
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular trapping of spiro radicals to produce unusual cyclization products from usual migration substrates†
Jingming
Zhang‡
a,
Chengkou
Liu‡
a,
Yaqi
Qiao
a,
Minghui
Wei
a,
Wenjing
Guan
a,
Ziren
Mao
a,
Hong
Qin
a,
Zheng
Fang
 *a and
Kai
Guo
*ab
*a and
Kai
Guo
*ab
aCollege of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, Nanjing, 211816, China. E-mail: fzcpu@njtech.edu.cn; guok@njtech.edu.cn
bState Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, Nanjing, 211816, China
First published on 2nd February 2023
Abstract
A conceptually new methodology to give unusual cyclization products from usual migration substrates was disclosed. The highly complex and structurally important and valuable spirocyclic compounds were produced through radical addition, intramolecular cyclization and ring opening instead of usual migration to the di-functionalization products of olefins. Furthermore, a plausible mechanism was proposed based on a series of mechanistic studies including radical trapping, radical clock, verification experiments of intermediates, isotope labeling and KIE experiments.
Introduction
The quest for novel synthetic methods to construct highly complex and unprecedented molecular scaffolds has increasingly attracted attention because of their utmost importance in both synthetic and medical industry, although only a very small fraction would be confirmed to be useful clinical candidates.1 The rearrangement reaction has been recognized as a practical and powerful synthesis methodology to produce unexpected products through controllable cleavage and reconstruction of chemical bonds.2 In the past century, many ionic rearrangement reactions have been reported including classical Smiles rearrangement, which features a key spirocyclic intermediate.3 Recently, radical chemistry has experienced a major resurgence because of its higher reactivity and better functional-group compatibility.4 In this context, radical reactions including radical-mediated rearrangements have attracted wide attention.5 Nevado and co-workers reported a first radical-mediated di-functionalization of alkenes by concomitant the aryl migration to give the amidyl radical intermediate through radical addition, aryl migration and desulfonylation, which also features a spirocyclic intermediate (Scheme 1a).6 The corresponding amides are obtained from hydrogen abstraction. It is noteworthy that oxindoles are produced through further cyclization. Furthermore, many studies to introduce different radicals including N3,6 phosphonyl,6b alkyl,7 aryl,8 sulfonyl9 and N-centered10 have been carried out. In addition, further cyclization cascades via N-amidyl radicals to many unexpected and highly functionalized heterocyclic scaffolds were investigated.11 Moreover, Zhu and co-workers disclosed a first difunctionalization of alkenes based on remote cyano migration (Scheme 1b).12 Immediately, a series of studies have been carried out to expand the scope of distal migration to numerous functional groups, including CN,13 heteroaryl,14 aryl,15 alkynyl,16 alkenyl,17 formyl,18 oximino,19 and alkoxyphosphine20 by Zhu and many other research groups including us.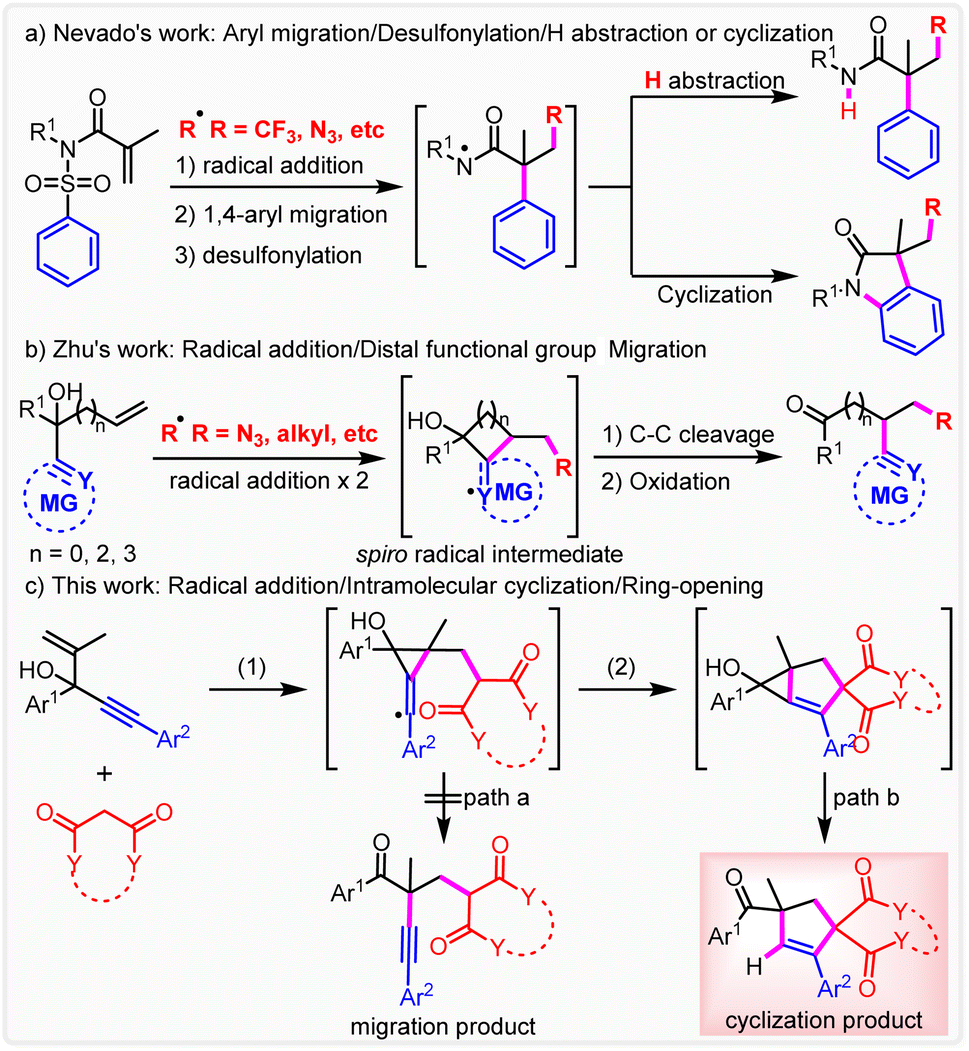 | ||
| Scheme 1 Design of radical-promoted addition/cyclization/ring-opening of 1,4-enynes. (a) Nevado's work: aryl migration/desulfonylation/H abstraction or cyclization; (b) Zhu's work: radical addition/distal functional group migration; (c) This work: radical addition/intramolecular cyclization/ring-opening. | ||
Attributed to the formation of a stabler carbonyl group, the di-functionalization of both activated and un-activated alkenes can be achieved. Besides, more abundant functional groups can be introduced into alkenes conveniently via the migration process, which is usually unreachable under traditional methodologies. Although remote functional group ipso-migration had been fully researched, the intercepting of the transient spiro radical intermediate by an intrinsic or extrinsic radical-trapping reagent to give a spirocyclic or subsequent ring-opening product has never been proposed (Scheme 1c).
In this context, this hypothesis was tested by a reaction between 1,4-enynes and activated methylene compounds, which can be transformed into the corresponding C-centered radical intermediate and also serves as an efficient radical acceptor and donor by radical addition and 1,n-HAT respectively. Herein, we report a first silver-promoted intramolecular trapping of spiro radicals to produce unusual cyclization products from usual migration substrates (Scheme 1c). This reaction involves radical addition, 5-exo-trig cyclization and final ring opening.
Results and discussion
The hypothesis was investigated based on a reaction between 3-(4-(tert-butyl)phenyl)-2-methyl-5-phenylpent-1-en-4-yn-3-ol 1 and 1,3-dimethylbarbituric acid 2 (Table 1). To our delight, 92% cyclization product 3 was generated using Ag2O as the oxidant and ACN as the solvent under 110 °C (entry 1). It was noteworthy that replacing ACN with MeOH led to a comparable yield (89%, entry 2). However, a dramatic decrease of the yield was obtained when other solvents including DMSO, DCE or toluene were involved (entry 2).|
|
||
|---|---|---|
| Entry | Variation from “standard conditions” | Yieldb (%) |
| a Reaction conditions: 1 (1 mmol, 304.2 mg), 2 (1.5 mmol, 234.1 mg), Ag2O (2 mmol, 463.5 mg), ACN (3 mL), 110 °C, and 6 h. b Yields were determined by 1H NMR using dibromomethane as the internal standard. c Isolated yield. d Ag2O (0.2 mmol, 46.4 mg), oxidant (2 mmol), PIDA: (diacetoxyiodo)benzene, and TBPB: tert-butyl peroxybenzoate. | ||
| 1 | None | 92c |
| 2 | MeOH, DMSO, DCE or toluene instead of ACN | 89, 61, 43, 56 |
| 3 | Cu(OAc)2, Cu(AcAc)2, Fe3O4, MnO2, NiO instead of Ag2O | 17, 6, 0, trace, trace |
| 4 | AgOAc, Ag2CO3, AgNO3, AgSbF6 instead of Ag2O | 65, 89, 31, trace |
| 5 | 90, 100 or 120 °C | 58, 78, 94 |
| 6 | 1.2, 1.5 or 1.8 eq. of Ag2O | 75, 83, 88 |
| 7 | 1.1 or 1.3 eq. of 2 | 79, 84 |
| 8d | K2S2O8, PIDA or TBPB | 18, 14, 15 |
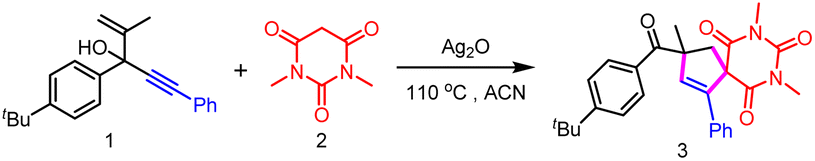
No or only trace amounts of the desired product were detected when Ag2O as replaced by an Fe, Mn or Ni salt (entry 3). Moreover, further optimization using a Cu salt as the oxidant showed that the yield of 3 decreased to 17% or 6% (entry 3). Notably, other Ag salts, such as AgOAc and AgNO3, also showed poor reactivity (entry 4). Using Ag2CO3 could also ensure efficient product formation (entry 4). Oddly, almost no product was detected when AgSbF6 was used (entry 4). Further lowering the reaction temperature to below 110 °C led to the decrease of the formation of the product (entry 5). The screening of the equivalent of Ag2O and 2 revealed that 2 eq. Ag2O or 1.5 eq. 2 was optimal (entries 6 and 7). It was found that significant inhibition of the product formation was observed when other oxidants including K2S2O8, PIDA and TBPB were used in the presence of catalytic amounts of Ag2O (entry 8).
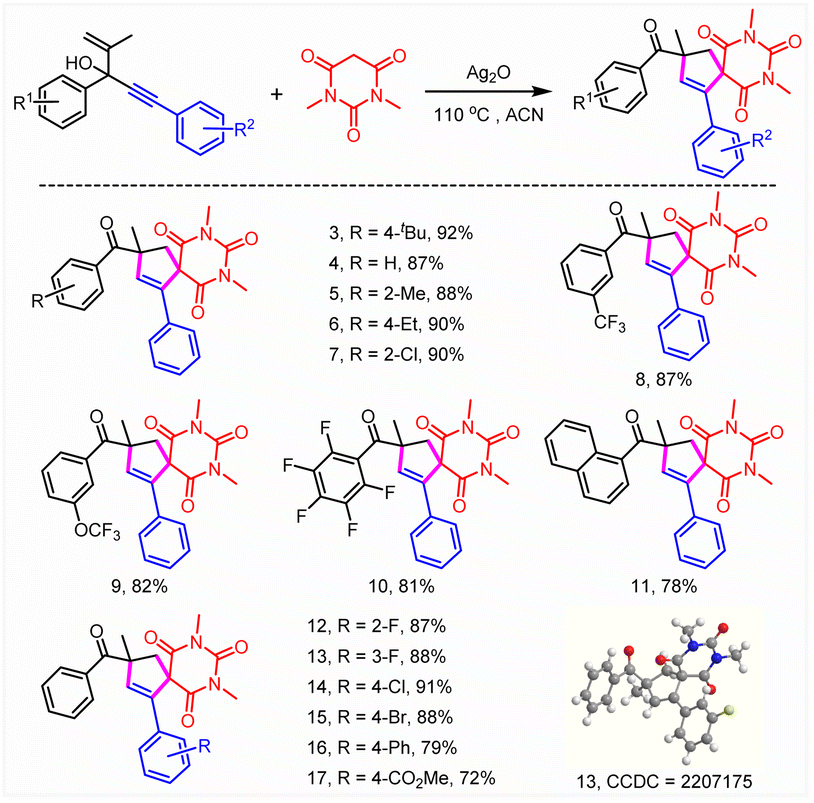
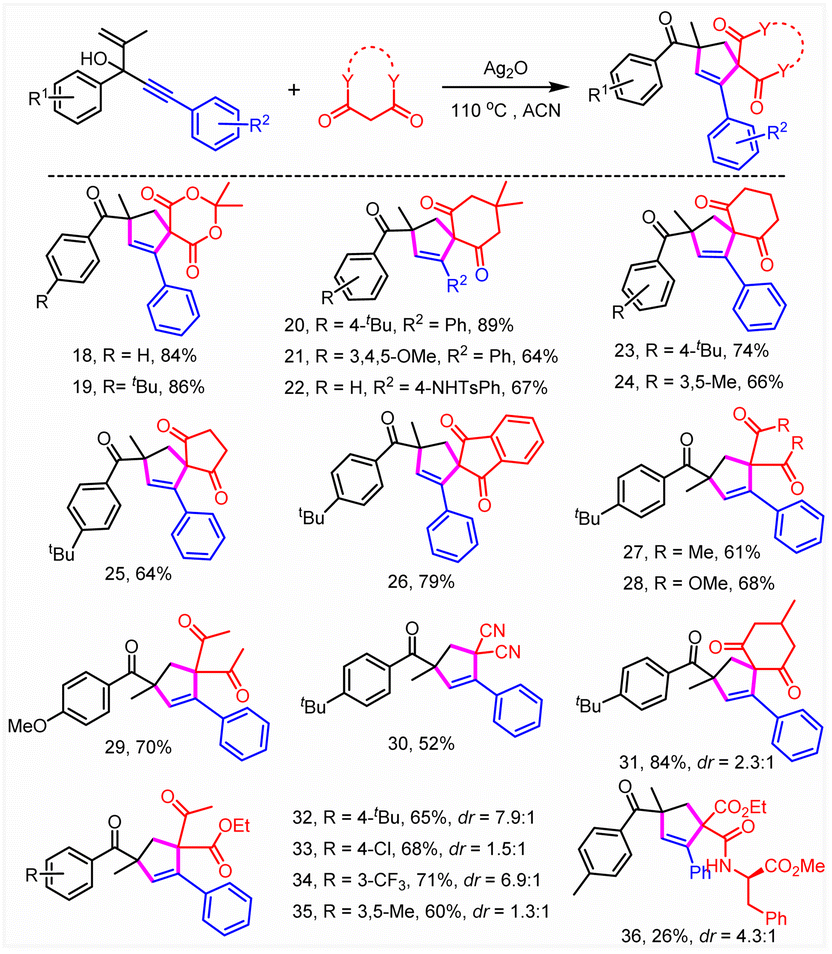
Subsequently, the substrate scope was investigated (Tables 2 and 3). To our delight, this reaction was broadly compatible with a series of functional groups including H (4), alkyl groups (3, 5, and 6), Ph (16), halogens (7, 10, and 12–15), CF3 (8), OCF3 (9) and CO2Me (17), which could also be introduced into the different positions of the phenyl ring. The structure of 13 was confirmed by X-ray crystallographic analysis (CCDC = 2207175). The good halogen tolerance made it easy to realize further derivatization through cross coupling. Moreover, no obvious electronic and steric effects were observed (8–10). A good yield was obtained when a perfluoro substituted phenyl ring or naphthalene relative to the tertiary alcohol moiety of substrates 1 was involved (10 and 11). Afterwards, the reaction scope of this unusual construction of spirocyclic compounds was explored by varying active methylene compounds (Table 3). To our delight, active methylenes including malonic amide (3–17), malonic esters (18, 19, and 28), diketones (20–27, 29, and 31), malononitrile (30) and keto esters (32–35) showed good to excellent reaction compatibility. Moreover, both cyclic and acyclic active methylene compounds led to the formation of the desired product in good to excellent yields, although acyclic substrates exhibited slightly poor reactivity. A slightly lower yield (64%, 25) was obtained when 1,3-cyclopentanedione was involved compared with 1,3-cyclohexanedione (74%, 23). Interestingly, the isolated yield could be boosted to 79% through expanding the conjugate field of 1,3-cyclopentanedione (26). Furthermore, no obvious difference in reactivity was observed when symmetrical or asymmetric substrates were involved (18–36). It was found that 26% isolated yield was obtained using chiral amide (36). Moreover, no selectivity for asymmetric construction of the target product was obtained, which might be attributed to the fact that the chiral amine moiety was far away from the reactive site. In addition, N,N-dimethyl-3-oxobutanamide completely abolished the target product formation. The results above further revealed that acyclic substrates especially amides exhibited poor reactivity.
Intrigued by this intramolecular trapping of the spiro radicals to produce unusual spirocyclic products, gram-scale synthesis and mechanistic studies were performed (see ESI,†Schemes 2–4). To our delight, no obvious scaling effect was observed with 89% isolated yield generated when the reaction was applied on a gram scale. It was found that the formation of the desired product was completely abolished in the presence of the radical scavenger 1,1-diphenylethylene or BHT (Scheme 2a).
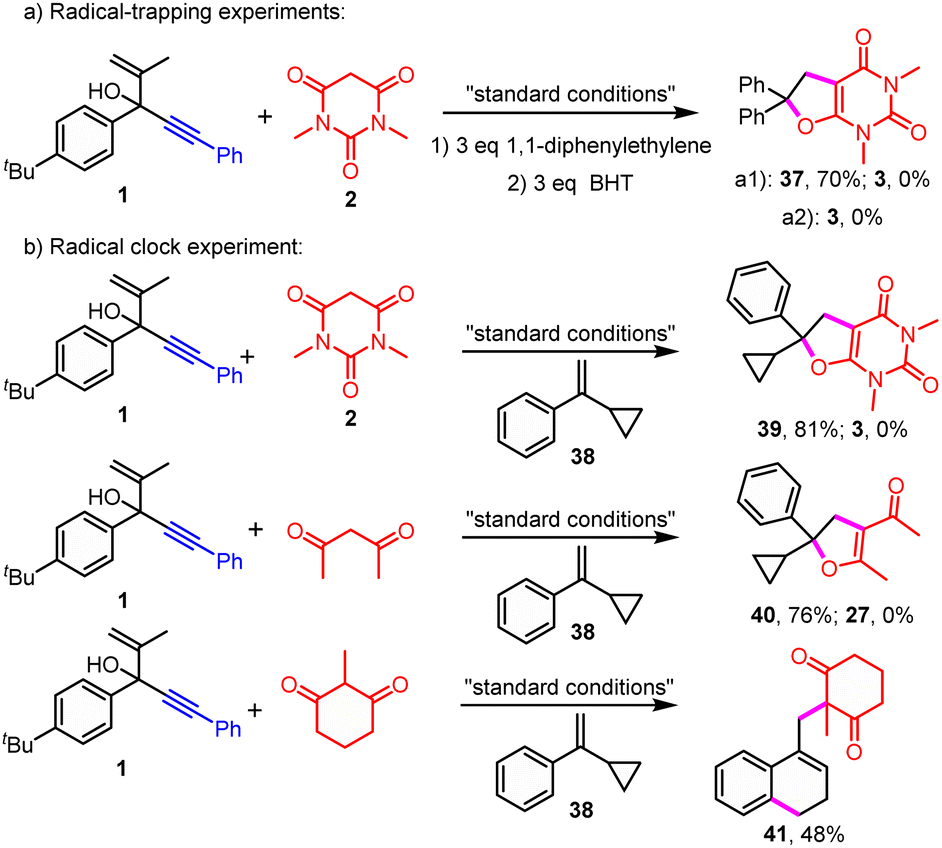 | ||
| Scheme 2 Radical-trapping and clock experiments. (a) Radical-trapping experiments; (b) clock experiments. BHT: butylated hydroxytoluene. | ||
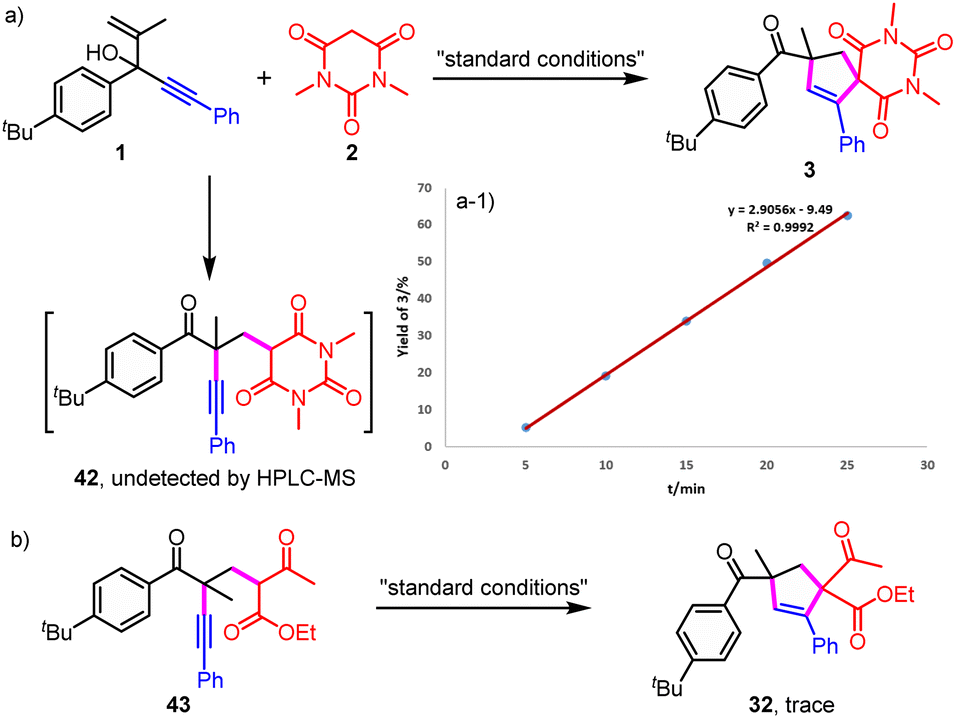 | ||
| Scheme 3 Verification experiments of intermediates. (a) The monitoring of 42 and the desired product 3; (b) verification experiment of intermediate 43. | ||
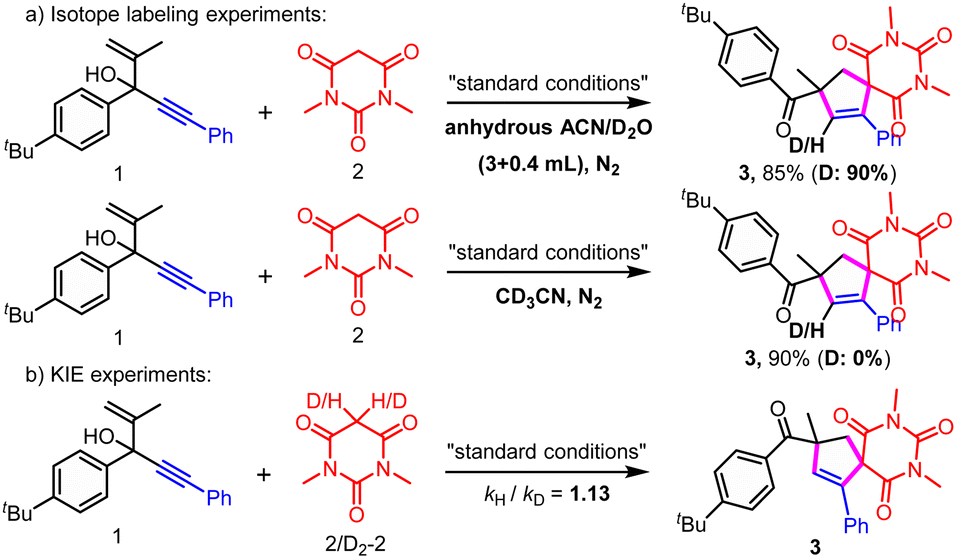 | ||
| Scheme 4 Isotope labeling and KIE experiments. (a) Isotope labeling experiments; (b) KIE experiments. | ||
In addition, the radical addition and cyclization product 37 between 1,1-diphenylethylene and 1,3-dimethylbarbituric acid was generated in 70% isolated yield (Scheme 2a). Furthermore, the radical clock experiments in the presence of radical clock substrate 38 were carried out (Scheme 2b). Interestingly, no normal radical addition/cyclization/migration product 3 or the radical addition/ring-opening product between 2 and 38 was observed. Nevertheless, we also isolated the corresponding radical addition/cyclization products 39 and 40 between active methylene compounds and a radical clock substrate (Scheme 2b). Remarkably, the usual ring-opening product between 2-methylcyclohexane-1,3-dione and radical clock substrate 38 was obtained in moderate yields (Scheme 2b, 41). These results implied the existence of the radical process. Besides, it revealed that active methylene motifs could serve as an efficient trapping reagent of radicals or in situ generated carbonium, which could block the usual spiro radical promoted migration process.
Then, verification experiments of intermediates were performed to investigate whether the normal migration product 42 was formed as a vital intermediate to give the final product from the further radical addition (Scheme 3). Firstly, it was found that no migration product was detected even through the reaction was proceeded in a relatively short time of 5, 10, 15, 20 or 25 minutes (Scheme 3a). In addition, no migration product was detected by HRMS at lower temperature including 90, 70 and 50 °C (Table S1†). Besides, the final product was obtained smoothly and the yield increased linearly (Scheme 3a-1). No delayed formation of the final product was observed, which ruled out the formation of a stable intermediate. Furthermore, almost no target product was detected from the radical addition/H abstraction when the migration product 43 was involved (Scheme 3b). Herein, 43 was used because 42 was hard to prepare. Therefore, the final product was not likely to be generated from the further radical addition of the in situ generated migration product.
On the other hand, 90% D-3 was formed when 0.4 mL D2O was added; meanwhile, using CD3CN as the solvent led to no D-3 generation, which revealed that the H most likely originated from the H cation not H radical (Scheme 4a). These results also indicated that no migration product was involved.
Kinetic isotopic effect (KIE) experiments indicated that C–H bond cleavage of active methylene compounds was likely not the rate-determining step (Scheme 4b, kH/kD = 1.13).
Based on a series of mechanistic studies and previous reports,6–12,21 a plausible mechanism was proposed (Scheme 5). The active methylene compound 2 is oxidized by an Ag salt to furnish a C-centered radical intermediate 2-1, which then undergoes radical addition with substrate 1 to produce the radical intermediate 3-1. The cyclic radical 3-2 is formed from the intramolecular radical addition, which is further oxidized to cation intermediate 3-3. Then, 5-exo-trig cyclization leads to the generation of spirocyclic intermediate 3-4, which is unstable and affords the final product by a ring-opening process. It was found that heating was necessary for this transformation, which might be used to overcome the energy barrier of the formation of metastable 3-4.
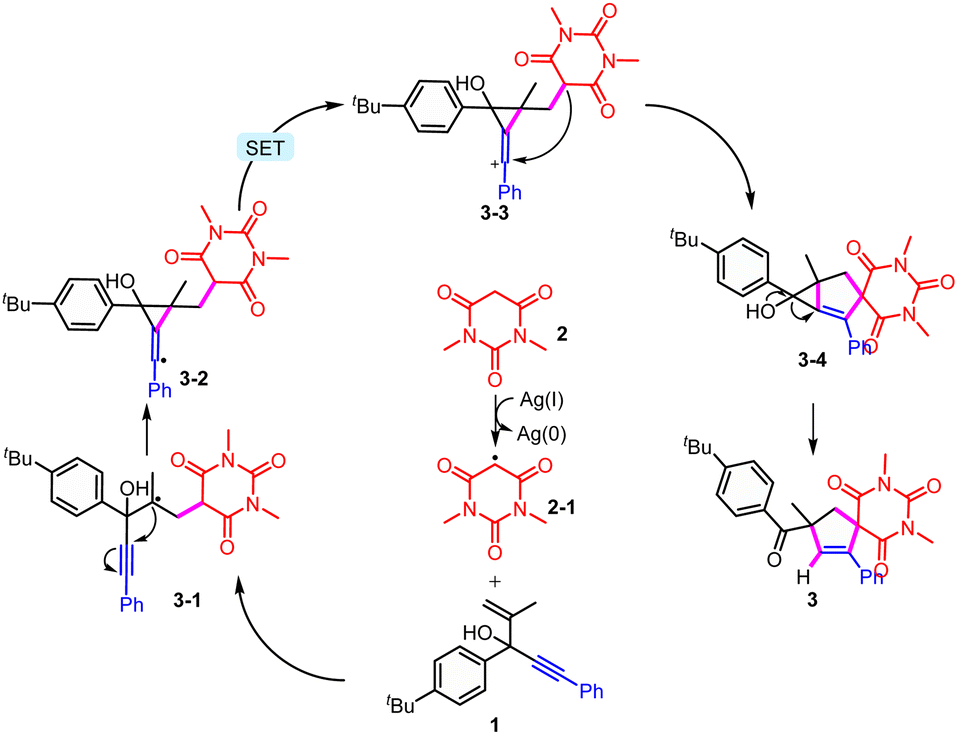 | ||
| Scheme 5 Proposed mechanism for this intramolecular trapping of the spiro radicals to produce unusual cyclization products from the usual migration substrates. | ||
Conclusions
In summary, an unprecedented silver-promoted intramolecular trapping of spiro radicals to produce unusual cyclization products from usual migration substrates was developed. A series of highly complex and structurally important and valuable spirocyclic products were produced in good to excellent yields. This reaction also features good functional group tolerance, high conversion efficiency, using easily available starting materials and good scalable potential, which make it a powerful synthetic tool. Furthermore, a possible mechanism was proposed based on a series of mechanistic studies including radical-trapping, radical clock, verification experiments of intermediates, isotope labeling and KIE experiments.Data availability
All experimental and characterization data including NMR spectra are available in the ESI.† Crystallo-graphic data for compound 13 has been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC 2207175.Author contributions
Chengkou Liu, Zheng Fang and Kai Guo designed the experiments. Jingming Zhang performed the experiments. Yaqi Qiao, Minghui Wei, Wenjing Guan, Ziren Mao, and Hong Qin analysed the data. Chengkou Liu drafted the manuscript. All authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science Foundation of China (22008118); Natural Science Foundation of Jiangsu Province, Frontier Project (BK20212003); Nanjing International Joint R&D Project (202002037); Jiangsu Province Industrial Prospects and Key Core Technologies-Competitive Projects (BE2021083).Notes and references
- (a) S. Caille, Science, 2019, 364, 635 CrossRef CAS PubMed; (b) C. J. Gerry and S. L. Schreiber, Nat. Rev. Drug Discovery, 2018, 17, 333–352 CrossRef CAS PubMed; (c) R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. S. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Nat. Rev. Drug Discovery, 2011, 10, 188–195 CrossRef CAS PubMed; (d) S. Dandapani and L. A. Marcaurelle, Nat. Chem. Biol., 2010, 6, 861–863 CrossRef CAS PubMed.
- (a) C. M. Rojas, Molecular Rearrangements in Organic Synthesis, Wiley-VCH, New York, 2015 CrossRef; (b) M. Korb and H. Lang, Chem. Soc. Rev., 2019, 48, 2829–2882 RSC; (c) I. Colomer, M. Velado, R. Fernandez de la Pradilla and A. Viso, Chem. Rev., 2017, 117, 14201–14243 CrossRef CAS PubMed; (d) A. A. Tabolin and S. L. Ioffe, Chem. Rev., 2014, 114, 5426–5476 CrossRef CAS PubMed; (e) E. Leemans, M. D'Hooghe and N. De Kimpe, Chem. Rev., 2011, 111, 3268–3333 CrossRef CAS PubMed.
- (a) A. Ramazani, F. Moradnia, H. Aghahosseini and I. Abdolmaleki, Curr. Org. Chem., 2017, 21, 1612–1625 CAS; (b) T. J. Snape, Chem. Soc. Rev., 2008, 37, 2452–2458 RSC.
- (a) M.-M. Wang, T. V. T. Nguyen and J. Waser, Chem. Soc. Rev., 2022, 51, 7344–7357 RSC; (b) Y. Zhang, J.-J. Chen and H.-M. Huang, Angew. Chem., Int. Ed., 2022, 61, e202205671 CAS; (c) J.-Z. Li, L. Mei, X.-C. Yu, L.-T. Wang, X.-E. Cai, T. Li and W.-T. Wei, Org. Chem. Front., 2022, 9, 5726–5757 RSC; (d) E. Nobile, T. Castanheiro and T. Besset, Angew. Chem., Int. Ed., 2021, 60, 12170–12191 CrossRef CAS PubMed; (e) S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Chem. Sci., 2020, 11, 12974–12993 RSC.
- (a) Y. Zhang, J.-J. Chen and H.-M. Huang, Angew. Chem., Int. Ed., 2022, 61, e202205671 CAS; (b) A. R. Allen, E. A. Noten and C. R. J. Stephenson, Chem. Rev., 2022, 122, 2695–2751 CrossRef CAS PubMed; (c) X. Wu, Z. Ma, T. Feng and C. Zhu, Chem. Soc. Rev., 2021, 50, 11577–11613 RSC; (d) I. Allart-Simon, S. Gerard and J. Sapi, Molecules, 2016, 21, 878 CrossRef PubMed; (e) R. Banerjee, Chem. Rev., 2003, 103, 2083–2094 CrossRef CAS PubMed; (f) W. Adam and T. Heidenfelder, Chem. Soc. Rev., 1999, 28, 359–365 RSC.
- (a) W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480–14483 CrossRef CAS PubMed; (b) W. Kong, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2014, 53, 5078–5082 CrossRef CAS PubMed.
- (a) J. Lan, K. Lin, X. Zhang and T. Zhu, Green Chem., 2022, 24, 6138–6144 RSC; (b) M. Hu, L.-Y. Guo, Y. Han, F.-L. Tan, R.-J. Song and J.-H. Li, Chem. Commun., 2017, 53, 6081–6084 RSC; (c) F.-L. Tan, R.-J. Song, M. Hu and J.-H. Li, Org. Lett., 2016, 18, 3198–3201 CrossRef CAS PubMed; (d) Z. He, P. Tan, C. Ni and J. Hu, Org. Lett., 2015, 17, 1838–1841 CrossRef CAS PubMed; (e) Y. Zhao, N. Sharma, U. K. Sharma, Z. Li, G. Song and V. d. E. V. Eycken, Chem.–Eur. J., 2016, 22, 5878–5882 CrossRef CAS PubMed.
- Z. Ni, X. Huang and Y. Pan, Org. Lett., 2016, 18, 2612–2615 CrossRef CAS PubMed.
- M. Zhang, X. Ding, A. Lu, J. Kang, Y. Gao, Z. Wang, H. Li and Q. Wang, Org. Chem. Front., 2021, 8, 961–967 RSC.
- (a) J.-L. Wang, M.-L. Liu, J.-Y. Zou, W.-H. Sun and X.-Y. Liu, Org. Lett., 2022, 24, 309–313 CrossRef CAS PubMed; (b) C. Cai, Y. Lu, C.-C. Yuan, Z. Fang, X. Yang, C.-K. Liu and K. Guo, ACS Sustainable Chem. Eng., 2021, 9, 16989–16996 CrossRef CAS.
- (a) W. Kong, N. Fuentes, A. Garcia-Dominguez, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 2487–2491 CrossRef CAS PubMed; (b) N. Fuentes, W. Kong, L. Fernandez-Sanchez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2015, 137, 964–973 CrossRef CAS PubMed; (c) J. Yan, H. W. Cheo, W. K. Teo, X. Shi, H. Wu, S. B. Idres, L.-W. Deng and J. Wu, J. Am. Chem. Soc., 2020, 142, 11357–11362 CrossRef CAS PubMed.
- Z. Wu, R. Ren and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 10821–10824 CrossRef CAS PubMed.
- (a) A. C. Seastram, M. D. Hareram, T. M. B. Knight and L. C. Morrill, Chem. Commun., 2022, 58, 8658–8661 RSC; (b) M. Ji, Z. Wu and C. Zhu, Chem. Commun., 2019, 55, 2368–2371 RSC.
- (a) Y. Wei, X. Wu and C. Zhu, Synlett, 2022, 33, 1017–1028 CrossRef CAS; (b) Z. Wu, D. Wang, Y. Liu, L. Huan and C. Zhu, J. Am. Chem. Soc., 2017, 139, 1388–1391 CrossRef CAS PubMed; (c) X. Duan, Q. Sun, X. Yuan, L.-Z. Qin, X.-P. Zhang, J. Liu, M.-Y. Wu, S.-S. Zhu, C.-L. Ma, J.-K. Qiu and K. Guo, Green Chem., 2021, 23, 8916–8921 RSC; (d) M. Ji, X. Wang, J. Liu, X. Wu and C. Zhu, Sci. China: Chem., 2021, 64, 1703–1708 CrossRef CAS; (e) C.-K. Liu, Q. Jiang, Y. Lin, Z. Fang and K. Guo, Org. Lett., 2020, 22, 795–799 CrossRef CAS PubMed; (f) M.-W. Zheng, X. Yuan, Y.-S. Cui, J.-K. Qiu, G. Li and K. Guo, Org. Lett., 2018, 20, 7784–7789 CrossRef CAS PubMed; (g) Z. Cao, H. Zhang, X. Wu, Y. Li and C. Zhu, Org. Chem. Front., 2021, 8, 6395–6399 RSC.
- (a) A. Bunescu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3132–3135 CrossRef CAS PubMed; (b) X. Liu, F. Xiong, X. Huang, L. Xu, P. Li and X. Wu, Angew. Chem., Int. Ed., 2013, 52, 6962–6966 CrossRef CAS PubMed; (c) Z.-M. Chen, W. Bai, S.-H. Wang, B.-M. Yang, Y.-Q. Tu and F.-M. Zhang, Angew. Chem., Int. Ed., 2013, 52, 9781–9785 CrossRef CAS PubMed.
- (a) Y. Xu, Z. Wu, J. Jiang, Z. Ke and C. Zhu, Angew. Chem., Int. Ed., 2017, 56, 4545–4548 CrossRef CAS PubMed; (b) Y. Gao, H. Mei, J. Han and Y. Pan, Chem.–Eur. J., 2018, 24, 17205–17209 CrossRef CAS PubMed; (c) J. Liu, W. Li, J. Xie and C. Zhu, Org. Chem. Front., 2018, 5, 797–800 RSC.
- X. Tang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 814–817 CrossRef CAS PubMed.
- (a) Z.-L. Li, X.-H. Li, N. Wang, N.-Y. Yang and X.-Y. Liu, Angew. Chem., Int. Ed., 2016, 55, 15100–15104 CrossRef CAS PubMed; (b) Y. He, X. Dan, Y. Tang, Q. Yang, W. Wang and Y. Cai, Green Chem., 2021, 23, 9577–9582 RSC.
- J. Yu, D. Wang, Y. Xu, Z. Wu and C. Zhu, Adv. Synth. Catal., 2018, 360, 744–750 CrossRef CAS.
- D.-T. Xie, H.-L. Chen, D. Wei, B.-Y. Wei, Z.-H. Li, J.-W. Zhang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2022, 61, e202201027 Search PubMed.
- (a) W. Zhou, T. Zhou, M. Tian, Y. Jiang, J. Yang, S. Lei, Q. Wang, C. Zhang, H. Qiu, L. He, Z. Wang, J. Deng and M. Zhang, J. Am. Chem. Soc., 2021, 143, 19975–19982 CrossRef CAS PubMed; (b) H. J. Gutke and D. Spitzner, Tetrahedron, 1999, 55, 3931–3936 CrossRef CAS; (c) V. Bagutski, N. Moszner, F. Zeuner, U. K. Fischer and A. de Meijere, Adv. Synth. Catal., 2006, 348, 2133–2147 CrossRef CAS; (d) G. Zhan, M.-L. Shi, Q. He, W.-J. Lin, Q. Ouyang, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2016, 55, 2147–2151 CrossRef CAS PubMed; (e) P. D. Wilson, D. Friedrich and L. A. Paquette, J. Chem. Soc., Chem. Commun., 1995, 13, 1351–1352 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2207175. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05768a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |