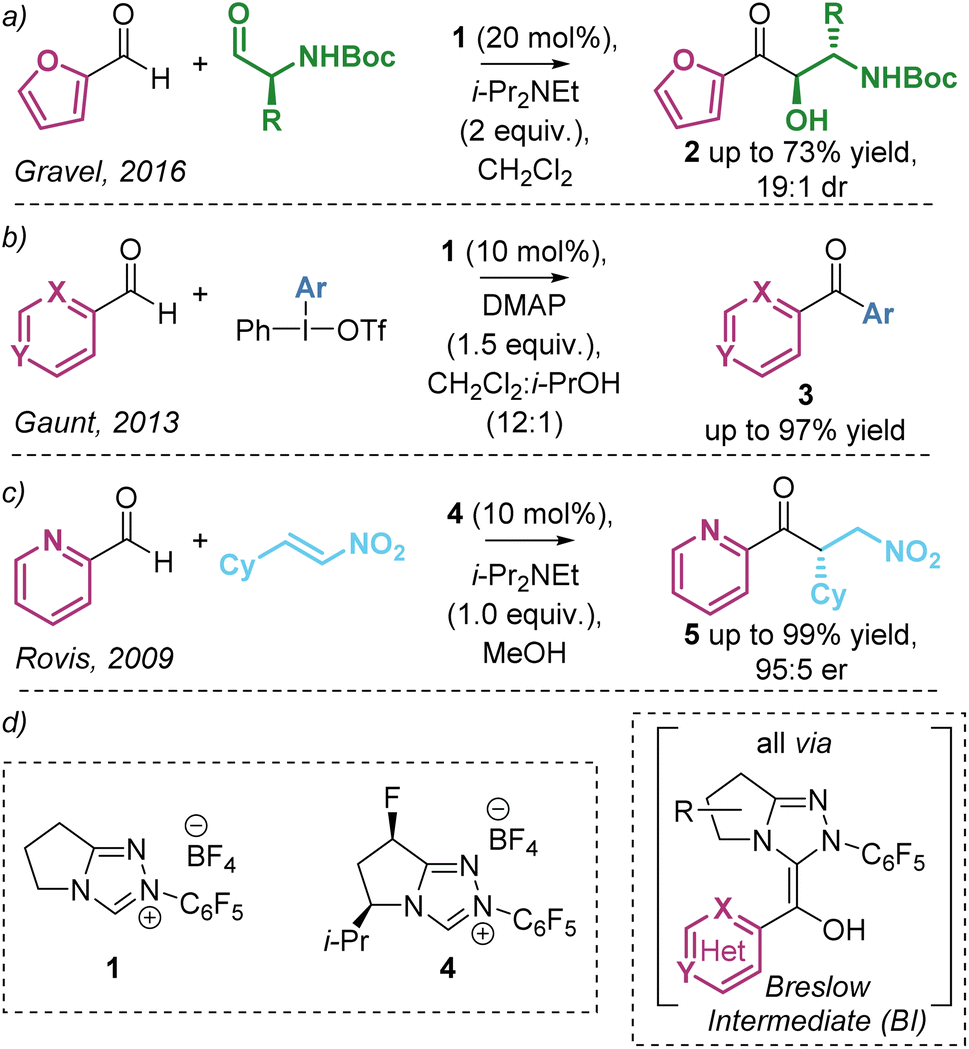 | ||
| Scheme 1 NHC-catalysed addition of heteroaromatic aldehydes to electrophiles via acyl anion equivalent Breslow intermediates. | ||
Despite significant interest in the use of NHCs as organocatalysts, complete and rigorous mechanistic studies of processes proposed to involve the BI are still limited, with significant discussion of the role and character of the BI still ongoing.8 The original mechanism proposed by Breslow in 1958 (ref. 9) was based on Lapworth's mechanism for cyanide-catalysed benzoin formation10 and proceeds through a tetrahedral adduct of the NHC and aldehyde followed by proton transfer to form the reactive aminoenol (Scheme 2a). Reaction with aldehyde and elimination of the catalyst gives the homo-benzoin product. In recent studies, evidence of radical intermediates in the benzoin reaction has been shown computationally and experimentally, particularly in the presence of molecular oxidants, indicating that radical intermediates are feasible and should be considered.11 Under the polar protic conditions explored in this manuscript, an ionic proton-transfer mechanism is expected to dominate due to the rapidity of proton exchange under the methanolic reaction conditions. Furthermore, no radical derived side products have been observed under our reaction conditions.
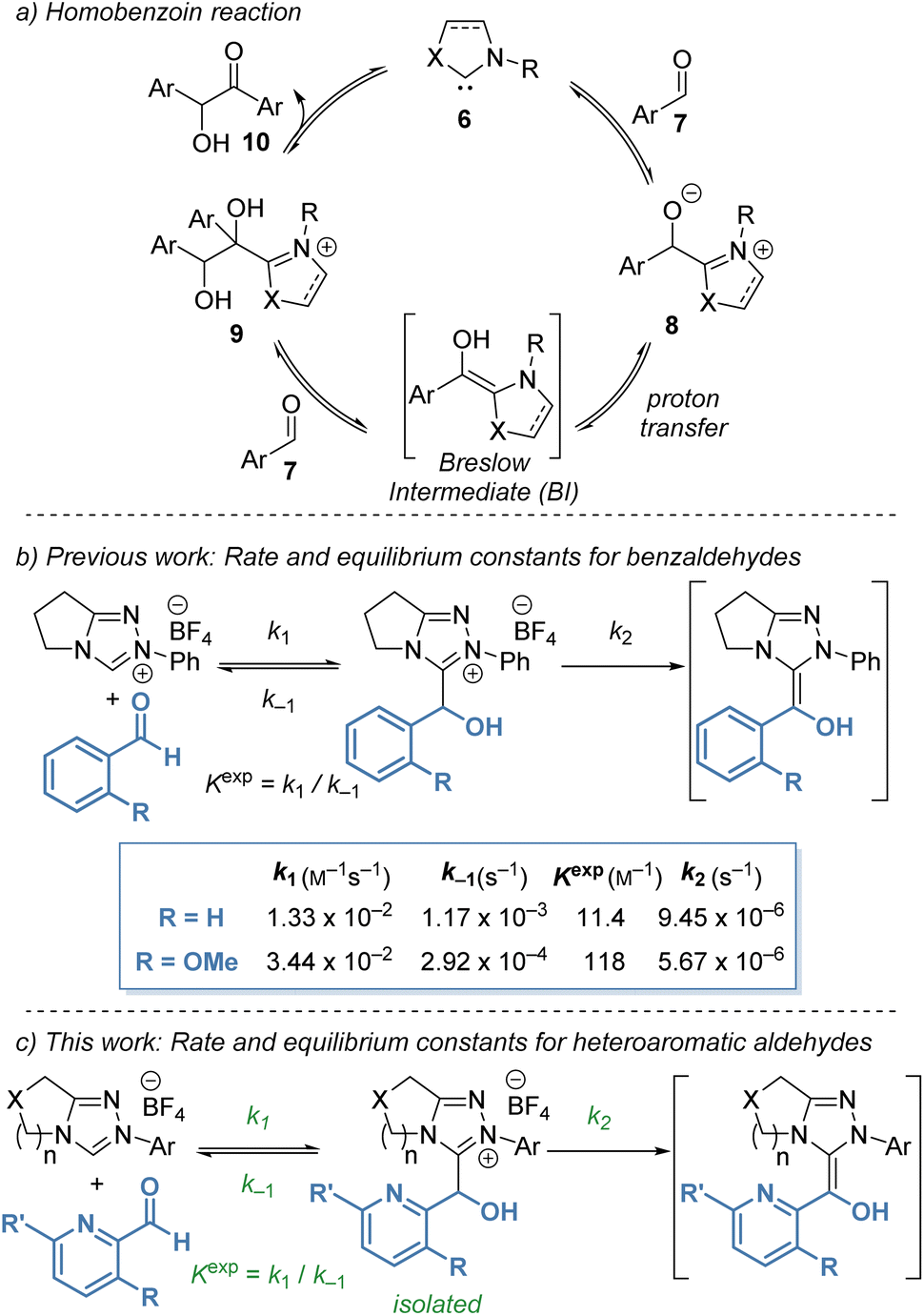 | ||
Scheme 2 Mechanistic investigation of Breslow intermediate formation from b: substituted benzaldehydes and c: heteroaromatic aldehydes (this work). Starting concentrations for b: aldehyde (0.04 M), NHC precatalyst (0.04 M) in CD3OD and 0.18 M Et3N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et3N·HCl (2:1) buffer at 25 °C. Starting concentrations for c: aldehyde (0.02 M), NHC precatalyst (0.02 M) in CD3OD and 0.09 M Et3N:Et3N·HCl (2:1) buffer at 25 °C. :Et3N·HCl (2:1) buffer at 25 °C. Starting concentrations for c: aldehyde (0.02 M), NHC precatalyst (0.02 M) in CD3OD and 0.09 M Et3N:Et3N·HCl (2:1) buffer at 25 °C. | ||
In previous work, our groups investigated the rate and equilibrium constants for the reaction of NHCs derived from triazolium salts with benzaldehydes in triethylamine buffered methanol (CD3OD and 0.18 M Et3N:Et3N·HCl (2:1) buffer at 25 °C).12 Benzaldehydes bearing heteroatomic 2-substituents exhibited significantly higher Kexp than the parent benzaldehyde (Scheme 2b). For example, 2-methoxybenzaldehyde (R = OMe) gave a ten-fold increase in Kexp when compared with benzaldehyde. Further kinetic investigation of the reaction of various aldehydes with triazolium salts revealed that the increase in Kexp for 2-substituted aldehydes was a compound result of an increase in k1 and a reduction in k−1 for formation and decay of tetrahedral hydroxyaryl adduct, respectively. Despite the high Kexp, 2-substituted aldehydes do not react selectively as nucleophilic partners in cross benzoin reactions. Instead, they are highly selective electrophilic partners, with this selectivity ascribed to onwards reactivity post BI formation. Also identified in this study, was an increased Kexp for 2-pyridyl carboxaldehyde in CH2Cl2 in comparison with benzaldehyde.
Intrigued by these observations, and the use of heteroaromatic aldehydes in NHC organocatalysis, we sought to investigate the kinetics of the reaction of heteroaromatic aldehydes with NHCs to determine if trends in reactivity could be identified. In this manuscript, the synthesis and isolation of a large range of tetrahedral adducts (20) formed from NHCs derived from triazolium salts and heteroaromatic aldehydes are reported (Scheme 2c).13 Kinetic analysis of tetrahedral adduct formation and decay was performed, and the effect of both catalyst structure (N-aryl substitution, variation of the fused ring-size (n) and incorporation of an oxygen atom within the fused ring) as well as heteroaromatic aldehyde structure on these rates is reported. Given the enormous interest in incorporating heteroaromatic groups into pharmaceutical and agrochemical compounds, understanding the reactivity of these heteroaromatic aldehydes in NHC-catalysed processes will be of significant interest.
Results and discussion
Synthesis and characterisation of tetrahedral adducts
To compare the rate and equilibrium constants for NHC addition to heteroaromatic aldehydes, characterisation of the corresponding tetrahedral adducts to allow unambiguous confirmation of their constitution was required (Scheme 3). Studies therefore began with the isolation of tetrahedral adducts through stoichiometric reaction of the NHCs derived from triazolium salts and heteroaromatic aldehydes in CH2Cl2 at room temperature. The reactions were allowed to reach equilibrium before direct purification by flash column chromatography to give the tetrahedral adducts in acceptable to good yields. A notable exception to this generalisation was observed when triazolium salts with 6- and 7-membered (n = 2, 3) auxiliary rings were used, with isolated yields generally lower, ranging from 27% to 45%. A selection of heteroaromatic aldehydes and triazolium salts were employed to facilitate the investigation of various steric and electronic aspects of each reaction component. For the heteroaromatic aldehydes, pyridine-2-carboxaldehyde derivatives with substituents in the 3-position (ortho to the aldehyde, R) or 6-position (ortho to the pyridine nitrogen, R′) as well as furfural were employed. For the triazolium salts, structural changes included variation of the N-aryl substituent on the triazolium nitrogen (Ar), variation of fused ring-size (n) and incorporation of an oxygen atom within the fused ring (X). Unfortunately, tetrahedral intermediates arising from the use of N-C6F5 substituted azolium salt 1 were not stable to purification and could not be isolated, although N-Ph, N-Mes and N-C6Cl3H2 substituted variants proved isolable.14 X-ray crystallographic analysis of 13 allowed unambiguous confirmation of product constitution.15 Due to their propensity to onward reactions under these conditions, 3- and 4-pyridyl, and quinolyl aldehydes led to complex mixtures from which the tetrahedral adducts 31, 32 and 33 could not be isolated but were identified spectroscopically in the crude reaction mixtures.16 | ||
| Scheme 3 Preparation and characterization of tetrahedral intermediates. | ||
Kinetic analysis
Under reaction conditions first reported by Leeper and since adapted by us and others to give consistent and comparable experimental results, the reaction of the NHCs with heteroaromatic aldehydes were monitored by 1H NMR in buffered methanol (triazolium salt 20 mM, aldehyde 20 mM, Et3N:Et3N·HCl (2:1) 90 mM in CD3OD at 25 °C).17 The pseudo second order rate constant of tetrahedral adduct formation was measured (k1) as well as the equilibrium constant for adduct formation (Kexp) (Table 1). The pseudo-first order rate constant for tetrahedral adduct decay (k−1) was calculated based on the measured values for Kexp and k1. The pseudo-first order reaction constant for forward reaction of the tetrahedral adduct (k2) was also estimated from the rate of decay of the tetrahedral intermediate once equilibrium for the intermediate formation had been established.16 It is assumed that this forward reaction proceeds via the corresponding BI. Although this intermediate cannot be observed directly under these polar protic conditions, the observation of H/D-exchange at Cα implicates a carbanionic intermediate. Furthermore, the increases in k2 observed in studies to date with electron withdrawing substituents on catalysts or aldehyde supports this proton transfer mechanism via a delocalised carbanion (cf. the diaminoenol or BI).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Tetrahedral adduct | Aldehyde | Triazolium salt | k 1 (×10−2 M−1 s−1) | k 1 fit (×10−2 M−1 s−1) | k −1 (×10−5 s−1) | k −1 fit (×10−5 s−1) | K exp (×102 M−1) | K fit (×102 M−1) | k 2 (×10−6 s−1) |
|
a Conditions: triazolium salt 20 mM, aldehyde 20 mM, Et3N:Et3N·HCl (2:1) 90 mM in CD3OD. k1, k−1, Kexp and k2 obtained from 1H NMR analysis, kfit1, k−1fit and Kfit obtained using Berkeley Madonna fitting software based on experimental data. See ESI for details.
b Competitive benzoin reaction prevented the experimental reaction from reaching equilibrium over the reaction course and so experimental data could not be calculated. The rate and equilibrium constants were extracted from experimental data through global fitting using Berkeley Madonna.
c The reaction proceeded rapidly and individual rate constants could not be calculated or extracted from global fitting using Berkeley Madonna.
d
k
2 could not be measured as no decrease in adduct concentration was observed.
|
||||||||||
| 1 | 11 | 34 | 39 | —b | 58.8 | —b | 29.4 | —b | 20.0 | 115 |
| 2 | 12 | 35 | 39 | 19.3 | 19.1 | 4.38 | 4.51 | 43.6 | 42.4 | 10.8 |
| 3 | 13 | 36 | 39 | 366 | 366 | 9.09 | 9.11 | 403 | 402 | 87.9 |
| 4 | 14 | 37 | 39 | —b | 253 | —b | 26.9 | —b | 94.1 | 344 |
| 5 | 15 | 38 | 39 | —b | 42.6 | —b | 21.6 | —b | 19.7 | 81.4 |
| 6 | 17 | 34 | 42 | —b | 77.7 | —b | 3.19 | —b | 244 | 105 |
| 7 | 18 | 35 | 42 | 35.0 | 33.9 | 0.725 | 0.710 | 482 | 477 | 5.65 |
| 8 | 19 | 36 | 42 | 491 | 424 | 1.30 | 1.11 | 3770 | 3810 | 9.00 |
| 9 | 20 | 37 | 42 | —b | 294 | —b | 3.28 | —b | 896 | 254 |
| 10 | 21 | 38 | 42 | —b | 79.1 | —b | 3.50 | —b | 226 | 64.3 |
| 11 | 23 | 34 | 44 | —c | —c | —c | —c | 481 | —c | 6190 |
| 12 | 47 | 35 | 44 | —c | —c | —c | —c | 1040 | —c | 1040 |
| 13 | 24 | 34 | 40 | —b | 5.06 | —b | 8.53 | —b | 5.93 | 3.94 |
| 14 | 25 | 34 | 41 | —b | 4.30 | —b | 15.4 | —b | 2.79 | —d |
| 15 | 26 | 35 | 40 | 1.86 | 1.92 | 4.11 | 4.12 | 4.53 | 4.66 | 2.28 |
| 16 | 27 | 35 | 41 | 1.27 | 1.21 | 7.99 | 7.37 | 1.59 | 1.65 | —d |
| 17 | 28 | 35 | 43 | 8.90 | 8.19 | 1.13 | 1.04 | 78.7 | 78.8 | 3.61 |
| 18 | 29 | 35 | 45 | 24.3 | 23.5 | 27.9 | 27.3 | 8.61 | 8.72 | 14.5 |
| 19 | 30 | 35 | 46 | 115 | 117 | 7.37 | 7.53 | 156 | 155 | 18.6 |
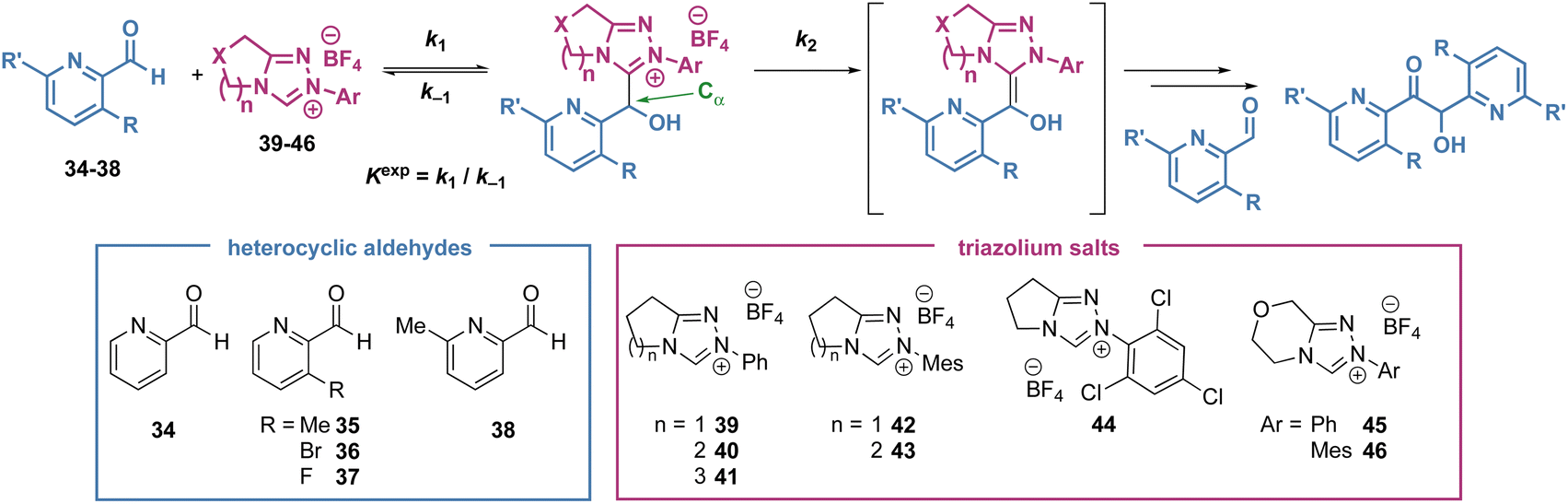
Equilibrium constants for the decay of selected isolated adducts to their respective aldehydes and NHCs (Kdiss) under identical reaction conditions were also measured. While performing the experiments, competitive generation of benzoin product in some cases limited the data that could be measured experimentally. For example, with furfural no useable data could be extracted despite 16 and 22 being isolable and so kinetic analysis was not feasible. With pyridyl aldehyde 34, the limited data measured by experiments could be fitted using Berkeley Madonna global fitting software to access the rate constants. This fitting using Berkeley Madonna was also performed for reactions where the experimental data could be used directly, with the parameters obtained from fitting in good agreement with the experimental data in all cases.
An initial observation comparing pyridine-2-carboxaldehyde with benzaldehyde (under slightly modified conditions, Scheme 2), revealed a significant increase in forward rate constants for the pyridyl aldehyde (k1 = 5.88 × 10−1 M−1 s−1, k2 = 1.15 × 10−4 s−1) and decrease in k−1 (2.94 × 10−4 s−1) reflected in an almost 200-fold increase in Kexp (2.00 × 103 M−1) (Table 1, entry 1 and Scheme 2b). Further trends in the reaction of pyridyl aldehydes with triazolium salts were observed and are discussed below.
Tetrahedral adduct formation (k1, k−1, Kexp)
The observed increase in k1 for 36 (R = Br) and 37 (R = F) can in part be attributed to an increase in electrophilicity of aldehyde when an electron-withdrawing group is ortho to the aldehyde. Given Br- and F- substituents have closely similar electron withdrawing substituent effects (e.g. Hammett σm = +0.39 (Br), +0.34 (F))18 the substantially enhanced k1 and Kexp for 36 (R = Br) points to an additional steric influence in this case. By twisting out of conjugation with the aldehyde and diminishing the donating effect of the heteroatom lone pair, sterically bulky 2-substituents (such as when R = Br) can additionally increase aldehyde reactivity towards nucleophiles.19 Steric effects are also revealed in a comparison of k−1 values. The decrease in k−1 for 35 and 36 (R = Me and R = Br) compared with 34 and 37 (R = H and R = F) parallels the observed trend of bulky substituents disfavouring reformation of starting materials from the tetrahedral intermediate.
Overall, these results clearly demonstrate that large 2-substituent effects are also observed for heteroaromatic aldehydes, which further enhance the effect of the 2-heteroatom in favouring formation of the tetrahedral intermediate. The largest combined effect is observed for the combination of the pyridyl nitrogen and ortho-bromo substituents with Kexp for 36 and triazolium 39 almost 4000-fold larger than for benzaldehyde (Table 1, entry 3 and Scheme 2).
In our recent study of the Cα-H/D exchange reactions of a large series of twenty bicyclic triazolium salts,22 a similar trend was observed with higher key rate constants for the 5-fused series (n = 1) and similar, smaller values for the 6- and 7-fused salts (n = 2 and n = 3). Based on X-ray structural data obtained for the twenty triazolium salts and DFT computational evaluation, these changes were attributed to several structural effects including changes in internal triazolyl NCN angle and positioning of the most proximal CH2 with variation in fused ring size. In the case of Cα-H/D exchange reactions, subtle changes were observed with kex up to 2.2 times larger for n = 1 compared with n = 2. In this study, the effect is magnified with k1 and Kexp in the range of four to ten times larger for n = 1 vs. n = 2 (39vs.40 and 42vs.43), and smaller but significant differences observed comparing n = 2 with n = 3 (40vs.41). This enhanced effect highlighted by our present results is likely a result of the greater steric demand for the addition to an aldehyde compared to relatively undemanding H/D exchange process.
Forward reaction of tetrahedral adduct (k2)
For a given aldehyde, the rate constant of the onward reaction of the tetrahedral adduct, k2, is generally in the same order of magnitude for 39 (n = 1, Ar = Ph) and 42 (n = 1, Ar = Mes) and, in the cases where it could be measured, was significantly higher for 44 (n = 1, Ar = C6Cl3H2) (entries 1, 6 and 11; 2, 7 and 12). Taking pyridine-2-carboxaldehyde 34 to demonstrate (entries 1, 6 and 11), the measured k2 values for 39 and 42 were roughly similar (k2 = 1.05 × 10−4 s−1 (39) and 1.15 × 10−4 s−1 (42)) but significantly larger for 44 (k2 = 6.19 × 10−3 s−1), consistent with electron withdrawing N-aryl substituents facilitating deprotonation. Although the trend is maintained for most aldehydes (35 (R = Me), 37 (R = F) and 38 (R′= Me)) in reactions with 39 and 42 (entries 2 and 7; 4 and 9; 5 and 10), using aldehyde 36 (R = Br) a significantly smaller k2 is observed for 42 (Ar = Mes, k2 = 9.00 × 10−6 s−1, entry 13) compared with 39 (Ar = Ph, k2 = 87.9 × 10−6 s−1, entry 12). The electron withdrawing electronic effect of an Br substituent ortho to the aldehyde would be expected to favour this deprotonation step, thus the observed ∼10-fold decrease in k2 is attributed to steric factors when 42 is used in combination with aldehydes bearing bulky 2-substituents. This provides another example of additional steric effects present in heteroaromatic aldehydes with ortho-bromo substituents, with the magnitude also dependent on the N-Ar substituent.For the size and constitution of the fused ring, decrease in ring size leads to a substantial decrease in k2 (e.g. 1.15 × 10−4 s−1 (39, n = 1) vs. 3.94 × 10−6 s−1 (40, n = 2) and negligible (41, n = 3) in reaction with 34, entries 1, 13 and 14). Inclusion of oxygen in the fused ring leads to a slight increase in k2 with either N-Ph (2.28 × 10−6 s−1 (40) vs. 1.45 × 10−5 s−1 (45), entries 15 and 18) or N-Mes catalysts (k2 = 3.61 × 10−6 s−1 (43) vs. 1.86 × 10−5 s−1 (46, entries 17 and 19).
The effect of aldehyde structure on k2 was also assessed. In reactions with both 39 (Ar = Ph) and 42 (Ar = Mes), significant decreases in k2 were observed for 35 (R = Me) when compared with 34 (R = H) (entries 1 and 2; 6 and 7), while increases in k2 were observed for 37 (R = F) when compared with 34 (R = H) (entries 1 and 4; 5 and 9). Using 39, a slight decrease in k2 is observed for 36 (R = Br) compared with 34 (R = H) (1.15 × 10−4 s−1 (34) vs. 8.79 × 10−5 s−1 (36), entries 1 and 3) but in reaction with 42, a more significant 10-fold decease is observed (1.05 × 10−4 s−1 (34) vs. 9.00 × 10−6 s−1 (36), entries 6 and 8) further highlighting the importance of steric effects in reaction of the tetrahedral adducts. Again, only a slight decrease in k2 is observed for 38 (R′= Me) vs.34 irrespective of triazolium (39 (entries 1 and 5) or 42 (entries 6 and 10)) indicating the minimal effect of 6-substitution on the pyridyl aldehyde.
Tetrahedral adduct dissociation (Kdiss)
To further validate these rate and equilibrium constants, the decay of selected tetrahedral adducts towards equilibrium was studied (Table 2). Analysis of the 1H NMR reaction profiles for dissociation of selected adducts of aldehyde 35 allowed rate and equilibrium constants of dissociation to be measured (kd, s−1 and Kdiss, M−1) and rate constants for association (ka, M−1 s−1) to be calculated. In this analysis, although ka = k1 and kd = k−1 a distinction has been made to differentiate between the two methods of measurement for either the forward (addition of NHC to aldehyde) or reverse (dissociation of tetrahedral adduct) processes. The observation of closely similar values for ka and k1, or kd and k−1, through starting from different ends of the equilibrium gives confidence in the absolute and relative magnitudes of these values. For example, the dissociation rate constant kd (4.54 × 10−5 s−1) of tetrahedral adduct 12 (Table 2, entry 1) is close to the reverse rate constant k−1 (4.38 × 10−5 s−1) of tetrahedral adduct 12 formation (Table 1, entry 2).|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Triazolium salt | Tetrahedral adduct | k d (×10−5 s−1) | k d fit (×10−5 s−1) | k a (×10−2 M−1 s−1) | k a fit (×10−2 M−1 s−1) | K diss (×10−5 M−1) | K fit (×10−5 M−1) | 1/Kdiss (×102 M−1) |
|
a Conditions: tetrahedral adduct 20 mM, Et3N:Et3N·HCl (2:1) 90 mM in CD3OD at 25 °C.
|
|||||||||
| 1 | 39 | 12 | 4.54 | 4.06 | 20.7 | 16.6 | 21.9 | 24.5 | 45.7 |
| 2 | 42 | 18 | 0.882 | 0.879 | 41.8 | 43.5 | 2.11 | 2.02 | 474 |
| 3 | 45 | 29 | 25.6 | 26.1 | 23.3 | 24.8 | 11.0 | 10.5 | 9.09 |
| 4 | 46 | 30 | 8.46 | 8.57 | 135 | 136 | 6.27 | 6.30 | 160 |

The rate constant ka (2.07 × 10−1 M−1 s−1) and the reciprocal (4.57 × 103 M−1) of equilibrium constant Kdiss of tetrahedral adduct 12 dissociation were also respectively similar to rate constant k1 (1.93 × 10−1 M−1 s−1) and equilibrium constant Kexp (4.36 × 103 M−1) of adduct 12 formation (kd ≈ k−1, ka ≈ k1, 1/Kdiss ≈ Kexp). The same trend was also observed comparing the corresponding rate and equilibrium constants of adduct formation and dissociation for other triazolium salts (entries 2–4 in Table 2vs. entries 7, 18, 19 in Table 1).23
Conclusions
In conclusion, measurements of equilibrium and rate constants for the reaction of triazolium NHC precatalysts with heteroaromatic aldehydes to give tetrahedral 3-(hydroxybenzyl)azolium adducts under stoichiometric conditions have been made. Twenty tetrahedral adducts were isolated and fully characterised including X-ray crystallographic analysis for 13. The results obtained from kinetic analysis and fitting data show that nucleophilic addition into pyridyl aldehydes is fast compared with existing data for addition to benzaldehydes. Notably, the combined effect of the heteroatom of the heterocyclic aldehyde and an additional substituent both ortho to the aldehydic position result in exceptionally large enhancements in equilibrium constants for tetrahedral adduct formation. In particular, large effects are observed with both 3-halogen (Br, F) and 3-methyl substituted pyridine-2-carboxaldehydes, which together substantially favour formation of the tetrahedral intermediate relative to benzaldehyde derivatives. Given this initial adduct-forming step is common to all NHC-catalysed transformations of aldehydes involving the BI, the accelerated formation of adduct clearly must underpin the preferential reactivity of heteroaromatic aldehydes. Catalyst and aldehyde structure affect the observed kinetics of adduct formation, decay to starting materials and onward reaction to BI. Key observations indicate that increased steric hindrance lead to a reduction in both k2 and k−1 for large (2,6-disubstituted)-N-Ar groups and sterically demanding aldehyde substituents in the 3-position, but not in the 6-position of the pyridine-2-carboxaldehyde derivatives. A decrease in the forward reaction constants (k1, k2) for larger fused rings (n = 2, 3) on the NHC is observed, but an increase in the forward reaction constants is observed upon incorporation of heteroatom in the fused ring. Further work from our laboratories will extend this type of analysis to alternative aldehyde and NHC-catalyst families.Data availability
The research data supporting this publication can be accessed at https://doi.org/10.17630/4e21c331-2fa6-440f-8636-f9ed563bda3c.Author contributions
ADS and AOD designed the project. ZD, CMY and JZ carried out the experimental work and analysed the data. AMZS carried out X-ray crystallographic analysis. CMY, ADS and AOD wrote the paper with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (JZ and CMY, EP/S020713/1 and EP/S019359/1) and ZD (CSC St Andrews PhD studentship) for funding.Notes and references
- (a) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 CrossRef CAS; (b) N. Gaggero and S. Pandini, Org. Biomol. Chem., 2017, 15, 6867 RSC.
- For selected examples, see: (a) D. Enders, A. Grossman, J. Fronert and G. Raabe, Chem. Commun., 2010, 46, 6282 RSC; (b) D. Enders, A. Henseler and S. Lowins, Synthesis, 2009, 24, 4125 Search PubMed; (c) Y. Bai, S. Xiang, M. L. Leow and X.-W. Liu, Chem. Commun., 2014, 50, 6168 RSC; (d) J. A. Murry, D. E. Frantz, A. Soheili, R. Tillyer, E. J. J. Grabowski and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 9696 CrossRef CAS PubMed; (e) W. Bi, Y. Yang, S. Ye and C. Wang, Chem. Commun., 2021, 57, 4452 RSC.
- (a) M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247 RSC; (b) A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611 RSC.
- P. Haghshenas and M. Gravel, Org. Lett., 2016, 18, 4518 CrossRef CAS PubMed.
- P. Haghshenas, J. Wilson Quail and M. Gravel, J. Org. Chem., 2016, 81, 12075 CrossRef CAS.
- Q. Yan Toh, A. McNally, S. Vera, N. Erdmann and M. J. Gaunt, J. Am. Chem. Soc., 2013, 135, 3772 CrossRef.
- (a) D. DiRocco, K. M. Oberg, D. M. Dalton and T. Rovis, J. Am. Chem. Soc., 2009, 131, 10872 CrossRef CAS PubMed; (b) D. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402 CrossRef CAS PubMed.
- (a) M. Pareek, Y. Reddi and R. B. Sunoj, Chem. Sci., 2021, 12, 7973 RSC; (b) A. Berkessel, S. Elfert, K. Etzenbach-Effers and J. H. Teles, Angew. Chem., Int. Ed., 2010, 49, 7120 CrossRef CAS; (c) A. Berkessel, S. Elfert, R. Yatham, J. Neudörfl, N. Schlörer and J. H. Teles, Angew. Chem., Int. Ed., 2012, 51, 12370 CrossRef CAS; (d) A. Berkessel, V. R. Yatham, S. Elfert and J.-M. Neudörfl, Angew. Chem., Int. Ed., 2013, 52, 11158 CrossRef CAS; (e) M. Paul, M. Breugst, J.-M. Neudörfl, R. B. Sunoj and A. Berkessel, J. Am. Chem. Soc., 2016, 138, 5044 CrossRef CAS PubMed; (f) M. Paul, P. Sudkaow, A. Wessels, N. E. Schlörer, J.-M. Neudörfl and A. Berkessel, Angew. Chem., Int. Ed., 2018, 57, 8310 CrossRef CAS; (g) M. Paul, J.-M. Neudörfl and A. Berkessel, Angew. Chem., Int. Ed., 2019, 58, 10596 CrossRef CAS PubMed; (h) M. Paul, K. Peckelsen, T. Thomulka, J. Martens, G. Berden, J. Oomens, J.-M. Neudörfl, M. Breugst, A. J. H. M. Meijer, M. Schäfer and A. Berkessel, Chem.–Eur. J., 2021, 27, 2662 CrossRef CAS PubMed; (i) A. Wessels, M. Klußmann, M. Breugst, N. E. Schlörer and A. Berkessel, Angew. Chem., Int. Ed., 2022, 61, e202117682 CAS.
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS.
- (a) A. Lapworth, J. Chem. Soc., Trans., 1903, 83, 995 RSC; (b) A. Lapworth, J. Chem. Soc., Trans., 1904, 85, 1206 RSC.
- (a) J. Rehbein, S.-M. Ruser and J. Phan, Chem. Sci., 2015, 6, 6013 RSC; (b) S. Alwarsh, Y. Xu, S. Y. Qian and M. C. McIntosh, Angew. Chem., Int. Ed., 2016, 55, 355 CrossRef CAS; (c) M.-H. Hsieh, G.-T. Huang and J.-S. K. Yu, J. Org. Chem., 2018, 83, 15202 CrossRef CAS; (d) M.-H. Hsieh and J.-S. K. Yu, Phys. Chem. Chem. Phys., 2021, 23, 27377 RSC; (e) R. Machín Rivera, N. R. Burton, L. D. Call, M. A. Tomat and V. N. G. Lindsay, Org. Lett., 2022, 24, 4275 CrossRef PubMed . For reviews on radical couplings postulated to form via the enolate form of the Breslow intermediate see; (f) T. Ishii, K. Nagao and H. Ohmiya, Chem. Sci., 2020, 11, 5630 RSC.
- (a) C. J. Collett, R. S. Massey, J. E. Taylor, O. R. Maguire, A. C. O'Donoghue and A. D. Smith, Angew. Chem., Int. Ed., 2015, 54, 6887 CrossRef CAS; (b) C. J. Collett, R. S. Massey, O. R. Maguire, A. S. Batsanov, A. C. O'Donoghue and A. D. Smith, Chem. Sci., 2013, 4, 1514 RSC.
- For previous examples of isolable tetrahedral intermediates derived from imidazolium derived NHCs see (a) A. Miyashita, A. Kurachir, Y. Matsuoka, N. Tanabe, Y. Suzuki, K.-I. Iwamoto and T. Higashimo, Heterocycles, 1997, 44, 417–426 CrossRef CAS; (b) S. Ohta, S. Hayakawa, K. Nishimura and M. Okamoto, Chem. Pharm. Bull., 1987, 35, 1058–1069 CrossRef; (c) V. K. Aggarwal, I. Emme and A. Mereu, Chem. Commun., 2002, 1612–1613 RSC; (d) L.-W. Xu, Y. Gao, J.-J. Yin, L. Li and C.-G. Xia, Tetrahedron Lett., 2005, 46, 5317–5320 CrossRef CAS . For those derived from thiazolium derived NHCs see; (e) R. Breslow and R. Kim, Tetrahedron Lett., 1994, 35, 699–702 CrossRef CAS; (f) Y.-T. Chen, G. L. Barletta, K. Haghjoo, J. T. Cheng and F. Jordan, J. Org. Chem., 1994, 59, 7714–7722 CrossRef CAS; (g) M. J. White and F. J. Leeper, J. Org. Chem., 2001, 66, 5124–5131 CrossRef CAS PubMed; (h) W. Schrader, P. P. Handayani, C. Burstein and F. Glorius, Chem. Commun., 2007, 716–718 RSC; (i) M. B. Doughty, G. E. Risinger and S. J. Jungk, Bioorg. Chem., 1987, 15, 15–30 CrossRef CAS; (j) J. Kiss, R. D'Souza and H. Spiegelberg, Helv. Chim. Acta, 1968, 51, 325–339 CrossRef; (k) J. J. Mieyal, G. Bantle, R. G. Votaw, I. A. Rosner and H. Z. Sable, J. Biol. Chem., 1971, 246, 5213–5219 CrossRef CAS; (l) A. Takamizawa, Y. Hamashima and H. Sato, J. Org. Chem., 1968, 33, 4038–4045 CrossRef CAS; (m) Y. Kageyama and S. Murata, J. Org. Chem., 2005, 70, 3140–3147 CrossRef CAS PubMed . For those derived from triazolium derived NHCs see ref. 12 and; (n) J. Henrique Teles, J.-P. Melder, K. Ebel, R. Schneider, E. Gehrer, W. Harder, S. Brode, D. Enders, K. Breuer and G. Raabe, Helv. Chim. Acta, 1996, 79, 61–83 CrossRef.
- Syntheses of tetrahedral intermediates using 1 were attempted but found to be unstable. See ESI† for further details and full characterisation of materials.
- CCDC deposition number 2209647†.
- See ESI† for details.
- (a) M. J. White and F. J. Leeper, J. Org. Chem., 2001, 66, 5124 CrossRef CAS PubMed; (b) R. S. Massey, J. Murray, C. J. Collett, J. Zhu, A. D. Smith and A. C. O'Donoghue, Org. Biomol. Chem., 2021, 19, 387 RSC; (c) C. J. Collett, C. M. Young, R. S. Massey, A. C. O'Donoghue and A. D. Smith, Eur. J. Org. Chem., 2021, 3670 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- One referee queried the use of σm over σp or σp+ as a measure of the electron withdrawing capability of F and Br substituents. Our reasoning for using σm is that for the halogens, inductive effects outweigh mesomeric effects, and so σm is larger in absolute value than σp (owing to the strong electron-withdrawing effect of a halogen and weaker donating effect of the lone pair). There are no σ-values for ortho-substituents, thus we considered σm to be a better measure of the electron withdrawing effect of a halogen than σp as the former is closer to the aldehydic position. We appreciate that this could still underestimate the electron-withdrawing effect as it is further away from the aldehyde than an ortho-substituent. We did consider using σp values, but decided they would only further underestimate the inductive electron withdrawing substituent effect. Although not accounting for ortho-inductive effects, a comparison of σp+ values (Br, 0.15; F, −0.07) does highlight than mesomeric donation from p-F is more favoured than from p-Br, which may also partially contribute to the decreased reactivity of aldehyde 37 (R = F) compared to 36 (R = Br).
- The reaction proceeded rapidly and individual rate constants for reactions with 44 could not be calculated or extracted from global fitting using Berkeley Madonna.
- The O-alkylation of tetrahedral adducts is particularly challenging irrespective of alkylation conditions supporting the proposal of general steric crowding around OH for all tetrahedral adducts; see ref. 8c and C. J. Collett, R. S. Massey, O. R. Maguire, A. S. Batsanov, A. C. O'Donoghue and A. D. Smith, Chem. Sci., 2013, 4, 1514 RSC.
- For an in-depth discussion of the effect of auxiliary ring size, see J. Zhu, I. Moreno, P. Quinn, D. S. Yufit, L. Song, C. M. Young, Z. Duan, A. R. Tyler, P. G. Waddell, M. J. Hall, M. R. Probert, A. D. Smith and A. C. O'Donoghue, J. Org. Chem., 2022, 87, 4241 CrossRef CAS.
- The research data supporting this publication can be accessed at https://doi.org/10.17630/4e21c331-2fa6-440f-8636-f9ed563bda3c.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2209647. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05704b |
| This journal is © The Royal Society of Chemistry 2023 |