
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermally activated delayed fluorescence with dual-emission and pressure-induced bidirectional shifting: cooperative effects of intramolecular and intermolecular energy transfer†
Chenyue
Zhao
a,
Zhipeng
Ding
a,
Yibin
Zhang
a,
Zhigang
Ni
 a,
Shijun
Li
a,
Shaolong
Gong
*b,
Bo
Zou
c,
Kai
Wang
*c and
Ling
Yu
*a
a,
Shijun
Li
a,
Shaolong
Gong
*b,
Bo
Zou
c,
Kai
Wang
*c and
Ling
Yu
*a
aCollege of Material, Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou, 311121, People's Republic of China. E-mail: lingyu@hznu.edu.cn
bHubei Key Lab on Organic and Polymeric Optoelectronic Materials, Department of Chemistry, Wuhan University, Wuhan, 430072, People's Republic of China. E-mail: slgong@whu.edu.cn
cState Key Laboratory of Superhard Materials, Jilin University, Qianjin Street 2699, Changchun 130012, People's Republic of China. E-mail: kaiwang@jlu.edu.cn
First published on 22nd December 2022
Abstract
Different from the conventional piezochromic materials with a mono-redshift of single emission, our well-designed molecule demonstrates a sensitive turn-on and color-tunable piezochromic luminescence in response to the hydrostatic pressure. The molecule PXZ-W-SOF possesses dual-emission and pressure-induced bidirectional shifting characteristics. On the basis of in-depth experimental studies, on one hand, it is confirmed that the origin of the dual-emission behavior is the intramolecular charge transfer, namely thermally activated delayed fluorescence (TADF), and the intermolecular excimer; on the other hand, the emission of the excimer exhibits three-step variations with increasing pressure, which is mainly attributed to the molecular structure and its crystal packing state. The remarkable color change of PXZ-W-SOF from sky-blue to green to deep-blue during the whole process of boosting and releasing pressure is a result of intramolecular and intermolecular energy-transfer interactions. The PXZ-W-SOF molecular model is an extremely rare example of highly sensitive fluorescence tuning driven by TADF and excimer conversion under mechanical stimulation, thus providing a novel mechanism for the field of piezochromism. The unique molecular design also offers a new idea for rare deep-blue and ultraviolet TADF materials.
Introduction
In the past decade, thermally activated delayed fluorescence (TADF) materials have flourished because they can utilize the up-conversion process of triplet excitons to achieve 100% internal quantum efficiency without the participation of heavy metals.1–6 Nevertheless, compared with TADF materials (such as sky-blue, green and red) in the visible light region, the development of deep-blue (λ < 460 nm) and ultraviolet (UV) (λ ≤ 400 nm) TADF materials is seriously backward,7,8 although it has broad application prospects in high-density information storage, biological/chemical sensing, excitation light sources, etc.9,10 The reason may be that in order to endow TADF materials with the intramolecular charge transfer (ICT) characteristic to trigger efficient reverse intersystem crossing (RISC), donors and acceptors with relatively strong electron-donating and electron-withdrawing ability, respectively are often employed as building blocks in a conjugated system. However, this generally results in TADF materials with relatively narrow energy band gaps (Egs), leading to long-wavelength emission away from the deep-blue and UV region.11 Thus, it is still highly challenging to develop TADF materials in the high-energy excited state.Piezochromic luminescent materials, a class of smart materials, exhibit significant emission color changes in response to external force stimuli (e.g., pressing, stretching and grinding), and they have been widely explored due to their great potential for applications in pressure sensors, security ink, optical data storage and photoelectronic devices.12–15 However, all the reported piezochromic materials are largely limited to a mono-redshift of single emission with increasing pressure or grinding, and reports on piezochromic materials with bidirectional shifting of dual-emission are extremely rare,16–18 let alone the TADF system.
In order to form high-contrast luminescence recordings, excellent piezochromic materials require significant two-color luminescence switching that is difficult to achieve with conventional piezochromic materials.19–22 Dual-emission and pressure-induced bidirectional shifting have not been reported in TADF materials. To achieve the above goals, there is no doubt that higher requirements are placed on the design of luminescent materials.
Herein, we break through the traditional design idea of directly conjugated “donor–acceptor (D–A)” or “D–π–A” type TADF and adopt the concept of “unconjugated D–A” to design high-energy excited state TADF materials. Constructing TADF materials in this intramolecular unconjugated way has the potential to combine the deep-blue or UV emission with multipath exciton harvesting. It is worth mentioning that two novel deep-blue and UV TADF molecules (Fig. 1 and Scheme S1†) have been successfully synthesized, one of which possesses remarkable dual-emission and pressure-induced bidirectional shifting properties, and meanwhile, its complex luminescence mechanism under pressure has been clearly elucidated through rigorous experiments.
 | ||
| Fig. 1 (a) Configuration and molecular structures of DMAC-W-SOF and PXZ-W-SOF and (b and c) HOMO and LUMO distributions, and calculated singlet (S1) and triplet (T1) energy levels of DMAC-W-SOF and PXZ-W-SOF based on TD-DFT at the B3LYP/6-311G(d) level in an implicit toluene atmosphere. | ||
Results and discussion
Design, synthesis and characterization
The key molecular design strategy based on the selection of a rigid and high-energy band-gap skeleton and a high-energy band-gap acceptor group is proposed, which is the basis for guaranteed deep-blue or UV emission. Fluorene derivatives have developed into an important class of blue luminescent materials due to their rigid planar aromatic biphenyl structure with some outstanding properties, such as a high-energy band gap, good thermal stability and easy modification of the molecular structure.23–25 The 9-position carbon of fluorene has an sp3 hybrid orbital (Fig. 1a), resulting in the intramolecular noncovalent interaction between the D/A units, so strong electron-donating ability is required to facilitate the charge-transfer interaction with the acceptor moiety. Diphenyl sulfone has emerged as the star acceptor group for designing blue and deep blue TADF molecules over the past decade.26 The introduction of the fluorine atom is used to enhance intramolecular and intermolecular electron interactions.27–29 9,9-Dimethyl-9,10-dihydroacridine (DMAC) and 10H-phenoxazin (PXZ) are commonly employed as donor units in TADF systems due to their rigid structure, controllable electron-donating ability and easy implementation,30–34 but they are used in UV TADF materials for the first time.The two novel TADF materials were synthesized through efficient three-step reactions (Scheme S1†). The key intermediate, I–W–SOF, was prepared from 9,9-bis(4-iodophenyl)fluorene and 4-fluorothiophenol successively by an Ullmann coupling reaction and an oxidation reaction. Then, the syntheses of DMAC-W-SOF and PXZ-W-SOF were achieved by the palladium catalyzed C–N coupling reaction between I–W–SOF and the corresponding donor units. Chemical structures of the two target compounds were fully confirmed by 1H NMR, 13C NMR and mass spectrometry (see the ESI† for details). X-ray single crystal analyses further confirmed the structures of DMAC-W-SOF and PXZ-W-SOF (Table S1†).
DFT calculations and electrochemical properties
The frontier molecular orbital (FMO) arrangement and energy levels of DMAC-W-SOF and PXZ-W-SOF can be predicted by using the B3LYP/6-311G(d) level. As shown in Fig. 1b, the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) are mainly distributed in the donor unit and the diphenyl sulfone acceptor fraction, respectively. As is known, the value of the singlet–triplet energy gap (ΔEST) is proportional to the exchange interaction integral between the HOMO and the LUMO wavefunction in a molecule.35 Therefore, the unconjugated D–A construct is beneficial to obtain small ΔEST, and their theoretical ΔESTs are about 0.11 eV for DMAC-W-SOF and 0.13 eV for PXZ-W-SOF. In addition, their experimental HOMO levels were investigated by cyclic voltammetry (CV) (Fig. S1† and Table 1), and the HOMO level increases accordingly with the enhancement of the electron-donating ability in the order of −5.36 eV (DMAC-W-SOF) < −5.10 eV (PXZ-W-SOF).| Compound | λ abs [nm] | λ Fl,max [nm] | λ Ph,max [nm] | ΔESTd[eV] | ΔEge[eV] | HOMO/LUMO [eV] |
|---|---|---|---|---|---|---|
| a Measured in toluene. b Measured in toluene/powder at room temperature. c Measured in 2-methyl-tetrahydrofuran at 77 K. d ΔEST = ES − ET for the fluorescence and phosphorescence data. e Calculated from the absorption edge of the UV-vis absorption spectra in toluene. f Theoretically calculated. i Determined from the onset of the oxidation potential. g Deduced from the HOMO and ΔEg. | ||||||
| DMAC-W-SOF | 289, 308 | 374/409 | 471 | 0.69/0.11f | 3.67 | −5.36i, −1.69g |
| PXZ-W-SOF | 287, 309, 325 | 404/438, 486 | 470 | 0.43/0.13f | 3.41 | −5.10i, −1.69g |
Photophysical properties
DMAC-W-SOF and PXZ-W-SOF were analyzed by UV-vis absorption spectra and photoluminescence (PL) measurements in organic solvent (Fig. S2†). In the absorption spectra, both DMAC-W-SOF and PXZ-W-SOF show the same absorption as I–W–SOF at about 280–300 nm. However, it should be noted that PXZ-W-SOF has an additional intramolecular charge transfer (ICT) transition band near 325 nm, displaying strong absorption between 320 and 380 nm (Fig. S3†), and indicating a non-trivial level of interaction between the D/A moieties. Then, the corresponding optical band gaps (Egs) can be calculated from the absorption edge to be 3.67 eV for DMAC-W-SOF and 3.41 eV for PXZ-W-SOF. Consistent with the results of wide Egs, their fluorescence spectra exhibit structureless emission at 374 nm for DMAC-W-SOF and 404 nm for PXZ-W-SOF (Fig. S2†), and PXZ-W-SOF is redshifted 30 nm than DMAC-W-SOF with the increase of electron-donating ability. Similar phosphorescence emission profiles were observed in 2-methyl-tetrahydrofuran at 77 K, indicating that the donor unit has little effect on phosphorescence emission in such molecular design. The experimental ΔESTs of DMAC-W-SOF and PXZ-W-SOF are calculated to be 0.69 and 0.43 eV (Table 1), respectively.The luminescence behaviors of the aggregation state for DMAC-W-SOF and PXZ-W-SOF also were monitored (Fig. S4†). As expected, they both show a clear red-shift phenomenon compared to their fluorescence emission in toluene. Surprisingly, however, unlike the mono-emission peak of DMAC-W-SOF, PXZ-W-SOF exhibits a dual-emission behavior with the peaks of 438 and 486 nm (Fig. S4a and b†), which are neither from PXZ, nor from I–W–SOF (Fig. S4c†).
Luminescence mechanism
Then, what are the emission mechanisms with peaks at 438 nm and 486 nm for the dual-emission behavior of PXZ-W-SOF? According to the UV-vis absorption spectra in toluene solution, PXZ-W-SOF has a strong ICT transition band. Therefore, one of the dual-emission peaks should come from the CT state emission. Yet now, which is the emission peak originating from the CT state, 438 nm or 486 nm? Where does the other fluorescence emission peak come from? To understand the photophysical behaviors, fluorescence spectra of different concentrations of PXZ-W-SOF in toluene solution were recorded. As shown in Fig. 2a and S5†, the short waveband emission of PXZ-W-SOF exhibits a red-shift and the fluorescence peak at around 500 nm appeared as well as gradually occupied the dominant position with the increase of its concentration. In particular, when the concentration increased to 0.01 M, the emission peak of the long waveband is significantly enhanced, and when it further increased to 0.05 M, the emission profile of PXZ-W-SOF is basically consistent with that of its aggregation state. The more detailed concentration distinction is shown in Fig. 2b. The above phenomena reveal that the excimer of PXZ-W-SOF will be formed when its concentration in toluene solution exceeds the 0.0001 M level. Therefore, it can be inferred that the emission peak at the short waveband comes from the CT state of PXZ-W-SOF and the fluorescence peak of the long waveband is derived from its excimer. | ||
| Fig. 2 Normalized fluorescence spectra of different concentrations (a) 0.0001–0.05 M and (b) 0.001–0.03 M for PXZ-W-SOF in toluene solution. | ||
The excimer of DMAC-W-SOF was not observed in the same experiment and why? We therefore conducted an in-depth analysis of their molecular conformation and crystal packing information. Interestingly, although only one single bond changed from DMAC-W-SOF to PXZ-W-SOF, the packing arrangements are completely different from each other. The two DMAC-W-SOF molecules with parallel diphenylsulfone units form a flatly spread dimer with an anti conformation to minimize the dipolar interactions (Fig. 3a).36 The intermolecular C–H⋯π, C–H⋯H–C and C–H⋯O short forces hold the dimers, showing a one-dimensional supramolecular chain along the crystallographic c-axis direction (Fig. S6†). Unlike DMAC-W-SOF, which has only one “face-to-face” arrangement based on the diphenyl sulfone units, PXZ-W-SOF has two kinds of oriented arrangements to form a “sandwich” structure (Fig. 3d), indicating the presence of strong electron exchange between the donor and the acceptor. The existence of short C–H⋯F forces in the directional arrangement of PXZ-W-SOF but not found in DMAC-W-SOF can also prove the above-mentioned point (Fig. S6†). In consequence, PXZ-W-SOF exhibits more kinds of short-range intermolecular forces in the crystal configuration, undoubtedly providing a favorable basic environment for its multi-type fluorescence emission (Fig. 3b and e). Their packing modes are displayed in Fig. 3c and f.
 | ||
| Fig. 3 Molecular packing modes of (a–c) DMAC-W-SOF and (d–f) PXZ-W-SOF. | ||
TADF and AIE characterization studies
Their TADF characteristics were verified by the transient photoluminescence (PL) spectra test in 10−4 M toluene solution and in powder. As exemplified in Fig. 4, when oxygen is present, both compounds only show prompt fluorescence emission, typically PL quenching due to oxygen.37 After degassing with argon in toluene, they distinctly display the bi-exponential decay PL spectra with prompt and delayed components. The photoluminescence quantum yields (PLQYs) of DMAC-W-SOF and PXZ-W-SOF before and after degassing with argon in 10−4 M toluene solution are 10%/15% and 21%/27%, respectively, indicating their sensitive behavior to oxygen. The TD-B3LYP/6-311G (d) horizontal simulation of the UV-vis spectrum shows that the maximum oscillation intensity of DMAC-W-SOF and PXZ-W-SOF is 0.295 for the S19 state and 0.499 for the S43 state, respectively. According to Kasha's rule, the S1 state will be populated through the internal conversion process. Therefore, it is easy to understand that the PLQY of PXZ-W-SOF is better than that of DMAC-W-SOF. For a deeper comparison, the transient behaviors of the two compounds in high concentration toluene solution (0.05 M) were detected. As shown in Fig. S7,† the delayed lifetimes of DMAC-W-SOF (7.68 μs) and PXZ-W-SOF (6.53 μs) are significantly increased compared with the case in 10−4 M toluene solution (280 ns for DMAC-W-SOF and 189 ns for PXZ-W-SOF), regardless of whether excimer association is generated. After bubbling argon in 0.05 M toluene, the PLQYs of DMAC-W-SOF and PXZ-W-SOF also decreased sharply to 4% and 11%, respectively. The reason for the above phenomena may be that aggregation is easy to occur in high concentration solution and affects the luminous properties. The aggregation behavior of organic molecules can reduce the luminescence efficiency of materials, and one of the reasons is the formation of the excimer. In particular for PXZ-W-SOF, the essential reasons are: on the one hand, the charge-transfer effect and forbidden symmetry for the excitation state of the exciplex lead to the greatly reduced radiation transition rate, and on the other hand, the dynamic behavior, the crossed potential energy surfaces, energy-transfer and other factors of the excited state structure of the exciplex result in a great increase for the non-radiative transition rate, which ultimately results in a serious luminescence quenching problem. In powder, their transient PL curves also exhibit a bi-exponential decay. Moreover, taking PXZ-W-SOF as an example, the delayed components are gradually intensified with the increase of ambient temperature from 100 to 300 K (Fig. 4c), definitely demonstrating its typical thermally activated nature,26,38 and its time-resolved spectral 3D map is shown in Fig. 4d.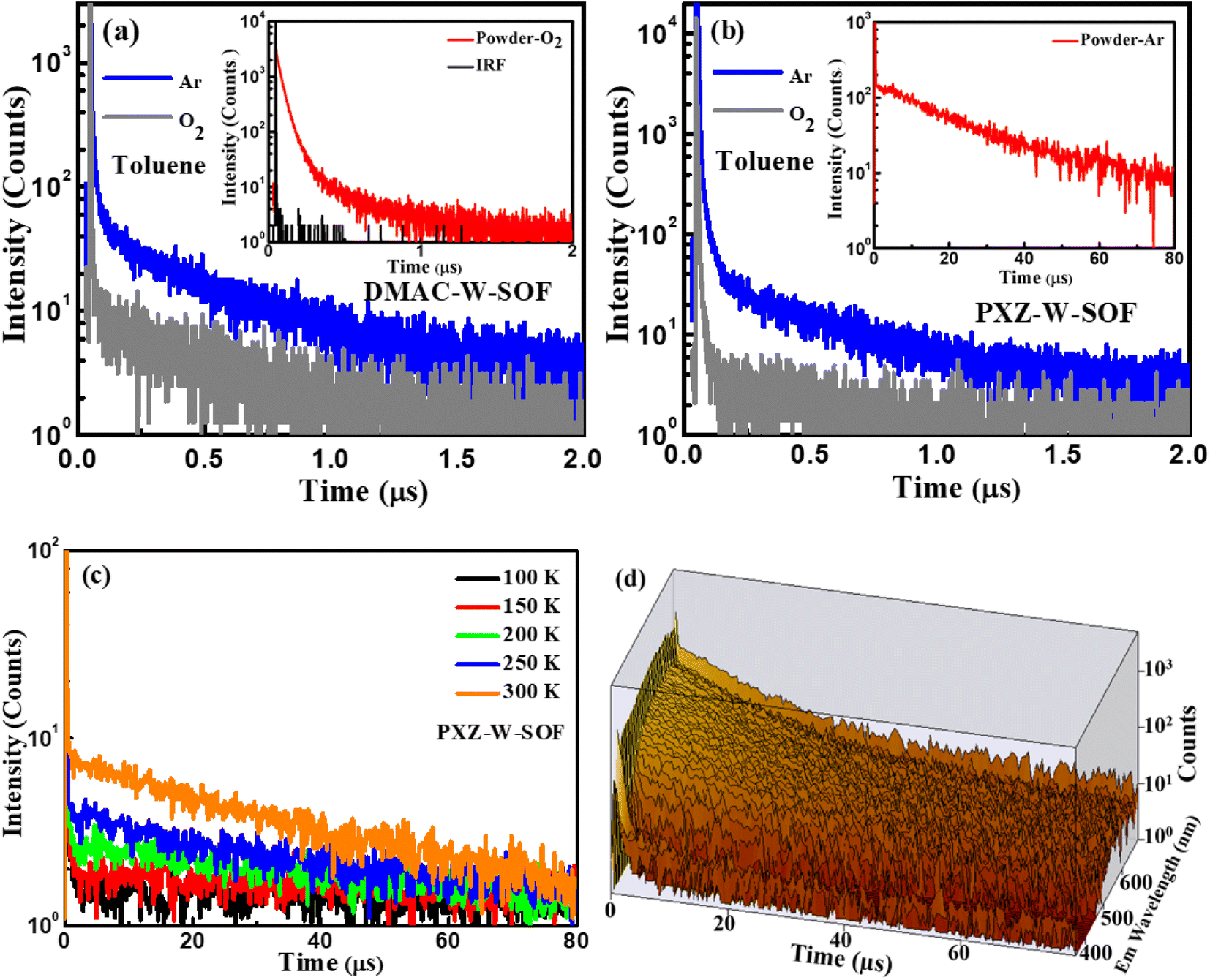 | ||
| Fig. 4 Transient PL characteristics of (a) DMAC-W-SOF and (b) PXZ-W-SOF in toluene (10−4 M) under oxygen and argon conditions at room temperature, and the temperature-dependent transient PL decays of (c) PXZ-W-SOF from 100 to 300 K in powder and (d) the time-resolved spectral 3D map of PXZ-W-SOF in powder at room temperature. The insets respectively show the transient PL decay of the corresponding compounds in powder. IRF: the instrument response function. | ||
In addition, the AIE nature of PXZ-W-SOF was confirmed by its PL behavior in THF/water with various water fractions from 0 to 99%. As shown in Fig. S8,† on one hand, when the water fraction (fw) is less than 70% in mixed solvents, the PL intensities decrease rapidly and meanwhile the emission is redshifted, which is attributed to the typical emission properties induced by twisted ICT (TICT) aggregation.39,40 On the other hand, when fw > 70%, the PL intensities increase significantly even more than those in pure organic solvent, and the emission peak is blue-shifted, indicating a prominent AIE feature. However, it can be seen from the previous PLQY detection that the AIE characteristic cannot completely resist the quenching effect of high concentration.
Isotropic hydrostatic pressure measurement
Thus, PXZ-W-SOF not only has remarkable dual-emission action, but also TADF and AIE properties, and its special luminescence behavior can be deeply analyzed by isotropic hydrostatic pressure measurement. Isotropic hydrostatic pressure measurement from a diamond anvil cell (DAC) is an effective way to explore the molecular structure–property relationship.41 Here, the isotropic hydrostatic pressure was directly applied to the PXZ-W-SOF crystal via the DAC, and the two emission peaks showed significant differentiation (Fig. 5). As mentioned above (Fig. S4b†), at 1 atmosphere, the PXZ-W-SOF crystal shows two emission peaks around 440 nm (herein called the high-energy emission band for clarity) and 490 nm (derived from the excimer, also called the low-energy emission band). As the pressure gradually increased, the fluorescence peak at 440 nm diminishes rapidly, but obvious three-step variations can be observed in the low-energy emission band (Fig. 5a). When the pressure is increased from 1 atm to 3.08 GPa, the low-energy emission band shows a gradual red-shift from blue (490 nm) to green (517 nm), together with decreasing intensity. Once pressure increased above 3.08 GPa, the peak of the low-energy emission band is continuously blue-shifted from 517 nm to 508 nm, which is contrary to most observations in the piezochromic luminescence field. With further compression beyond 8.11 GPa, the low-energy emission band demonstrates a significant red-shift (from 508 nm to 537 nm) again.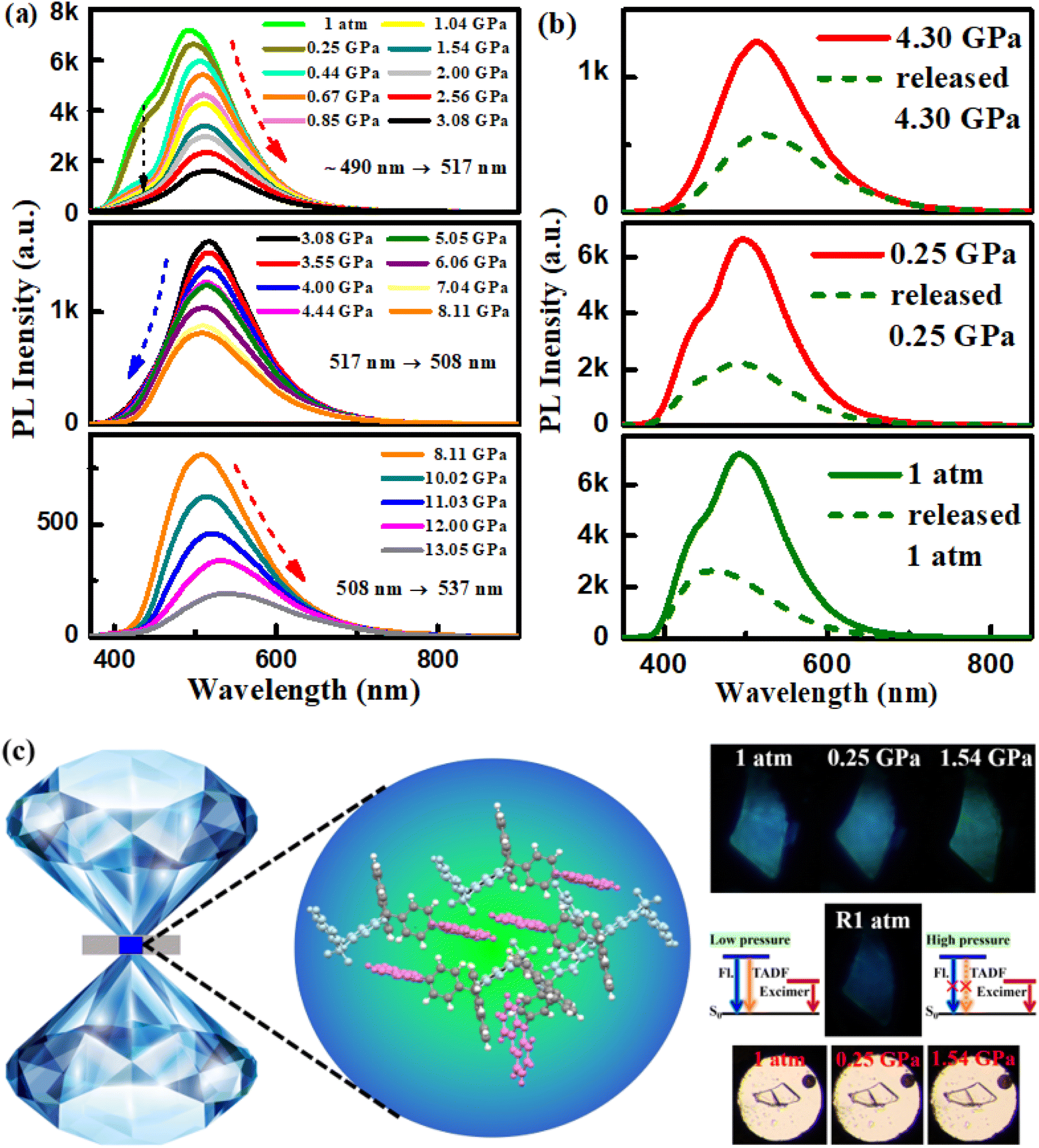 | ||
| Fig. 5 (a) Emission spectra and (b) verification of the reversibility of the PXZ-W-SOF crystal released to 4.30 GPa, 0.25 GPa and 1 atm from 13.05 GPa, respectively. (c) Schematic illustration of the DAC apparatus, molecular packing, fluorescent photographs, and visible photographs and the schematic diagram of pressure-induced emission of the PXZ-W-SOF crystal. | ||
With the increase of pressure, the PXZ-W-SOF unit aggregates to form a tighter state, leading to a sharp weakening of the ICT behavior and thus the rapid disappearance of the high-energy emission band. Pressure-induced three-step variations of the low-energy emission band during PXZ-W-SOF crystal compression may be explained as follows. As the pressure further increased, the intramolecular vibrations and rotations of PXZ-W-SOF can be effectively restricted due to the combination of more interactions. Therefore, the blue-shift emission caused by the reduced Stokes shift may be derived from the suppressed geometric relaxation. As for the redshift and quenching of the low-energy emission band, the energy gap between the excited state of the excimer and the ground state of the dimer becomes dramatically narrower due to the decrease of the π–π distance with increasing pressure. Therefore, according to the energy gap law, the non-radiative internal conversion (IC) rate will be aggravated. This bidirectional shifting upon application of pressure may be the difference between excimers formed spontaneously and excimers formed by external force stimuli. When the pressure is continuously released to 1 atm, the high-energy emission band (i.e., the TADF emission peak) is reversible, but the low-energy emission band from the excimer is not fully recovered (Fig. 5b). This effect may stem from the difficulty of the PXZ-W-SOF unit to promptly restore its geometry to the original state due to structural deformation under pressure. Thus, the high-contrast luminescence with continuous variation of the emission color from sky blue to green to deep-blue is attributed to the dual-emission of PXZ-W-SOF. The schematic diagram of the DAC device, fluorescence photos, and visible photos and the pressure-induced emission schematic diagram of the PXZ-W-SOF crystal are shown in Fig. 5c.
Conclusions
In conclusion, our work demonstrates that deep-blue and UV-emissive TADF materials can be successfully obtained through rational molecular design, which can also achieve significant dual-emission and pressure-induced bidirectional shifting characteristics. This phenomenon is an extremely rare case in the field of piezochromic luminescence. Detailed and rigorous experiments have proved that the high-energy emission band is derived from the emission of ICT, namely TADF, and the low-energy emission band originates from the intermolecular excimer. With increasing pressure, the three-step variation emission of the excimer is anomalous to the common mono-redshift phenomenon, which is mainly attributed to the molecular structure and its crystal packing state. Throughout the process of pressurization and depressurization, PXZ-W-SOF shows a remarkable color change of sky-blue to green to deep-blue as a result of intramolecular and intermolecular interactions. This work reports a novel principle for high-contrast piezochromic luminescent materials formed by combining TADF and excimers. This study may also advance the understanding of molecular design for dual-emission in fundamental photophysics.Data availability
The datasets supporting this article have been uploaded as part of the ESI material.†Author contributions
L. Y. conceived the project. C. Z, Z. D., Y. Z., B. Z. and K. W. performed the experiments. Z. N conducted the DFT calculations. L. Y., C. Z., S. G. and S. L. analyzed the data and wrote the paper. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 52103204, 22071040 and 22003014), the Natural Science Foundation of Zhejiang Provincial (No. LQ19E030015), and the Science & Technology Innovation Program of Zhejiang Province (2018R52051). L. Y. and Z. N. are also supported by the Scientific Research Startup Foundation of Hangzhou Normal University (No. 4095C50222204125 and 4095C50220006). We thank Dr Zhong Chen from the Instrumentation and Service Center for Molecular Sciences at Westlake University for the temperature-dependent transient PL decay measurement of PXZ-W-SOF and also thank Jiyong Liu (Department of Chemistry, Zhejiang University, Hangzhou 310027, P. R. China) for the single-crystal characterization of DMAC-W-SOF and PXZ-W-SOF.Notes and references
- B. S. Kim and J. Y. Lee, Adv. Funct. Mater., 2014, 24, 3970–3977 CrossRef.
- P. Li, H. Chan, S. L. Lai, M. Ng, M. Y. Chan and V. W. Yam, Angew. Chem., Int. Ed., 2019, 58, 9088–9094 CrossRef PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef PubMed.
- H. J. Tan, G. X. Yang, Y. L. Deng, C. Cao, J. H. Tan, Z. L. Zhu, W. C. Chen, Y. Xiong, J. X. Jian, C. S. Lee and Q. X. Tong, Adv. Mater., 2022, 34, e2200537 CrossRef PubMed.
- L. Yu, Z. Wu, G. Xie, W. Zeng, D. Ma and C. Yang, Chem. Sci., 2018, 9, 1385–1391 RSC.
- L. Yu and C. Yang, J. Mater. Chem. C, 2021, 9, 17265–17286 RSC.
- R. Komatsu, T. Ohsawa, H. Sasabe, K. Nakao, Y. Hayasaka and J. Kido, ACS Appl. Mater. Interfaces, 2017, 9, 4742–4749 CrossRef CAS.
- Y. Luo, S. Li, Y. Zhao, C. Li, Z. Pang, Y. Huang, M. Yang, L. Zhou, X. Zheng, X. Pu and Z. Lu, Adv. Mater., 2020, 32, 2001248 CrossRef CAS.
- M. Berggren, M. Granströim, O. Inganas and M. Andersson, Adv. Mater., 1995, 7, 900–903 CrossRef CAS.
- T.-C. Chao, Y.-T. Lin, C.-Y. Yang, T. S. Hung, H.-C. Chou, C.-C. Wu and K.-T. Wong, Adv. Mater., 2005, 17, 992–996 CrossRef CAS.
- S. Xue, X. Qiu, S. Ying, Y. Lu, Y. Pan, Q. Sun, C. Gu and W. Yang, Adv. Opt. Mater., 2017, 5, 1700747 CrossRef.
- Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS PubMed.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- H. Liu, Y. Gu, Y. Dai, K. Wang, S. Zhang, G. Chen, B. Zou and B. Yang, J. Am. Chem. Soc., 2020, 142, 1153–1158 CrossRef CAS.
- Y. Liu, Q. Zeng, B. Zou, Y. Liu, B. Xu and W. Tian, Angew. Chem., Int. Ed., 2018, 57, 15670–15674 CrossRef CAS PubMed.
- Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef PubMed.
- K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef PubMed.
- S. Zhang, Y. Dai, S. Luo, Y. Gao, N. Gao, K. Wang, B. Zou, B. Yang and Y. Ma, Adv. Funct. Mater., 2017, 27, 1602276 CrossRef.
- W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. Feng, H. H. Y. Sung, Y. Lu, I. D. Williams, J. Z. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef PubMed.
- M. S. Kwon, J. Gierschner, S.-J. Yoon and S. Y. Park, Adv. Mater., 2012, 24, 5487–5492 CrossRef PubMed.
- Y. Gong, Y. Tan, J. Liu, P. Lu, C. Feng, W. Z. Yuan, Y. Lu, J. Z. Sun, G. He and Y. Zhang, Chem. Commun., 2013, 49, 4009–4011 RSC.
- Y. Zhang, J. Sun, G. Zhuang, M. Ouyang, Z. Yu, F. Cao, G. Pan, P. Tang, C. Zhang and Y. Ma, J. Mater. Chem. C, 2014, 2, 195–200 RSC.
- B. Liu, W.-L. Yu, Y.-H. Lai and W. Huang, Chem. Mater., 2001, 13, 1984–1991 CrossRef CAS.
- S. L. Tao, Z. K. Peng, X. H. Zhang, P. F. Wang, C.-S. Lee and S.-T. Lee, Adv. Funct. Mater., 2005, 15, 1716–1721 CrossRef CAS.
- Z. Zhao, C. Zeng, X. Peng, Y. Liu, H. Zhao, L. Hua, S. J. Su, S. Yan and Z. Ren, Angew. Chem., Int. Ed., 2022, 61, e202210864 CAS.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- Y. Liu, L. Hua, Z. Zhao, S. Ying, Z. Ren and S. Yan, Adv. Sci., 2021, 8, 2101326 CrossRef CAS.
- X. Zheng, R. Huang, C. Zhong, G. Xie, W. Ning, M. Huang, F. Ni, F. B. Dias and C. Yang, Adv. Sci., 2020, 7, 1902087 CrossRef CAS.
- Q. Xue and G. Xie, Adv. Opt. Mater., 2021, 9, 2002204 CrossRef CAS.
- L. Yu, Z. Wu, G. Xie, C. Zhong, Z. Zhu, H. Cong, D. Ma and C. Yang, Chem. Commun., 2016, 52, 11012–11015 RSC.
- L. Yu, Z. Wu, C. Zhong, G. Xie, K. Wu, D. Ma and C. Yang, Dyes Pigm., 2017, 141, 325–332 CrossRef CAS.
- L. Yu, Z. Wu, C. Zhong, G. Xie, Z. Zhu, D. Ma and C. Yang, Adv. Opt. Mater., 2017, 5, 1700588 CrossRef.
- L. Yu, Z. Wu, G. Xie, J. Luo, Y. Zou, D. Ma and C. Yang, J. Mater. Chem. C, 2020, 8, 12445–12449 RSC.
- L. Zhou, F. Ni, N. Li, K. Wang, G. Xie and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202203844 CAS.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- H. Wang, L. Xie, Q. Peng, L. Meng, Y. Wang, Y. Yi and P. Wang, Adv. Mater., 2014, 26, 5198–5204 CrossRef CAS.
- T. Nakagawa, S. Y. Ku, K. T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580–9582 RSC.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS.
- Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef PubMed.
- A. Nicol, W. Qin, R. T. K. Kwok, J. M. Burkhartsmeyer, Z. Zhu, H. Su, W. Luo, J. W. Y. Lam, J. Qian, K. S. Wong and B. Z. Tang, Chem. Sci., 2017, 8, 4634–4643 RSC.
- P. F. McMillan, Chem. Commun., 2003, 919–923 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including the synthesis procedure, X-ray structural analysis as well as the thermal, photophysical, and electrochemical data. CCDC 2164967 and 2164968. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05792a |
| This journal is © The Royal Society of Chemistry 2023 |