
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective Suzuki cross-coupling of 1,2-diboryl cyclopropanes†
Javier
Teresa
 a,
Marina
Velado
b,
Roberto
Fernández de la Pradilla
b,
Alma
Viso
b,
Blanca
Lozano
a and
Mariola
Tortosa
*ac
a,
Marina
Velado
b,
Roberto
Fernández de la Pradilla
b,
Alma
Viso
b,
Blanca
Lozano
a and
Mariola
Tortosa
*ac
aOrganic Chemistry Department, Center for Innovation in Advanced Chemistry (ORFEO-CINQA), Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain. E-mail: mariola.tortosa@uam.es
bInstituto de Química Orgánica General (IQOG), CSIC, Juan de la Cierva 3, 28006 Madrid, Spain
cInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain
First published on 13th January 2023
Abstract
Herein, we describe the catalytic enantioselective cross-coupling of 1,2-bisboronic esters. Prior work on group specific cross coupling is limited to the use of geminal bis-boronates. This desymmetrization provides a novel approach to prepare enantioenriched cyclopropyl boronates with three contiguous stereocenters, that could be further derivatized through selective functionalization of the carbon–boron bond. Our results suggest that transmetallation, which is the enantiodetermining step, takes place with retention of stereochemistry at carbon.
Introduction
The Suzuki–Miyaura cross-coupling reaction is one of the main tools used in the pharmaceutical industry to forge carbon–carbon bonds.1 A large variety of libraries of compounds are synthesized every year using this cross-coupling as a key point of diversification. In fact, a recent analysis showed that this transformation is the second most commonly used reaction in pharma, placed after amide coupling.2 The robustness of the Suzuki cross-coupling has also made possible its use in automated settings3 and in the synthesis of DNA encoded libraries.4As the pharmaceutical industry is shifting from compounds with a strong sp2 character to libraries of compounds with increased three-dimensionality,5 the need to develop robust stereoselective Suzuki cross-couplings to provide compounds with an increased sp3 character becomes apparent.6 In this context, the development of enantioselective Suzuki–Miyaura cross-coupling reactions using prochiral bis-boronates remains largely unexplored.7,8 Morken9 and Hall10 reported the enantioselective Suzuki cross-coupling reaction of symmetric geminal bis-boronates to prepare enantiomerically enriched boronic esters (Fig. 1a). However, despite the elegant strategies developed to selectively functionalize 1,2-bis-boronates,11,12 the enantioselective desymmetrization of these species is still an unmet challenge (Fig. 1b). The degree of complexity in the products, relative to those prepared from geminal bis-boronates, would be higher as two contiguous stereocenters are generated in the process.
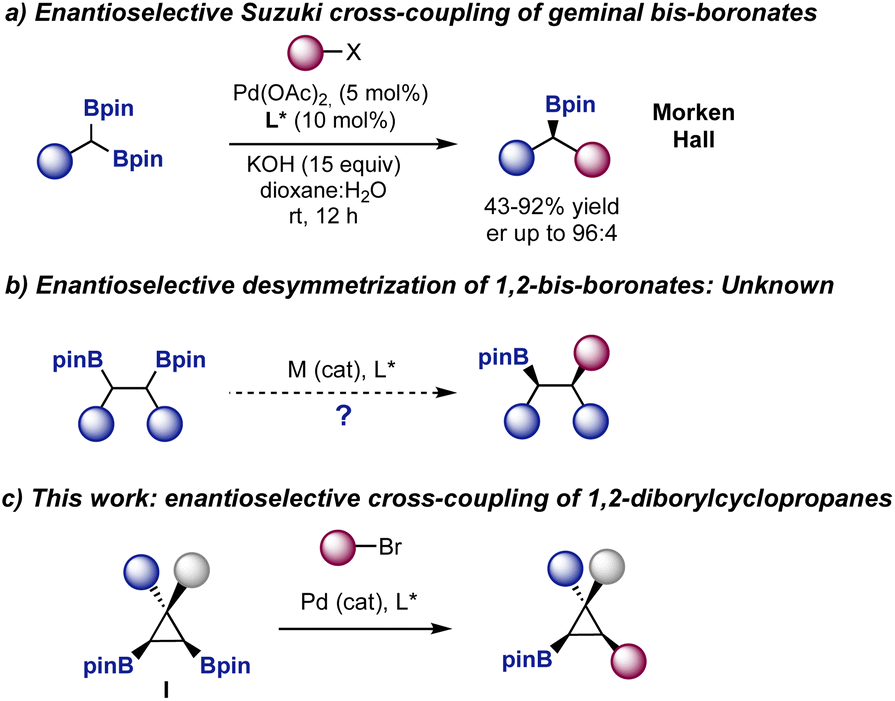 | ||
| Fig. 1 Enantioselective Suzuki cross-coupling of prochiral bis-boronates. | ||
Following our interest in the functionalization of small rings13 and inspired by the relevance of cyclopropanes in synthetic methodology and pharmaceutical industry,14 we envisioned that symmetric bis-boryl cyclopropanes I could offer an ideal scenario to test this transformation (Fig. 1c). The products would be enantiomerically enriched cyclopropyl boronates,15 with three substituents in a hindered cis orientation and a handle for further stereospecific C–B functionalization.16
We realized from the outset that the proposed desymmetrization was a challenging transformation from the point of view of asymmetric catalysis (Fig. 2). Under the basic conditions needed for the Suzuki reaction, the bis-boronic ester I is likely to be in equilibrium with the racemic mixture of monoacids II and the prochiral bis-boronic acid III. If hydrolysis is a prerequisite for transmetallation, as it was proposed for geminal bis-boronates,9,10 the chiral L*PdAr(X) complex formed after oxidative addition would be exposed to mixtures of II and III. Although a dynamic kinetic resolution (DKR) could not be discarded, the reaction of a L*PdAr(X) complex with racemic mixture II would likely provide racemic products, having a detrimental effect on the overall enantiocontrol. Therefore, we hypothesized that to maximize a potential ligand-controlled enantioselectivity the equilibriums in Fig. 2 should be displaced towards bis-boronic acid III.
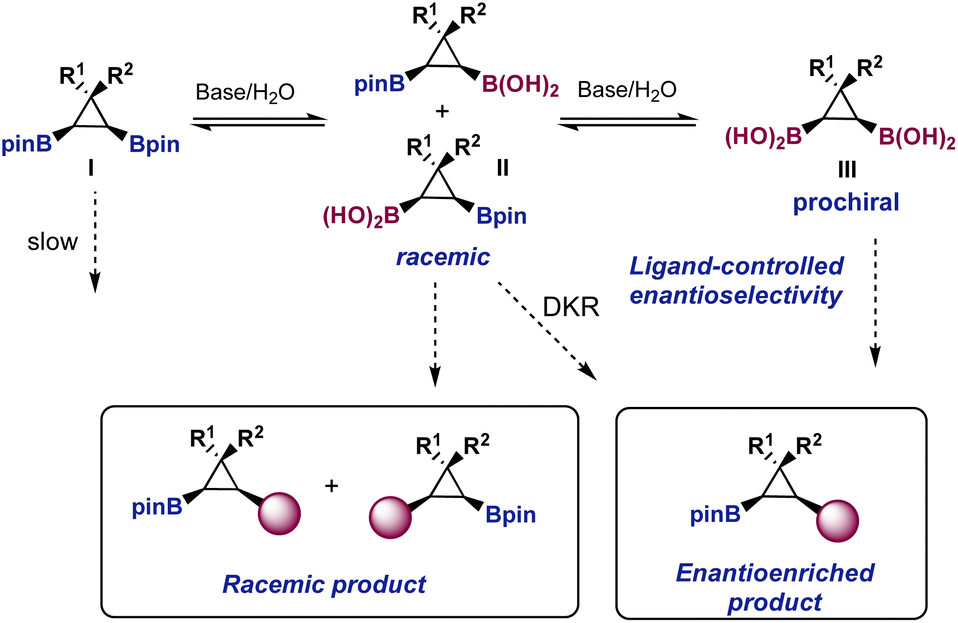 | ||
| Fig. 2 Challenges involved in the proposed desymmetrization. | ||
Results and discussion
When we started our study, we realized that bis-boryl cyclopropanes such as 2 had not been prepared in the literature before.17,18 We envisioned that diboration of readily available cyclopropenes 1 could provide easy access to these intermediates. Our first attempts using Pt-catalyzed diboration conditions19,20 afforded a complex crude product from which we isolated a 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35 mixture of diborylated diastereomers 2a/2a′ in 11% yield. Although we did not identify any other products, the dimerization of the cyclopropene under the reaction conditions could be a potential undesired reaction.13b,21 Then, we turned our attention to the use of transition-metal free borylation conditions. We were pleased to find that heating cyclopropanes 1 in MeOH, in the presence of MeONa and B2pin2, provided exclusively 1,2-syn-diboryl cyclopropanes 2a–2k in good yields as single diastereomers (Scheme 1).22,23
:35 mixture of diborylated diastereomers 2a/2a′ in 11% yield. Although we did not identify any other products, the dimerization of the cyclopropene under the reaction conditions could be a potential undesired reaction.13b,21 Then, we turned our attention to the use of transition-metal free borylation conditions. We were pleased to find that heating cyclopropanes 1 in MeOH, in the presence of MeONa and B2pin2, provided exclusively 1,2-syn-diboryl cyclopropanes 2a–2k in good yields as single diastereomers (Scheme 1).22,23
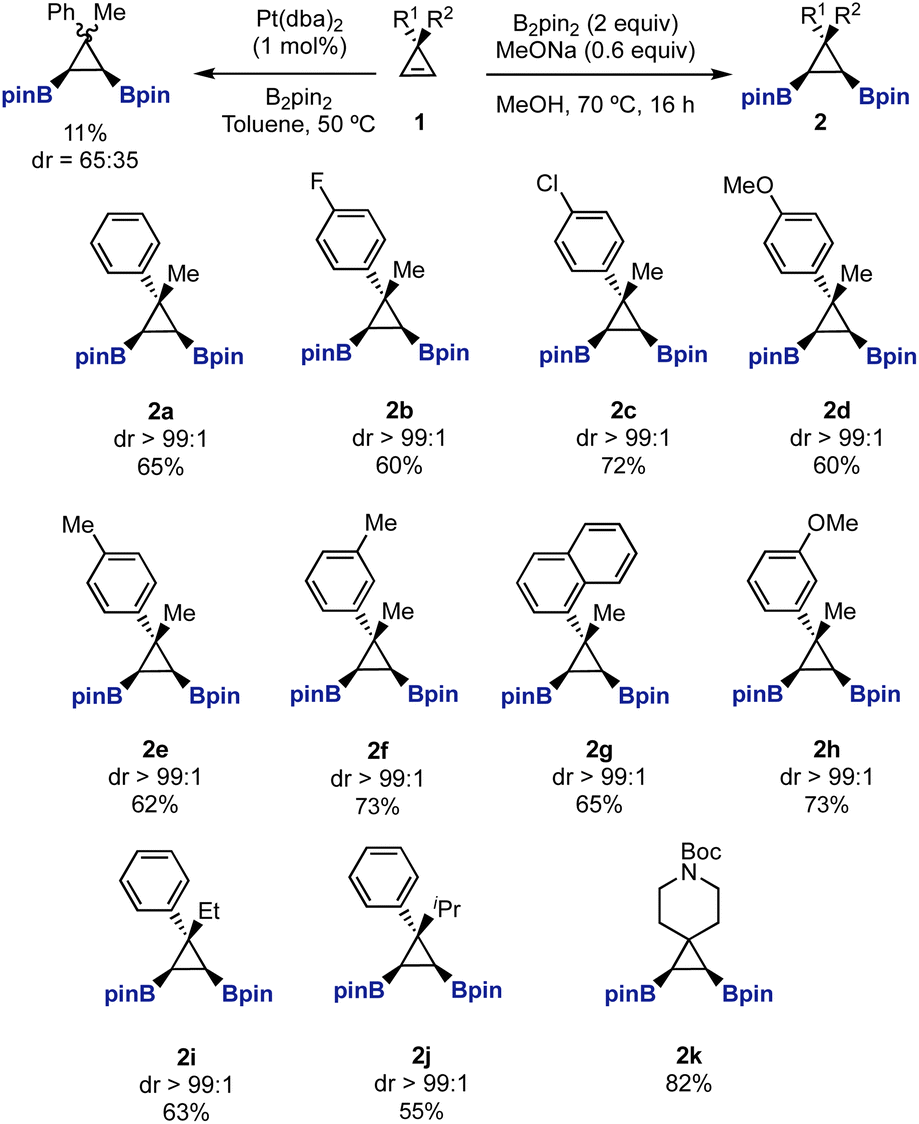 | ||
| Scheme 1 Base-promoted diboration of cyclopropenes. | ||
With a method to prepare diboryl cyclopropanes in hand, we started to explore the feasibility of the enantioselective Suzuki cross-coupling using cyclopropane 2a, bromobenzene, and NaOH as the base, in the presence of 5 mol% of Pd(OAc)2. Chiral bidentate ligands commonly used in palladium-catalyzed asymmetric transformations (not shown) consistently provided a racemic mixture of 3a in variable yields.24 A key observation was that in the absence of an added ligand, we observed exclusive formation of the protodeboronation product (Table 1, entry 1).25 Indeed, we soon realized that the inhibition of this background reaction was one of the main challenges in this cross-coupling reaction. We then moved to explore the reactivity of monodentate ligands, hoping that three-coordinate LPdAr(X) complexes would show enhanced selectivity.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | L | Equiv. NaOH | Equiv. KHF2 | T | Yield 3ab (%) | erc |
|
a Reaction conditions: 2a (0.1 mmol), PhBr (1.2 equiv.), Pd(OAc)2 (5 mol%), L* (10 mol%), NaOH (8 equiv.), KHF2 (0–6 equiv.), THF:H2O (10:1, 0.1 M), and 16 h.
b Yield calculated by 1H NMR using an internal standard.
c Enantiomeric ratio determined by chiral-phase HPLC.
d 20 mol% of L1 used.
e 5 mol% L1 used.
f PhI was used instead of PhBr.
g Reaction conditions [a] except for the equiv. of NaOH.
|
||||||
| 1 | — | 8 | — | 60 | — | — |
| 2a | L1 | 8 | — | 60 | 66 | 83:17 |
| 3a | L1 | 8 | 3 | 60 | 78 | 82:18 |
| 4a | L1 | 8 | 6 | 60 | 85 | 83:17 |
| 5a | L2 | 8 | 6 | 60 | 72 | 70:30 |
| 6a | L3 | 8 | 6 | 60 | 65 | 79:21 |
| 7a | L4 | 8 | 6 | 60 | 54 | 80:20 |
| 8a | L5 | 8 | 6 | 60 | 59 | 73:27 |
| 9a | L6 | 8 | 6 | 60 | 74 | 83:17 |
| 10a | L7 | 8 | 6 | 60 | 66 | 84:16 |
| 11a | L1 | 8 | 6 | 40 | 75 | 86:14 |
| 12 | L1 | 8 | 6 | 40 | 89 |
87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :13 :13
|
| 13e | L1 | 8 | 6 | 40 | 68 | 84:16 |
| 14f | L1 | 8 | 6 | 40 | 46 | 72:28 |
| 15g | L1 | 1 | 6 | 40 | NR | — |
| 16g | L1 | 4 | 6 | 40 | Traces | — |
| 17g | L1 | 15 | 6 | 40 | 65 | 88:12 |
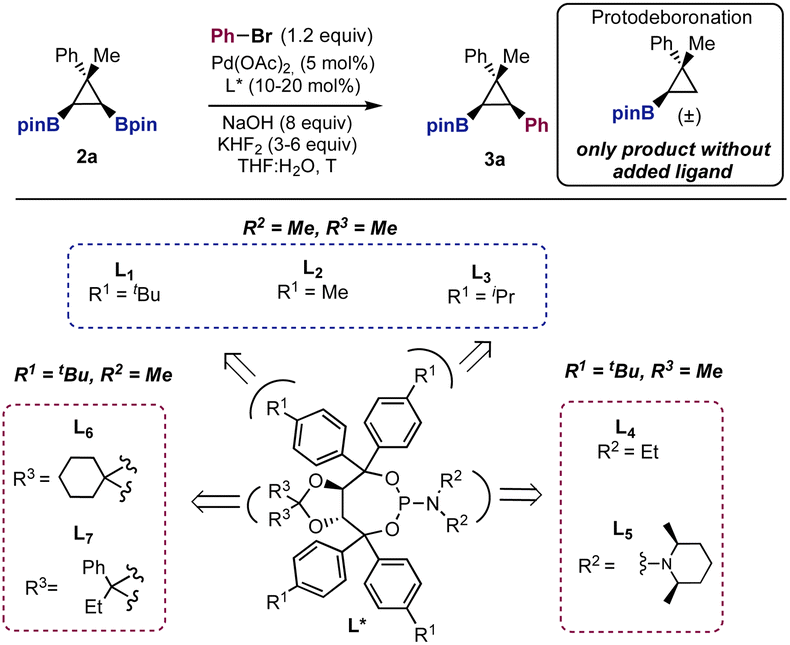
After extensive experimentation, we found that TADDOL derived phosphoramidites (L1–L7, Table 1) showed encouraging results. Using bromobenzene, 5 mol% of Pd(OAc)2, NaOH (8 equiv.) and chiral ligand L1, at 60 °C, cross-coupling product 3a was obtained in 66% yield with a promising enantiomeric ratio (er = 83:17, Table 1, entry 2). It is known that the addition of a fluoride source helps to increase the rate of the Suzuki cross-coupling reaction.26 In our case, the addition of KHF2 (Table 1, entry 3) had a positive effect on controlling the protodeboronation and, therefore, increasing the yield up to 78%. Using 6 equiv. of KHF2 the yield was further improved to 85% without reducing the enantiomeric ratio (er = 83:17). Less bulky groups in the para position of the aromatic rings of the ligands provided lower stereocontrol (L2–L3, Table 1, entries 5–6). Tuning of the substituents at nitrogen (L4–L5, Table 1, entries 7–8) and the acetal backbone (L6–L7, Table 1, entries 9–10) did not improve the results obtained with L1. While at room temperature the cross-coupling did not occur, at 40 °C compound 3a was obtained with higher enantioselectivity (er = 86:14, Table 1, entry 11). Additionally, the use of 20 mol% of L1 had a beneficial effect on both the yield and the enantiomeric ratio (89%, er = 87:13, Table 1, entry 12). This is probably due to the inhibition of the background protodeboronation. According to this result, the use of 5 mol% of L1 was detrimental to the yield and the enantioselectivity (Table 1, entry 13). Aryl iodides (Table 1, entry 14) afforded compound 3a with poorer results. Finally, entries 15–17 show that the use of a large excess of base was necessary. Using 1 and 4 equiv. of NaOH we did not observe product formation. Increasing the amount of base to 15 equiv. maintained the level of stereocontrol (er = 88:12) but also increased the protodeboronation, lowering the yield of 3a to 65% (Table 1, entry 17). The use of other bases, Pd precatalysts or solvents were not found to be beneficial.24
It should be highlighted that compound 3a was obtained as a single syn-diastereomer. Assuming that the reductive elimination step occurs with retention, this result suggests that the transmetallation step takes place with retention of stereochemistry at carbon. This is in contrast with the results observed in the enantioselective cross-coupling with 1,1-diboronates, in which the transmetallation seems to proceed with inversion.9
We next studied the structural scope of the enantioselective Suzuki cross-coupling reaction (Scheme 2). We were pleased to find that the reaction was quite general for different aryl bromides (3b–3t) and cyclopropanes (3u–3ab). Importantly, in many cases the enantioselectivity was higher than that observed for bromobenzene. Aryl bromides with electron donating groups in the para and ortho positions provided the cross-coupling products (3b–3d and 3h–3l) in good yields and high enantiomeric ratios (up to 95:5 for 3h). Aryl bromides with electron-withdrawing substituents afforded arylated boryl cyclopropanes with similar stereoselectivites (3e–3g). Electrophiles with substituents in the meta position (3k–3m), sterically hindered aryl bromides (3j), as well as naphthalene (3n) and methylenedioxy derivatives (3o) were well tolerated. Heterocycles such as indole (3p), benzofuran (3q), thiobenzofuran (3r), chromane (3s) and tetrahydroquinoline (3t) worked particularly well, providing cross-coupling products in good yields and enantiomeric ratios higher to that observed for bromobenzene. Finally, the method allowed structural modifications on the aryl and methyl groups of the diboryl cyclopropane framework (3u–3ab). In those cases, the cross-coupling products were prepared with comparable efficiencies to those shown with the Ph/Me substitution pattern.
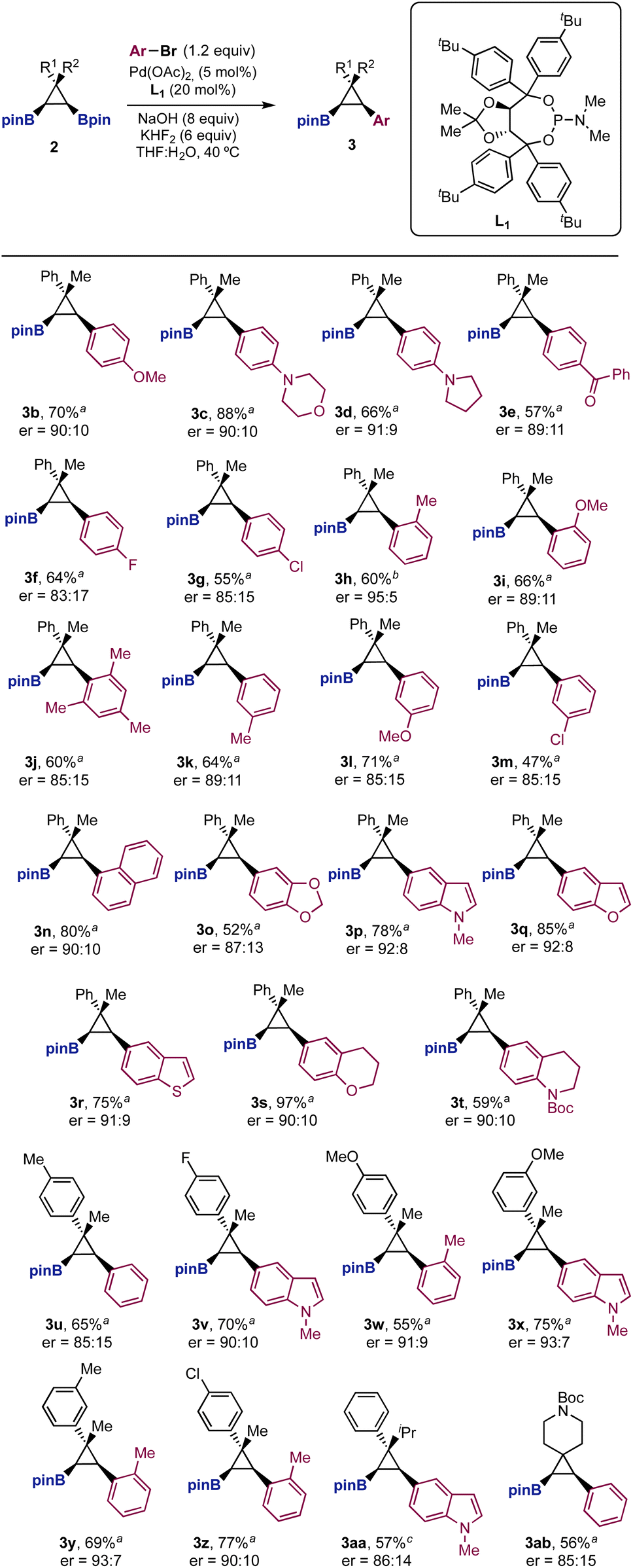 | ||
| Scheme 2 Scope of the enantioselective desymmetrization. aReaction conditions: 2 (0.2 mmol), PhBr (1.2 equiv.), Pd(OAc)2 (5 mol%), L* (20 mol%), NaOH (8 equiv.), KHF2 (6 equiv.), THF:H2O (10:1, 0.1 M), 40 °C, and 16 h. b15 equiv. of NaOH were used. c8 equiv. of NaOH were used at 60 °C. | ||
To gain insight into the boron species formed under the reaction conditions, we mixed pinacol ester 2a with (+)-pinanediol with KHF2 (6 equiv.) and NaOH (8 equiv.) at 40 °C, in the absence of Pd(OAc)2 and bromobenzene (Scheme 3). We observed immediate disappearance of compound 2a by TLC and transesterified pinanediol derivative 2l was obtained in 86% yield. It is known that pinacol boronic esters form potassium trifluoroborate salts in the presence of KHF2, and that these salts are hydrolyzed to boronic acids27 in the basic aqueous media used in Suzuki cross-coupling reactions. Therefore, the experiment in Scheme 3 supports the in situ formation of bis-boronic acids but does not rule out the formation of transient trifluoroborate salts that could also participate in the equilibriums shown in Fig. 2. According to our results, the use of KHF2 has a positive effect on controlling the protodeboronation without compromising the enantioselectivity. Although the exact role of KHF2 is not clear at this point, the in situ formation of trifluoroborate salts could provide a slower release of the bis-boronic acid28 and fluoride26 under the basic aqueous conditions, minimizing the protodeboronation and increasing the rate of the cross-coupling. Bis-ester 2l did not react under the optimized conditions. Since pinanediol boronic esters are more robust towards hydrolysis, this result further supports the participation of a bis-boronic acid III (Fig. 2) as an intermediate in the reaction.
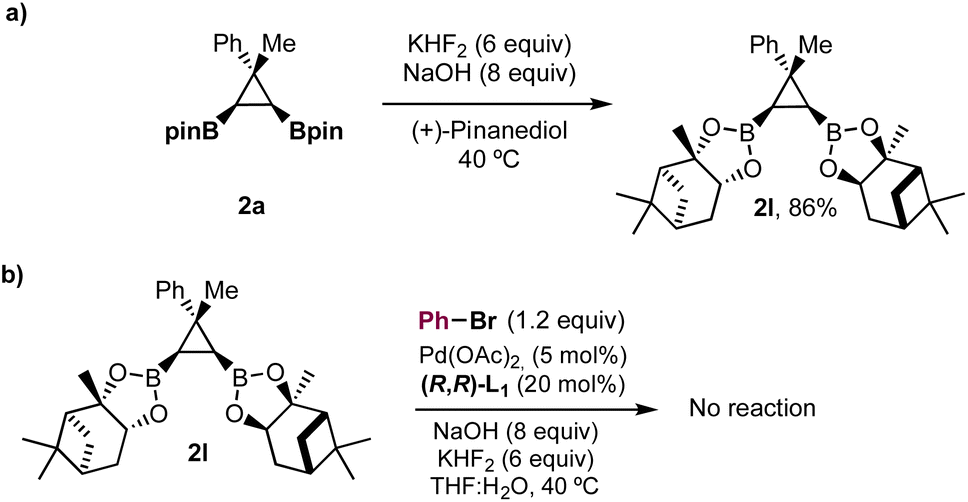 | ||
| Scheme 3 Support for the in situ hydrolysis of diboronic ester 2a. | ||
Another question that emerged from our study was the role played by the second boryl moiety. Morken has reported that the presence of a vicinal pinacol boronic ester may have an activating effect towards transmetalation in Suzuki cross-coupling reactions.12g More recently, Morken has also shown that terminal 1,2-diboronic esters, in the presence of potassium methoxide, form a 5-membered heterocycle with a single oxygen atom bridging both boron atoms.29 This chelated cyclic ate complex seemed to play a key role in the transmetallation with a copper complex. We were intrigued to compare the behavior of our 1,2-bisboryl cyclopropanes with that observed by Morken with terminal 1,2-bisboronates. We checked the reactivity of a monoborylated derivative (Scheme 4a) under the optimized reaction conditions and no product was observed after 16 hours. This result indicates that the presence of the adjacent boryl moiety is necessary for the transmetallation to take place. Additionally, we studied by 11B NMR spectroscopy the alkoxide complexation of mono- and diborylated cyclopropane (Scheme 4b). The treatment of the monoborylated derivative (32.9 ppm) with 1 equivalent of KOMe resulted in complete conversion to a compound with an upfield shifted resonance (7.4 ppm), which is in agreement with the formation of a sp3 hybridized borate complex. When we treated diborylated cyclopropane 2a (32.4 ppm) with 1 equivalent of KOMe, we observed an almost quantitative upfield shift of the 11B NMR peak to 10.4 ppm. The smaller peak at 7.4 ppm corresponds to the borate complex of the residual protodeboronation product. Considering that we are using 0.5 equivalents of KOMe relative to the total amount of boron, the signal at 10.4 ppm suggests a chelation similar to that proposed by Morken for acyclic terminal 1,2-diboronates. Although the conditions used for the 11B NMR experiments (THF and KOMe) are not those used in the Suzuki cross coupling reaction (THF:H2O, NaOH, and KHF2), these results suggest that chelated cyclic ate complexes could potentially participate in our catalytic cycle. These observations could open the door to the design of further desymmetrizations.
 | ||
| Scheme 4 Role of the vicinal boryl moiety. | ||
The reaction was scaled up to 1 mmol with diboryl cyclopropane 2a and 5-bromoindole, affording cyclopropane 3p in similar yield and enantioselectivity to those observed before (Scheme 5). From boronate 3p, we performed the oxidation of the C–B bond followed by benzoylation to obtain benzoate 4. The recrystallization of 4 provided crystals of high enantiopurity (er = 97:3), which allowed us to assign the absolute configuration of the products in Table 1 and Scheme 2.30
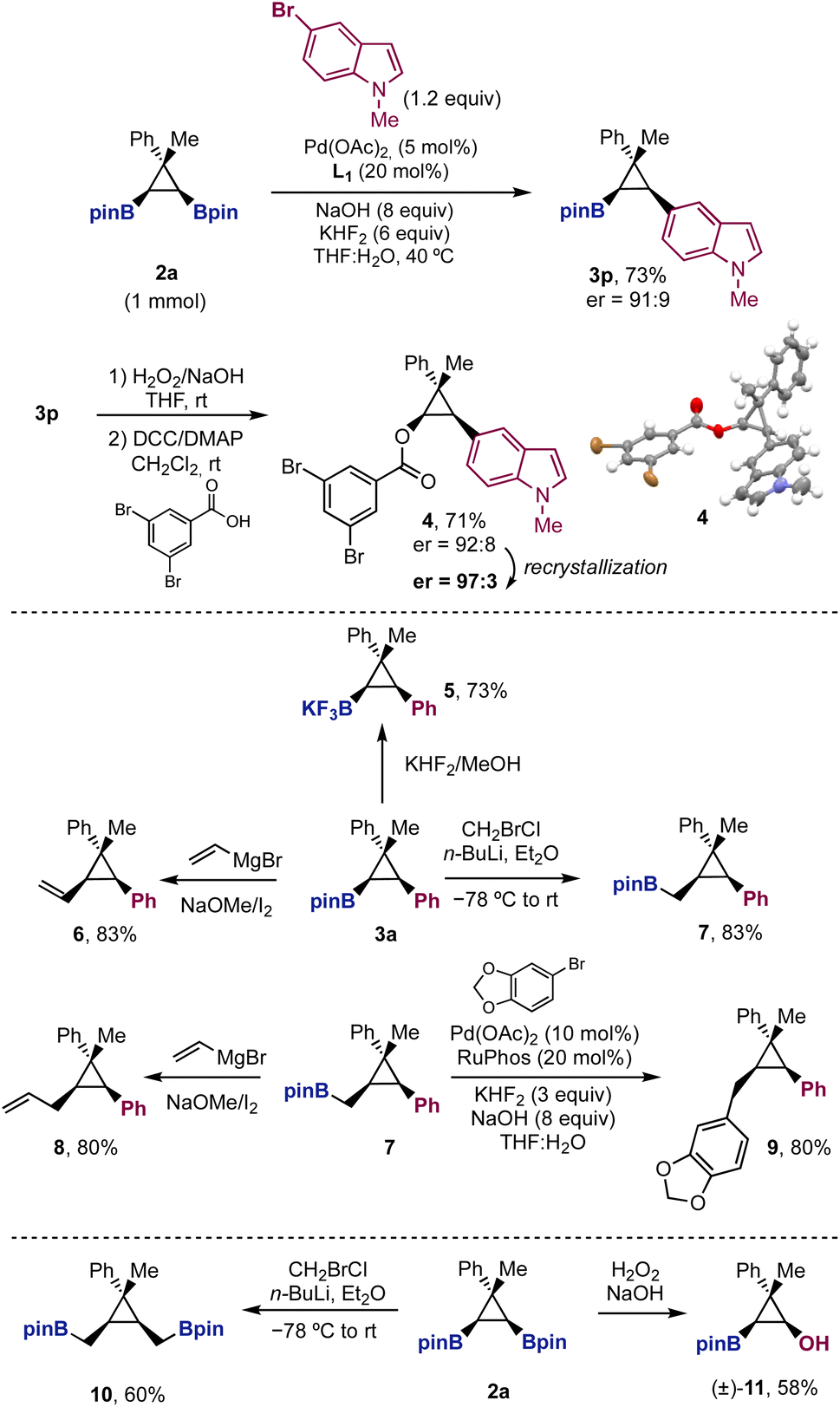 | ||
| Scheme 5 Assignment of the absolute stereochemistry and C–B bond functionalizations. | ||
Finally, we showed the synthetic potential of the Suzuki cross-coupling products 3 with further stereospecific functionalization of the remaining boryl unit (Scheme 5). The transformation of the pinacol ester in the potassium trifluoroborate salt took place in high yield. Zweifel and Matteson homologations provided the desired products (6 and 7) in excellent yields, even though the boryl unit is placed in a crowded environment, surrounded by two substituents in syn relative distribution. The Matteson homologation product 7 is especially interesting as it still preserves the boryl unit for further transformations. A second sp2–sp3 Suzuki cross-coupling from 7 afforded trisubstituted cyclopropane 9 in 80% yield. Additionally, Zweifel homologation from 7 provided allylcyclopropane 8 in high yield. Finally, we briefly explored the synthetic potential of diboryl cyclopropanes 2 with reactions different than the Suzuki cross-coupling. Selective monooxidation of 2a afforded boryl cyclopropanol 11 as a single diastereomer. Moreover, double Matteson homologation provided symmetric bisboronate 10, which could be used to explore further desymmetrizations.
Conclusions
In summary, we have developed the first enantioselective desymmetrization of a 1,2-bisboronic ester. Prior work on group specific cross coupling has focused on geminal bis-boronates. Therefore, our study might pave the road to develop further desymmetrizations of prochiral 1,2-bis-boronates. This strategy allows for the preparation of highly functionalized enantioenriched boryl cyclopropanes with three stereocenters, one of them being quaternary. The products still preserve one boryl unit that can be used to design further stereospecific transformations. We believe that this methodology provides a useful tool for industrial and medicinal chemists to introduce functionalized three-membered rings into libraries of compounds.Data availability
All the data supporting the findings of this study are available in the ESI.† Crystallographic data for compound 4 has been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC 21910290.Author contributions
J. T., M. V., R. F., A. V. and B. L. performed the experiments, wrote the ESI† and participated in discussions. M. T. conceived and directed the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank MICINN (PID2019-107380GB-I00) for financial support. J. T. thanks MICINN for a predoctoral fellowship. We acknowledge Dr Josefina Perles (UAM) for X-ray structure analysis. We thank Elio Moya for his help during the preparation of compound (±)-4 and Leon Gerken and Gonzalo Delgado for their help in the initial borylation experiments.Notes and references
- (a) B. S. Takale, F.-Y. Kong and R. R. Thakore, Organics, 2022, 3, 1–21 CrossRef CAS; (b) M. J. Blanco and M. J. Buskes, Molecules, 2020, 25, 3493–3515 CrossRef PubMed; (c) J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- (a) D. J. Blair, S. Chitti, M. Trobe, D. M. Kostyra, H. M. S. Haley, R. L. Hansen, S. G. Ballmer, T. J. Woods, W. Wang, V. Mubayi, M. J. Schmidt, R. W. Pipal, G. F. Morehouse, A. M. E. Palazzolo Ray, D. L. Gray, A. L. Gill and M. D. Burke, Nature, 2022, 604, 92–97 CrossRef CAS PubMed; (b) J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. E. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221–1226 CrossRef CAS PubMed.
- (a) F. Lovering, MedChemComm, 2013, 515–519 RSC; (b) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- (a) J. Li and H. Huang, Bioconjugate Chem., 2018, 29, 3841–3846 CrossRef CAS PubMed; (b) Y. Ding and M. A. Clark, ACS Comb. Sci., 2015, 17, 1–4 CrossRef CAS PubMed.
- Recent reviews on enantioselective and stereospecific Suzuki cross-coupling: (a) X. Ma, B. Murray and M. R. Biscoe, Nat. Rev. Chem., 2020, 4, 584–599 CrossRef CAS PubMed; (b) J. P. G. Rygus and C. M. Crudden, J. Am. Chem. Soc., 2017, 139, 18124–18137 CrossRef CAS PubMed; (c) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS PubMed.
- For a pioneering desymmetrization involving distal boranes in an intramolecular Suzuki cross-coupling, see: S. Y. Cho and M. Shibasaki, Tetrahedron: Asymmetry, 1998, 9, 3751–3754 CrossRef CAS.
- Recent reviews on enantioselective metal-catalyzed transformations of geminal bisboronates: (a) Y. Lee, S. Han and S. H. Cho, Acc. Chem. Res., 2021, 54, 3917–3929 CrossRef CAS PubMed; (b) C. Zhang, W. Hu and J. P. Morken, ACS Catal., 2021, 11, 10660–10680 CrossRef CAS PubMed.
- (a) C. Sun, B. Potter and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 6534–6537 CrossRef CAS PubMed; (b) B. Potter, A. A. Szymaniak, E. K. Edelstein and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 17918–17921 CrossRef CAS PubMed.
- H. Sun, K. Kubota and D. G. Hall, Chem. –Eur. J., 2015, 21, 19186–19194 CrossRef CAS PubMed.
- Recent review on selective functionalization of 1,2-bis-boronates: A. Viso, R. Fernández de la Pradilla and M. Tortosa, ACS Catal., 2022, 12, 10603–10620 CrossRef CAS.
- Selected recent examples of selective functionalization of 1,2-bis-boronates: (a) N. Xu, Z. Kong, J. Z. Wang, G. J. Lovinger and J. P. Morken, J. Am. Chem. Soc., 2022, 144, 17815–17823 CrossRef CAS PubMed; (b) H. Wang, W. Han, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2022, 61, e202207988 CAS; (c) H. Wang, J. Wu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2022, 61, e202202061 CAS; (d) D. Kaiser, A. Noble, V. Fasano and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 14104–14109 CrossRef CAS PubMed; (e) A. Fawcett, D. Nitsch, M. Ali, J. M. Bateman, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2016, 55, 14663–14667 CrossRef CAS PubMed; (f) C. M. Crudden, C. Ziebenhaus, J. P. G. Rygus, K. Ghozati, P. J. Unsworth, M. Nambo, S. Voth, M. Hutchinson, V. S. Laberge, Y. Maekawa and D. Imao, Nat. Commun., 2016, 7, 11065 CrossRef PubMed; (g) S. N. Mlynarski, C. H. Schuster and J. P. Morken, Nature, 2014, 505, 386–390 CrossRef CAS PubMed.
- (a) L. Nóvoa, L. Trulli, A. Parra and M. Tortosa, Org. Lett., 2021, 23(19), 7434–7438 CrossRef PubMed; (b) L. Nóvoa, L. Trulli, A. Parra and M. Tortosa, Angew. Chem., Int. Ed., 2021, 60, 11763–11768 CrossRef PubMed; (c) L. Amenós, L. Trulli, L. Nóvoa, A. Parra and M. Tortosa, Angew. Chem., Int. Ed., 2019, 58, 3188–3192 CrossRef PubMed; (d) M. Guisan-Ceinos, A. Parra, V. Martin-Heras and M. Tortosa, Angew. Chem., Int. Ed., 2016, 55, 6969–6972 CrossRef CAS PubMed; (e) A. Parra, L. Amenós, M. Guisan-Ceinos, A. López, J. L. Garcia-Ruano and M. Tortosa, J. Am. Chem. Soc., 2014, 136, 15833–15836 CrossRef CAS PubMed.
- (a) M.-R. Sun, H.-L. Li, M.-Y. Ba, W. Cheng, H.-L. Zhu and Y.-T. Duan, Mini-Rev. Med. Chem., 2021, 21, 150–170 CrossRef CAS PubMed; (b) T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS PubMed; (c) Y. Cohen and I. Marek, Acc. Chem. Res., 2022, 55, 2848–2868 CrossRef CAS PubMed.
- Recent selected stereoselective synthesis of cyclopropylboronates: (a) A. U. Augustin, S. Di Silvio and I. Marek, J. Am. Chem. Soc., 2022, 144, 16298–16302 CrossRef CAS PubMed; (b) Y. Shi, Y. Yang and S. Xu, Angew. Chem., Int. Ed., 2022, 61, e202201463 CAS; (c) H. Iwamoto, Y. Ozawa, Y. Hayashi, T. Imamoto and H. Ito, J. Am. Chem. Soc., 2022, 144, 10483–10494 CrossRef CAS PubMed; (d) N. Hanania, M. Nassir, N. Eghbarieh and A. Masarwa, Chem. –Eur. J., 2022, e202202748 CAS; (e) G. Benoit and A. B. Charette, J. Am. Chem. Soc., 2017, 139, 1364–1367 CrossRef CAS PubMed; (f) B. Tian, Q. Liu, X. Tong, P. Tian and G.-Q. Lin, Org. Chem. Front., 2014, 1, 1116–1122 RSC; (g) C. Zhong, S. Kunii, Y. Kosaka, M. Sawamura and H. Ito, J. Am. Chem. Soc., 2010, 132, 11440–11442 CrossRef CAS PubMed; (h) M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2003, 125, 7198–7199 CrossRef CAS PubMed.
- D. G. Hall, Boronic Acids: Preparation and Application in Organic Synthesis, Medicine and Materials, Wiley-VCH, Boston, 2nd edn, 2011 Search PubMed.
- Synthesis of related 1,2-diboryl cyclopropanes, see: (a) F.-P. Wu, X. Luo, U. Radius, T. B. Marder and X.-F. Wu, J. Am. Chem. Soc., 2020, 142, 14074–14079 CrossRef CAS PubMed; (b) M. Mali, G. V. Sharma, S. Ghosh, T. Roisnel, B. Carboni and F. Berrée, J. Org. Chem., 2022, 87, 7649–7657 CrossRef CAS PubMed.
- (a) For an early review on transition metal catalyzed diboration, see: T. B. Marder and N. C. Norman, Top. Catal., 1998, 5, 63–73 CrossRef CAS; (b) For a review on the reactivity of diboron compounds, see: E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed.
- For pioneering work on Pt-catalyzed diboration: (a) T. Ishiyama, M. Yamamoto and N. Miyaura, Chem. Commun., 1997, 689–690 RSC; (b) C. N. Iverson and M. R. Smith III, Organometallics, 1997, 16, 2757–2759 CrossRef CAS . For relevant work on Pt-catalyzed 1,2-diboration of alkenes using chiral diboron reagents:; (c) T. B. Marder, N. C. Norman and C. R. Rice, Tetrahedron Lett., 1998, 39, 155–158 CrossRef CAS; (d) L. T. Kliman, S. N. Mlynarski and J. P. Morken, J. Am. Chem. Soc., 2009, 131, 13210–13211 CrossRef CAS PubMed; (e) L. T. Kliman, S. N. Mlynarski, G. E. Ferris and J. P. Morken, Angew. Chem., Int. Ed., 2012, 51, 521–524 CrossRef CAS PubMed; (f) J. R. Coombs, F. Haeffner, L. T. Kliman and J. P. Morken, J. Am. Chem. Soc., 2013, 135, 11222–11231 CrossRef CAS PubMed; (g) J. R. Coombs, L. Zhang and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 16140–16143 CrossRef CAS PubMed.
- For the first metal-catalyzed 1,2-diboration of alkenes, see: (a) R. T. Baker, P. Nguyen, T. B. Marder and S. A. Westcott, Angew. Chem., Int. Ed., 1995, 34, 1336 CrossRef CAS ; for a pioneering example of Rh(I) catalyzed 1,2-diboration of alkenes with B2cat2, see:; (b) C. Dai, E. G. Robins, A. J. Scott, W. Clegg, D. S. Yufit, J. A. K. Howard and T. B. Marder, Chem. Commun., 1998, 1983 RSC . For enantioselective Rh(I) catalyzed 1,2-diboration of alkenes, see:; (c) J. B. Morgan, S. P. Miller and J. P. Morken, J. Am. Chem. Soc., 2003, 125, 8702 CrossRef CAS PubMed; (d) S. Trudeau, J. B. Morgan, M. Shrestha and J. P. Morken, J. Org. Chem., 2005, 70, 9538 CrossRef CAS PubMed; (e) K. Toribatake and H. Nishiyama, Angew. Chem., Int. Ed., 2013, 52, 11011 CrossRef CAS PubMed.
- W. M. Sherrill and M. Rubin, J. Am. Chem. Soc., 2008, 130, 13804 CrossRef CAS PubMed.
- Base-promoted 1,2-diboration of alkenes: (a) A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158–7161 CrossRef CAS PubMed; (b) T. P. Blaisdell, T. C. Caya, L. Zhang, A. Sanz-Marco and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 9264–9267 CrossRef CAS PubMed; (c) A. Bonet, C. Sole, H. Gulyás and E. Fernández, Org. Biomol. Chem., 2012, 10, 6621–6623 RSC; (d) L. Fang, L. Yan, F. Haeffner and J. P. Morken, J. Am. Chem. Soc., 2016, 138, 2508–2511 CrossRef CAS PubMed; (e) A. J. Vendola, C. Allais, A.-M. R. Dechert-Schmitt, J. T. Lee, R. A. Singer and J. P. Morken, Org. Lett., 2021, 23, 2863–2867 CrossRef CAS PubMed.
- S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem. –Eur. J., 2015, 21, 7082–7098 CrossRef CAS PubMed.
- For optimization full details see the ESI.†.
- (a) P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2016, 138, 9145–9157 CrossRef CAS PubMed; (b) H. L. D. Hayes, R. Wei, M. Assante, K. J. Geogheghan, N. Jin, S. Tomasi, G. Noonan, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2021, 143, 14814–14826 CrossRef CAS PubMed.
- C. Amatore, A. Jutand and G. Le Duc, Angew. Chem., Int. Ed., 2012, 51, 1379–1382 CrossRef CAS PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2012, 134, 7431–7441 CrossRef CAS PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Isr. J. Chem., 2010, 50, 664–674 CrossRef CAS.
- N. Xu, Z. Kong, J. Z. Wang, G. J. Lovinger and J. P. Morken, J. Am. Chem. Soc., 2022, 144, 17815–17823 CrossRef CAS PubMed.
- The absolute configuration of the cross-coupling products was established from compound 4 by single crystal X-ray crystallography. CCDC 2191020 contains the supplementary crystallographic data for 4.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2191020. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05789a |
| This journal is © The Royal Society of Chemistry 2023 |