DOI:
10.1039/D2SC06145G
(Edge Article)
Chem. Sci., 2023,
14, 1732-1741
Activation of perfluoroalkyl iodides by anions: extending the scope of halogen bond activation to C(sp3)–H amidation, C(sp2)–H iodination, and perfluoroalkylation reactions†
Received
7th November 2022
, Accepted 29th December 2022
First published on 24th January 2023
Abstract
A simple, efficient, and convenient activation of perfluoroalkyl iodides by tBuONa or KOH, without expensive photo- or transition metal catalysts, allows the promotion of versatile α-sp3 C–H amidation reactions of alkyl ethers and benzylic hydrocarbons, C–H iodination of heteroaryl compounds, and perfluoroalkylations of electron-rich π bonds. Mechanistic studies show that these novel protocols are based on the halogen bond interaction between perfluoroalkyl iodides and tBuONa or KOH, which promote homolysis of perfluoroalkyl iodides under mild conditions.
Introduction
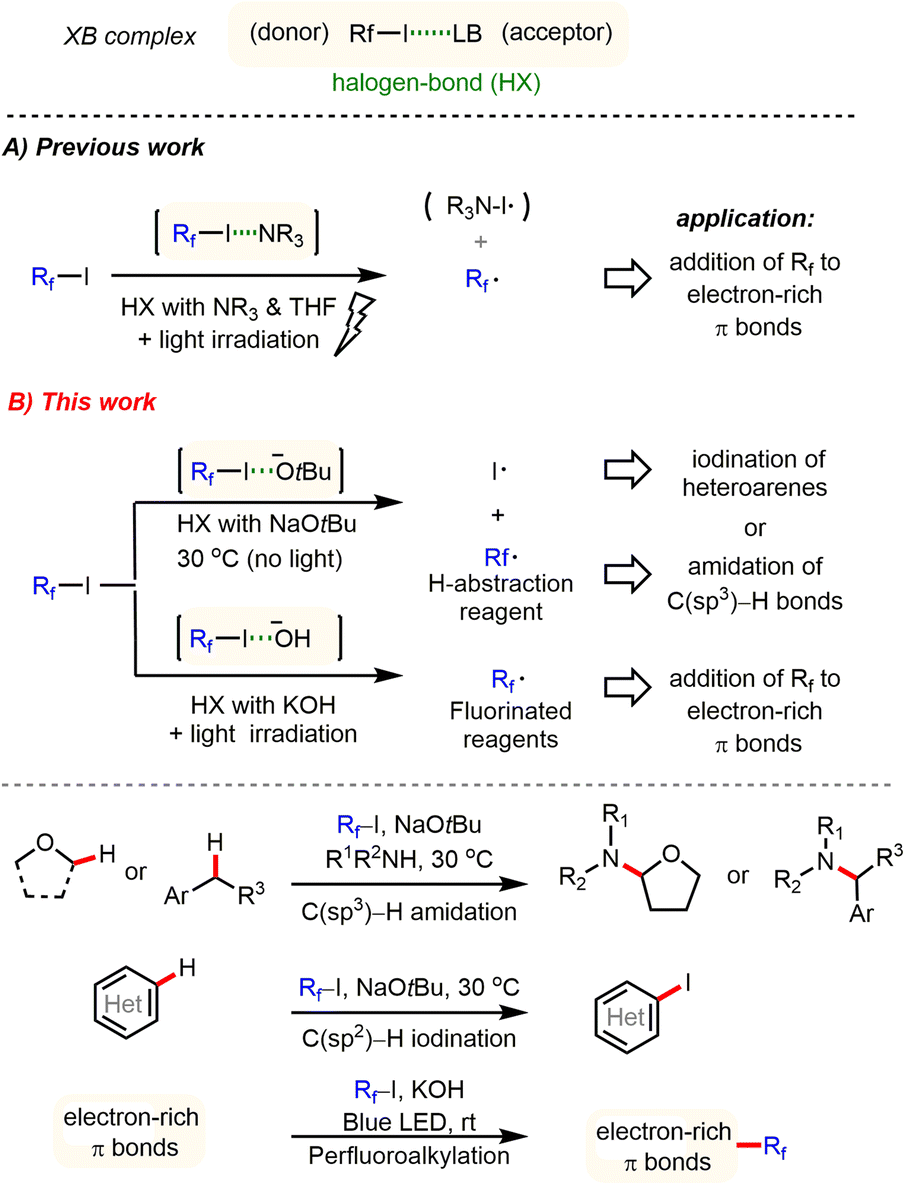
Developing new activation modes for carbon–halogen bonds of fluoroalkyl halides offers interesting possibilities for the synthesis of pharmaceuticals, agrochemicals, and new materials.1,2 Unlike typical alkyl halides, halogen atoms linked to strongly electron-withdrawing groups possess an electrophilic character, the so-called σ hole, which is opposite to that for R–X bonds. This type of organic halide (donor) can form an additional halogen bond (HX) with a Lewis base (acceptor) through an n → σ* charge-transfer interaction.3 Despite its importance in crystal engineering4 and supramolecular chemistry,5 utilization of HX interactions is still in its infancy for the development of new synthetic methodologies. In their pioneering studies, Bolm,6 Huber7 and others8 demonstrated that the σ* acidity of HX donors can be used to activate several types of HX acceptors, including N-heteroarenes, carbonyl groups, and halides. Recently, Chen,9 Melchiorre,10 Yu, and other groups11 showed that perfluoroalkyl iodides (Rf–I) represent valuable H–X donors, which can be activated by amines (HX acceptor) under light irradiation. Thereby perfluoroalkyl radicals are generated, which subsequently can be added to various electron-rich π bonds to give perfluoroalkylated products (Scheme 1A). Most recently, Niu and co-workers also discovered that allyl glycosyl sulfones can form halogen bond complexes with perfluoroalkyl iodides, which, by means of visible light irradiation, fragment via radical intermediates to give synthetically valuable glycosyl iodides.12 Despite these interesting results, activation of fluoroalkyl halides by different anions has been largely ignored in organic synthesis.13
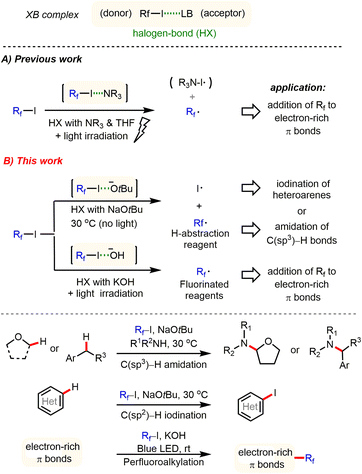 |
| | Scheme 1 General principles of halogen bond-promoted activation of perfluoroalkyl iodides: selected known examples and this work. | |
Herein, we report a general method for activation of perfluoroalkyl iodides via halogen bond interaction with tBuONa or KOH at ambient temperature. This novel activation mode enables several interesting synthetic applications to be carried out, including C(sp3)–H amidation of ethers and benzylic hydrocarbons, C–H iodination of heteroarenes, and perfluoroalkylation of electron-rich π bonds under very mild conditions (Scheme 1B).
Results and discussion
Discovery of selective amidation reactions of ethers and benzylic compounds
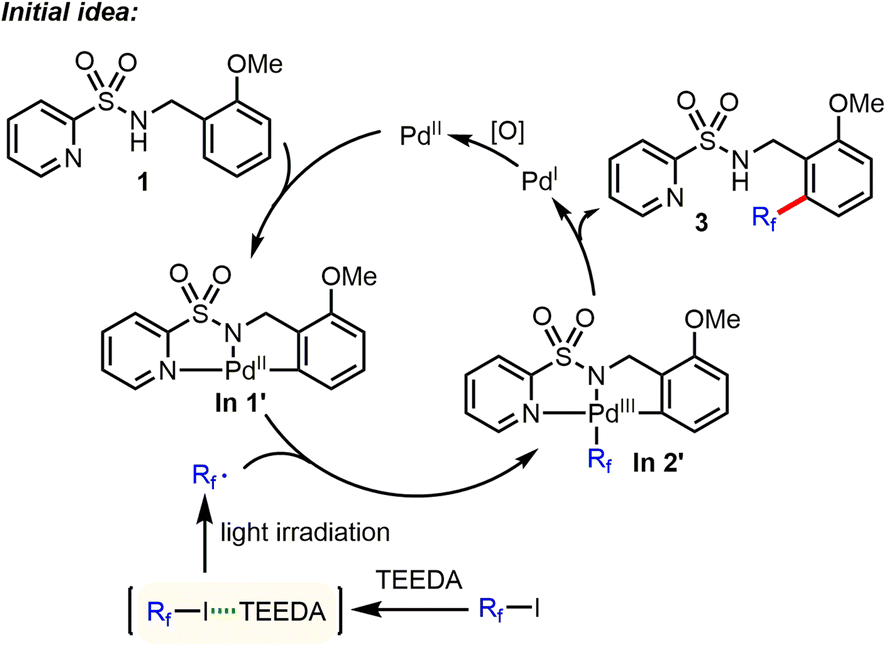
Recently, some of us discovered that perfluoroalkyl iodides could be efficiently activated by light irradiation in the presence of amine (such as N,N,N′,N′-tetraethylethylenediamine (TEEDA)) to generate the corresponding Rf radicals.9 Based on this work, we had the idea of directed Pd-catalyzed, selective C(sp2)–H perfluoroalkylation of substrate 1 (Scheme 2). Here, the palladacycle intermediate In 1′, which is generated by Pd-catalyzed C–H activation, would react with perfluoroalkyl radicals produced via photochemical activation of the halogen bond complex (Rf⋯TEEDA) to form the PdIII intermediate In 2′, which can undergo reductive elimination to form selective C(sp2)–H perfluoroalkylation products.
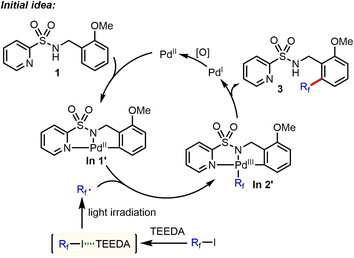 |
| | Scheme 2 Initial idea of Pd-catalyzed, selective C(sp2)–H perfluoroalkylation. *TEEDA: N,N,N′,N′-tetraethylethylenediamine. | |
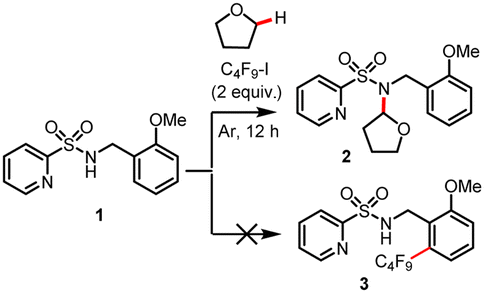
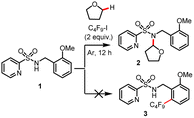
We began to evaluate this hypothesis by using N-benzyl pyridylsulfonamide 1 as substrate. However, performing the model reaction of N-benzyl pyridylsulfonamide 1 with C4F9–I, in the presence of TEEDA and palladium acetate, did not give the desired product 3. Instead, unexpectedly, a small amount of the C–H amidated product of tetrahydrofuran (THF) solvent was generated (Table 1, entry 1). Surprisingly, using the inorganic base Cs2CO3 gave 2 in 62% yield (Table 1, entry 2). Even more astonishing, the reaction in the absence of Pd also gave 2 in 65% yield (Table 1, entry 3). While the activation of perfluoroalkyl iodides (Rf–I) by Cs2CO3 has been reported,14 the underlying mechanism is still unknown. We suspected that the carbonate ligand of Cs2CO3 might activate Rf–I via the halogen bond under the specific conditions applied. Thus, we examined the effect of different bases. Interestingly, NaOtBu facilitated the reaction at 30 °C in near quantitative yield (Table 1, entry 11). In contrast, the use of KOtBu gave little product (Table 1, entry 10). Next, we studied the influence of different reaction parameters. Notably, the amidation reaction proceeded well with and without light (Table 1, entry 11 vs. entry 14); however, the reaction yield diminished to 28% when the reaction was carried out under an atmosphere of air (Table 1, entry 16). In addition, the activation of Rf–I is also feasible by halide anions (Cl−, Br−, I−) (Table 1, entries 23–25).
Table 1 Optimization of conditions for C(sp3)–H amidation of THF using N-benzyl pyridylsulfonamide (1) and nonafluoro iodobutane (C4F9I)a
|
|
| Entry |
Reagents (equiv.)/temperature |
Solvent |
Yielda (%), 2 |
|
Yields are based on 1H NMR analysis on a 0.2 mmol scale in 4 mL glass vial.
CFL light (25 W) irradiation.
Isolated yield.
In darkness.
|
| 1b |
Pd(OAc)2 (0.1), TEEDA (3), 60 °C |
THF |
23 |
| 2 |
Pd(OAc)2 (0.1), Cs2CO3 (3), 60 °C |
THF |
62 |
| 3 |
Cs2CO3 (3), 60 °C |
THF |
65 (56c) |
| 4 |
Cs2CO3 (3), 30 °C |
THF |
<2 |
| 5 |
K2CO3 (3), 60 °C |
THF |
10 |
| 6 |
Na2CO3 (3), 60 °C |
THF |
<2 |
| 7 |
NaOCH3 (3), 60 °C |
THF |
33 |
| 8 |
KOtBu (3), 60 °C |
THF |
<2 |
| 9 |
NaOtBu (3), 60 °C |
THF |
>99 (90c) |
| 10 |
KOtBu (3), 30 °C |
THF |
<2 |
| 11 |
NaOtBu (3), 30 °C
|
THF
|
>99 (90c)
|
| 12 |
NaOtBu (2), 30 °C |
THF |
81 |
| 13 |
NaOtBu (1), 30 °C |
THF |
58 |
| 14d |
NaOtBu (3), 30 °C |
THF |
>99 (90c) |
| 15 |
NaH (3), 30 °C |
THF |
<2 |
| 16 |
NaOtBu (3), 30 °C, air |
THF |
28 |
| 17 |
NaOtBu (3), THF (10), 30 °C |
CCl4 |
71 |
| 18 |
NaOtBu (3), THF (10), 30 °C |
PhCF3 |
67 |
| 19b |
TEEDA (3), 30 °C |
THF |
21 |
| 20b |
Et3N (3), 30 °C |
THF |
18 |
| 21d |
Et3N (3), 30 °C |
THF |
<2 |
| 22 |
Bu4NCl or Bu4NBr or Bu4NI (3), 30 °C |
THF |
<2 |
| 23 |
Bu4NCl (3), NaH (1), 30 °C |
THF |
65 |
| 24 |
Bu4NBr (3), NaH (1), 30 °C |
THF |
63 |
| 25 |
Bu4NI (3), NaH (1), 30 °C |
THF |
64 |
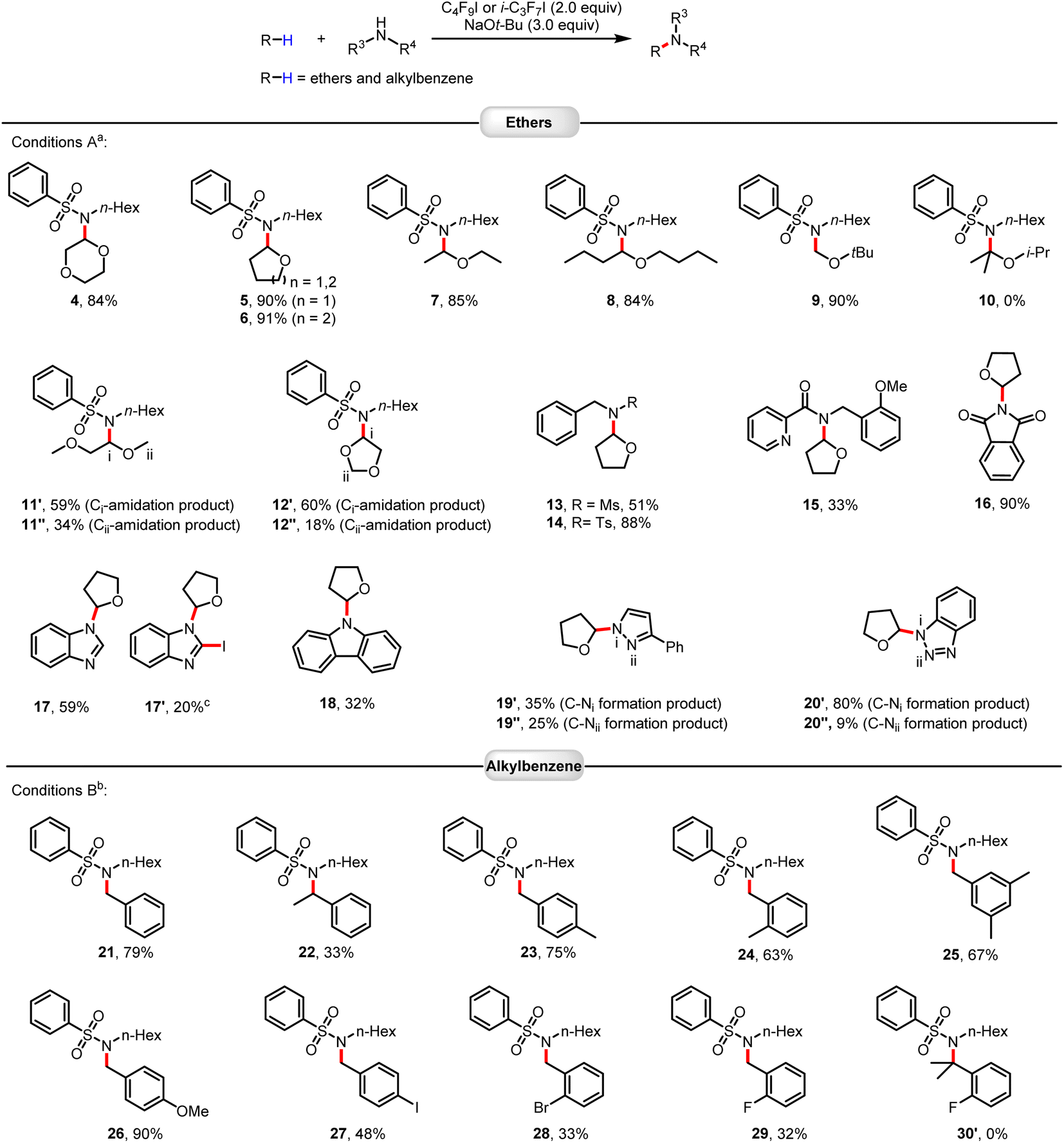
With optimized conditions in hand, we explored the scope of this amidation of ethers.15 As shown in Fig. 1, a variety of cyclic and linear ethers worked well (4–12). The regioselectivity of these transformations was very high, with C–H activation occurring at a specific C–H bond in the ortho-position to the oxygen atom. This can be explained by the relatively lower bond dissociation energy of this C–H bond compared to the other bonds and the stability of the resulting carbon radicals. It should be noted that the electron density of the substrate ethers influences the reaction (12′vs.12′′). However, this reaction system was not applicable to sterically hindered substituted ethers, such as 2-isopropoxypropane (10). Substrates containing functional groups, including sulfonamides, Ms, Ts, amide, imide, and nitrogen-containing heteroarenes, can be well applied in this protocol. Apart from ethers, selective sp3 C–H amidation of benzylic hydrocarbons could also be achieved smoothly using the same strategy. Here, activation of the halogen bond of heptafluoroisopropyl iodide with NaOtBu allowed for smooth functionalization with high efficiency and good functional group tolerance. Again, the steric hindrance had a strong impact on the outcome of the amidation reactions (21vs.22). In general, benzylic hydrocarbons with electron-donating groups performed better than benzylic hydrocarbons containing electron-withdrawing groups (24vs.29).
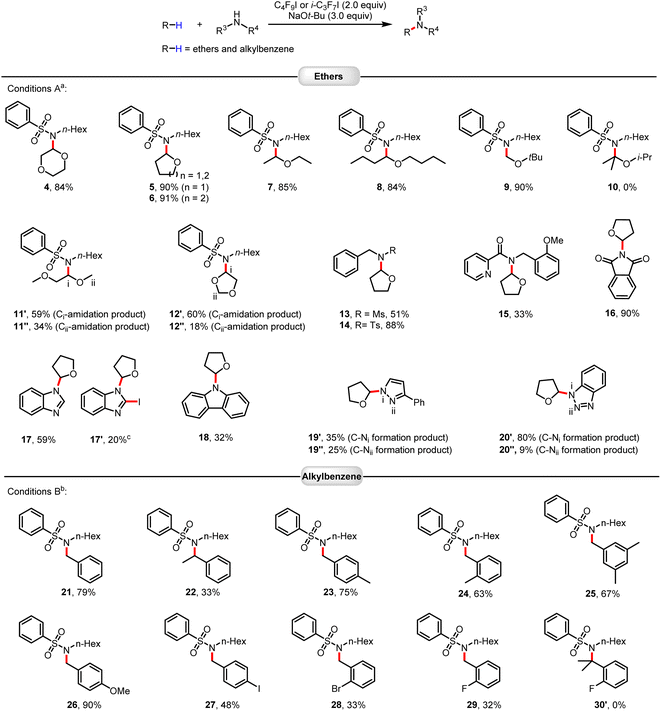 |
| | Fig. 1 Scope of C(sp3)–H amidation of ethers and alkylbenzenes. Reactions were performed on a 0.2 mmol scale. Isolated yields are given. aConditions A: C4F9I (2.0 equiv.), NaOtBu (3.0 equiv.), T = 30 °C. bConditions B: i-C3F7I (2.0 equiv.), NaOtBu (3.0 equiv.), Ar–H (10.0 equiv.), T = 60 °C, solvent PhCF3 (1 mL). cIodination product 17′ of compound 17 was obtained (see ESI†). | |
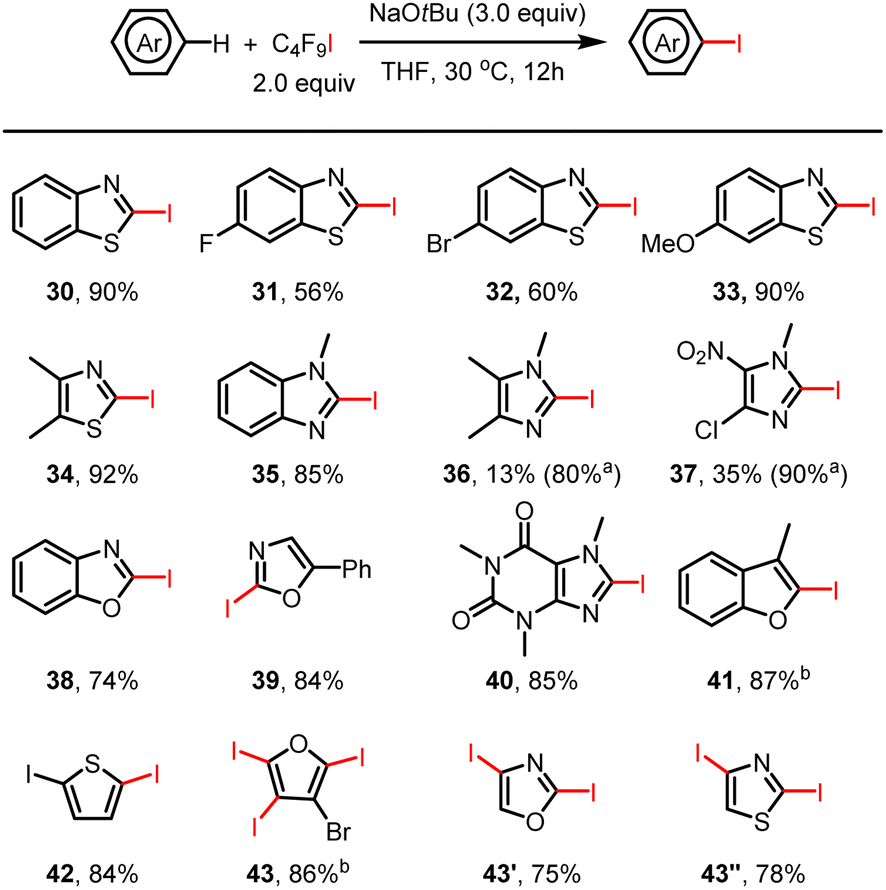
Discovery and substrate scope of selective C–H iodination reactions
Much to our surprise, the amination of THF with benzimidazole under standard reaction conditions not only gave the desired aminated product 17 in 59% isolated yield but also led to the iodinated product of compound 17 (Fig. 1, 17′). Apparently, in addition to the perfluoroalkyl radical, an iodine radical could be also be produced in our reaction system. This should allow selective iodination of heteroarenes with perfluoroalkyl iodides under similarly mild conditions to be achieved. Indeed, as shown in Fig. 2, a wide variety of heteroarenes could be successfully reacted with C4F9–I in the presence of NaOtBu with good to excellent yields and regioselectivity. More specifically, thiazole, oxazole, imidazole, benzothiazole, benzoxazole, and benzimidazole derivatives were all efficiently iodinated with high selectivity in the 2-position (30–39). Moreover, when 4- and 5-unprotected oxazole and thiazole were examined for the iodination system, di-iodinated products were obtained in high isolated yield (43′ and 43′′). The observed regioselectivity in these reactions agrees with iodination reactions of heterocycles using other iodination reagents.16 As an example of a bioactive compound, caffeine offered the iodinated product in an excellent yield of 85% (40). In all cases, good to excellent isolated product yields were achieved; however, in the case of 4-chloro-1-methyl-5-nitro-imidazole and 1,4,5-trimethylimidazole isolation of the products was more difficult, leading to a lower isolated yield. Finally, it should be mentioned that it is possible to obtain multiple iodinated products, as shown in the case of triple iodination of 3-bromofuran.
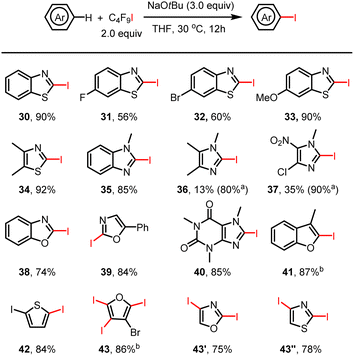 |
| | Fig. 2 Selective heteroaryl C(sp2)–H iodination by halogen bond-promoted activation. Reactions were performed on a 0.2 mmol scale. Isolated yields are given. aCrude 1H NMR yield. b4.0 equiv. of C4F9I and NaOtBu were used. | |
Discovery of perfluoroalkylation reactions: synthesis of perfluoroalkylated phenanthridines and isoquinolines
Clearly, the interaction of perfluoroalkyl iodides with the halide bond acceptor leads to activation of the iodide and the perfluoroalkyl residue. Hence, following this concept, it should be possible to perform perfluoroalkylation reactions as well. Indeed, when using other substrate classes, such as isocyanides, olefins, alkynes, and electron-rich arenes or heteroarenes, under previously developed standard conditions, we observed perfluoroalkylation reactions. As shown in Scheme S7 (see ESI†), the synthesis of 6-perfluoroalkylated phenanthridine 44 was achieved from 4′,5-dichloro-2-isocyano-1,1′-biphenyl and C4F9–I in the presence of tBuONa. While 44 was obtained in 23% yield when the reaction was performed in the dark, the product yield increased in the presence of light. More specifically, under irradiation with a blue light-emitting diode (LED), and utilizing 1.5 equiv. of tBuONa, provided 44 in 72% yield in THF. Notably, using irradiation with blue LED without tBuONa present, the reaction gave only a poor yield (see ESI† for details). Interestingly, under LED irradiation a variety of other anions act as halide bond acceptors and could promote this reaction (see ESI, Scheme S7†). To our surprise, even simple potassium hydroxide was an effective additive and gave excellent results, providing 44 in 87% isolated yield. With KOH as additive, the reaction also gave high yields of the product under irradiation from a low-intensity Hg UV lamp (254 nm, 25 W) or sunlight. For halide anion additives, the lighter, more charge dense halides were more effective in this reaction (F− > Cl− > Br− > I−), which is in line with the trend of reported values for the association constant (Ka) for interactions of anions with C4F9I.13
Next, the scope of the phenanthridine and isoquinoline synthesis was explored under the optimized conditions with KOH additive and blue LED light irradiation (Fig. 3). 2-Isocyanobiphenyls (44–56) and vinyl isocyanide (66–76) bearing various substitution patterns reacted smoothly with C4F9–I with good to excellent yield and regioselectivity. Isocyanide substrates containing a pyridine, pyrrole, indole, dioxolane, or naphthalene ring were tolerated (see 57–59, 77, and 78). Furthermore, reactions of both linear and branched perfluoroalkyl iodides of various lengths provided excellent results, to give products 60–65 and 81–85.
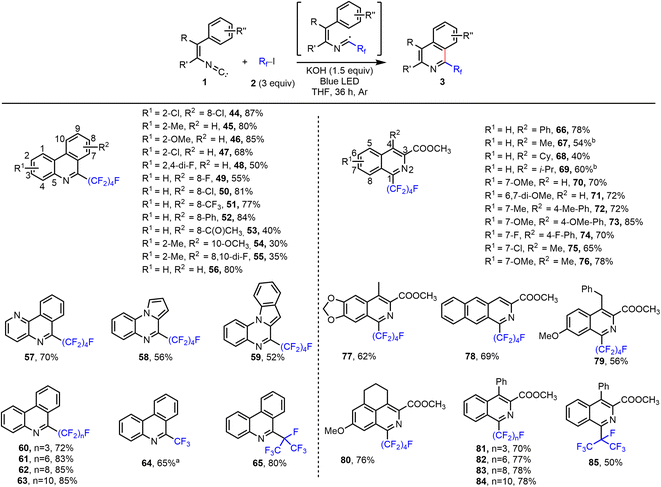 |
| | Fig. 3 Halogen bond-promoted reaction system for perfluoroalkylated phenanthridine and isoquinoline synthesis. Reactions were performed on a 0.2 mmol scale. Isolated yields are given. aExcess of CF3I was used (sparging THF solution with CF3I gas; see the ESI†). b6 equiv. C4F9I and 3 equiv. KOH were used. | |
Addition reactions of perfluoroalkyl iodides to alkenes and alkynes
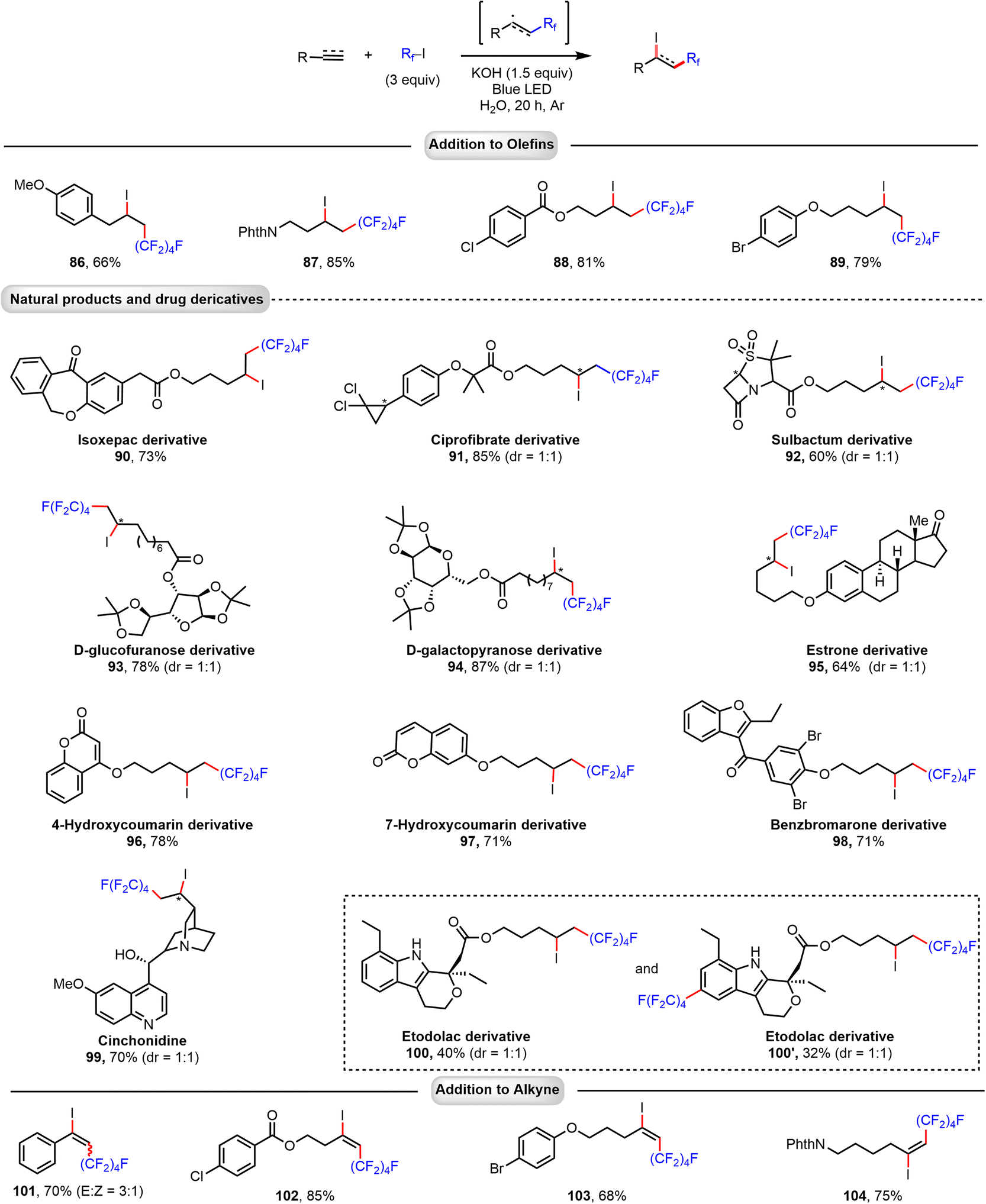
Applying the same conditions as above (KOH as additive and blue LED light irradiation) for the reaction of C4F9–I with terminal alkenes, a 1,2-addition process to give iodoperfluoroalkylated products is observed. Notably, using water as solvent, excellent regioselectivity is achieved (Fig. 4). Functional groups, such as phthalimide (PhthN), benzoate ester, phenyl ether, and ketone, were tolerated. Various alkene substrates derived from natural products and drugs were also surveyed. For example, terminal alkenes bearing isoxepac (90), ciprofibrate (91), sulbactum (92), D-glucofuranose (93), D-galactopyranose (94), estrone (95), coumarin (96, 97), benzbromarone (98), and etodolac (100) can be conveniently applied to give the corresponding products in good yields (60–87%). Notably, subjecting quinine, which contains hydroxyl and pyridine moieties, to the reaction provided 99 in good yield and selectivity. Perfluoroalkyl iodides also reacted, under similar conditions, with terminal alkynes to give iodoperfluoroalkylation products in good yield and excellent regioselectivity, and halogen (Cl, Br), benzoate ester, phenyl ether, and PhthN substituents were well tolerated. In all these reactions, we assume that perfluoroalkyl radicals generated via photochemical activation of the halogen bond complex (C4F9I…−OH) add directly to the alkene or alkyne substrate to form a secondary alkyl or vinyl radical intermediate. The resulting radical could then abstract an iodine atom from a perfluoroalkyl iodide molecule to give the difunctionalized product and propagate the radical chain.
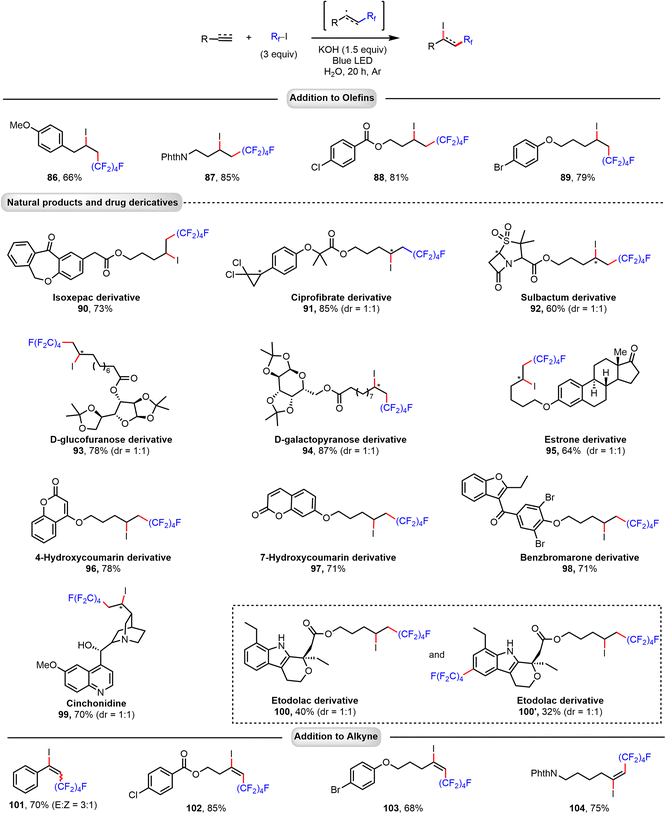 |
| | Fig. 4 Addition of perfluoroalkyl iodide to alkenes and alkynes. Reactions were performed on a 0.2 mmol scale. Isolated yields are given. | |
C–H perfluoroalkylation of electron-rich arenes and heteroarenes
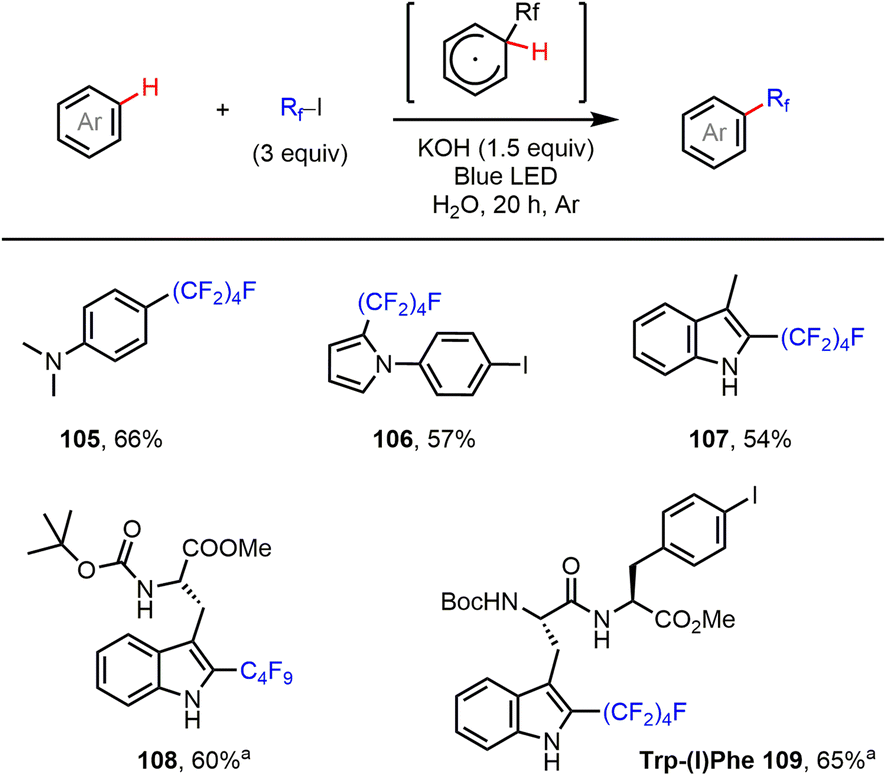
Finally, the C–H perfluoroalkylation of electron-rich arenes and heteroarenes with perfluoroalkyl iodides was investigated. Such transformations have been achieved using several different radical initiation systems.9,17 As shown in Fig. 5, reactions of aniline and pyrrole, as well as indole, using our halogen bond-promoted method (C4F9I…−OH) provided perfluoroalkylated products (105–107) in moderate to good yields. Furthermore, we were delighted to find that tryptophan (Trp)-containing short peptides could be perfluoroalkylated at the C2 position of the Trp residue, in good yield and with excellent selectivity, under the standard reaction conditions. This reaction might offer a valuable orthogonal strategy for peptide labeling based on under-explored Trp chemistry.
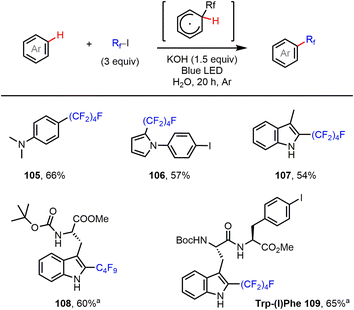 |
| | Fig. 5 C–H perfluoroalkylation of aniline and electron-rich heteroarenes. Reactions were performed on a 0.2 mmol scale. Isolated yields are given. aCH2Cl2 was used as solvent. | |
Mechanistic investigations and studies
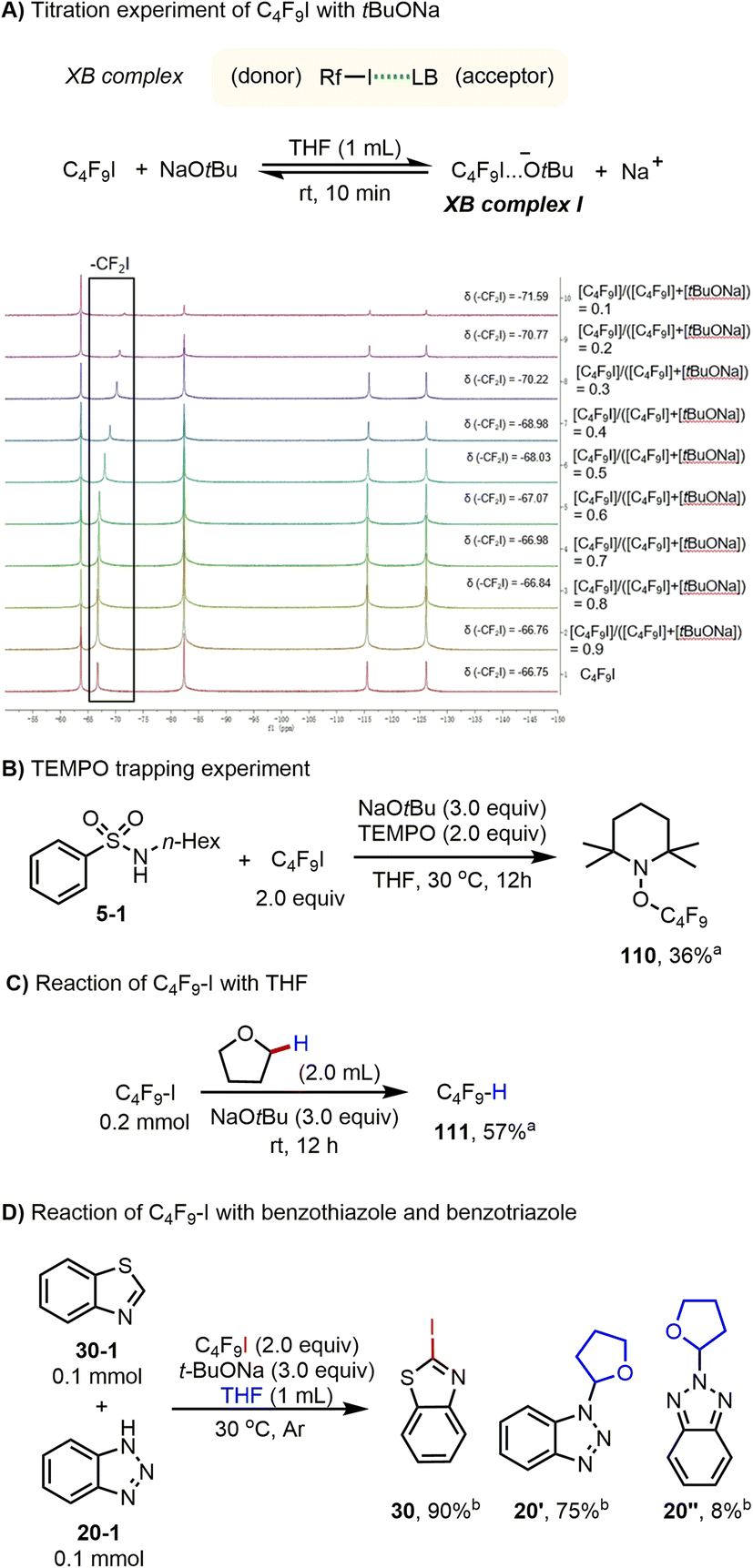
Having explored the synthetic utility of this novel activation mode for perfluoroalkyl iodide, we turned our attention to the mechanism, to explain the different outcomes of the amidation, iodination, and perfluoroalkylation reactions. In 2013, the interactions of iodoperfluoroarenes and iodoperfluoroalkanes with anions in organic solvent were studied by Taylor and co-workers, and their data indicated that favorable halogen bonding interactions exist between halide anions and iodoperfluoroalkanes.13b,c,e Based on this and our previous work,9 we speculated that, in the present reaction system, oxoanions (tBuO− and OH−) should have a halogen bonding interaction with perfluoroalkyl iodides under mild conditions. To verify the existence of the proposed halogen bonding interaction between tBuO− and Rf–I, a series of NMR experiments were performed using a solution of NaOtBu and C4F9I in the reaction solvent THF. The formation of a halogen bonding complex between NaOtBu and C4F9I is supported by the 19F NMR titration experiments. The resonance corresponding to the F of the CF2I group was shifted to the upfield region when NaOtBu was added to the THF solution of C4F9I (Scheme 3A).18–20 It is worth mentioning that the molar ratio (donor vs. acceptor) and the binding constant of the halogen bond complex (Rf–I…−OtBu) could not be accurately measured because NaOtBu was not completely soluble in the solvent, and the substrate Rf–I is converted to perfluorobutane over time. For C4F9I and KOH, a similar halogen bonding interaction was observed, which is also supported by the 19F NMR titration experiments (see ESI†). Moreover, from 19F NMR titration experiments of C4F9I with tBuONa and C4F9I with KOH, it was found that tBuONa has a stronger interaction with perfluoroalkyl iodides than does KOH (Table S2 vs. S3†).
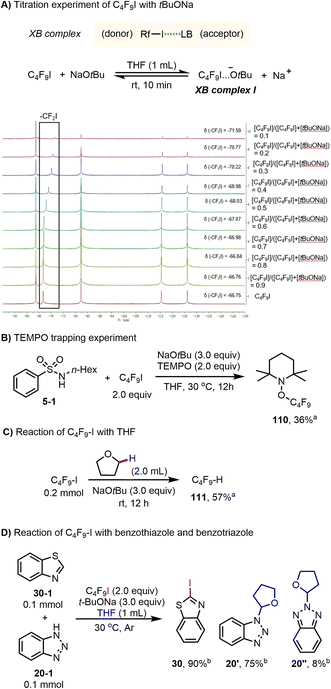 |
| | Scheme 3 Mechanistic experiments. aCrude 19F NMR yield. bIsolated yield. | |
Moreover, when a radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) was present in a reaction mixture containing C4F9–I and NaOtBu, formation of 5 was completely inhibited; instead, the corresponding product of radical trapping 110 (ref. 21 and 22) was obtained in 36% yield (Scheme 3B). Furthermore, perfluorobutyl iodide and tBuONa were dispersed in THF and the reaction mixture was stirred at 30 °C for 12 h. After adding PhCF3 as an internal standard for 19F NMR analysis, the formation of C4F9H was confirmed in 57% yield (Scheme 3C). These studies confirmed that: (1) C4F9I and NaOtBu have halogen bonding interactions in the present reaction system; (2) the halogen bonding complex between C4F9I and NaOtBu could produce perfluorobutyl radicals; and (3) the perfluorobutyl radical could serve as an H abstractor to activate a specific C(sp3)–H bond of THF. Finally, amidation and iodination reactions could proceed simultaneously under the standard reaction conditions (Scheme 3D), suggesting that perfluorobutyl and iodine radicals are produced. For the iodination reaction of benzothiazole, we noted that when 3 equivalents of TEMPO were added to the reaction mixture, product formation was suppressed (see ESI, Scheme S17†).
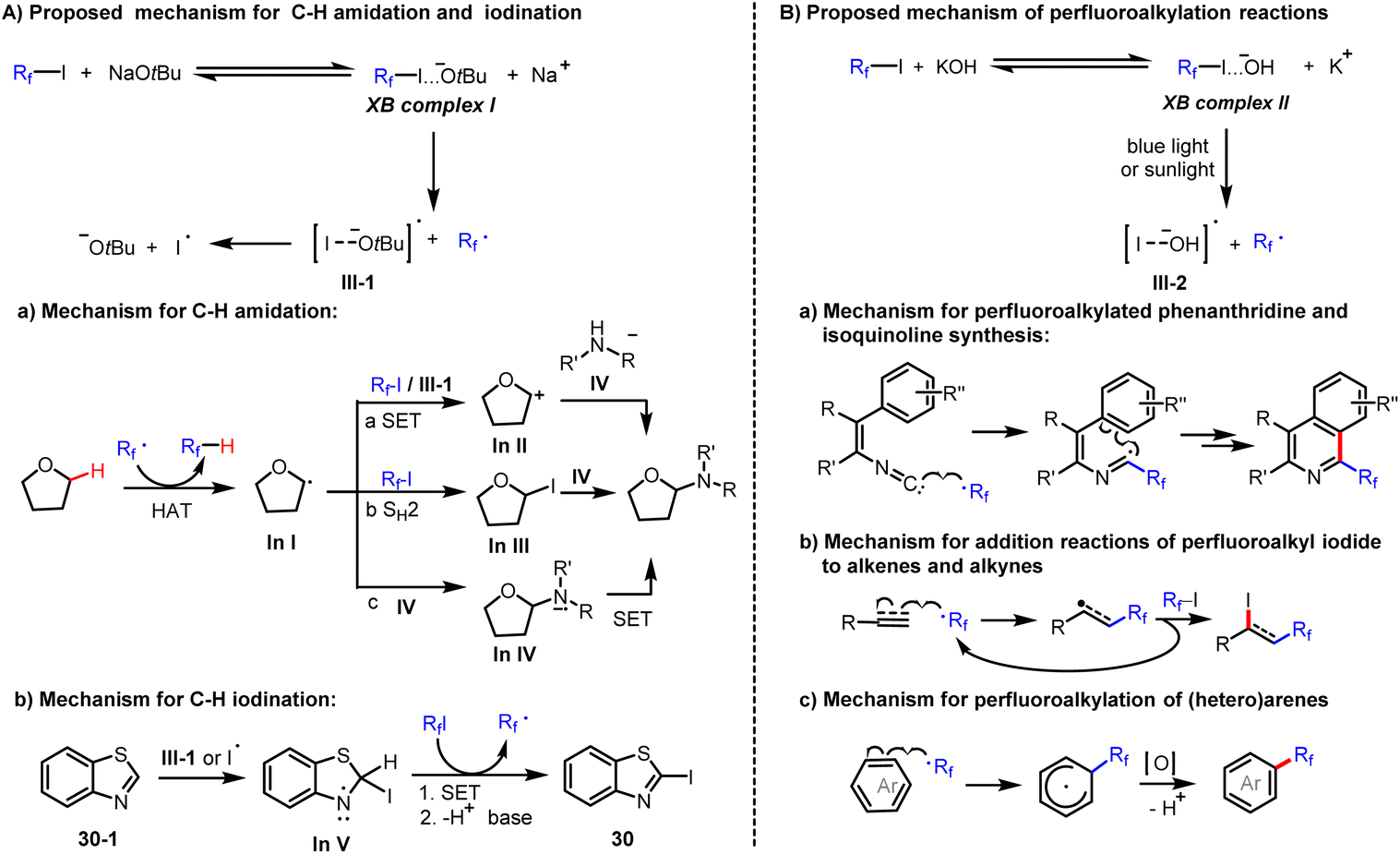
Based on the above results, we propose the following mechanism (Scheme 4). Reaction of perfluoroalkyl iodides with tBuONa leads to the formation of the corresponding halogen bonding complexes (XB complex I). Due to this activation, the weak C–I bond of XB complexes I presumably undergo homolytic cleavage to form both a perfluoroalkyl radical and an iodine radical under mild conditions (Scheme 4). In the observed amidation reactions, the generated perfluoroalkyl radical acts as a hydrogen abstractor to convert the C–H substrate to a carbon-centered radical, In I. For the transformation of In I to the observed product, different reaction paths are feasible. In path (a), In I is further oxidised by single electron transfer (SET) to obtain the carbocation In II, which reacts with amide anion to produce the amidation product. In path (b), In I reacts with Rf–I by SH2 (homolytic bimolecular substitution) to give In III, which again reacts with amide anion by SN2 (bimolecular nucleophilic substitution) to produce the amidation product. Finally, in path (c), the carbon-centered radical In I reacts with amide anion to form the corresponding radical anion (In IV), which is easily oxidized by perfluoroalkyl iodides to obtain the amidation product and generates perfluoroalkyl radical as well as iodide anion during the process (Scheme 4). In contrast, in the observed iodination reactions, an iodine radical attacks the 2-position of heterocycles to form iodinated In V. Then, the iodinated In V undergoes a SET and deprotonation to generate the observed iodination product (Scheme 4). Similarly, perfluoroalkyl iodides form halogen bonding complexes (XB complex II) with KOH (Scheme 4). Under irradiation with blue light or sunlight, the C–I bond of XB complexes II undergoes homolytic cleavage to form both a perfluoroalkyl radical and an iodine radical (III-2). The former radical reacts with isonitriles or electron-rich π bonds of alkenes, alkynes, and (hetero)arenes to accomplish perfluoroalkylation addition reactions.
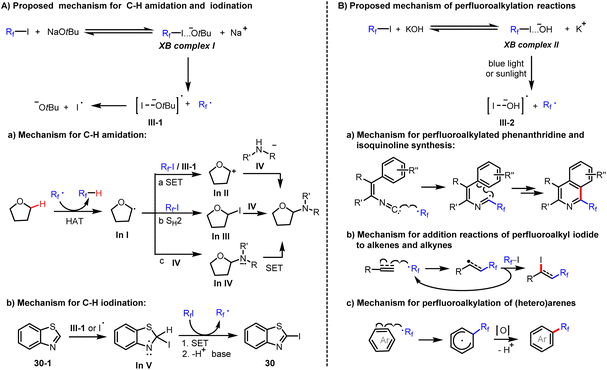 |
| | Scheme 4 Proposed mechanism. | |
Conclusions
In conclusion, we have developed versatile reaction systems for the amidation of alkyl ethers and benzylic hydrocarbons, for α-C(sp3)–H bond amidation, (hetero)arenes C–H iodination, and addition of perfluoroalkyl iodides to electron-rich π bonds. All these different synthetic protocols rely on activation of perfluoroalkyl iodides by simple inorganic bases, such as sodium butoxide or potassium hydroxide. Mechanistic studies suggest that halogen bond interactions between perfluoroalkyl iodides and the inorganic base promote the homolysis of perfluoroalkyl iodides. Notably, there is no requirement for expensive photoredox catalysts or transition metals, and the generated perfluoroalkyl radicals act as a hydrogen abstractor, iodine source, or perfluoroalkyl source for the synthesis of hemiaminal ethers, N-benzyl-arenesulfonamides, heteroaryl iodides, and perfluoroalkyl-substituted, and potentially bioactive, molecules. In addition, the practicality, easy scale up, and mild reaction conditions make these synthetic transformations attractive and valuable for organic synthesis.
Author contributions
Yaxin Wang formulated the initial ideas of this work, supervised the project, carried out most of the reaction optimization, C(sp3)–H amidation reaction study, C(sp2)–H iodination reaction study, structural determination of products, and prepared manuscript and the ESI. Zehui Cao and Qin He developed perfluoroalkylation reactions and take part in the mechanism study. Xin Huang prepared some amide substrates and take part in the C(sp3)–H amidation reaction study and Jiaxi Liu prepared some isonitrile substrates and take part in perfluoroalkylation reactions study. Helfried Neumann provide fund for the project and revised the manuscript and the ESI. Gong Chen provide fund for the project and gave suggestion for the project and manuscript. Matthias Beller provide fund, gave suggestion for the project and prepared most of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the National Science Foundation of China (no. 22001128), the National Science Foundation of Jiangsu Province (no. BK20200847), the National Science Foundation of Nanjing University of Chinese Medicine (no. NZY22001128) and CSC scholarship.
Notes and references
- Activation of carbon-halogen bond of alkyl halides:
(a) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed;
(b) P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119–6122 CrossRef CAS PubMed;
(c) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed;
(d) N. Iqbal, S. Choi, E. Ko and E. J. Cho, Tetrahedron Lett., 2012, 53, 2005–2008 CrossRef CAS;
(e) H. Jiang, Y. Z. Cheng, R. Z. Wang, M. M Zheng, Y. Zhang and S. Y. Yu, Angew. Chem., Int. Ed., 2013, 52, 13289–13292 CrossRef CAS PubMed;
(f) E. Kim, S. Choi, H. Kim and E. J. Cho, Chem.–Eur. J., 2013, 19, 6209–6212 CrossRef CAS PubMed;
(g) N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539–542 CrossRef CAS PubMed;
(h) N. J. W. Straathof, H. P. L. Gemoets, X. Wang, J. C. Schouten, V. Hessel and T. Noël, ChemSusChem, 2014, 7, 1612–1617 CrossRef CAS PubMed;
(i) S. Tang, Y. L. Deng, J. Li, W. X. Wang, G. L. Ding, M. W. Wang, Z. P. Xiao, Y. C. Wang and R. L. Sheng, J. Org. Chem., 2015, 80, 12599–12605 CrossRef CAS PubMed;
(j) C. Bottecchia, X. J. Wei, K. P. L. Kuijpers, V. Hessel and T. Noël, J. Org. Chem., 2016, 81, 7301–7307 CrossRef CAS PubMed;
(k) A. Postigo, Eur. J. Org. Chem., 2018, 6391–6404 CrossRef CAS;
(l) S. Zhang, N. Rotta-Loria, F. Weniger, J. Rabeah, H. Neumann, C. Taeschler and M. Beller, Chem. Commun., 2019, 55, 6723–6726 RSC;
(m) Y. Li, H. Neumann and M. Beller, Chem.–Eur. J., 2020, 26, 6784 CrossRef CAS PubMed;
(n) K. P. S. Cheung, D. Kurandina, T. Yata and V. Gevorgyan, J. Am. Chem. Soc., 2020, 142, 9932–9937 CrossRef PubMed;
(o) D. E. Yerien, S. Barata-Vallejo, E. W. M. Floresa and A. Postigo, Catal. Sci. Technol., 2020, 10, 5113–5128 RSC;
(p) M. Lubbesmeyer, E. G. Mackay, M. A. R. Raycroft, J. Elfert, D. A. Pratt and A. Studer, J. Am. Chem. Soc., 2020, 142, 2609–2616 CrossRef PubMed.
- Activation of carbon–halogen bond of aryl halides:
(a) E. Shirakawa, K. I. Itoh, T. Higashino and A. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537–15539 CrossRef CAS PubMed;
(b) L. Zhang, H. Yang and L. Jiao, J. Am. Chem. Soc., 2016, 138, 7151–7160 CrossRef CAS PubMed;
(c) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607–610 CrossRef CAS PubMed;
(d) S. Zhou, E. Doni, G. M. Anderson, R. G. Kane, S. W. MacDougall, V. M. Ironmonger, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2014, 136, 17818–17826 CrossRef CAS PubMed;
(e) J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410 CrossRef CAS PubMed;
(f) S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673–4676 CrossRef CAS PubMed;
(g) W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. W. Lei, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS PubMed;
(h) Y. Qiu, Y. Liu, K. Yang, W. K. Hong, Z. Li, Z. Y. Wang, Z. Y. Yao and S. Jiang, Org. Lett., 2011, 13, 3556–3559 CrossRef CAS PubMed;
(i) B. S. Bhakuni, A. Kumar, S. J. Balkrishna, J. A. Sheikh, S. Konar and S. Kumar, Org. Lett., 2012, 14, 2838–2841 CrossRef CAS PubMed;
(j) B. S. Bhakuni, A. Yadav, S. Kumar, S. Patel, S. Sharma and S. Kumar, J. Org. Chem., 2014, 79, 2944–2954 CrossRef CAS PubMed;
(k) C. L. Sun, Y. F. Gu, W. P. Huanga and Z. J. Shi, Chem. Commun., 2011, 47, 9813–9815 RSC;
(l) C. L. Sun, H. Li, D. G. Yu, M. Yu, X. Zhou, X. Y. Lu, K. Huang, S. F. Zheng, B. J. Li and Z. J. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed;
(m) J. Chen and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 3951–3955 CrossRef CAS PubMed;
(n) H. Q. Zhao, J. Shen, C. L. Ren, W. Zeng and H. Q. Zeng, Org. Lett., 2017, 19, 2190–2193 CrossRef CAS PubMed.
-
(a) R. S. Mulliken, J. Am. Chem. Soc., 1950, 72, 600–608 CrossRef CAS;
(b) O. Hassel, Science, 1970, 170, 497–502 CrossRef CAS PubMed;
(c) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed;
(d) M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC;
(e) S. M. Huber, E. Jimenez-Izal, J. M. Ugaldeb and I. Infante, Chem. Commun., 2012, 48, 7708–7710 RSC;
(f) M. P. Mitoraj and A. Michalak, J. Mol. Model., 2013, 19, 4681–4688 CrossRef CAS PubMed;
(g) L. P. Wolters, P. Schyman, M. J. Pavan, W. L. Jorgensen, F. M. Bickelhaupt and S. Kozuch, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 523–540 CAS;
(h) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
-
(a) S. C. Blackstock, J. P. Lorand and J. K. Kochi, J. Org. Chem., 1987, 52, 1451–1460 CrossRef CAS;
(b) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed;
(c) A. C. Legon, Phys. Chem. Chem. Phys., 2010, 12, 7736–7747 RSC;
(d) G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772–3783 RSC;
(e) T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC;
(f) G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS;
(g) A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
-
(a) A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef;
(b) E. Corradi, S. V. Meille, M. T. Messina, P. Metrangolo and G. Resnati, Tetrahedron Lett., 1999, 7519–7523 CrossRef CAS;
(c) P. Metrangolo, W. Panzeri, F. Recupero and G. Resnati, J. Fluorine Chem., 2002, 114, 27–33 CrossRef CAS;
(d) P. Metrangolo, T. Pilati, G. Resnati and A. Stevenazzi, Chem. Commun., 2004, 1492–1493 RSC;
(e) T. Caronna, R. Liantonio, T. A. Logothetis, P. Metrangolo, T. Pilati and G. Resnati, J. Am. Chem. Soc., 2004, 126, 4500–4501 CrossRef CAS PubMed;
(f) A. Casnati, R. Liantonio, P. Metrangolo, G. Resnati, R. Ungaro and F. Ugozzoli, Angew. Chem., Int. Ed., 2006, 45, 1915–1918 CrossRef CAS PubMed;
(g) P. Metrangolo, T. Pilati, G. Terraneo, S. Biella and G. Resnati, CrystEngComm, 2009, 11, 1187–1196 RSC;
(h) F. Meyer and P. Dubois, CrystEngComm, 2013, 15, 3058–3071 RSC;
(i) A. PriiMagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed;
(j) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed;
(k) B. Li, S. Q. Zanga, L. Y. Wang and T. C. W. Mak, Coord. Chem. Rev., 2016, 308, 1–21 CrossRef CAS;
(l) A. Abate, R. Dehmel, A. Sepe, N. Linh Nguyen, B. Roose, N. Marzari, J. K. Hong, J. M. Hook, U. Steiner and C. Neto, J. Mater. Chem. A, 2019, 7, 24445–24453 RSC.
- A. Bruckmann, M. A. Pena and C. Bolm, Synlett, 2008, 6, 900–902 Search PubMed.
-
(a) S. M. Walter, F. Kniep, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2011, 50, 7187–7191 CrossRef CAS PubMed;
(b) S. M. Huber, E. Jimenez-Izal, J. M. Ugalde and I. Infante, Chem. Commun., 2012, 48, 7708–7710 RSC;
(c) F. Kniep, S. H. Jungbauer, Q. Zhang, S. M. Walter, S. Schindler, I. Schnapperelle, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2013, 52, 7028–7032 CrossRef CAS PubMed;
(d) S. H. Jungbauer, S. M. Walter, S. Schindler, L. Rout, F. Kniep and S. M. Huber, Chem. Commun., 2014, 50, 6281–6284 RSC;
(e) S. H. Jungbauer and S. M. Huber, J. Am. Chem. Soc., 2015, 137, 12110–12120 CrossRef CAS PubMed;
(f) R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
-
(a) S. Dordonne, B. Crousse, D. Bonnet-Delpon and J. Legros, Chem. Commun., 2011, 47, 5855–5857 RSC;
(b) W. He, Y. C. Ge and C. H. Tan, Org. Lett., 2014, 16, 3244–3247 CrossRef CAS PubMed;
(c) Y. Takeda, D. Hisakuni, C. H. Lin and S. Minakata, Org. Lett., 2015, 17, 318–321 CrossRef CAS PubMed;
(d) F. Sladojevich, E. McNeill, J. Bçrgel, S. L. Zheng and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3712–3716 CrossRef CAS PubMed;
(e) M. Saito, N. Tsuji, Y. Kobayashi and Y. Takemoto, Org. Lett., 2015, 17, 3000–3003 CrossRef CAS PubMed.
- Y. X. Wang, J. H. Wang, G.-X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442–1445 CrossRef CAS PubMed.
-
(a) M. Nappi, G. Bergonzini and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 4921–4925 CrossRef CAS PubMed;
(b) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
-
(a) Y. Z. Cheng, X. A. Yuan, J. Ma and S. Y. Yu, Chem.–Eur. J., 2015, 21, 8355–8359 CrossRef CAS PubMed;
(b) Y. Z. Cheng and S. Y. Yu, Org. Lett., 2016, 18, 2962–2965 CrossRef CAS PubMed;
(c) X. Y. Sun, Y. Y. He and S. Y. Yu, J. Photochem. Photobiol., C, 2018, 355, 326–331 CrossRef CAS;
(d) X. Tang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 814–817 CrossRef CAS PubMed;
(e) J. Cao, G. Wang, L. Gao, H. Chen, X. Liu, X. Cheng and S. Li, Chem. Sci., 2019, 10, 2767–2772 RSC.
- C. Zhang, H. Zuo, G. Y. Lee, Y. Zou, Q. Dang, K. N. Houk and D. Niu, Nat. Chem., 2022, 14, 686–694 CrossRef CAS PubMed.
-
(a) A. E. Feiring, J. Org. Chem., 1985, 50, 3269–3274 CrossRef CAS;
(b) E. Dimitrijevic, O. Kvak and M. S. Taylor, Chem. Commun., 2010, 46, 9025–9027 RSC;
(c) M. S. Taylor, Top. Curr. Chem., 2014, 27–48 CrossRef PubMed , Halogen Bonding II;;
(d) L. K. Morka, V. Tata, D. J. Ulnessa, B. G. Ericksonb and M. W. Gealy, J. Mol. Liq., 2018, 271, 647–654 CrossRef;
(e) M. G. Sarwar, B. Dragisić, E. Dimitrijević and M. S. Taylor, Chem.–Eur. J., 2013, 19, 2050–2058 CrossRef CAS PubMed.
-
(a) Y. Aihara, M. Tobisu, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2014, 136, 15509–15512 CrossRef CAS PubMed;
(b) B. Zhang and A. Studer, Org. Lett., 2014, 16, 3990–3993 CrossRef CAS PubMed;
(c) S. Zhang, N. R. Loria, F. Weniger, J. Rabeah, H. Neumann, C. Taeschlerc and M. Beller, Chem. Commun., 2019, 55, 6723–6726 RSC;
(d) Y. Li, H. Neumann and M. Beller, Chem.–Eur. J., 2020, 26, 6784–6788 CrossRef CAS PubMed.
-
(a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed;
(b) M. Daz-Requejo and P. Prez, Chem. Rev., 2008, 108, 3379–3394 CrossRef PubMed;
(c) W. B. Parker, Chem. Rev., 2009, 109, 2880–2893 CrossRef CAS PubMed;
(d) P. Merino, T. Tejero and I. Delso, Curr. Med. Chem., 2008, 15, 954–967 CrossRef CAS PubMed;
(e) R. Garg, S. P. Gupta, H. Gao, M. S. Babu, A. K. Debnath and C. Hansch, Chem. Rev., 1999, 99, 3525–3602 CrossRef CAS PubMed.
-
(a) L. Tanwar, J. Börgel, J. Lehmann and T. Ritter, Org. Lett., 2021, 23, 5024–5027 CrossRef CAS PubMed;
(b) Q. Shi, S. Zhang, J. Zhang, V. F. Oswald, A. Amassian, S. R. Marder and S. B. Blakey, J. Am. Chem. Soc., 2016, 138, 3946–3949 CrossRef CAS PubMed;
(c) H. Do and O. Daugulis, Org. Lett., 2009, 11, 421–423 CrossRef CAS PubMed.
-
(a) S. Barata-Vallejo, M. M. Flesia, B. Lantano, J. E. Arguello, A. B. Penenory and A. Postigo, Eur. J. Org. Chem., 2013, 2013, 998–1008 CrossRef CAS;
(b) N. Iqbal, S. Choi, E. Ko and E. J. Cho, Tetrahedron Lett., 2012, 53, 2005–2008 CrossRef CAS;
(c) N. J. W. Straathof, H. P. L. Gemoets, X. Wang, J. C. V. Schouten, V. Hessel and T. Noel, ChemSusChem, 2014, 7, 1612–1617 CrossRef CAS PubMed.
- For details of the 19F NMR titration experiments, see the ESI.†.
- For 19F NMR analysis of XB complexes of Rf–I, see:
(a) P. Job, Justus Liebigs Ann. Chem., 1928, 9, 113–118 CAS;
(b) M. T. Messina, P. Metrangolo, W. Panzeri, E. Ragg and G. Resnati, Tetrahedron Lett., 1998, 39, 9069–9072 CrossRef CAS;
(c) D. V. G. Tiers, J. Fluorine Chem., 2000, 102, 175–184 CrossRef;
(d) D. D. Burton, F. Fontana, P. Metrangolo, T. Pilati and G. Resnati, Tetrahedron Lett., 2003, 44, 645–648 CrossRef CAS.
- M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646–1653 CrossRef CAS PubMed.
-
(a) A. Foris, Magn. Reson. Chem., 2004, 42, 534–555 CrossRef CAS PubMed;
(b) M. Hanack and J. Ullmann, J. Org. Chem., 1989, 54, 1432–1435 CrossRef CAS;
(c) X. Wang, H. Song, S. Wang, J. Yang, H. Qin, X. Jiang and C. Zhang, Tetrahedron, 2016, 72, 7606–7612 CrossRef CAS.
- D. Jiang, C. Liu, Y. Guo, J. Xiao and Q. Chen, Eur. J. Org. Chem., 2014, 2014, 6303–6309 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Gong
Chen
*b,
Gong
Chen