
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHysteresis behavior in the unfolding/refolding processes of a protein trapped in metallo-cages†
Takahiro
Nakama
 a,
Anouk
Rossen
a,
Risa
Ebihara
a,
Maho
Yagi-Utsumi
bcd,
Daishi
Fujita
e,
Koichi
Kato
bcd,
Sota
Sato
a and
Makoto
Fujita
*ab
a,
Anouk
Rossen
a,
Risa
Ebihara
a,
Maho
Yagi-Utsumi
bcd,
Daishi
Fujita
e,
Koichi
Kato
bcd,
Sota
Sato
a and
Makoto
Fujita
*ab
aDepartment of Applied Chemistry, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: mfujita@appchem.t.u-tokyo.ac.jp
bInstitute for Molecular Science (IMS), 5-1 Higashiyama, Myodaiji, Okazaki, Aichi 444-8787, Japan
cExploratory Research Center on Life and Living Systems (ExCELLS), 5-1 Higashiyama, Myodaiji, Okazaki, Aichi 444-8787, Japan
dGraduate School of Pharmaceutical Sciences, Nagoya City University, 3-1 Tanabe-dori, Mizuho-ku, Nagoya 467-8603, Japan
eInstitute for Integrated Cell-Material Sciences (iCeMS), Institute for Advanced Study, Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan
First published on 22nd February 2023
Abstract
Confinement of molecules in a synthetic host can physically isolate even their unstable temporary structures, which has potential for application to protein transient structure analysis. Here we report the NMR snapshot observation of protein unfolding and refolding processes by confining a target protein in a self-assembled coordination cage. With increasing acetonitrile content in CD3CN/H2O media (50 to 90 vol%), the folding structure of a protein sharply denatured at 83 vol%, clearly revealing the regions of initial unfolding. Unfavorable aggregation of the protein leading to irreversible precipitation is completely prevented because of the spatial isolation of the single protein molecule in the cage. When the acetonitrile content reversed (84 to 70 vol%), the once-denatured protein started to regain its original folded structure at 80 vol%, showing that the protein folding/unfolding process can be referred to as a phase transition with hysteresis behavior.
Introduction
Protein folding is a fundamental process of life, in which proteins gain a specific structure and function.1 Unfolding and refolding of proteins are highly associated with homeostasis in cells, and the aggregation of unfolded proteins can cause serious disorders such as Alzheimer's disease.2 Although protein unfolding and refolding have been structurally studied,3 the transient structures of proteins are difficult to analyze. This is often because they are temporary structures that irreversibly result in insoluble aggregates.4The confined space in a synthetic host can isolate guest molecules that are reactive or unstable in bulk solution.5 We expect this spatial isolation of molecules to be applied to the analysis of unstable transient structures of proteins. Proteins have previously been encapsulated in various synthetic hosts, such as micelles,6 vesicles,7 polymers,8 coacervates,9 DNA nanostructures,10 protein cages,11 and porous solid materials,12 and reverse micelles have been utilized for NMR analysis of protein folding.13 None of them, however, have a cavity suitable for detailed structural analysis of protein unfolding/refolding processes induced by environmental changes. The host should provide a well-defined space that isolates a single protein but allows for the free diffusion of denaturants through its large openings. NMR study of a protein confined in such a host would enable the unfolding and refolding analysis at higher resolution than existing single-molecule techniques.14 We report here the detailed analysis of protein unfolding and refolding processes by confining a protein in a self-assembled coordination cage (Fig. 1). 1H–15N HSQC NMR was used for analyzing the transient structures of the caged protein. An M12L24 self-assembled spherical complex 1 with a 6 nm diameter15,16 was used as the synthetic host. Because its large apertures offer free access of denaturants to the protein in the well-defined cavity,16 the complex allows us to investigate transiently unfolded protein structures in bulk solution. We have already reported the one-pot preparation of a caged protein complex, CLE@1, where CLE is a cutinase-like enzyme.16,17 We show that, with increasing content of an organic solvent in aqueous media, the protein is denatured not gradually but sharply into a molten globule state at a certain organic content. Interestingly, the once-denatured CLE shows hysteresis behavior when regaining its original folded structure upon adjusting back to more aqueous conditions.
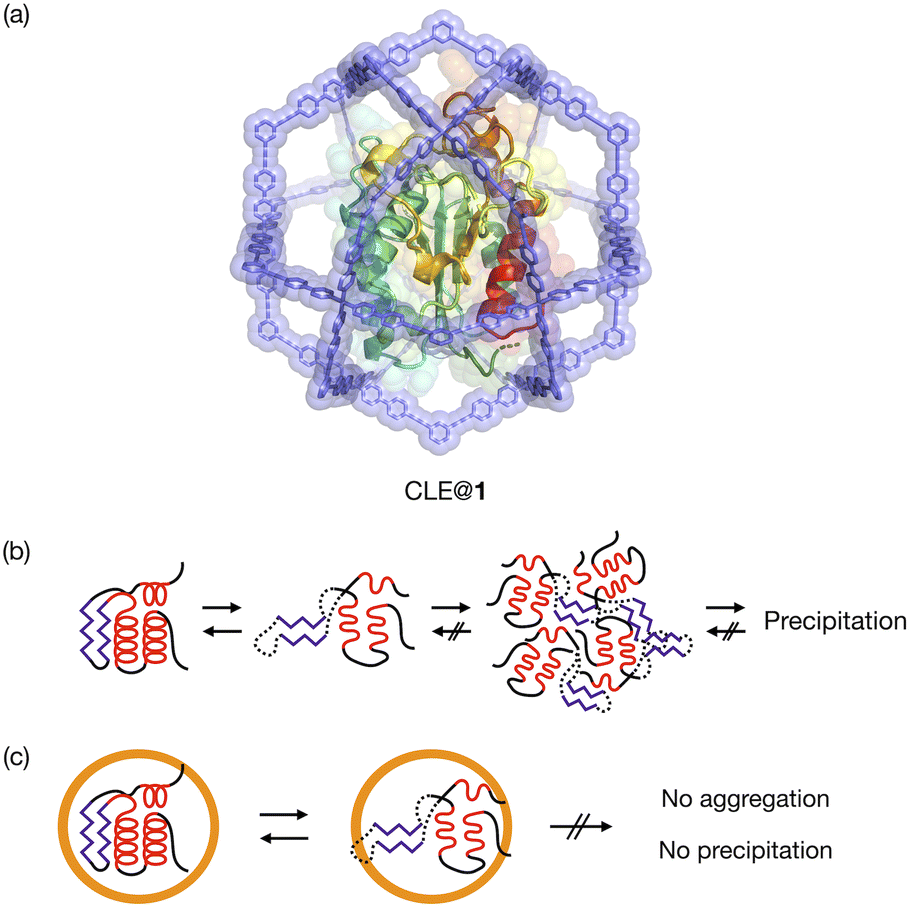 | ||
| Fig. 1 Schematic representation of analysis of protein unfolding/refolding by confinement in a metallo-cage. (a) Molecular modeling of CLE in an M12L24 coordination cage 1 (CLE@1). (b) The aggregation of an unfolded protein hampers the observation of the intermediate structures. (c) The spatial isolation in the cage suppresses protein aggregation and precipitation, thus allowing NMR study of the unfolding and refolding of a caged protein. | ||
Results and discussion
In our previous study, the caged CLE was shown to retain its native folded structure even in organic media (up to ∼80% acetonitrile/water (v/v) contents).16 Under more forcing conditions (i.e., at 90% acetonitrile content), the caged CLE denatured to a molten globule state. For a detailed analysis, we first prepared CLE@1 in 80% acetonitrile/water by dialysis and then changed the acetonitrile content in 1% increments (Fig. 2).18 The 1H–15N HSQC NMR spectra indicated that in up to 82% acetonitrile, the caged CLE almost completely retained its native structure with some minor structure changes (Fig. 2b and S1†). However, upon raising the ratio to 83%, many signals became faint or disappeared. Furthermore, more signals were lost at 84%, giving an HSQC NMR profile almost identical to that at 90%.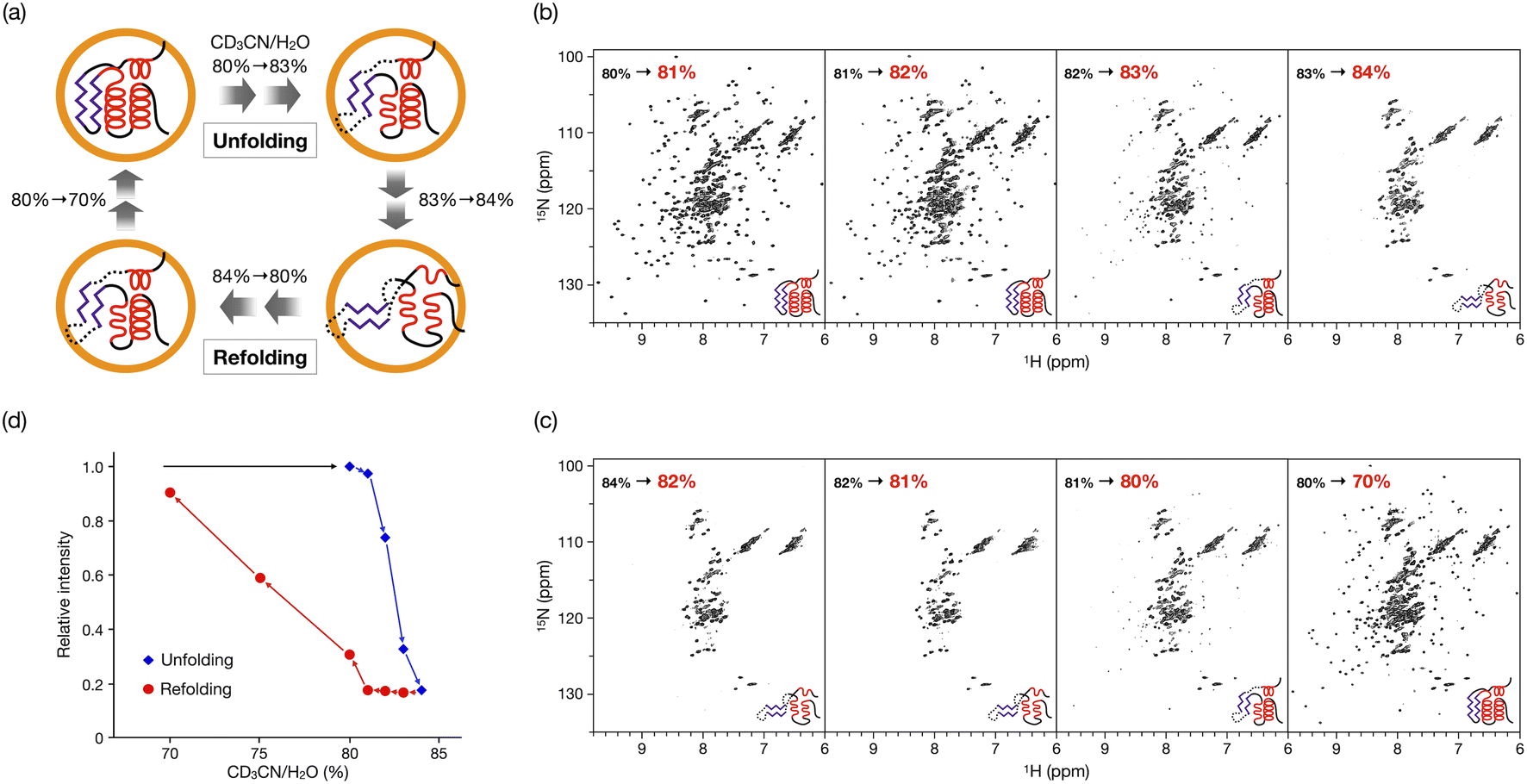 | ||
| Fig. 2 Hysteretic unfolding and refolding of CLE@1. (a) The caged CLE structure was monitored by NMR with varying acetonitrile/water (v/v) ratio. The acetonitrile content was increased stepwise from 80% to 84% and then decreased back to 70%. (b and c) 1H–15N HSQC spectra of (b) the unfolding and (c) refolding of caged CLE 1 (800 MHz, 300 K). See also Fig. S1 and S9.† (d) Hysteresis in the unfolding/refolding of caged CLE 1. Each point represents the average relative cross-peak intensities for all the assigned residues. The acetonitrile–water ratio are apparent values without taking into account density changes in the titrant solutions. The corrected actual values for both the unfolding and refolding processes are given in Table S1,† by which the systematic errors are shown to be negligible. | ||
These results clearly indicated that there is a critical transition point at 83%, at which CLE is sharply denatured into a partially unfolded structure. The NMR snapshot observation for the transient process cannot be applied to uncaged CLE at any acetonitrile ratio because, once partially denatured, uncaged CLE immediately precipitates. In fact, similar dialysis experiments with uncaged CLE showed complete disappearance of all the signals at high acetonitrile content due to the heavy precipitation of CLE (Fig. S2†). Dynamic light scattering (DLS) and 1H diffusion-ordered NMR spectroscopy (DOSY) also demonstrated that caged CLE is prevented from aggregating and remains as a monomer while the uncaged protein forms aggregates (Fig. S3 and S4†).
The HSQC NMR spectrum at 83% reveals the CLE structure at the moment it starts unfolding. We thus carefully analyzed the spectrum to study which regions trigger the denaturation of the CLE tertiary structure. From 80 to 82% acetonitrile ratio, the cross-peak intensities remained almost unchanged. At 83%, however, we observed a sharp decrease in the cross-peak intensities (Fig. 3a and S5†). The large intensity reduction and peak loss were observed in some helices (α1, α5, α7, and α9), sheets (β5 and β6), and a loop (residues 161–170) around the catalytic center (Fig. 3a,b, and S6†). Chemical shift perturbations (CSP) were also found in the C-terminal regions (residues 171–181) (Fig. S7–S9†). These results suggested that the CLE unfolding starts at the flexible domains close to the catalytic site (Fig. 3b, green) along with the adjacent helices and sheets in a cooperative manner. The structure further denatured at 84% acetonitrile content, where most of the regions, except for the α2 helix, lost the native structure (Fig. S5–S9†). Thus, the intermediate structure in the unfolding process, which is not accessible by conventional NMR studies, can be analyzed in detail by spatially isolating individual protein molecules in the cage.
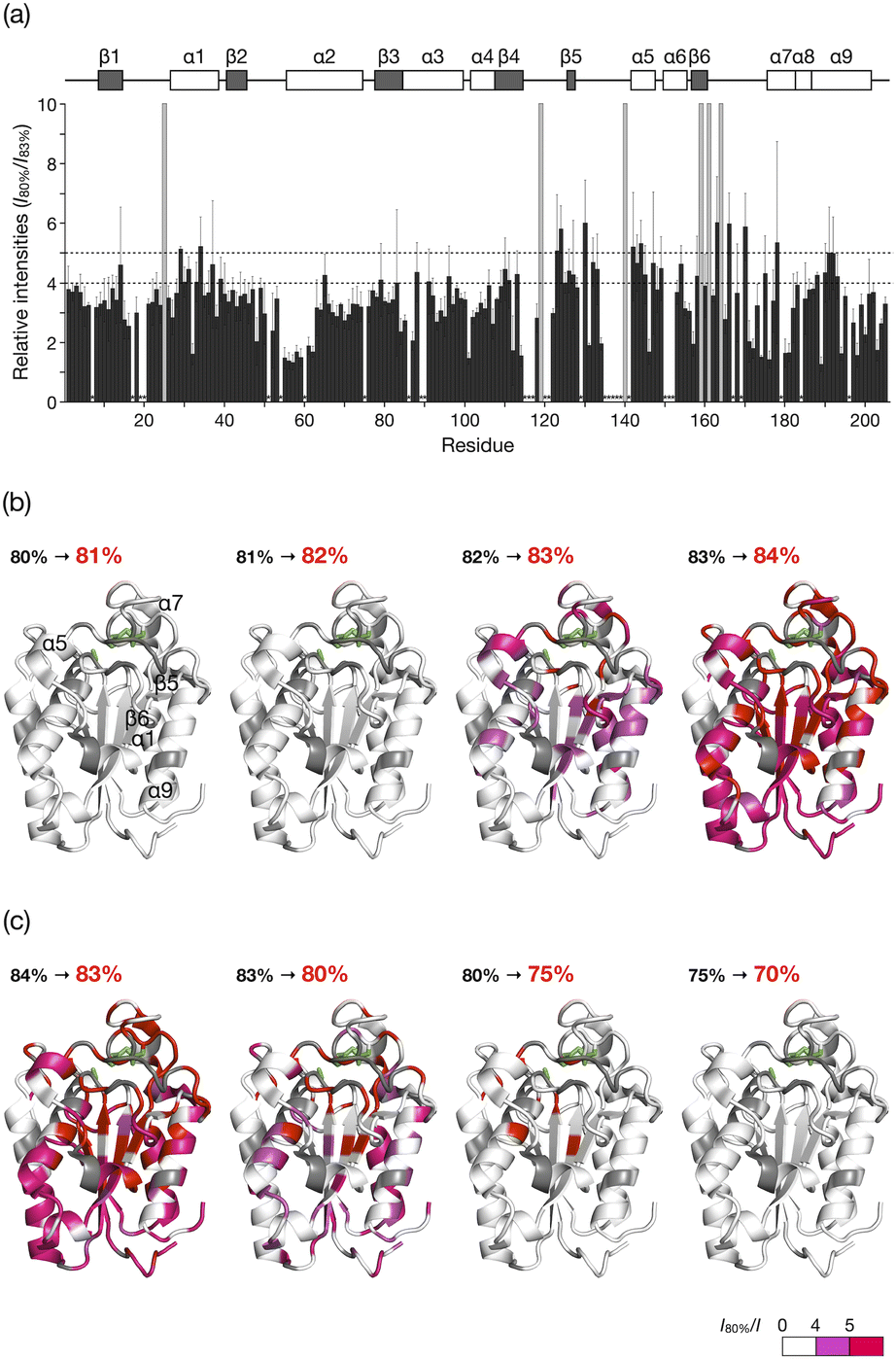 | ||
| Fig. 3 Unfolding and refolding CLE structures estimated from cross-peak intensity change. (a) Resonance intensities for each residue of caged CLE 1 in the unfolding at 83% acetonitrile content, relative to 80%. Error bars represent standard deviation (n = 3). Peaks that disappeared at 83% are indicated by gray bars. The unassigned residues are denoted with asterisks. (b and c) Mapping of the intensity change onto the CLE structure (PDB: 2CZQ) in (b) the unfolding and (c) refolding. The catalytic triad (S85, D165, and H180) is highlighted in green. The residues assigned to the disappeared peaks and unassigned residues are shown in red and gray, respectively. See also Fig. S4 and S11.† | ||
CD measurements showed that most of the secondary structure remained unchanged during the transition (80 to 84% acetonitrile content), indicative of the formation of a molten globular structure,19 where only the tight packing of the protein domains is loosened, rather than the formation of a largely unfolded random structure (Fig. S10†). It is therefore noteworthy that the domain structures of caged CLE are tolerant even of 90% acetonitrile.
When the acetonitrile content was changed back to more aqueous conditions in 1% increments, we observed the refolding of CLE into its native structure with hysteresis behavior (Fig. 2c,d and S11†). The HSQC NMR showed that the structure of caged CLE 1 remained denatured when the acetonitrile content was decreased from 84% to 82%, at which CLE existed in a native form during the forward unfolding process (Fig. 2b and c). Presumably, the partially unfolded structure is kinetically trapped because of interactions between exposed hydrophobic residues and the organic solvent. The protein began refolding at 80%, and the native structure was fully restored at 70%, as indicated by the peak intensity recovery (Fig. 2c,d, and S11†). The intensity change at each residue showed the refolding pathway in which some C-terminal domains recovered sooner than other unfolded regions (Fig. 3c, S12, and S13†). The result suggests that the refolding pathway might be different from that of the unfolding. Hysteretic folding has often been discussed in single-molecule analysis under non-equilibrium conditions.14,20,21 In contrast, the observation of a hysteresis in protein refolding in bulk solution has been limited to complex protein structures such as multidomain and knotted proteins and has rarely been observed in simple small ones.20,22
The unfolding and refolding processes of CLE in a larger cavity were investigated with metallo-cage 2 to clarify the spatial isolation effect of the cage (Fig. 4, S14–17, and Schemes S1, S2†). The CLE@2 encapsulation complex was prepared similarly to CLE@1. As confirmed by 1H DOSY NMR, caged CLE 2 showed a smaller diffusion coefficient D than CLE@1 and free CLE (Fig. S18†).15,16 Even though CLE can fluctuate more freely in the larger cavity of 2 (Fig. 4 and S19†), we observed nearly identical hysteresis behavior for the unfolding/refolding processes of CLE (Fig. S20–S22†). Namely, the native structure of caged CLE 2 was retained in up to 82% acetonitrile content and partially denatured at 83% in the larger inner space as confirmed by 1H–15N HSQC. The hysteretic refolding occurred in cage 2 when the acetonitrile content was lowered to 80% and further. CD spectra and the enzymatic activity assay also supported that CLE structures in cages 1 and 2 are identical at all acetonitrile contents (Fig. S23–S25†). These observations clearly reveal that the CLE stabilization and its hysteresis behavior are ascribed not to the tight packing effect but to the spatial isolation effect of the cage framework that completely suppresses the unfavorable protein aggregation and isolates transient unfolding structures.
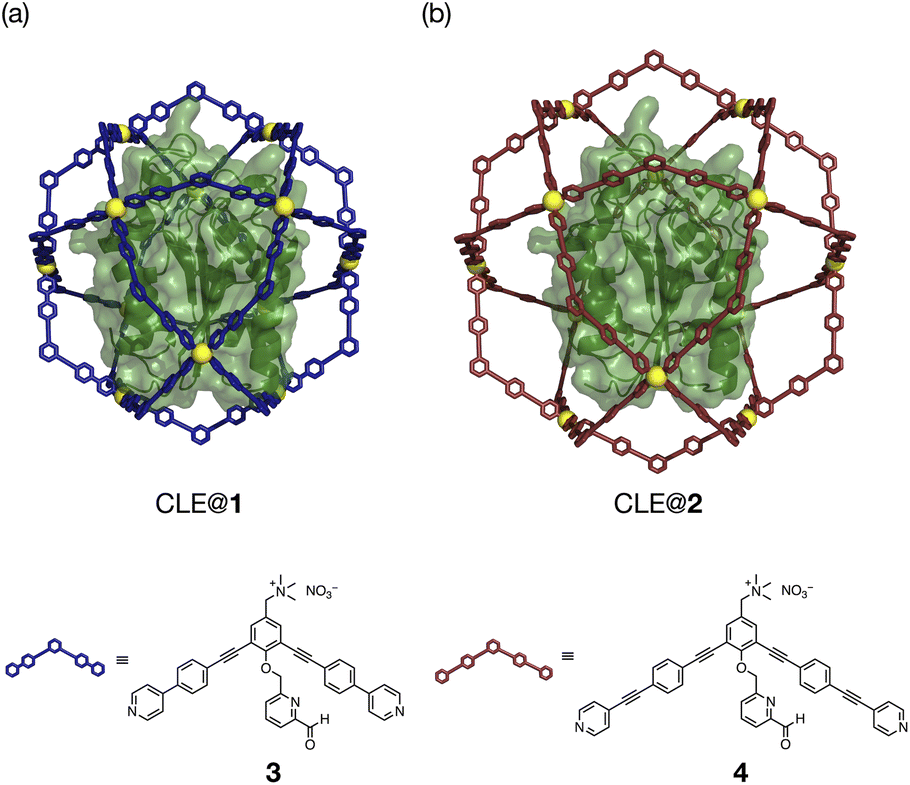 | ||
| Fig. 4 CLE in metallo-cages with different sizes. (a and b) Molecular modeling of CLE in Pd(II)-complexes (a) 1 and (b) 2, self-assembled from ligands 3 and 4, respectively. | ||
Conclusions
In conclusion, we succeeded in the snapshot observation of protein unfolding and refolding processes in a gigantic metallo-cage by NMR studies, where unprecedented hysteresis behavior was monitored stepwise by precise solvent exchange experiments. The encapsulation in the cage protected the protein from aggregation while maintaining its interactions with denaturants and visualized the cooperative unfolding and hysteretic refolding. This study demonstrated that the spatial isolation in our coordination cage is a powerful strategy for the analysis of unstable protein structures that temporarily exist in solution. We envision that our method can reveal transient protein structures such as weak protein–ligand complexes, intermediates of large protein assemblies, and oligomeric structures in amyloid fibrillation.Data availability
The authors declare that all data supporting the findings of this study are available within the article and ESI,† and raw data files are available from the corresponding author upon reasonable request.Author contributions
T. N., M. Y.-U., D. F., and M. F. conceived and designed the project. T. N., A. R., and R. E. carried out experiments and analyzed the data with assistance from M. Y.-U. T. N. and M. F. wrote the manuscript. All authors discussed the results and commented on the manuscript. T. N. and A. R. are co-first authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by JSPS Grants-in-Aid for Specially Promoted Research (JP19H05461 to M.F.) and JSPS Grant-in-Aid for Early-Career Scientists (JP21K14640 to T. N.), JST PRESTO (JPMJPR22AC to M. Y.-U.), and AMED-Science and Technology Platform Program for Advanced Biological Medicine (to D. F.). A. R. thanks SPRING-GX for financial support. The research was also supported by Joint Research of the Exploratory Research Center on Life and Living Systems (ExCELLS program No. 22EXC319).Notes and references
- (a) M. J. Gething and J. Sambrook, Nature, 1992, 335, 33–45 CrossRef; (b) C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS.
- (a) D. J. Selkoe, Nature, 2003, 426, 900–904 CrossRef CAS; (b) D. Balchin, M. Hayer-Hartl and F. U. Hartl, Science, 2016, 353, aac4354 CrossRef.
- (a) D. Neri, M. Billeter, G. Wider and K. Wüthrich, Science, 1992, 257, 1559–1563 CrossRef CAS; (b) H. Roder, G. A. Elöve and S. W. Englander, Nature, 1988, 335, 700–704 CrossRef CAS; (c) H. J. Dyson and P. E. Wright, Chem. Rev., 2004, 104, 3607–3622 CrossRef CAS; (d) P. Neudecker, P. Robustelli, A. Cavalli, P. Walsh, P. Lundström, A. Zarrine-Afsar, S. Sharpe, M. Vendruscolo and L. E. Kay, Science, 2012, 336, 362–366 CrossRef CAS; (e) D.-H. Chen, D. Madan, J. Weaver, Z. Lin, G. F. Schröder, W. Chiu and H. S. Rye, Cell, 2013, 153, 1354–1365 CrossRef CAS; (f) S. W. Englander, L. Mayne, Z.-Y. Kan and W. Hu, Annu. Rev. Biophys., 2016, 45, 135–152 CrossRef CAS; (g) A. Bax and G. M. Clore, J. Magn. Reson., 2019, 306, 187–191 CrossRef CAS.
- (a) M. Silow and M. Oliveberg, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6084–6086 CrossRef CAS PubMed; (b) C. M. Dobson, Semin. Cell Dev. Biol., 2004, 15, 3–16 CrossRef CAS; (c) T. R. Alderson and L. E. Kay, Curr. Opin. Struct. Biol., 2020, 60, 39–49 CrossRef CAS.
- (a) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS; (b) A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC; (c) A. B. Grommet, M. Feller and R. Klajn, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS.
- P. L. Luisi, M. Giomini, M. P. Pileni and B. H. Robinson, Biochim. Biophys. Acta, 1988, 947, 209–246 CrossRef CAS.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- B. Panganiban, B. Qiao, T. Jiang, C. DelRe, M. M. Obadia, T. D. Nguyen, A. A. A. Smith, A. Hall, I. Sit, M. G. Crosby, P. B. Dennis, E. Drockenmuller, M. Olvera de la Cruz and T. Xu, Science, 2018, 359, 1239–1243 CrossRef CAS.
- W. C. Blocher McTigue and S. L. Perry, Small, 2020, 16, 1907671 CrossRef CAS.
- Z. Zhao, J. Fu, S. Dhakal, A. Johnson-Buck, M. Liu, T. Zhang, N. W. Woodbury, Y. Liu, N. G. Walter and H. Yan, Nat. Commun., 2016, 7, 10619 CrossRef CAS.
- B. Wörsdörfer, K. J. Woycechowsky and D. Hilvert, Science, 2011, 331, 589–592 CrossRef.
- (a) S. Hudson, J. Cooney and E. Magner, Angew. Chem., Int. Ed., 2008, 47, 8582–8594 CrossRef CAS PubMed; (b) C. Wang and K. Liao, ACS Appl. Mater. Interfaces, 2021, 13, 56752–56776 CrossRef CAS.
- (a) R. W. Peterson, K. Anbalagan, C. Tommos and A. J. Wand, J. Am. Chem. Soc., 2004, 126, 9498–9499 CrossRef CAS; (b) C. R. Babu, V. J. Hilser and A. J. Wand, Nat. Struct. Mol. Biol., 2004, 4, 352–357 CrossRef.
- (a) B. Schuler and W. A. Eaton, Curr. Opin. Struct. Biol., 2008, 18, 16–26 CrossRef CAS; (b) R. Petrosyan, A. Narayan and M. T. Woodside, J. Mol. Biol., 2021, 433, 167207 CrossRef CAS.
- (a) K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC; (b) D. Fujita, K. Suzuki, S. Sato, M. Yagi-Utsumi, Y. Yamaguchi, N. Mizuno, T. Kumasaka, M. Takata, M. Noda, S. Uchiyama, K. Kato and M. Fujita, Nat. Commun., 2012, 3, 1093 CrossRef.
- D. Fujita, R. Suzuki, Y. Fujii, M. Yamada, T. Nakama, A. Matsugami, F. Hayashi, J.-K. Weng, M. Yagi-Utsumi and M. Fujita, Chem, 2021, 7, 2672–2683 CAS.
- (a) K. Masaki, N. R. Kamini, H. Ikeda and H. Iefuji, Appl. Environ. Microbiol., 2005, 71, 7548–7550 CrossRef CAS; (b) Y. Kodama, K. Masaki, H. Kondo, M. Suzuki, S. Tsuda, T. Nagura, N. Shimba, E. Suzuki and H. Iefuji, Proteins, 2009, 77, 710–717 CrossRef CAS.
- In the preparation of mixed solvents, changes in partial specific volumes were not accounted for. To avoid confusions, the v/v (%) values were converted into molar ratios, mole/mole (%): v/v (%) [mole/mole (%)] are 80 [56.2], 81 [57.8], 82 [59.4], 83 [61.0], 84 [62.7].
- O. B. Ptitsyn, R. H. Pain, G. V. Semisotnov, E. Zerovnik and O. I. Razgulyaev, FEBS Lett., 1990, 262, 20–24 CrossRef CAS.
- B. T. Andrews, D. T. Capraro, J. I. Sulkowska, J. N. Onuchic and P. Jennings, J. Phys. Chem. Lett., 2013, 4, 180–188 CrossRef CAS.
- (a) A. Minajeva, M. Kulke, J. M. Fernandez and W. A. Linke, Biophys. J., 2001, 80, 1442–1451 CrossRef CAS; (b) C. He, C. Hu, X. Hu, X. Hu, A. Xiao, T. T. Perkins and H. Li, Angew. Chem., Int. Ed., 2015, 54, 9921–9925 CrossRef CAS.
- (a) C. G. Benítez-Cardoza, A. Rojo-Domínguez and A. Hernández-Arana, Biochemistry, 2001, 40, 9049–9058 CrossRef; (b) S. Singh and A. Zlotnick, J. Biol. Chem., 2003, 278, 18249–18255 CrossRef CAS; (c) D. T. Capraro and P. A. Jennings, Biophys. J., 2016, 110, 1044–1051 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, full 1H–15N NMR spectra and their analysis, CD spectra, and synthetic details of ligand 4. See DOI: https://doi.org/10.1039/d2sc05879k |
| This journal is © The Royal Society of Chemistry 2023 |