
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed defluorinative arylboration of vinylarenes with polyfluoroarenes†
Fu-Peng
Wu
a,
Xing-Wei
Gu
a,
Hui-Qing
Geng
a and
Xiao-Feng
Wu
 *ab
*ab
aLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: xiao-feng.wu@catalysis.de
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023, Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
First published on 6th February 2023
Abstract
An unprecedented but challenging defluorinative arylboration has been achieved. Enabled by a copper catalyst, an interesting procedure on defluorinative arylboration of styrenes has been established. With polyfluoroarenes as the substrates, this methodology offers flexible and facile access to provide a diverse assortment of products under mild reaction conditions. In addition, by using a chiral phosphine ligand, an enantioselective defluorinative arylboration was also realized, affording a set of chiral products with unprecedented levels of enantioselectivity.
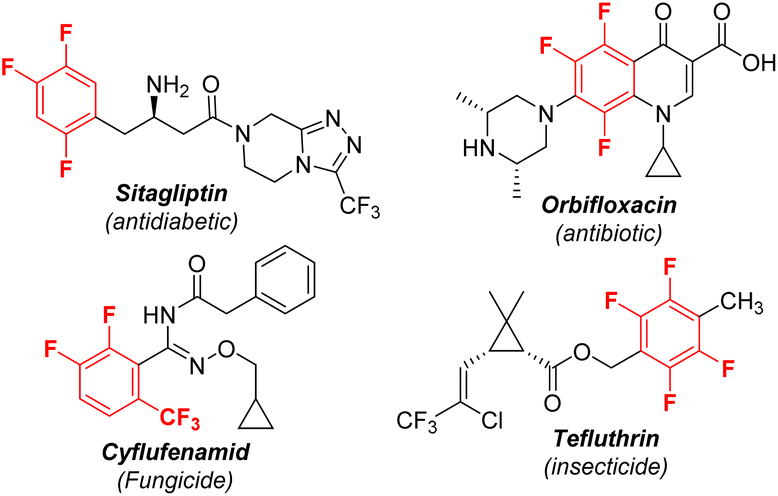
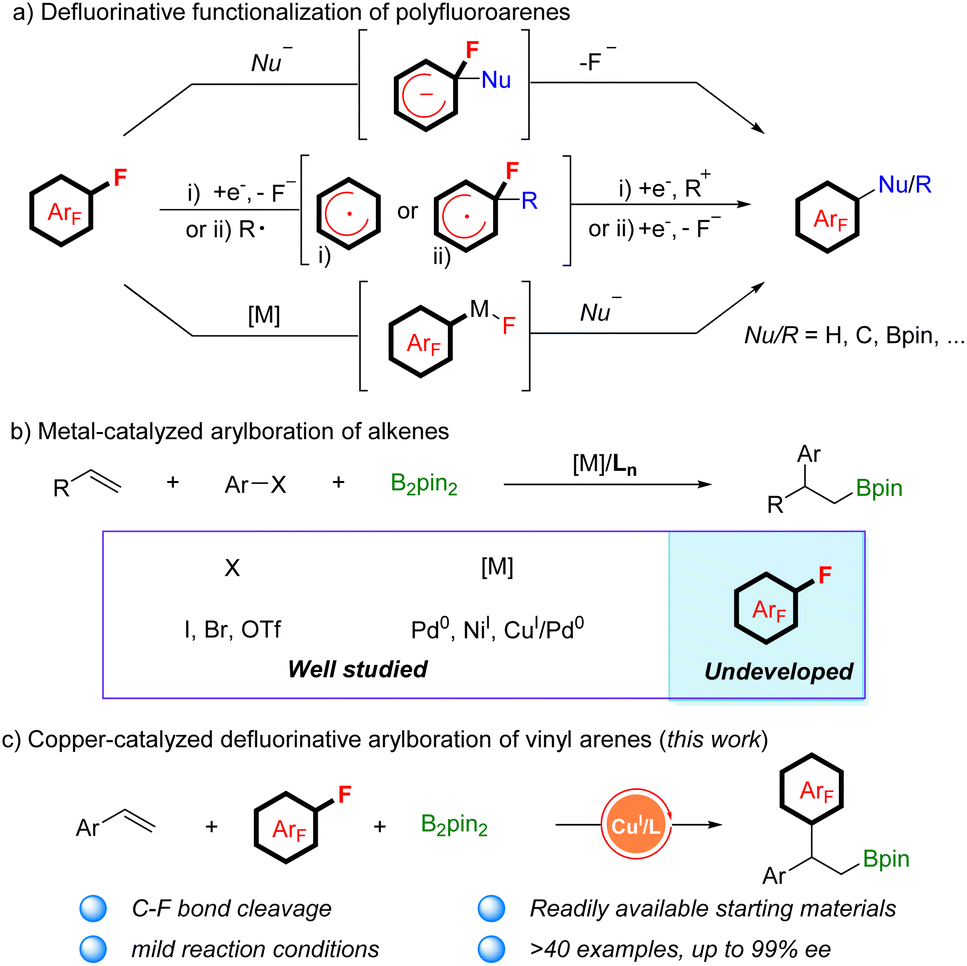
Polyfluorinated aromatics are highly prized molecules in pharmaceutical chemistry1 and materials science2 due to their special properties such as metabolic stability and intermolecular π–πF-interactions (Scheme 1).3 Two new polyfluorinated aromatic-containing drugs were approved by the FDA in 2021.4 During the past decades, the dominant fluorination approaches were the introduction of a single fluorine atom into an aromatic ring via C–H activation5 or C–X substitution.6 While these methods are more applicable for the preparation of mono-fluorinated arenes, they are unsuitable for polyfluoroarenes due to the requirement of the preinstallation of multiple functional groups or unique directing groups. Actually, polyfluorinated aromatics were produced by the substitution of a fluorine atom (defluorinative functionalization) of easily available simple polyfluoroarenes, including nucleophilic aromatic substitution (SNAr), reaction via a radical polyfluoroaryl intermediate, and metal-catalyzed C–F bond functionalization (Scheme 2a).7 For example, a simple and robust hydrodefluorination of polyfluoroarenes (HDF) has been well established.8 In addition, defluorinative borylation of polyfluoroarenes has also been realized in recent years.9
 | ||
| Scheme 1 Selected examples of important poylfluoroaryl compounds. | ||
 | ||
| Scheme 2 Defluorinative functionalization of polyfluoroarenes. | ||
Among the synthetic transformations, carbon–carbon cross-coupling of simple polyfluoroarenes is one of the most widespread applications of defluorinative functionalization, offering a general approach for synthesizing more complex polyfluoroaryl-containing products. However, the reactions are usually limited to carbon nucleophiles and using stoichiometric amounts of organometallic reagents such as alkyl- or arylmetallic reagents (metal: lithium, magnesium, and zinc), which is a great challenge for complex molecules because of the poor compatibility with diverse functional groups.10 To avoid the use of stoichiometric organometallic regents, Ritter's group reported a photocatalytic decarbonylative polyfluoroarylation of aliphatic carboxylic acid via radical addition to polyfluoroarenes and then elimination of a fluoride.11 Additionally, several examples of transition-metal-catalyzed carbon–carbon cross-coupling of polyfluoroarenes have also been disclosed in the absence of organometallic reagents.12 Radius and co-workers13 described a nickel-catalyzed C–F bond arylation of polyfluoroarenes with aryl boronic acid as the nucleophilic reagent in 2006. Subsequently, such reactions were extended to palladium based catalytic systems.14 Recently, Xiong and co-workers reported a defluorinative hydroarylation of alkenes with polyfluoroarenes by in situ catalyst formation.15
Additionally, the metal-catalyzed arylboration of alkenes has recently emerged as a general method to access diverse alkyl boronic esters with good regioselectivity.16 In 2014, Semba and Nakao17 reported their results on arylboration of alkenes by cooperative Pd/Cu catalysis. The reaction was generally initiated by alkene migratory insertion into a Cu–Bpin species, which leads to a β-boryl alkylcopper(I) intermediate. The intermediate reacts with an aryl electrophile under the assistance of palladium catalysis to give 1,2-arylboration products. Subsequently, great advances have been achieved in alkene carboboration with Pd0 or NiI catalysis by research groups of Brown,18 Engle,19 Yin,20 and Liao.21 However, most of the aryl electrophile leaving groups are restricted to I, Br, or OTf (Scheme 2b). The arylboration of aryl fluorides with alkenes has not been realized because of the strength of the C–F bond. Inspired by their creative achievements and our continual interest in borylation of alkenes,22 we attempted to develop a new catalyst system for the simultaneous borylation and defluorinative C–C cross coupling. We speculated that the β-boryl alkylcopper(I) complex, which is generated in situ, might attack the C–F bond of polyfluoroarenes through the SNAr mechanism, bypassing the metal oxidative addition step. In addition, stereospecific transformation of the chiral β-boryl alkylcopper(I) complex could be realized in the presence of a chiral ligand.23 Herein, we report a copper-catalyzed defluorinative arylboration of vinylarene with polyfluoroarenes to access β-polyfluoroaryl boronates with excellent reactivity and regioselectivity. With slight modifications of the reaction conditions, an enantioselective defluorinative arylboration of alkenes was developed as well, producing a set of chiral β-polyfluoroaryl boronates with unprecedented levels of enantioselectivity (Scheme 2c).
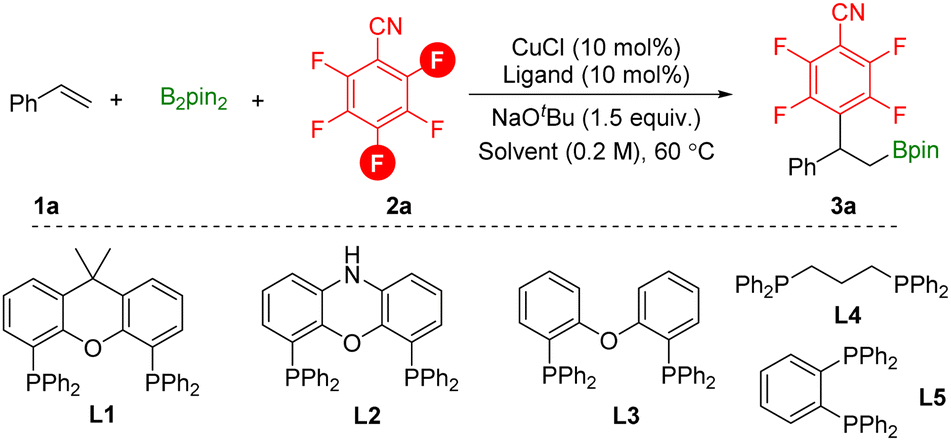
To study this defluorinative arylboration transformation, styrene 1a, pentafluorobenzonitrile 2a, and B2pin2 were selected as the model substrates. As shown in Table 1, by using low valent CuCl as the catalyst, xantphos L1 as the ligand, and NaOtBu as the base at 60 °C for 16 h, para-defluorinative β-polyfluoroaryl boronates 3a were successfully obtained in 35% yield and a small amount of ortho-defluorinative product was detected (Table 1, entry 1). In the testing of solvents, non-polar solvent n-heptane decreased the reaction efficiency and regioselectivity (Table 1, entry 2). When the reaction was performed in coordinative solvents such as 1,4-dioxane or THF, product 3a was obtained in moderate yield with excellent regioselectivity. Then bidentate phosphine ligands were examined, and smaller bite angle DPEphos L3 delivered the desired product 3a in 64% yield, while side product β-boryl styrene was detected (Table 1, entry 7). Increased selectivity was observed by decreasing the temperature (Table 1, entry 10). Finally, the yield can be further improved to 90% by increasing the amount of base applied (Table 1, entry 11).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Yield (%) | Ratio (p![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :o) :o) |
| a Reaction conditions: styrene (0.2 mmol), pentafluorobenzonitrile (1.5 equiv.), B2pin2 (1.5 equiv.), CuCl (10 mol%), ligand (10 mol%), NaOtBu (1.5 equiv.), solvent (0.2 M), 60 °C, 16 h. b Stirred at room temperature (23 °C). c NOtBu (2.0 equiv.). d Isolated yield. | ||||
| 1 | L1 | Toluene | 35 | 80/20 |
| 2 | L1 | n-Heptane | 16 | 75/25 |
| 3 | L1 | 1,4-Dioxane | 45 | 86/14 |
| 4 | L1 | THF | 57 | 93/7 |
| 5 | L1 | DMAc | 0 | n.d. |
| 6 | L2 | THF | 33 | 94/6 |
| 7 | L3 | THF | 64 | 93/7 |
| 8 | L4 | THF | 25 | 94/6 |
| 9 | L5 | THF | 0 | n.d. |
| 10b | L3 | THF | 65 | 95/5 |
| 11c | L3 | THF | 90 (75)d | 95/5 |
With optimized reaction conditions in hand, we investigated various vinylarenes for this transformation. As shown in Scheme 3, styrenes bearing electron-donating groups can be utilized successfully and deliver the desired products in moderate to good yields with excellent regioselectivity (3b–3g). meta-, ortho-Substituted or disubstituted styrenes underwent this transformation smoothly to give the target products (3i–3n). Electron-withdrawing groups such as F, acetyl, and highly lipophilic OCF3 groups (3o–3q) on the styrenes were suitable as well. Furthermore, functional groups including borates, indole, pyrrole, morpholine, methylthio, amino, and furan groups (3r–3y) are well compatible, offering the corresponding products in moderate to good yields with excellent regioselectivity. In addition, the substrate containing terminal alkene (3z) was tolerated and selectively transformed. It is noteworthy that when 1,2-dihydronaphthalene was used, the corresponding anti-addition product (3aa) can be produced with good regioselectivity and diastereoselectivity which could benefit from the benzo fused system. ortho-Defluorinative products were obtained when styrenes contain sterically bulky groups or strong electron-withdrawing groups (3bb–3dd). More complex vinylarenes were successfully transformed under our defluorinative arylboration conditions, delivering the corresponding products in moderate to good yields. However, no reaction occurred with aliphatic alkenes (both internal and terminal).
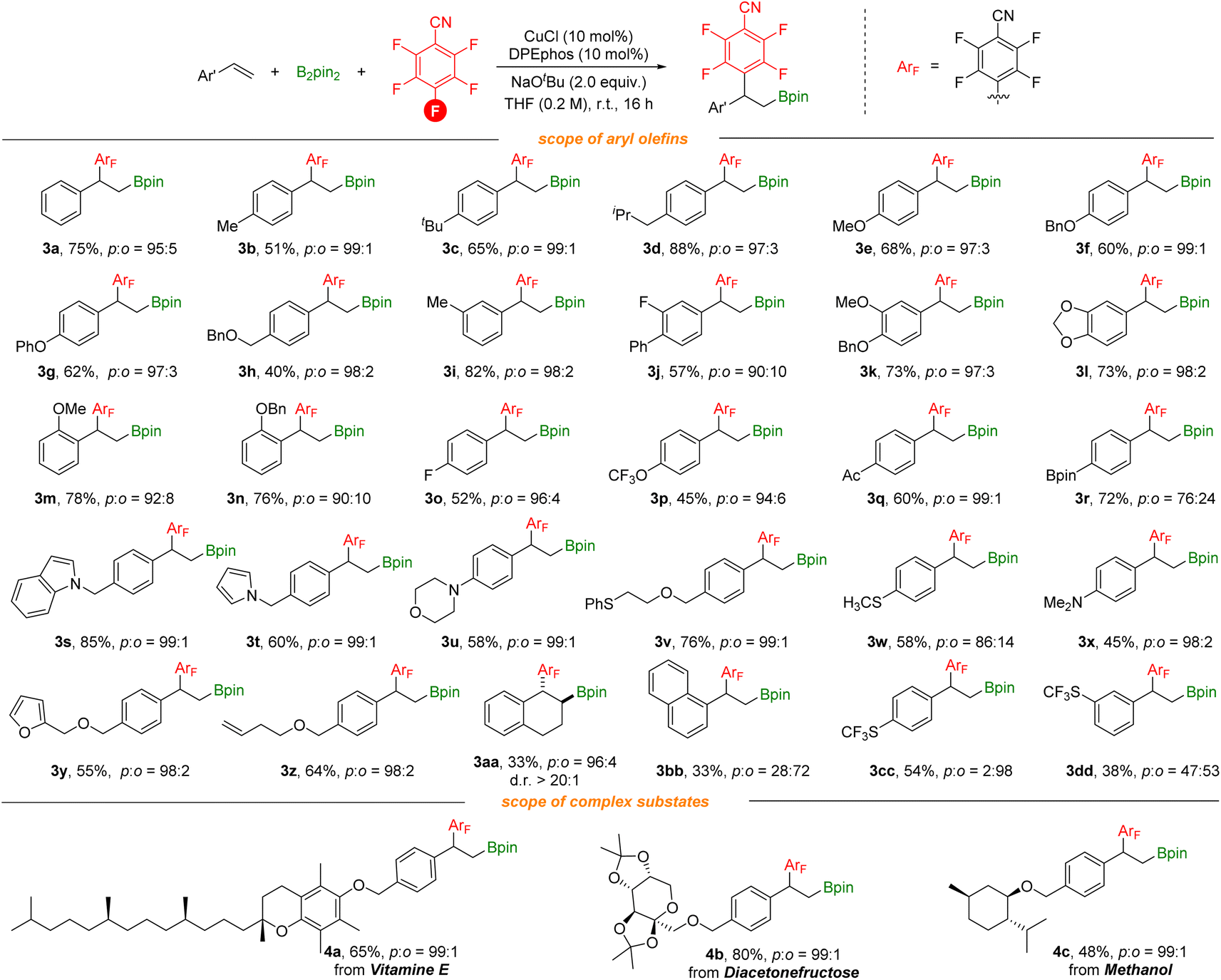 | ||
| Scheme 3 Reaction conditions: styrene (0.2 mmol), polyfluoroarenes (1.5 equiv.), B2pin2 (1.5 equiv.), CuCl (10 mol%), DPEphos (10 mol%), NaOtBu (2.0 equiv.), THF (0.2 M), stirred at room temperature (23 °C) for 16 h, isolated yields. Site selectivity and ratio were determined by 1H NMR, 19F NMR and GC analysis. | ||
To further explore the substrate scope of this defluorinative arylboration reaction, we further evaluated a series of polyfluoroarenes (Scheme 4). Tetrafluoro-substituted benzonitriles were competent coupling partners, providing the arylboration product 5a in moderate yield with poor regioselectivity and 5b with excellent regioselectivity. 3,4,5-Trifluorobenzonitrile was also assessed, and meta-defluorinative arylboration product 5c was obtained in moderate yield. Moreover, p-fluorobenzonitrile was tested as well, and a 15% yield of the corresponding product was detected by 1H NMR. CF3-substituted polyfluoroarenes and polyfluoropyridine were tested as well and corresponding products (5e and 5f) were obtained in moderate to good yields. However, this transformation is unsuitable with electron-rich polyfluoroarenes. The results obtained here are due to the joint effects from electronic and steric influences. Additionally, besides as the activating group, the nitrile group could also coordinate with the copper catalyst to facilitate C–F bond activation.
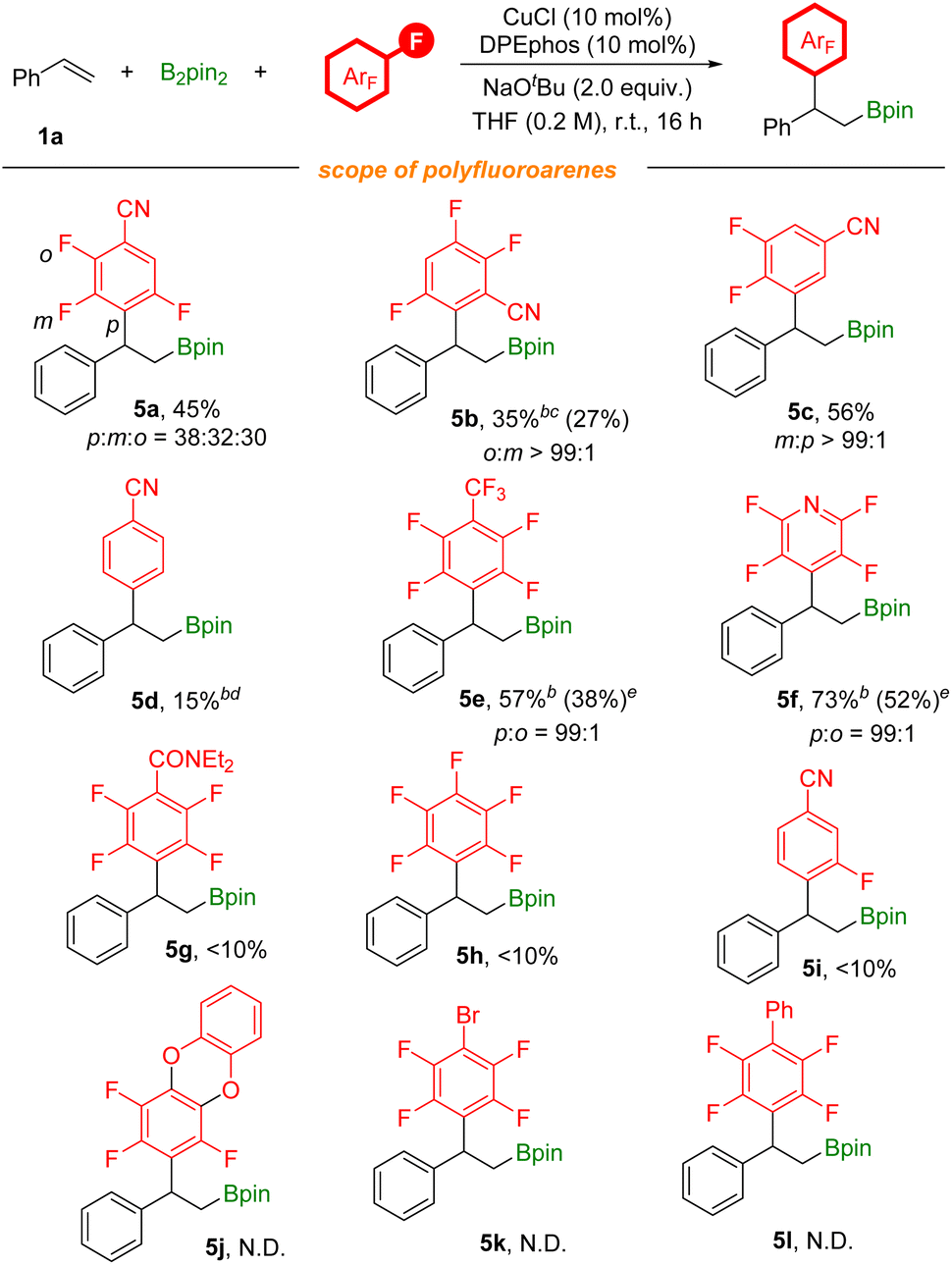 | ||
| Scheme 4 [a] Reaction conditions: styrene (0.2 mmol), polyfluoroarenes (1.5 equiv.), B2pin2 (1.5 equiv.), CuCl (10 mol%), DPEphos (10 mol%), NaOtBu (2.0 equiv.), THF (0.2 M), stirred at room temperature (23 °C) for 16 h, isolated yields. Site selectivity and ratio were determined by 1H NMR, 19F NMR and GC analysis. [b] NMR yields using 1,3,5-trimethoxybenzene as the internal standard. [c] Xantphos (10 mol%), n-heptane (0.2 M), stirred at 60 °C. [d] Xantphos (10 mol%). [e] 5e and 5f were oxidized to the corresponding alcohols before isolation. | ||
In order to further demonstrate the synthetic value of these defluorinative arylboration reactions, synthetic transformation of product 3a was carried out (Scheme 5). A gram-scale reaction was performed, and 3a was obtained in 60% yield. The C–B bond can be easily converted into a hydroxyl group by NaBO3 oxidation, providing the corresponding β-hydroxy polyfluoroarene (6a). Furthermore, high-value potassium borate salt (6b) was obtained in a simple step with KHF2. Subsequently, functional groups including bromo and vinyl were produced through bromination (6c) and vinylation (6d). Additionally, the product 3a and also the potassium borate salt (6b) are suitable for palladium-catalyzed C–C bond formation reactions based on reported procedures.24
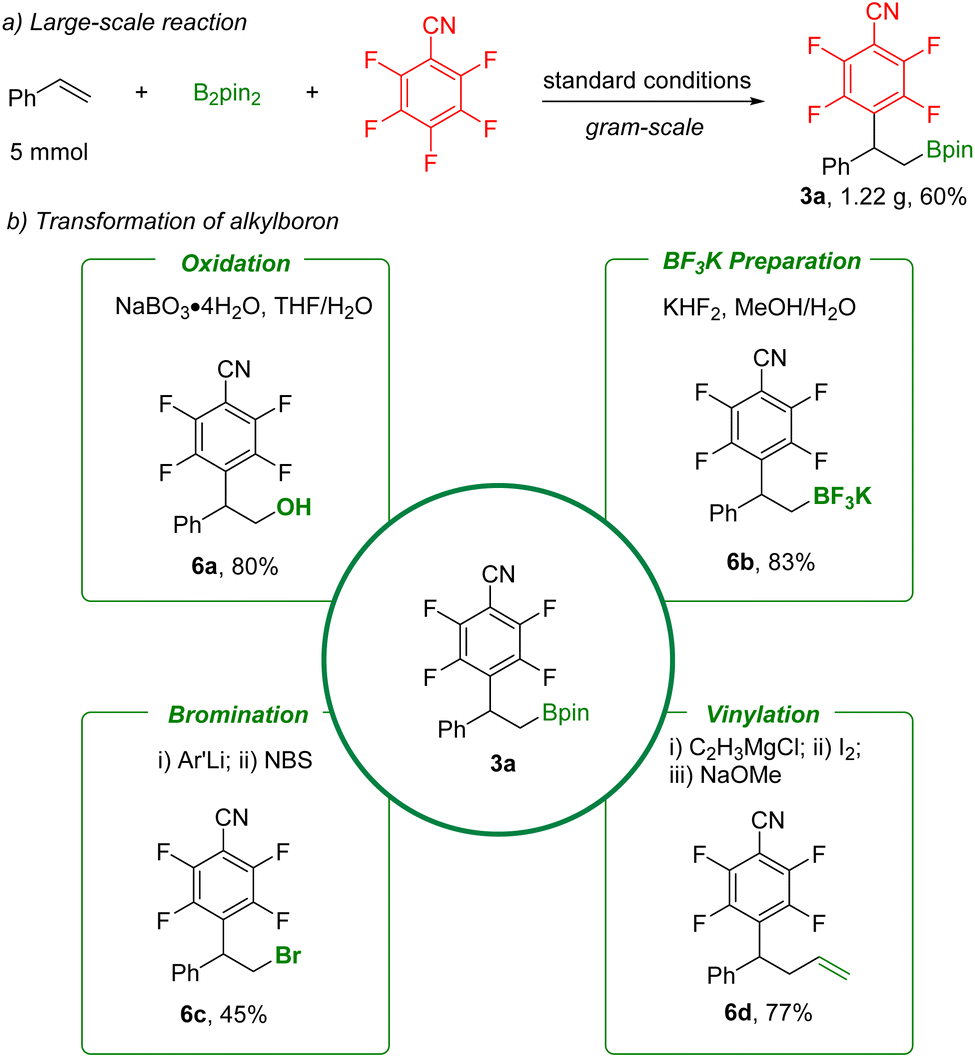 | ||
| Scheme 5 Large-scale reaction and transformations of organoboranes. | ||
We subsequently set out to develop an enantioselective variant of the copper-catalyzed defluorinative arylboration of vinylarenes (Scheme 6). Styrene 1a and pentafluorobenzonitrile 2a were selected as model coupling partners. Copper complexes supported by chiral bidentate phosphine ligands were evaluated. In the presence of the chiral (S,S)-Ph-BPE L6*, the chiral 3a′ was afforded in 48% yield with 73% ee. Low conversions and enantioselectivity were measured with other commercially available chiral phosphine ligands L7*–L11*. Although, (R,Sp)-Josiphos ligand L12* effectively improves the yield of 3a′, enantioselectivity was very poor. Thus, with the L6* as the best ligand, we further evaluated other parameters. The temperature has little effect on enantioselectivity, but an increase in the chemical yield was achieved. To our surprise, the transformation was processed with LiOtBu as the base and delivered the 3a′ in moderate yield with excellent enantioselectivity. However, other bases, mixed bases, and solvents all failed to improve the yields since the formation of the side hydroboration products and vinyl boronate products could not be avoided (see ESI†).
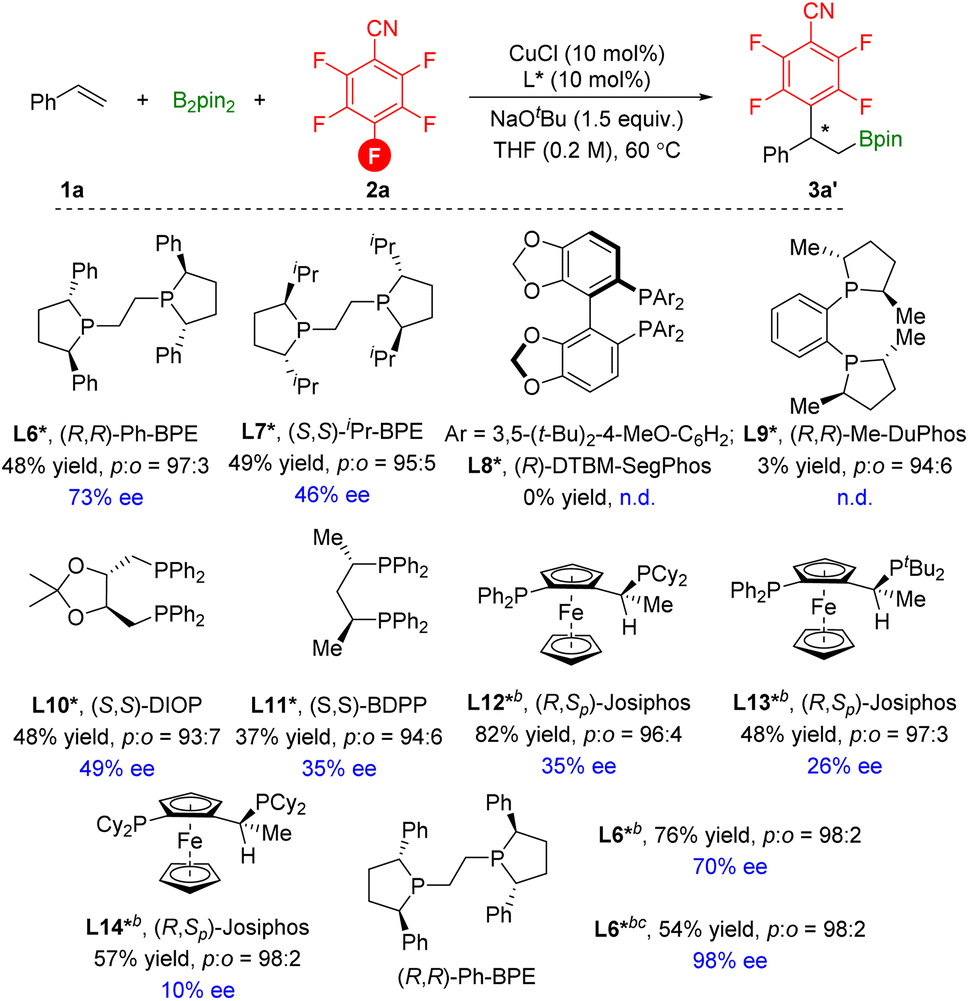 | ||
| Scheme 6 [a] Reaction conditions: styrene (0.2 mmol), pentafluorobenzonitrile (1.5 equiv.), B2pin2 (1.5 equiv.), CuCl (10 mol%), ligand (10 mol%), NaOtBu (1.5 equiv.), THF (0.2 M), 60 °C, 16 h. [b] stirred at room temperature (23 °C). [c] LiOtBu (2.0 equiv.) instead of NaOtBu. | ||
After obtaining the optimized asymmetric reaction conditions, we investigated the substrate scope of the reaction (Scheme 7). Overall, various styrene derivatives worked well under the catalytic system, leading to the corresponding chiral products in moderate yields with excellent enantioselectivities. Functional groups including boron, sulfur, and terminal alkene were all compatible to deliver the desired products with excellent enantioselectivities. The absolute configuration of 3c′ was clearly confirmed by X-ray crystallography.
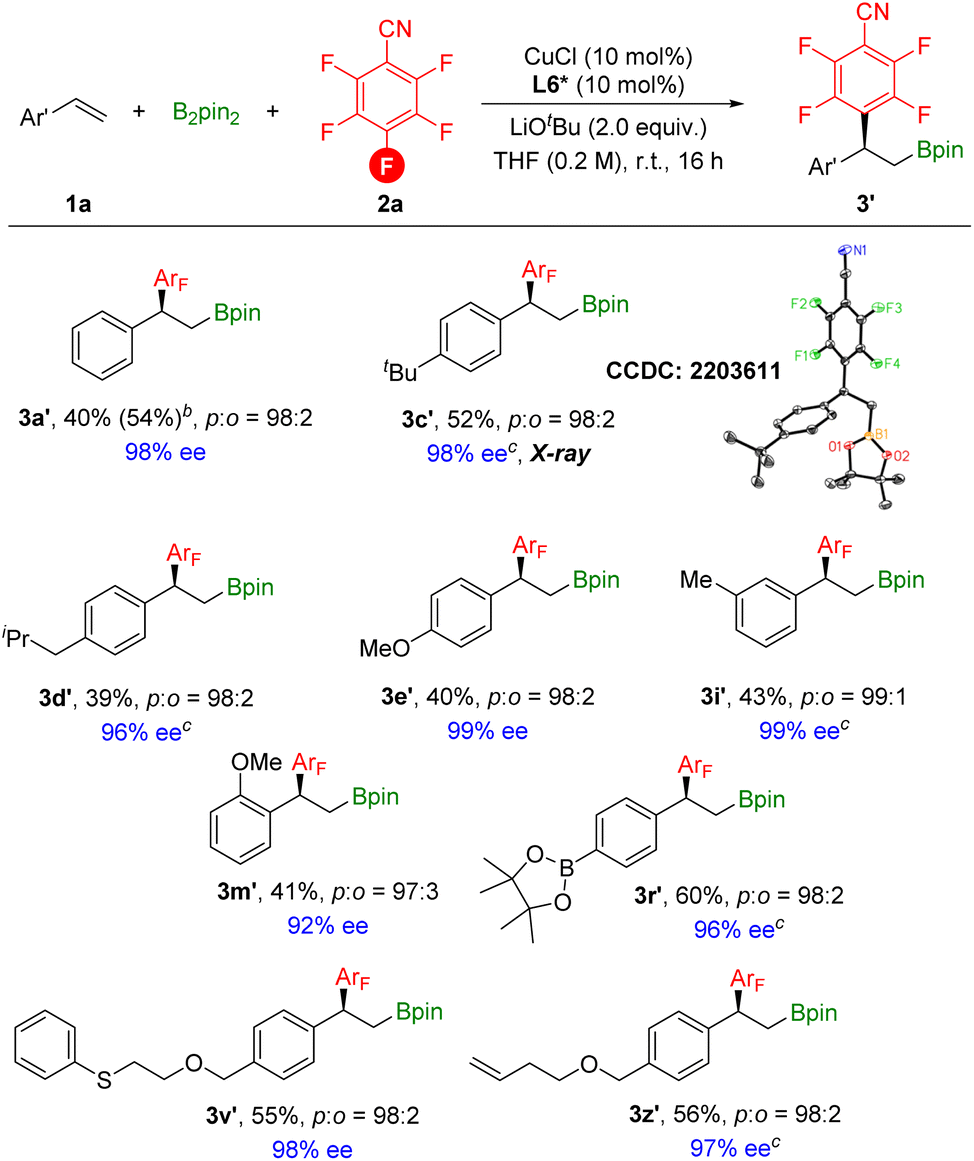 | ||
| Scheme 7 [a] Reaction conditions: styrene (0.2 mmol), 2,3,4,5,6-pentafluorobenzonitrile (1.5 equiv.), B2pin2 (1.5 equiv.), CuCl (10 mol%), L6* (10 mol%), LiOtBu (2.0 equiv.), THF (0.2 M), stirred at room temperature (23 °C) for 16 h, isolated yields. Site selectivity and ratio were determined by 1H NMR, 19F NMR and GC analysis. Enantiomeric excess (ee) was determined by chiral-phase HPLC; displacement ellipsoid plot of 3c′ (30% probability level, without H and the second orientation of the disordered tBu group). [b] GC yield. [c] Ee was determined based on the corresponding alcohol. | ||
In summary, starting from readily available vinylarenes and polyfluorenes, a copper-catalyzed defluorinative arylboration has been described in this study. The transformation provides a direct approach for the synthesis of β-polyfluoroaryl boronates and displays a broad functional group tolerance. Synthetic transformations of the β-polyfluoroaryl boronates demonstrate their utility. Notably, by using (S,S)-Ph-BPE as the ligand, an enantioselective defluorinative arylboration was also achieved.
Author contributions
XFW directed this project and revised the manuscript. FPW, XWG and HQG performed all the experiments and prepared the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the China Scholarship Council for a PhD Scholarship. We appreciate the analytical department of the Leibniz-Institute for Catalysis at the University of Rostock for their assistance. The authors also thank Dr Anke Spannenberg for the X-ray crystal analysis.References
- (a) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed; (b) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- S. Bacchi, M. Benaglia, F. Cozzi, F. Demartin, G. Filippini and A. Gavezzotti, Chem.–Eur. J., 2006, 12, 3538–3546 CrossRef CAS PubMed.
- J. He, Z. Li, G. Dhawan, W. Zhang, A. E. Sorochinsky, G. Butler, V. A. Soloshonok and J. Han, Chin. Chem. Lett., 2023, 34, 107578 CrossRef CAS.
- (a) R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824–14848 CrossRef CAS PubMed; (b) K. Yamamoto, J. Li, J. A. O. Garber, J. D. Rolfes, G. B. Boursalian, J. C. Borghs, C. Genicot, J. Jacq, M. van Gastel, F. Neese and T. Ritter, Nature, 2018, 554, 511–514 CrossRef CAS PubMed; (c) P. S. Fier and J. F. Hartwig, Science, 2013, 342, 956–960 CrossRef CAS PubMed.
- P. A. Champagne, J. Desroches, J. D. Hamel, M. Vandamme and J. F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef CAS PubMed.
- (a) J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2017, 56, 7266–7270 CrossRef CAS PubMed; (b) A. Arora and J. D. Weaver, Acc. Chem. Res., 2016, 49, 2273–2283 CrossRef CAS PubMed; (c) S. Senaweera and J. D. Weaver, J. Am. Chem. Soc., 2016, 138, 2520–2523 CrossRef CAS PubMed; (d) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed; (e) A. Singh, J. J. Kubik and J. D. Weaver, Chem. Sci., 2015, 6, 7206–7212 RSC; (f) V. D. Shteingarts, J. Fluorine Chem., 2007, 128, 797–805 CrossRef CAS.
- (a) M. K. Cybulski, J. E. Nicholls, J. P. Lowe, M. F. Mahon and M. K. Whittlesey, Organometallics, 2017, 36, 2308–2316 CrossRef CAS; (b) K. Kikushima, M. Grellier, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2017, 56, 16191–16196 CrossRef CAS PubMed; (c) J. Lu, N. S. Khetrapal, J. A. Johnson, X. C. Zeng and J. Zhang, J. Am. Chem. Soc., 2016, 138, 15805–15808 CrossRef CAS PubMed; (d) S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS PubMed; (e) H. Lv, Y. B. Cai and J. L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 3203–3207 CrossRef CAS PubMed.
- (a) Y. M. Tian, X. N. Guo, M. W. Kuntze-Fechner, I. Krummenacher, H. Braunschweig, U. Radius, A. Steffen and T. B. Marder, J. Am. Chem. Soc., 2018, 140, 17612–17623 CrossRef CAS PubMed; (b) J. Zhou, M. W. Kuntze-Fechner, R. Bertermann, U. S. Paul, J. H. Berthel, A. Friedrich, Z. Du, T. B. Marder and U. Radius, J. Am. Chem. Soc., 2016, 138, 5250–5253 CrossRef CAS PubMed; (c) S. I. Kalläne, M. Teltewskoi, T. Braun and B. Braun, Organometallics, 2015, 34, 1156–1169 CrossRef; (d) T. Niwa, H. Ochiai, Y. Watanabe and T. Hosoya, J. Am. Chem. Soc., 2015, 137, 14313–14318 CrossRef CAS PubMed; (e) W. H. Guo, Q. Q. Min, J. W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 9075–9078 CrossRef CAS PubMed; (f) M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew. Chem., Int. Ed., 2010, 49, 3947–3951 CrossRef CAS PubMed.
- (a) X. Yi, R. Mao, L. Lavrencic and X. Hu, Angew. Chem., Int. Ed., 2021, 60, 23557–23563 CrossRef CAS PubMed; (b) D. Yu, C. S. Wang, C. Yao, Q. Shen and L. Lu, Org. Lett., 2014, 16, 5544–5547 CrossRef CAS PubMed; (c) F. Zhu and Z. X. Wang, J. Org. Chem., 2014, 79, 4285–4292 CrossRef CAS PubMed.
- X. Sun and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 10557–10562 CrossRef CAS PubMed.
- (a) H. Saijo, H. Sakaguchi, M. Ohashi and S. Ogoshi, Organometallics, 2014, 33, 3669–3672 CrossRef CAS; (b) D. Yu, L. Lu and Q. Shen, Org. Lett., 2013, 15, 940–943 CrossRef CAS PubMed.
- (a) T. Schaub, M. Backes and U. Radius, J. Am. Chem. Soc., 2006, 128, 15964–15965 CrossRef CAS PubMed; (b) J. Zhou, J. H. Berthel, M. W. Kuntze-Fechner, A. Friedrich, T. B. Marder and U. Radius, J. Org. Chem., 2016, 81, 5789–5794 CrossRef CAS PubMed.
- M. Ohashi, H. Saijo, M. Shibata and S. Ogoshi, Eur. J. Org. Chem., 2013, 2013, 443–447 CrossRef CAS.
- X. Li, B. Fu, Q. Zhang, X. Yuan, Q. Zhang, T. Xiong and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23056–23060 CrossRef CAS PubMed.
- A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10, 11578–11622 CrossRef CAS.
- K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 7567–7570 CrossRef CAS PubMed.
- (a) G. L. Trammel, R. Kuniyil, P. F. Crook, P. Liu and M. K. Brown, J. Am. Chem. Soc., 2021, 143, 16502–16511 CrossRef CAS PubMed; (b) A. L. Lambright, Y. Liu, I. A. Joyner, K. M. Logan and M. K. Brown, Org. Lett., 2021, 23, 612–616 CrossRef CAS PubMed; (c) L. A. Chen, A. R. Lear, P. Gao and M. K. Brown, Angew. Chem., Int. Ed., 2019, 58, 10956–10960 CrossRef CAS PubMed; (d) K. M. Logan, S. R. Sardini, S. D. White and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 159–162 CrossRef CAS PubMed; (e) S. R. Sardini and M. K. Brown, J. Am. Chem. Soc., 2017, 139, 9823–9826 CrossRef CAS PubMed.
- Z. Liu, J. Chen, H. X. Lu, X. Li, Y. Gao, J. R. Coombs, M. J. Goldfogel and K. M. Engle, Angew. Chem., Int. Ed., 2019, 58, 17068–17073 CrossRef CAS PubMed.
- (a) W. Wang, C. Ding, Y. Li, Z. Li, Y. Li, L. Peng and G. Yin, Angew. Chem., Int. Ed., 2019, 58, 4612–4616 CrossRef CAS PubMed; (b) W. Wang, C. Ding and G. Yin, Nat. Catal., 2020, 3, 951–958 CrossRef CAS; (c) H. Pang, D. Wu, H. Cong and G. Yin, ACS Catal., 2019, 9, 8555–8560 CrossRef CAS.
- Y. Liao, X. Yin, X. Wang, W. Yu, D. Fang, L. Hu, M. Wang and J. Liao, Angew. Chem., Int. Ed., 2020, 59, 1176–1180 CrossRef CAS PubMed.
- (a) F.-P. Wu, Y. Yang, D. P. Fuentes and X.-F. Wu, Chem, 2022, 8, 1982–1992 CrossRef CAS; (b) F. P. Wu and X. F. Wu, Chem. Sci., 2022, 13, 4321–4326 RSC.
- (a) Q. Dherbassy, S. Manna, C. Shi, W. Prasitwatcharakorn, G. E. M. Crisenza, G. J. P. Perry and D. J. Procter, Angew. Chem., Int. Ed., 2021, 60, 14355–14359 CrossRef CAS PubMed; (b) J. Ye, Y. Liao, H. Huang, Y. Liu, D. Fang, M. Wang, L. Hu and J. Liao, Chem. Sci., 2021, 12, 3032–3038 RSC; (c) D. Fiorito, Y. Liu, C. Besnard and C. Mazet, J. Am. Chem. Soc., 2020, 142, 623–632 CrossRef CAS PubMed; (d) S. Manna, Q. Dherbassy, G. J. P. Perry and D. J. Procter, Angew. Chem., Int. Ed., 2020, 59, 4879–4882 CrossRef CAS PubMed; (e) F.-P. Wu, J. Holz, Y. Yuan and X.-F. Wu, CCS Chem., 2020, 2, 2643–2654 Search PubMed; (f) S. Zhang, J. d. Pozo, F. Romiti, Y. Mu, S. Torker and A. H. Hoveyda, Science, 2019, 364, 45–51 CrossRef CAS PubMed; (g) P. H. S. Paioti, J. Del Pozo, M. S. Mikus, J. Lee, M. J. Koh, F. Romiti, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2019, 141, 19917–19934 CrossRef CAS PubMed; (h) T. Itoh, Y. Kanzaki, Y. Shimizu and M. Kanai, Angew. Chem., Int. Ed., 2018, 57, 8265–8269 CrossRef CAS PubMed; (i) A. Whyte, K. I. Burton, J. Zhang and M. Lautens, Angew. Chem., Int. Ed., 2018, 57, 13927–13930 CrossRef CAS PubMed; (j) T. Jia, P. Cao, B. Wang, Y. Lou, X. Yin, M. Wang and J. Liao, J. Am. Chem. Soc., 2015, 137, 13760–13763 CrossRef CAS PubMed.
- (a) S. R. Chemler, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4544–4568 CrossRef CAS; (b) J. El-Maiss, T. M. E. Dine, C.-S. Lu, I. Karamé, A. Kanj, K. Polychronopoulou and J. Shaya, Catalysts, 2020, 10, 296 CrossRef CAS; (c) Y. Baumgartner and O. Baudoin, ACS Catal., 2020, 10, 10508–10515 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2203611. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06472c |
| This journal is © The Royal Society of Chemistry 2023 |