
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA decacationic ferrocene-based metallostar†
Susanne Margot
Rupf
 ,
Amina Leoni
Moshtaha
and
Moritz
Malischewski
*
,
Amina Leoni
Moshtaha
and
Moritz
Malischewski
*
Freie Universität Berlin, Fabeckstr. 34-36, 14195 Berlin, Germany. E-mail: moritz.malischewski@fu-berlin.de
First published on 28th December 2022
Abstract
Decacationic metallostars have been prepared by the reaction of permercurated ferrocene FeC10(HgO2CCF3)10 with superacidic (C5F5NH)(SbF6) (pKa = −11 estimated in H2O) in multigram scale. In the resulting compound, [FeC10Hg10(NC5F5)n][SbF6]10, the labile pentafluoropyridine ligands are readily displaced by acetonitrile (MeCN) or tetrahydrothiophene (THT). In the X-ray structure of [FeC10Hg10(THT)10][SbF6]10·24 MeCN no cation–anion contacts between mercury and fluorine were observed. Moreover, cyclic voltammetry measurements of [FeC10(Hg(MeCN))10]10+ and [FeC10(Hg(THT))10]10+ revealed a (quasi)reversible one-electron oxidation of Fe(II) to Fe(III). From the reaction of [FeC10(Hg(MeCN))10]10+ with MoF6 as oxidant the ferrocenium cation [FeC10(Hg(MeCN))10]11+ was obtained and characterized via single crystal XRD. These electrophilic metallostars are promising potential building blocks for the synthesis of dendritic architectures containing a robust, tenfold functionalized ferrocene core.
Introduction
The relevance of biological redox processes of metalloproteins for living organisms inspired chemists to design redox active macromolecular compounds.1 In this context the functionalization of dendrimers with redox active building blocks has received considerable attention as they are well defined macromolecules in terms of size and peripheral chemical functionality.2 In the last decades these dendrimers have found applications as multielectron redox catalysts as well as reservoirs, in the surface modification of electrodes or in electrochemical sensors.3,4 Their molecular architecture often resembles star-shaped structures with a core and different ligand building blocks which are not limited to organic moieties but also include examples of p-block and metal(organic) fragments.5 A widely used redox active building block is the ferrocenyl group due to the reversibility of the Fe+II/+III redox couple and the high thermal stability of the oxidized and the native form in particular.6 In many molecular stars the metallocene unit is located in the outskirts of the molecule7–22 despite its C5 symmetry rendering it as an ideal core building block. Examples of ferrocene centered metallostars are rare23,24 probably due to the synthetic difficulties associated with the poly- or perfunctionalization of ferrocene.25In 2021 we reported a simple perfunctionalization approach by treatment of ferrocene with mercury(II) carboxylates to obtain FeC10(HgX)10 (X = O2CC3H7, O2CCF3, O2CCCl3) in multigram scale.26 Although these compounds offer relatively weak Hg–C bonds allowing transmetallation27,28 and electrophilic substitution reactions29,30 they are chemically inert towards air and strong Brønsted acids (e.g. CF3CO2H pKa = −2.7 estimated in H2O31) caused by the low polarity of the Hg–C bond as well as the low Lewis acidity of the R–Hg–X units to form adducts with increased coordination number.32 Higher coordination numbers are usually observed in organomercury compounds with electron deficient substituents,33–35 as shown by the Gabbaï group for [o-(HgC6F4)3] which forms supramolecular architectures with aromatic hydrocarbons36,37 or weak Lewis acid/base adducts with a variety of O, N, P and S donors.38–40 In contrast to this, in polar organomercury compounds R–Hg–X the anionic groups can be displaced by neutral ligands, e.g. phosphines, to give [RHg(PR3)]X (Scheme 1) maintaining the linear two-fold coordination.41–43 The resulting cation [RHg(PR3)]+ is stable in presence of weakly coordinating anions (e.g. X = ClO4−) but dismutates in presence of halides (X = Cl− < Br− < I−).44 We envisioned that the displacement of anionic ligands in permercurated ferrocenes FeC10(HgX)10 would lead to the formation of highly charged species. Moreover, such polycations should be ideal precursors for ferrocene centered metallostars since they offer accessible coordination sites with predominantly linear coordination around the mercury atoms.
 | ||
| Scheme 1 Reactivity of cationic organomercury compounds towards neutral ligand like phosphine41–44 and pyridine derivatives (this work) in presence of weakly coordinating anions (WCA). WCA = ClO4−, NO3−.41–43 | ||
Results and discussion
FeC10(HgO2CCF3)10 was dissolved in tetrahydrofuran and an excess of 4-(dimethylamino)pyridine (dmap) was added (Scheme 1) yielding an orange precipitate. The solid was analysed by NMR and vibrational spectroscopy as well as elemental analysis. While the IR spectrum of the coordination compound exhibits bands which correspond only to the trifluoroacetate45 (TFA) as well as the nitrogen based ligand,46 the Raman spectrum features further information especially with respect to the metallocene framework. CCp–CCp deformation is found at![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 951 cm−1 and Hg–Cp vibrations at = 100 cm−1. Moreover, the Hg–OTFA band ( = 311 cm−1 in FeC10(HgTFA)10) vanished and new bands at = 193–158 cm−1 appeared, indicating the successful replacement of the carboxylic ligands by the pyridine analogues. 1H, 19F and 13C NMR signals show the intact ligands and anions but as in the starting material the 199Hg resonance is not visible. The coordination to the mercury atoms causes a low field shift of Δδ = 6 ppm of the CCp signals in the 13C NMR spectrum while all other signals are not significantly affected. The composition was determined by elemental analysis yielding the overall formula FeC10Hg10dmap10TFA10 (1).
= 951 cm−1 and Hg–Cp vibrations at = 100 cm−1. Moreover, the Hg–OTFA band ( = 311 cm−1 in FeC10(HgTFA)10) vanished and new bands at = 193–158 cm−1 appeared, indicating the successful replacement of the carboxylic ligands by the pyridine analogues. 1H, 19F and 13C NMR signals show the intact ligands and anions but as in the starting material the 199Hg resonance is not visible. The coordination to the mercury atoms causes a low field shift of Δδ = 6 ppm of the CCp signals in the 13C NMR spectrum while all other signals are not significantly affected. The composition was determined by elemental analysis yielding the overall formula FeC10Hg10dmap10TFA10 (1).
Single crystals were obtained by recrystallization from tetrahydrofuran. Compound 1 crystallized in the monoclinic space group P21/n containing one metallocene molecule in the asymmetric unit. All mercury atoms are almost linearly coordinated by one cyclopentadienyl and one dmap ligand with angles of 167.0(3)–178.6(4)° (Table 1), respectively. The distortion from linearity is caused by Hg–OTFA interactions within the sum of van der Waals radii of mercury (rvdW = 1.75 Å)47 and oxygen (rvdW = 1.52 Å).48 The Hg–O distances display a broad range of 2.24(1)–3.28(4) Å and are longer than in FeC10(HgO2CCF3)10 (Table 1). The TFA anions serve as bridging ligands between two to three metal centers (Fig. 1A) similarly to the structure of Hg(TFA)2·2 dmap which crystallizes as a dimer bridged by two TFA anions with a distorted octahedral coordination environment around the mercury atoms.46 This coordination motif causes an eclipsed conformation in 1a (Cptorsion = 3.6(1)°) in contrast to FeC10(HgO2CCF3)10 which exhibits a staggered conformation in solid state. As a result, one intramolecular Hg–Hg contact of 3.440(2) Å is observed. Taking all interactions into account the total coordination number of the mercury atoms is four to six. As only two non-coordinating (distorted) TFA units within the unit cell with an overall occupancy of 1.33 are observed, this metallocene is better described as a “monocation” [FeC10(Hgdmap)10(TFA)8.66][TFA]1.33.
| Compound | FeC10(HgO2CCF3)26 (X = OTFA) | 1a (X = OTFA) | 2b (X = STHT) | 2c[MoF6] (X = NMeCN) |
|---|---|---|---|---|
| Fe–Cpcenter | 1.660(1) | 1.672(2) | 1.651(1), 1.658(1) | 1.738(1) |
| Hg–C | 2.034(9)–2.040(8) | 2.009(10)–2.062(8) | 2.022(10)–2.046(9) | 2.023(6)–2.039(7) |
| Hg–X | 2.082(7)–2.115(6) | 2.241(17)–3.286(36) | 2.395(2)–2.405(2) | 2.050(6)–2.062(5) |
| Hg–Hgintramol. | — | 3.440(2) | — | — |
| C–Hg–X | 168.8(3)–176.5(3) | 167.0(3)–178.6(4) | 170.8(2)–176.7(2) | 171.2(3)–175.1(2) |
| Conformation | Staggered | Eclipsed | Staggered | Staggered |
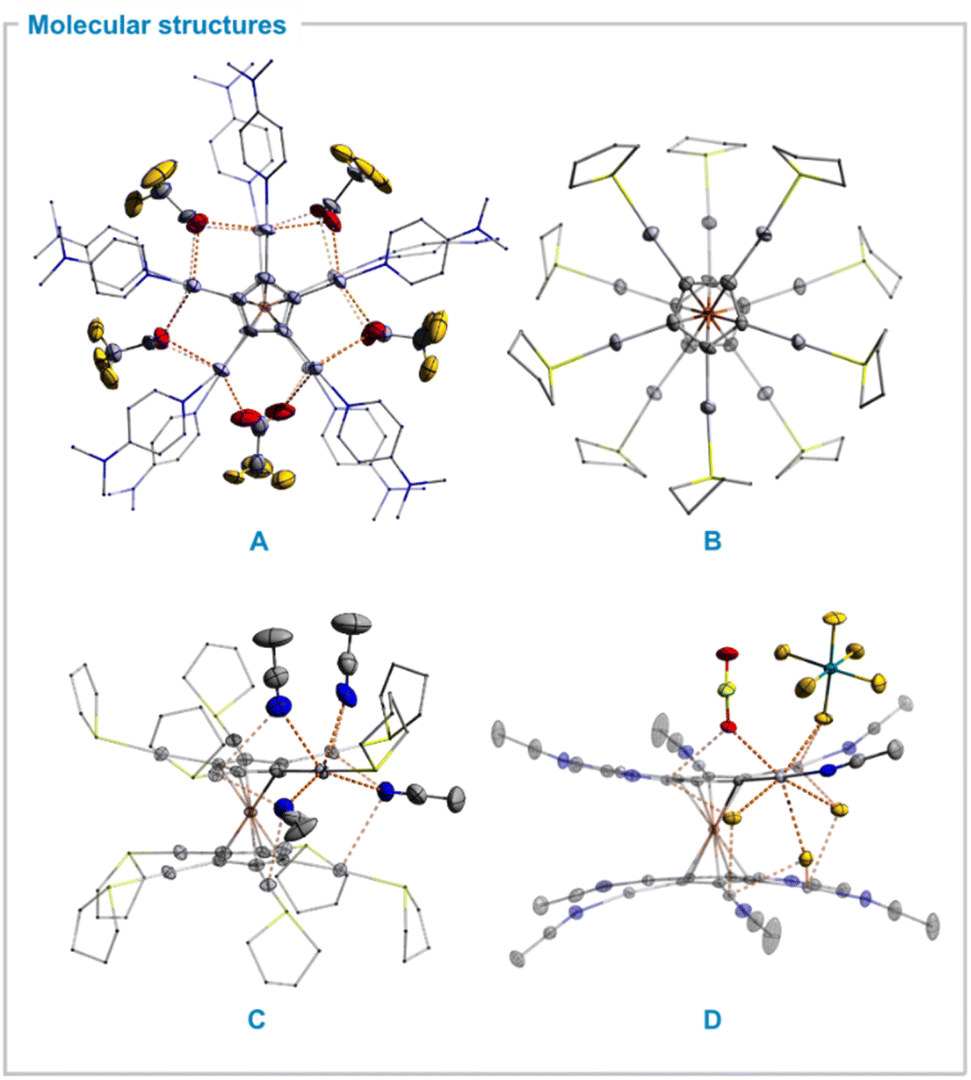 | ||
| Fig. 1 Molecular structures of selected star-shaped permercurated metallocene cations in solid state. Ellipsoids with 50% probability level. Color code: mercury – light grey, antimony – turquoise, iron – orange, sulfur – neon yellow, fluorine – yellow, oxygen – red, nitrogen – blue, carbon – dark grey. Metal-anion or solvent interactions are shown as dashed orange lines. Hydrogen atoms, selected solvents and counter anions as well as disorder is omitted for clarity. (A) Inner sphere coordination of bridging TFA anions in FeC10Hg10dmap10(O2CCF3)10. (B) [FeC10(HgTHT)10]10+ molecular fragment. (C) Solvent coordination motif in [FeC10(HgTHT)10]10+ structure. (D) [FeC10(HgMeCN)10]11+ molecular fragment as well as coordination of selected solvents and counter anion. | ||
In order to reduce cation anion interactions, we introduced more weakly coordinating anions by protonation of the TFA anions with protonated pentafluoropyridine [C5F5NH][SbF6] (pKa ≈ −11 estimated in H2O49) in liquid SO2 (Scheme 1) yielding [FeC10Hg10(NC5F5)n][SbF6]10 (2a) quantitatively as an insoluble orange solid whereas trifluoroacetic acid can be removed in vacuum. The completeness of the protonation was confirmed by the absence of TFA vibrations in the corresponding IR as well as Raman spectra (Fig. S7†). Instead, additional Sb–F bands are present at = 660 and 633 cm−1 in the infrared spectrum as well as C–C and C–F vibrations in the Raman and infrared spectrum which could be assigned to C5F5N50 indicating coordination to the metal center. Protodemercuration is negligible as neither ν(Cp–H) in the 3100 cm−1 region nor (intense) δ(Cp–H) vibrations in the 1100 cm−1 region are visible in both spectra which again confirms the stability of the Cp–Hg bonds. Unfortunately, the total composition could not be determined by elemental analysis due to the sensitivity of the compound.
Substitution of the C5F5N ligands improves the solubility of the corresponding compounds considerably and enables the full characterization of the compounds [FeC10(Hg solv.)10][SbF6]10 (solv. = tetrahydrothiophene (THT) 2b, MeCN 2c). Both metallocenes show similar features in the vibrational spectra as discussed for compound 2a. The CCp signals in the 13C NMR spectra (δ = 111.8 (2b) and 97.7 ppm (2c)) are again low field shifted in comparison to the signal of FeC10(HgTFA)10 as shown for compound 1, previously.
Single crystals of the THT-complex 2b·24 MeCN were obtained upon recrystallization from acetonitrile at −30 °C. It crystallized in the space group C2/c. The asymmetric unit contains a {Fe(CHgTHT)5} fragment with the iron position located at a center of inversion. The Cp ligands are aligned in a staggered conformation with torsion angles between 30.3(4) and 34.1(4)° (Fig. 1B). Again, the mercury atoms are linearly coordinated by a cyclopentadienyl and a THT ligand. In contrast to the structure of 1 no mercury anion contacts are visible (Fig. 1C). Instead, the decacationic fragment [FeC10(HgTHT)10]10+ is fully solvated by 20 acetonitrile molecules. Ten of those are coordinating systematically two to three different mercury atoms from the space within both Cp rings and the other ten from the outer-sphere of the metallocene framework yielding a distorted octahedral geometry for all mercury atoms. The Hg–N distances are in range of 2.941(8)–3.289(8) Å. The interactions between cations and anions are mainly a consequence of hydrogen bonding to the solvent as well as the THT ligand. A detailed discussion of the packing motif of the decacation (2b) can be found in the ESI.†
Since [FeC10(HgTHT)10][SbF6]10 can be regarded as a ferrocene with ten cationic [Hg(THT)]+ substituents the oxidation potentials of the metallocene are expected to be higher than in unsubstituted ferrocene as similar observations were reported for a series of acceptor substituted metallocenes.51 Herein, Sundermeyer et al. observed increasing oxidation potentials of up to ΔE = 474 mV per cationic group {SR2}+ group. The electrochemical properties of the compounds 1 and 2b,c were determined by cyclic voltammetry. Unfortunately, the metallocene 1 decomposed upon oxidation preventing the determination of the oxidation potentials. The measurements of the soluble decacationic THT and MeCN complexes 2b and 2c were performed in acetonitrile and referenced against the FcH/FcH+ redox couple (Fig. 2). Both compounds exhibit a reversible one-electron oxidation of the redox couple Fe+II/+III at E1/2THT = 0.522 V and E1/2MeCN = 0.713 V as well as irreversible oxidations above Ea > 1.3 V (Fig. S24 and S25, ESI†). The comparison of the Fe+II/+III potentials with the starting material FeC10(HgTFA)10 (E1/2 = 0.450 V) shows increasing oxidation potentials depending on the coordination strength of the ligand (TFA > THT > MeCN). Therefore, [FeC10Hg10(NC5F5)n][SbF6]10, is expected to exhibit the highest oxidation potential in the series TFA < THT < MeCN < NC5F5 although its insolubility prevented its electrochemical characterization. A similar trend was already observed in Ag(I) compounds which vary enormously in oxidation potentials depending on the ligands at the metal center or simply the solvation ability of the solvents.52 The surprisingly low potentials of the 10+/11+ redox couple are probably a consequence of the large size and effective solvation of these highly charged species as in multicationic ferrocenyl dendrimers.53
 | ||
| Fig. 2 Cyclic voltammograms of compounds FeC10(HgO2CCF3)10 in THF with 0.1 M TBAPF6 as supporting electrolyte (bottom),26 [FeC10(HgTHT)10][SbF6]10 (2b, middle) and [FeC10(HgMeCN)10][SbF6]10 (2c, top) in acetonitrile without supporting electrolyte at 100 mV s−1 scan rate and room temperature. | ||
To realise the chemical oxidation of the permercurated ferrocene decacations we used different moderate oxidants in liquid SO2 which allow solely oxidation to the desired ferrocenium compounds. The oxidations were tested with Ag[SbF6], NO[BF4], NO2[SbF6] as well as MoF6. Unfortunately, the presence of coordinating ligands seemed to reduce the reactivity of silver salts as no reaction of none of the compounds was observed with AgSbF6. Moreover, [FeC10Hg10(NC5F5)n][SbF6]10 showed no reaction with (NO)+ but with (NO2)+ indicating high oxidation potentials which perfectly fits to the correlation between ligand strength and redox potential. Reactions with (NO2)+ and MoF6 were successful in all cases.
Single crystals of 2c[MoF6]·10 HF·2 SO2 were obtained upon recrystallisation from HF/SO2 at −74 °C. The compound crystallized in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit consists of one {FeC5(HgMeCN)5} moiety with a center of inversion located at the iron atom. The oxidation of the iron center is indicated by an elongated iron-cyclopentadienyl distance of 1.738(1) Å which is in the same order of magnitude as other substituted ferrocenium salts but significantly longer than those of non-substituted ferrocenium cations.54 The metallocene cation is again fully solvated by 10 HF molecules with Hg–FHF distances of 2.817(5)−2.956(4) Å within the inner-sphere of the metallocene framework in a similar fashion as discussed for compound 2b triggering a staggered conformation with torsion angles in the range of 31.1(1)° to 35.9(1). However, the structure features several additional Hg–F cation–anion interactions of 2.824(5)–3.090(5) Å length yielding a coordination number of four to six for all mercury atoms (Fig. 1D). The Hg–F contacts to the [SbF6]− counter anions are present due to the low solvation of the undecacation in HF, in contrast to the THT complex 2b which was recrystallized from acetonitrile.
. The asymmetric unit consists of one {FeC5(HgMeCN)5} moiety with a center of inversion located at the iron atom. The oxidation of the iron center is indicated by an elongated iron-cyclopentadienyl distance of 1.738(1) Å which is in the same order of magnitude as other substituted ferrocenium salts but significantly longer than those of non-substituted ferrocenium cations.54 The metallocene cation is again fully solvated by 10 HF molecules with Hg–FHF distances of 2.817(5)−2.956(4) Å within the inner-sphere of the metallocene framework in a similar fashion as discussed for compound 2b triggering a staggered conformation with torsion angles in the range of 31.1(1)° to 35.9(1). However, the structure features several additional Hg–F cation–anion interactions of 2.824(5)–3.090(5) Å length yielding a coordination number of four to six for all mercury atoms (Fig. 1D). The Hg–F contacts to the [SbF6]− counter anions are present due to the low solvation of the undecacation in HF, in contrast to the THT complex 2b which was recrystallized from acetonitrile.
Both, the solid-state structure of [FeC10(HgTHT)10]10+ and [FeC10(HgMeCN)10]11+ are rare examples of isolated organometallic deca- and undecacations. Although the large variety of multicationic organic or organometallic compounds,55–60 only a few examples for crystallographically characterized octa-,61 nona-,62 deca-,63–65 dodeca66 and even hexadecacations67 have been reported in the past years. In such multications, the positive charges are kinetically stabilized by organic ligands via formation of large aggregates.68 These so-called “lipophilically wrapped polyion aggregates” are usually not perfectly spheric causing voids within the crystal packing. Therefore, the formation of a closest packing arrangements or typical structure types as observed for (pseudo-)spherical components of low charge is prevented. In the crystal structure of [FeC10Hg(MeCN)10][SbF6]10[MoF6]·2 SO2·10 HF the iron atoms produce a distorted hexagonal distorted arrangement along the [111] direction but with a ABC-stacking pattern similar to the face-centered cubic structure of copper causing a distorted cuboctahedral geometry around each iron position (Fig. 3, top). Considering only the centers of gravity for all molecular entities, no simple packing arrangement is observed for the anionic units. The undecacation is surrounded by 16 anions in a distorted square orthobicupolar geometry (Fig. 3, bottom). These polyhedra are edge-sharing with the same polyhedra of adjacent unit cells along a and b and corner-sharing along c resulting in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11 composition of cations and anions. Sulphur moieties from SO2 solvates are capping two adjacent faces on both apical sides of the bicupolas.
:11 composition of cations and anions. Sulphur moieties from SO2 solvates are capping two adjacent faces on both apical sides of the bicupolas.
 | ||
| Fig. 3 Packing motifs of anions and cations in the crystal structure of [FeC10Hg(MeCN)10][SbF6]10[MoF6] 2 SO2·10 HF. Top: asym. unit and closest iron positions forming a cuboctahedron. Bottom: four units cells containing heavy metal positions forming distorted square orthobicupolas around the undecacation. Ellipsoids drawn with 50% probability level. Color code: mercury – light grey, molybdenum – purple, antimony – turquoise, iron – orange, sulphur – neon yellow, fluorine – yellow, oxygen – red, carbon – dark – grey. | ||
Conclusions
In summary, we investigated the reactivity of permercurated ferrocene derivatives towards neutral ligands in standard organic solvents as well as in superacidic medium.69 We demonstrated that in the compound FeC10(HgTFA)10 all mercury atoms are accessible to Lewis bases. The Lewis acidity of the {FeC10Hg10}10+ framework was tuned by variation of the counter anions. While the TFA salt only binds to strong σ-donors like dmap, the presence of weakly coordinating anions allows to isolate solvent adducts [FeC10(HgL)10][SbF6]10 (with L = C5F5N, MeCN, THT). Surprisingly, in those compounds the Hg–C bonds are inert to Brønsted acids, such as [C5F5NH][SbF6]. Moreover, we report the preparation of undecacations [FeC10(HgMeCN)10]11+ and [FeC10(HgTHT)10]11+ as stable solids. Both, the solid-state structure of [FeC10(HgTHT)10]10+ and [FeC10(HgMeCN)10]11+ are rare examples of isolated organometallic deca- and undecacations.70 The combination of the accessible coordination sites, electrochemical properties as well as their reliable multigram synthesis renders these deca- and undecacations as promising precursors for even larger star-shaped molecular architectures (e.g. dendrimers).Data availability
Besides the ESI† and the crystal structures in CCDC we have not deposited any data in other repositories.Author contributions
The project was developed by S. M. R. and M. M. Both authors wrote the manuscript with main contributions from S. M. R. The synthetic work was carried out by S. M. R. and A. L. M, S. M. R. performed cyclic voltammetry and UV/vis experiments as well as measured and solved the SCXRD data. A. L. M. performed further NMR, IR and Raman experiments and prepared the single crystals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Gefördert durch Deutsche Forschungsgemeinschaft (DFG) – Projektnummer: 387284271 – SFB 1349. Funded by Deutsche Forschungsgemeinschaft (DFG) – project number: 387284271 – SFB 1349. Computing time was made available by High-Performance Computing at ZEDAT/FU Berlin. We would like to acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG. The authors thank Prof. Dr Florian Kraus (Philipps-Universität Marburg) and Dr Günther Thiele (Freie Universität Berlin) for helpful discussions.Notes and references
- D. Astruc, J. Leather Sci. Eng., 2020, 2, 1–17 CrossRef.
- C. Gorman, Adv. Mater., 1998, 10, 295–309 CrossRef CAS.
- D. Astruc, C. Ornelas and J. Ruiz, Acc. Chem. Res., 2008, 41, 841–856 CrossRef CAS PubMed.
- G. R. Newkome, E. He and C. N. Moorefield, Chem. Rev., 1999, 99, 1689–1746 CrossRef.
- H. Detert, M. Lehmann and H. Meier, Materials, 2010, 3, 3218–3330 CrossRef.
- D. Astruc, Eur. J. Inorg. Chem., 2017, 6–29 CrossRef.
- B. Kayser, J. Altman, H. Nöth, J. Knizek and W. Beck, Eur. J. Inorg. Chem., 1998, 1791–1798 CrossRef.
- M. Iyoda, T. Kondo, T. Okabe, H. Masuyama, S. Sasaki and Y. Kuwatani, Chem. Lett., 1997, 26, 35–36 CrossRef.
- R. Misra, R. Maragani, C. P. Singh and R. Chari, Dye Pigm., 2016, 126, 110–117 CrossRef.
- A. K. Diallo, C. Absalon, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2011, 133, 629–641 CrossRef.
- J. C. Santos, F. Madrid-Moliné, C. A. Cisternas, F. Paul, C. A. Escobar, P. Jara-Ulloa and A. Trujillo, Inorg. Chim. Acta, 2018, 486, 95–100 CrossRef.
- G. Erbland, S. Abid, Y. Gisbert, N. Saffon-Merceron, Y. Hashimoto, L. Anderoni, T. Guérin, C. Kammerer and G. Rapenne, Chem.–Eur. J., 2019, 25, 16328–16339 CrossRef PubMed.
- A. Hildebrandt, T. Rüffer, E. Erasmus, J. C. Swarts and H. Lang, Organometallics, 2010, 29, 4900–4905 CrossRef.
- G. Vives, A. Carella, J.-P. Launay and G. Rapenne, Chem. Commun., 2006, 2283–2285 RSC.
- J.-L. Fillaut, J. Lineares and D. Astruc, Angew. Chem., Int. Ed. Engl., 1994, 33, 2460–2462 CrossRef.
- V. Mamane, I. Ledoux-Rak, S. Deveau, J. Zyss and O. Riant, Synthesis, 2003, 3, 433–467 Search PubMed.
- V. Mamane, A. Gref and O. Riant, New J. Chem., 2004, 28, 585–594 RSC.
- Y. Yu, A. D. Bond, P. W. Leonard, K. P. C. Vollhardt and G. D. Whitener, Angew. Chem., Int. Ed., 2006, 45, 1794–1799 CrossRef PubMed.
- B. Alonso, I. Cuadrado, M. Morán and J. Losada, J. Chem. Soc., Chem. Commun., 1994, 2575–2576 RSC.
- Y. Yu, A. D. Bond, P. W. Leonard, U. J. Lorenz, T. V. Timofeeva, K. P. C. Vollhardt, G. D. Whitener and A. A. Yakovenko, Chem. Commun., 2006, 2572–2574 RSC.
- R. Misra, R. Sharma and S. M. Mobin, Dalton Trans., 2014, 43, 6891–6896 RSC.
- R. Misra, R. Maragani, B. Pathak, P. Gautam and S. M. Mobin, RCS Adv., 2015, 5, 71046–71051 Search PubMed.
- K. Rahimpour and R. Teimuri-Mofrad, Appl. Organomet. Chem., 2020, 34, e5943 Search PubMed.
- P. Jutzi, C. Batz, B. Neumann and H.-G. Stammler, Angew. Chem., Int. Ed. Engl., 1996, 35, 2118–2121 CrossRef.
- H. Butenschön, Synthesis, 2018, 50, 3787–3808 CrossRef.
- S. M. Rupf, G. Schröder, R. Sievers and M. Malischewski, Chem.–Eur. J., 2021, 27, 5125–5129 CrossRef PubMed.
- K. N. Seneviratne, A. Bretschneider-Hurley and C. H. Winter, J. Am. Chem. Soc., 1996, 118, 5506–5507 CrossRef.
- A. Bretschneider-Hurley and C. H. Winter, J. Am. Chem. Soc., 1994, 116, 6468–6469 CrossRef.
- S. M. Rupf, I. S. Dimitrova, G. Schröder and M. Malischewski, Organometallics, 2022, 41, 1261–1267 CrossRef.
- Y.-H. Han, M. J. Heeg and C. H. Winter, Organometallics, 1994, 13, 3009–3019 CrossRef.
- S. Saito, S. Saito, T. Ohwada and K. Shudo, Chem. Pharm. Bull., 1991, 39, 2718–2720 CrossRef.
- I. P. Beletskaya, K. P. Butin, A. N. Ryabtsev and O. A. Reutov, J. Organomet. Chem., 1973, 59, 1–44 CrossRef.
- T.-P. Lin, R. C. Nelson, R. Wu, J. T. Miller and F. P. Gabbaï, Chem. Sci., 2012, 3, 1128–1136 RSC.
- P. Nockemann, F. Schulz, D. Maumann and G. Meyer, Z. Anorg. Allg. Chem., 2005, 631, 649–653 CrossRef.
- M. Nolte, I. Patenburg and G. Meyer, Z. Anorg. Allg. Chem., 2007, 634, 362–368 CrossRef.
- M. Tsunoda and F. P. Gabbaï, J. Am. Chem. Soc., 2000, 122, 8335–8336 CrossRef.
- M. R. Haneline, M. Tsunoda and F. P. Gabbaï, J. Am. Chem. Soc., 2002, 124, 3737–3742 CrossRef.
- M. Fleischmann, J. S. Jones, G. Balázs and F. P. Gabbaï, Dalton Trans., 2016, 45, 13742–13749 RSC.
- M. R. Haneline, R. E. Taylor and F. P. Gabbaï, Chem.–Eur. J., 2003, 9, 5188–5193 CrossRef PubMed.
- T. J. Taylor, C. N. Burress and F. P. Gabbaï, Organometallics, 2007, 26, 5252–5263 CrossRef CAS.
- G. E. Coates and A. Lauder, J. Chem. Soc., 1965, 1857–1864 RSC.
- T. S. Lobana, M. K. Sandhu, D. C. Povey and G. W. Smith, J. Chem. Soc., Dalt. Trans., 1988, 2913–2914 RSC.
- T. S. Lobana and M. K. Sandhu, J. Chem. Soc., Dalt. Trans., 1989, 2339–2341 RSC.
- G. Schwarzenbach and M. Schellenberg, Helv. Chim. Acta, 1965, 48, 28–46 CrossRef CAS.
- A. J. Downs, E. A. V. Ebsworth and H. J. Emeléus, J. Chem. Soc., 1962, 1254–1260 RSC.
- W. Tyrra, D. Naumann and I. Pantenburg, J. Fluorine Chem., 2003, 120, 13–19 CrossRef CAS.
- P. Pyykkö and M. Straka, Phys. Chem. Chem. Phys., 2000, 2, 2489–2493 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- S. L. Bell, R. D. Chambers, K. R. Musgrave and J. G. Thorpe, J. Fluorine Chem., 1971, 1, 51–57 CrossRef CAS.
- K. Züchner, T. J. Richardson, O. Glemser and N. Bartlett, Angew. Chem., Int. Ed. Engl., 1980, 19, 944–945 CrossRef.
- A. Venker, T. Vollgraff and J. Sundermeyer, Dalton Trans., 2018, 47, 1933–1941 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- D. Astruc, New J. Chem., 2011, 35, 764–772 RSC.
- R. Martinez and A. Tiripicchio, Acta Cryst., 1990, C46, 202–205 CAS.
- D. Astruc, M.-C. Daniel and J. Ruiz, Chem. Commun., 2004, 2637–2649 RSC.
- D. Astruc, C. Ornelas and J. Ruiz, Chem.–Eur. J., 2009, 15, 8936–8944 CrossRef CAS.
- J. Chen, J. Shan, Y. Xu, P. Su, L. Tong, L. Yuwen, L. Weng, B. Bao and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 34455–34463 CrossRef CAS PubMed.
- K.-Y. Pu, K. Li and B. Liu, Adv. Mater., 2010, 22, 643–646 CrossRef CAS.
- C. McCusker, J. B. Caroll and V. M. Rotello, Chem. Commun., 2005, 996–998 RSC.
- L.-I. Rodríguez, O. Rossell, M. Seco and G. Muller, J. Organomet. Chem., 2007, 692, 851–858 CrossRef.
- D. M. L. Goodgame, S. Kealey, P. D. Lickiss and A. J. P. White, J. Mol. Struct., 2008, 890, 232–239 CrossRef CAS.
- M. A. Little, M. A. Halcrow, L. P. Harding and M. J. Hardie, Inorg. Chem., 2010, 49, 9486–9496 CrossRef CAS.
- C. S. Campos-Fernandez, R. Clerac, J. M. Koomen, D. H. Russell and K. R. Dunbar, J. Am. Chem. Soc., 2001, 123, 773–774 CrossRef CAS PubMed.
- C. S. Campos-Fernandez, B. L. Schottel, H. T. Chifotides, J. K. Bera, J. Bacsa, J. M. Koomen, D. H. Russell and K. R. Dunbar, J. Am. Chem. Soc., 2005, 127, 12909–12923 CrossRef CAS PubMed.
- I. D. Giles, H. T. Chifotides, M. Shatruk and K. R. Dunbar, Chem. Commun., 2011, 47, 12604–12606 RSC.
- R. Lavendomme, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 12147–12158 CrossRef CAS PubMed.
- J. Yang, X.-Y. Chang, K.-C. Sham, S.-M. Yui, H.-L. Kwong and C.-M. Che, Chem. Commun., 2016, 52, 5981–5984 RSC.
- H. Bock, A. John, C. Näther and Z. Havlas, J. Am. Chem. Soc., 1995, 117, 9367–9368 CrossRef CAS.
- A previous version of this manuscript has been deposited on a preprint server: S. M. Rupf, A. L. Moshtaha, and M. Malischewski, ChemRxiv, 2022, DOI: 10.26434/chemrxiv-2022-5rxlm Search PubMed.
- Deposition numbers CCDC 2214089 ([FeC10(HgDMAP)10][TFA]10), 2213962 ([FeC10(HgTHT)10][SbF6]10·24 MeCN) and 2213956 ([FeCp2(HgMeCN)10][SbF6]10[MoF6]·2SO2·10 HF) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.https://www.ccdc.cam.ac.uk/structures.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2214089, 2213962 and 2213956. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06151a |
| This journal is © The Royal Society of Chemistry 2023 |