
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of complex tricyclo[3.2.2.0]nonenes from racemic norcaradienes: kinetic resolution via Diels–Alder reaction†
Siyuan
Wang
,
Yuqiao
Zhou
 ,
Wanlong
Xiao
,
Zegong
Li
,
Xiaohua
Liu
* and
Xiaoming
Feng
*
,
Wanlong
Xiao
,
Zegong
Li
,
Xiaohua
Liu
* and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn
First published on 12th January 2023
Abstract
Herein, the enantioselective synthesis of complex tricyclo[3.2.2.0]nonenes through the Diels–Alder reaction is reported. Utilizing racemic norcaradienes prepared from the visible-light-mediated dearomative cyclopropanation of m-xylene as dienes and enone derivatives as dienophiles, the overall process represents a kinetic asymmetric transformation in the presence of a chiral cobalt(II) complex of chiral N,N′-dioxide. High diastereo- and enantioselectivity could be obtained in most cycloaddition processes and part racemization of norcaradiene is observed. The topographic steric maps of the catalysts were collected to rationalize the relationship between reactivity and enantioselectivity with the catalysts.
Introduction
Bridged bicyclo[2,2,2]octane structure represents an important class of polycyclic hydrocarbon scaffolds, and their rigidity allows functional groups encompassing three-dimensional shapes precisely.1 Such structure exists in numerous natural products and drugs, such as kingianin A2 and buprenorphine3 (Scheme 1a). Tecovirimat, the class of ST-246 orthodox antivirals, contains the tricyclo[3.2.2.0]nonene backbone as a variation.4 Additionally, chiral bicyclo[2,2,2]octene-based diene has been discovered as a class of privileged ligands for transition metal-catalyzed asymmetric reactions (Scheme 1a).5 The Diels–Alder reaction of 1,3-cyclohexadiene with dienophile is a straightforward route for synthesizing bicyclo[2.2.2] ring system with a bridgehead olefin,6 but the cyclic diene system exhibits more flexibility and competing aromatization to arene.7 Norcaradienes (NCDs) are useful diene partners bearing a cyclopropane bridge, rendering enhanced rigidity and predictable topographical selectivity8 accessible via arene cyclopropanation.8e,f,9 However, the existence of norcaradiene (NCD)-cycloheptatriene (CHT) equilibrating via ring opening and closure has an uncertain effect on the functionalization process.10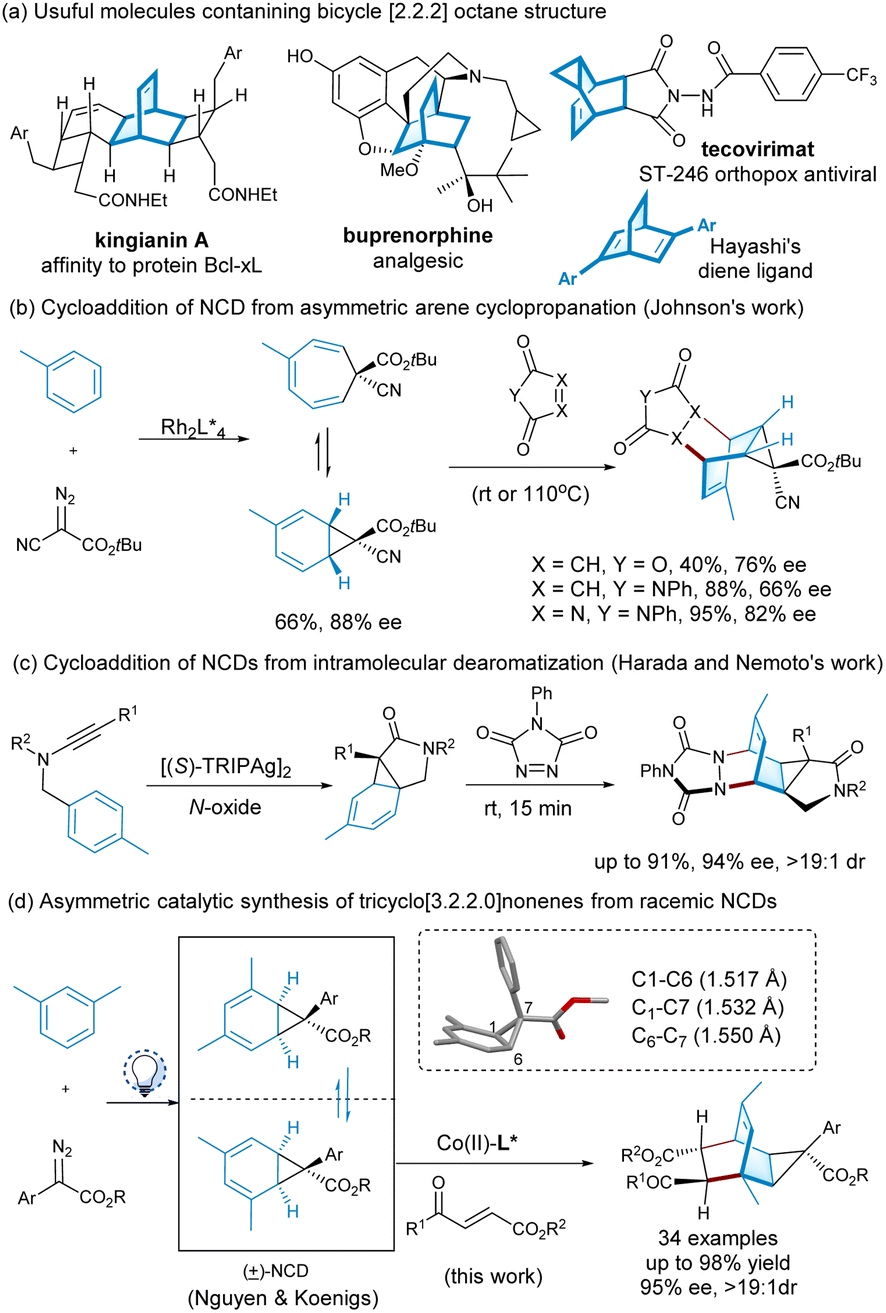 | ||
| Scheme 1 Importance of the bicyclo[2.2.2]octane core and [4 + 2] cycloadditions accessing tricyclic varieties from NCDs. | ||
Chiral transition metal complex-catalyzed arene cyclopropanations enable the in situ generation of enantiomerically enriched NCDs and their valence tautomers,11 which have been employed for rapid conversion to an array of complex bridge tricyclic structures and other cyclohexane building blocks. For instance, the Johnson group took advantage of chiral dirhodium complex-mediated regio- and stereoselective arene cyclopropanation using α-cyanodiazoacetate as a carbene procedure to affordd optically enriched NCDs, serving as tunable templates for the synthesis of complex cyclohexanes (Scheme 1b).12 The azo-Diels–Alder reaction with 1,2,4-triazole-3,5-dione performed well, but the cycloaddition of maleic anhydride or N-phenyl maleimide required high reaction temperature with the slight erosion of enantioselectivity. Very recently, Johnson's group also used enantioenriched cycloheptatriene from anisole and aryl-substituted α-diazoester for diastereoselective [4 + 2] cycloaddition with nitroethylene, enabling the preparation of chiral tricycle[2.2.2]nonene for late-stage functionalization into diene ligands.13 Harada et al. developed a method for the diazo-free generation of silver-carbene species from an ynamide to transiently generate chiral norcaradiene, which can be further decorated to complex molecules upon cycloaddition (Scheme 1c).14
We are interested in developing an asymmetric catalytic Diels–Alder reaction to construct the chiral tricyclo[3.2.2.0]nonenes, similar to the tecovirimat structure, which might be useful for bioactivity evaluation.4 In addition to typical transition-metal-mediated arene cyclopropanation, Nguyen and coworkers investigated the metal-free visible-light photolysis of aryl diazoacetates in aromatic solvents for NCD synthesis (Scheme 1d).8e We envision an alternative pathway to achieve complex tricyclononenes from racemic NCDs via Diels–Alder reaction with dienophile (Scheme 1d). As chiral NCD could locate the facial approach of the dienophile,12–15 three issues need to be considered for the process: (1) the discriminate enantiomers of the diene with a chiral catalyst in the cycloaddition; (2) diastereoselectivity controlled by chiral substrate or chiral catalyst; (3) tautomerization and racemization of the NCDs in the presence of Lewis acids. In this context, the choice of chiral Lewis acid catalyst is critical, and our endeavor in developing chiral Lewis acids of N,N′-dioxides16 now allows for the asymmetric catalytic Diels–Alder reaction of racemic norcaradienes with enones. Herein, we describe the enantioselective construction of tricyclo[3.2.2.0]nonenes with seven stereocenters from racemic NCDs based on the visible-light-mediated dearomative cyclopropanation of m-xylene.8e Kinetic resolution process in connection with diastereo- and enantioselective cycloaddition performs well with chiral cobalt(II) complex under mild reaction conditions.
The racemic norcaradiene synthesis followed the method reported by Nguyen and Koenigs via the visible-light-mediated cyclopropanation reaction of arenes.8e Considering the regioselectivity of the cyclopropanation and the optical property of the norcaradienes, the racemic exo-norcaradiene 1a from m-xylene and methyl 2-diazo-2-phenylacetate is prepared for the Diels–Alder reaction (Scheme 1d). In contrast to norcaradiene from cyanodiazoacetate,12,17 the related exo-ester norcaradiene 1a of m-xylene is stable and a favourable tautomer. From the X-ray crystallographic data of norcaradiene 1a,10c,18 it is found that the C1–C6 bond is shorter than the C1–C7 and C6–C7 bonds. This agrees with the molecular orbital explanation of the stabilization of norcaradiene bearing an electron-withdrawing group at the C7-position.19 This implies that the anti-bonding character of the C1–C6 bond is weakened and the electron density is drawn away from the cyclopropane into the vicinal π-system, benefiting the normal electron-demanding Diels–Alder reaction.
Electron-deficient ethyl (E)-4-oxo-4-phenylbutenoate 2a is selected as the dienophile. We investigate the feasibility of the reaction by identifying chiral Lewis acid catalysts using N,N′-dioxide L3-PiPr2 as the ligand (Fig. 1). Initially, the reaction is carried out by mixing an equal amount of norcaradiene 1a and ethyl (E)-4-oxo-4-phenylbutenoate 2a in CH2Cl2 at 30 °C (Table 1). Several metal salts were subjected to the reaction system, but only a minority of them could accelerate the cycloaddition to an extent, and representative results are listed in entry 1–4 in Table 1. Although Yb(OTf)3 could afford comparable reactivity and enantioselectivity, but diastereoselectivity is poor (entry 2). The use of Co(OTf)2 leads to the sole generation of one diastereoisomer 3aa with moderate results (entry 4). The next modification of the ring structure of the N,N′-dioxides yield gradually improved enantioselectivity (entries 4–7), orderly from L3-PiPr2, L3-PrPr2, L3-RaPr2, to octahedron-1H-indole-based L3-PePr2. The five-substituted tricyclo[3.2.2.0]nonene product 3aa could be isolated in a 35% yield, >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 89% ee in the presence of L3-PePr2 and Co(OTf)2 (entry 7). Further exploration of the reaction solvents reveals that 1,1,1,2-tetrachloroethane (TCE) increases enantioselectivity to 92% ee (Table 1, entry 7–9). Polar halohydrocarbons, which are good solvents for the solution of the chiral cobalt(II) complex of chiral N,N′-dioxides, were critical to the yield and enantioselectivity (see the ESI† for details). When the amount of norcaradiene increases into two equivalents, the yield of the product doubles (entry 10), with up to a 92% yield, >19:1 dr and 91% ee are afforded after the reaction time is prolonged (entry 11). The absolute configuration of product 3aa was determined to be (1R, 2R, 3S, 4R, 5S, 6R, 7S) by X-ray crystallography analysis.18
:1 dr and 89% ee in the presence of L3-PePr2 and Co(OTf)2 (entry 7). Further exploration of the reaction solvents reveals that 1,1,1,2-tetrachloroethane (TCE) increases enantioselectivity to 92% ee (Table 1, entry 7–9). Polar halohydrocarbons, which are good solvents for the solution of the chiral cobalt(II) complex of chiral N,N′-dioxides, were critical to the yield and enantioselectivity (see the ESI† for details). When the amount of norcaradiene increases into two equivalents, the yield of the product doubles (entry 10), with up to a 92% yield, >19:1 dr and 91% ee are afforded after the reaction time is prolonged (entry 11). The absolute configuration of product 3aa was determined to be (1R, 2R, 3S, 4R, 5S, 6R, 7S) by X-ray crystallography analysis.18
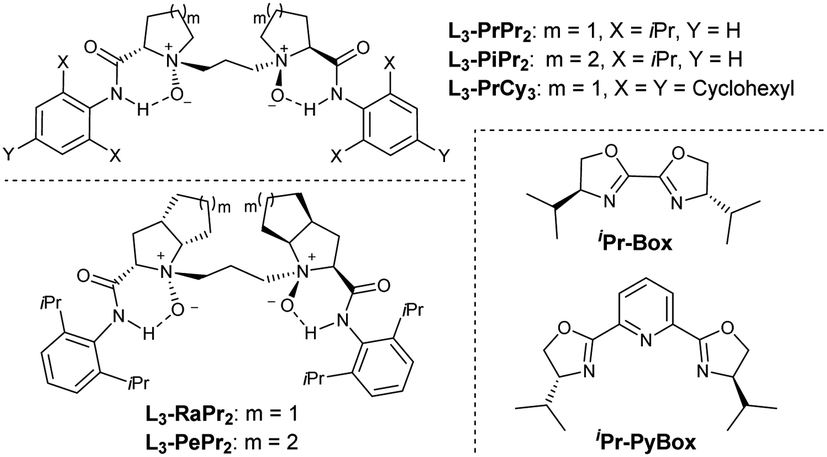 | ||
| Fig. 1 Chiral ligands used for the reaction. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal salt/ligand | Solvent | Yieldb (%) | drc | eed |
| a Unless otherwise noted, all reactions were carried out with metal salt/ligand (1:1, 10 mol%), 1a (1.0 equiv.), 2a (0.10 mmol) in DCM (1.0 mL) at 30 °C for 24 h. b Isolated yield of 3aa. c Determined by 1H NMR. d Determined by HPLC on a chiral stationary phase. e 1a (2.0 equiv.). f 48 h. DCE = 1,2-dichloroethane; TCE = 1,1,1,2-tetrachloethane. | |||||
| 1 | Sc(OTf)3/L3-PiPr2 | DCM | 27 | >19:1 |
27 |
| 2 | Yb(OTf)3/L3-PiPr2 | DCM | 30 | 1:3 |
30 |
| 3 | Zn(OTf)2/L3-PiPr2 | DCM | 38 | >19:1 |
17 |
| 4 | Co(OTf)2/L3-PiPr2 | DCM | 26 | >19:1 |
35 |
| 5 | Co(OTf)2/L3-PrPr2 | DCM | 28 | >19:1 |
65 |
| 6 | Co(OTf)2/L3-RaPr2 | DCM | 34 | >19:1 |
77 |
| 7 | Co(OTf)2/L3-PePr2 | DCM | 35 | >19:1 |
89 |
| 8 | Co(OTf)2/L3-PePr2 | DCE | 8 | >19:1 |
73 |
| 9 | Co(OTf)2/L3-PePr2 | TCE | 32 | >19:1 |
92 |
| 10e | Co(OTf)2/L3-PePr2 | TCE | 67 | >19:1 |
91 |
| 11e,f | Co(OTf)2/L3-PePr2 | TCE | 92 | >19:1 |
91 |
| 12e,f | Co(OTf)2/L3-TQPr2 | TCE | 20 | 3:1 |
0/12 |
| 13e,f | Co(OTf)2/iPrPyBox | TCE | Trace | — | — |
| 14e,f | Co(OTf)2/iPr-Box | TCE | Trace | — | — |
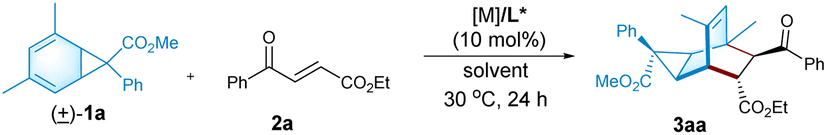
With the optimized set of conditions in hand, we evaluate the generality of this reaction. As illustrated in Table 2, first, various 4-oxo-4-arylbutenoates undergo cycloaddition with norcaradiene 1a to afford the desired products (3aa–3ar) as nearly one diastereomer (>19:1 dr) with good enantioselectivity (81–93% ee). In general, higher yields are observed with electron-withdrawing substitution at the benzoyl group than alkyl-substituted ones (3ak–3an), but para-nitro-bearing 3aj is an exception owing to the decomposition of the related substrate. Notably, the alkynyl group is tolerable in the reaction, and the desired product 3ao is isolated in 97% yield and 87% ee. In addition, the reaction is amenable to 2-naphthoyl-containing dienophile, affording the corresponding product 3as in a 90% yield with 87% ee. The ester group of 4-oxo-4-benzylbutenoates could be changed from ethyl to methyl, n-butyl, isopropyl and benzyl groups; the enantioselectivity could basically be maintained (3at−3aw, 79–92% yields, 88–94% ee).
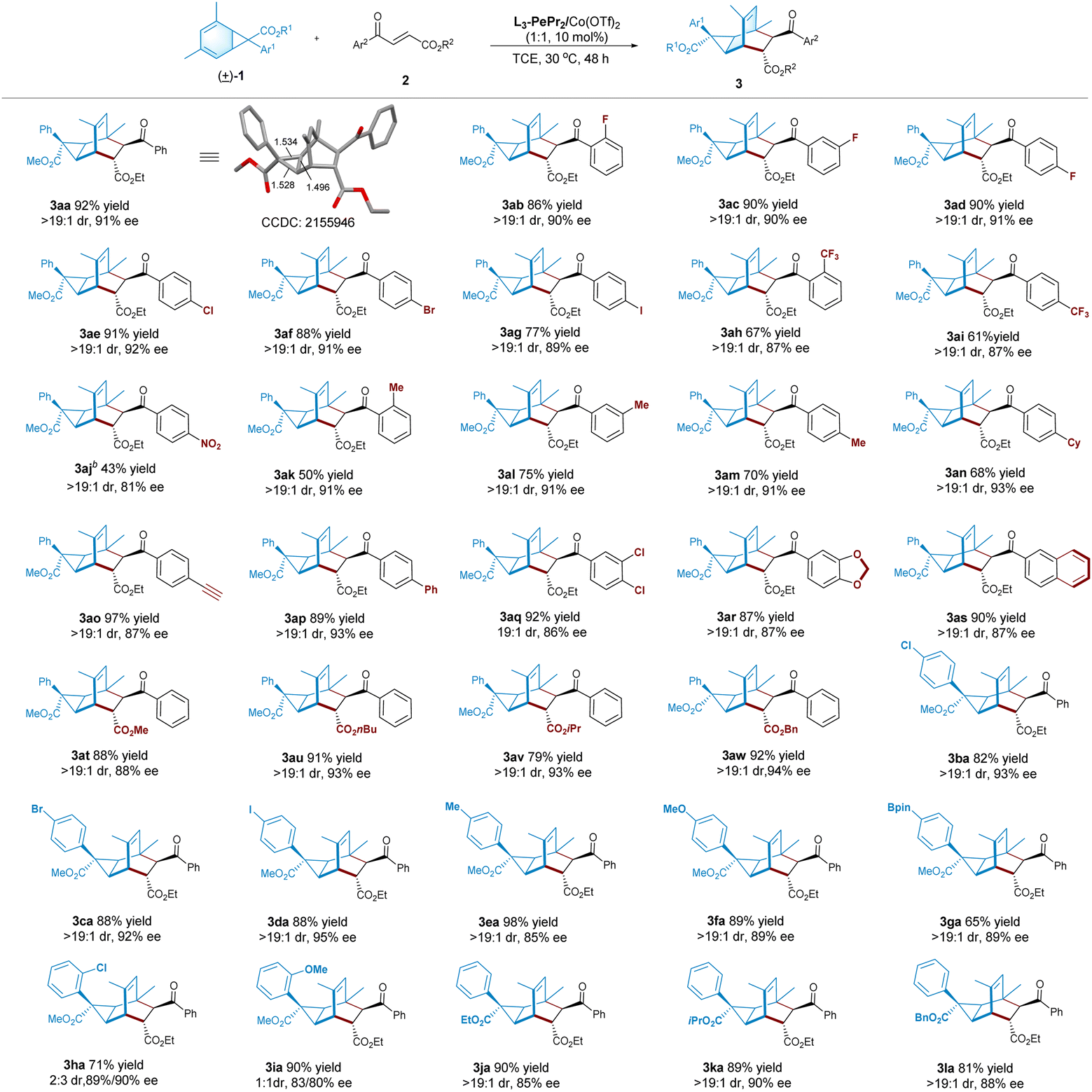
Norcaradienes synthesized from different aryl α-diazoestres are also suitable for this process. The reaction of 7-aryl norcaradienes bearing para-substitution as halo, methyl, methoxyl, or Bpin, under the standard conditions, performs well to provide the related tricyclo[3.2.2.0]nonenes (3ba–3ga) in good to excellent yields (65–98%), with up to 19:1 dr with satisfied enantioselectivity (85–95% ee). The reaction of norcaradienes with a 2-chlorophenyl or a 2-methoxyphenyl at the C7-position is clearly different from the other transformations in terms of diastereoselectivity. The related adducts (3ha and 3ia) are obtained in poor diastereoselectivity but comparable good enantioselectivity for both isomers. This might be due to the decreased split rate of the norcaradiene, as a result of steric hindrance, which led to the two enantioisomers of the norcaradienes having similar reaction rates. Low diastereoselectivity is also observed when 3-halophenyl-substituted norcaradienes are subjected to standard conditions (see the ESI† for details). In addition, the 7-ester group of norcaradienes could be changed from methyl to ethyl, iso-propyl or benzyl group, and all of the corresponding products (3ja–3la) could obtain good results (81–90% yields, >19:1 dr, 85–90% ee).
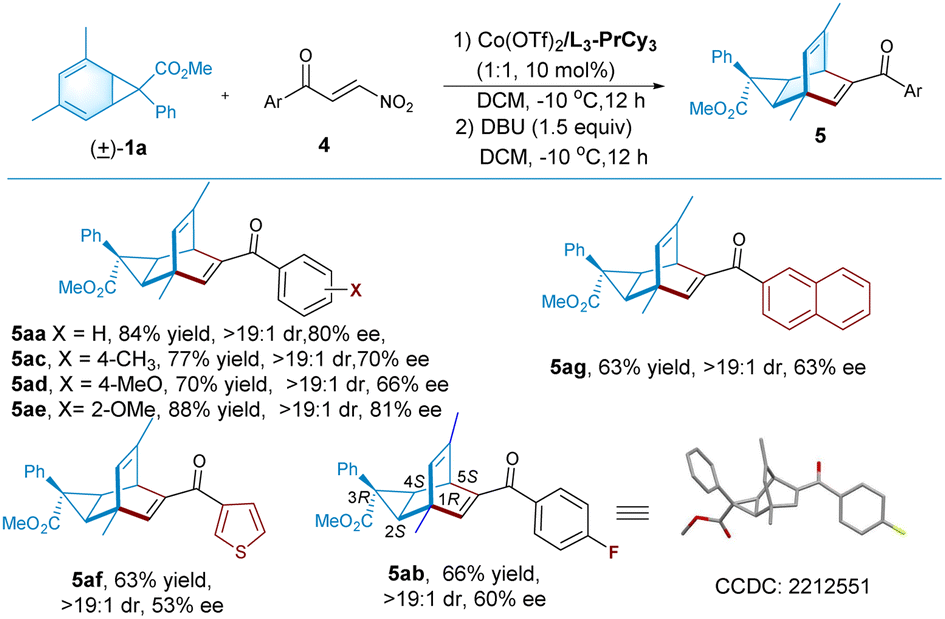
We next ivestigated 3-nitrophenylpropenones as the dienophiles to synthesize bicyclo[2,2,2]octene-based dienes that represent a type of interesting chiral ligands.5 As shown in Table 3, the reaction between racemic norcaradiene 1a and β-nitroenone 4a in the presence of the cobalt(II) complex of N,N′-dioxide L3-PrCy3, following the treatment with DBU, led to the formation of the corresponding diene 5aa in 84% yield, >19:1 dr, and 80% ee. Noteworthily, there is a reversed regioselectivity using β-nitroenone compared with 4-oxo-4-arylbutenoate as a dissymmetrical pull–pull olefin for cyclo-addition20 mainly because of the natural bond polarizations. Other β-nitroenones 4 bearing-substituted benzoyl groups, 2-naphthoyl group, or thiophene-3-carbonyl group, were tolerable to afford the desired dienes (5ab–5ag) in moderate to good yield and enantioselectivity. The absolute configuration of product 5ab was determined to be (1R, 2S, 3R, 4S, 5S) by X-ray crystallography analysis.18 This indicates that there is an antilogous preference for the enantiomer of norcaradiene and the activation of the electron-withdrawing group of dienophile.
|
a Unless otherwise noted, all reactions were carried out with 1a (0.20 mmol), 4 (0.1 mmol), and Co(OTf)2/L3-PrCy3 (1:1, 10 mol%) in DCM (1.0 mL) under nitrogen at −10 °C. After 12 hours, DBU (0.15 mmol) was added, and the mixture continued stirring for another 12 hours. Isolated yield. dr was determined by 1H NMR, and ee was determined by HPLC on a chiral stationary phase.
|
|---|
|
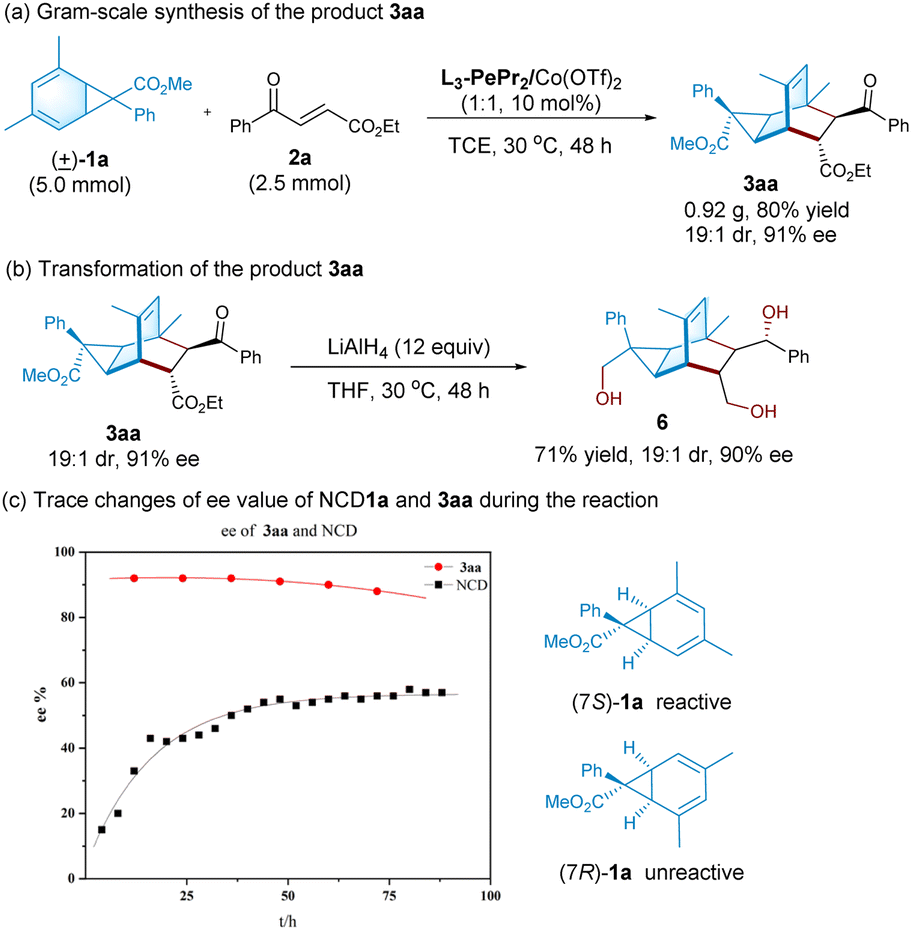
To illustrate the potential synthetic utility of the current catalytic system, a gram-scale synthesis was carried out with racemic norcaradiene 1a (5.0 mmol) and enone 2a (2.5 mmol) under the optimal catalytic system, which afforded the corresponding tricyclo[3.2.2.0]nonene 3aa in 80% yield with 19:1 dr and 91% ee (Scheme 2a). Sequential reduction by LiAlH4 led to the formation of the tri-hydroxyl-substituted derivative 6 in 71% yield, 19:1 dr and 90% ee (Scheme 2b).
 | ||
| Scheme 2 Other experiments. | ||
To probe into the stereocontrol of the process, we detected the enantioselectivity of product 3aa and unreacted norcaradiene 1a at various timescales. As shown in Scheme 2c, high enantioselectivity was detected during the whole process although it slightly dropped after around 40 hours. Simultaneously, the optical activity of norcaradiene gradually increased but did not exceed 60% ee at the end. From the absolute configuration of product 3aa, we can rationalize that (7S)-1a reacts more readily with 2a than (7R)-1a in the presence of the chiral catalyst of Co(II)/L3-PePr2. Given the high diastereoselectivity and enantioselectivity of 3aa but the moderate ee of the recovered NCD, it is proposed that the kinetic resolution and racemization of NCD might occur along with the cycloaddition.
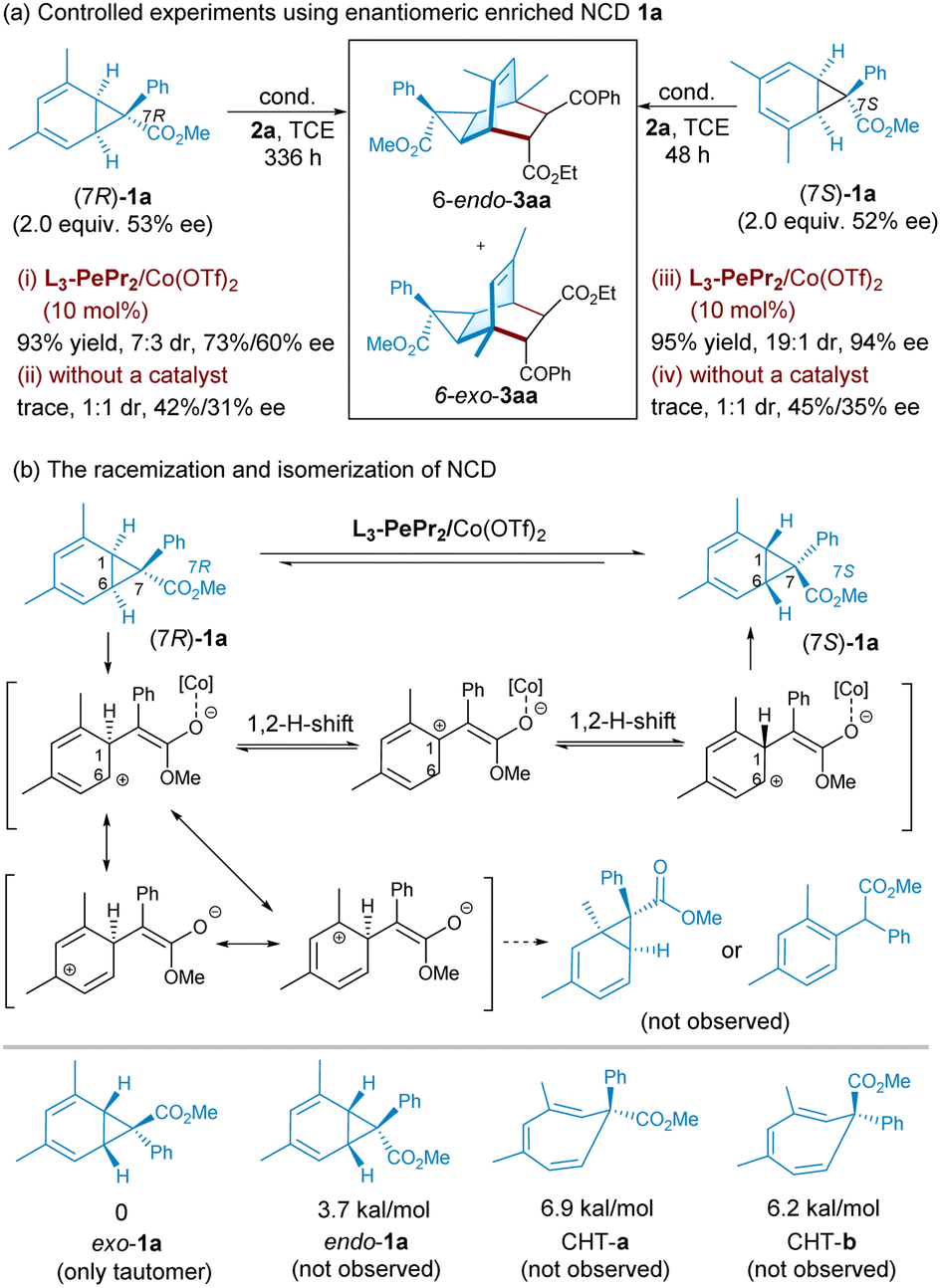
To probe into the stereo–selective profile of the cycloaddition, control experiments were performed using enantioenriched norcaradiene 1a (Scheme 3), which exhibited disparate reactivity and stereoselectivity. When the recovered norcaradiene (rationalized as 7R-configuration) with moderate ee value (53% ee) was put into the chiral catalyst system, the desired product was afforded a good yield after a long reaction time with moderate diastereo- and enantioselectivity (Scheme 3a, condition i). If there is no catalyst, the spontaneous process is weak, and only a trace amount of tricyclononene is detected in 1:1 dr and decreases enantioselectivity (condition ii). However, the reaction with (7S)-enriched norcaradiene 1a performed well in the standard reaction condition, with a 95% yield >19:1 dr and 94% ee (condition iii), confirming that the (7S)-isomer of 1a participates in the cycloaddition in priority. Equally, only trace adduct is detected with a poor dr value and reduced enantioselectivity without a catalyst after a two-day reaction time (condition iv).
 | ||
| Scheme 3 The control experiments and identification of the racemization of NCD. | ||
The erosion of the optical activity of norcaradiene in the presence of a chiral Lewis acid catalyst was observed (Scheme 3b). The stability of exo- and endo-1a, as well as its valence tautomer CHTs, was evaluated by applying the Gaussian 09 D.01 package at the level of M06-2X-D3/def-SVP//M06-2X-D3/def2-TZVP with the SMD solvent model of dichloromethane (see ESI† for more details).21Exo-1a exhibits the lowest Gibbs free energy compared to that of endo-1a (3.7 kcal mol−1), CHT-a (6.9 kcal mol−1) and CHT-b (6.2 kcal mol−1), and this reveals that exo-ester 1a is more stable than the others, which agrees with the experimental results. Next, the possible decreased enantioselectivity was rationalized via C6–C7 heterolysis, which is the longest bond of cyclopropane, assisted by Lewis acid to form dimethyl-cyclohex-2,4-dienylium ions (Scheme 3b). Neither the rearomatization byproduct nor other types of norcaradiene isomers are tracked two days after the reaction. As shown in Scheme 3b, the occurrence of the 1,2H shift of the zwitterionic intermediates between the C1 and C6 positions leads to the loss of stereochemistry at the C1-carbon center, and C6–C7 rebound may yield exo-ester norcaradiene with the opposite configuration.
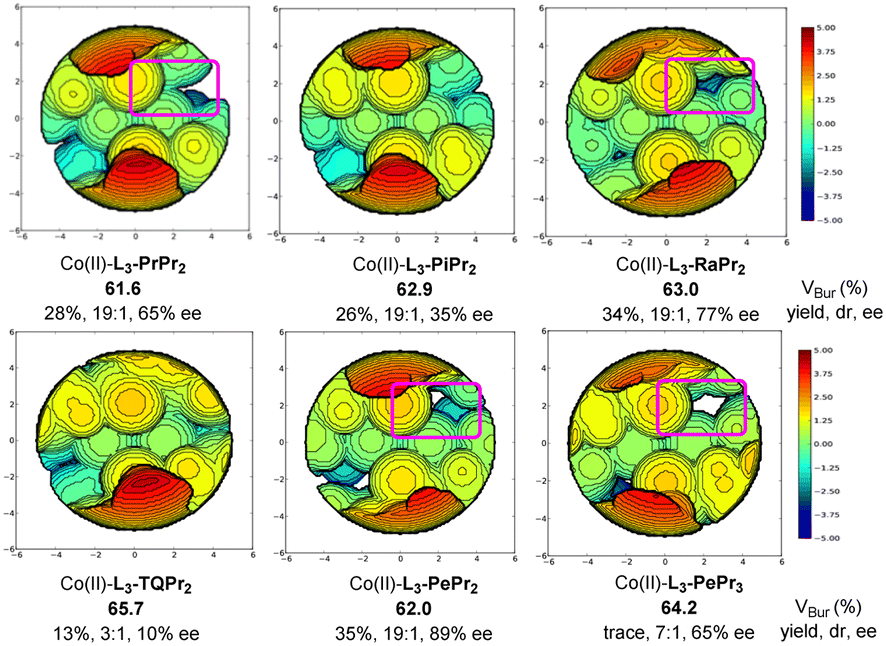
To understand the steric effects on the enantioselectivity of the ligand, we choose Cavallo's SambVca 2 Web tool22,23 to construct topographical steric maps (Scheme 4) based on the general structures of the metal complex of chiral N,N′-dioxide ligands.24 It is found that the percent buried volumes of the catalysts varied dramatically with the ring structure of the amino acid backbone of the ligands, following L3-PrPr2 < L3-PePr2 < L3-PiPr2 ≅ L3-RaPr2 < L3-PePr3 < L3-TQPr2. The significantly increased buried volume of L3-TQPr2 might arise from its distorted trigonal bipyramidal arrangement25 after coordination with cobalt salt, differing from the majority of the structures of the others in an octahedron configuration. In connection with the observed yield and enantioselectivity, it seems that if a crowded space exists around the metal center with a larger buried volume (L3-TQPr2 and L3-PePr3), the reactivity drops. However, if there are obvious grooves inside the pink box (less steric hindrance), the enantioselectivity of the reaction is higher.
 | ||
| Scheme 4 Steric map of cobalt(II) complexes of various ligands. | ||
The four transition states shown in Fig. 2 are rationalized to account for the generation of possible isomers of tricyclononene products. Albeit there are seven stereocenters in the adduct, due to the conformation-limitation of NCD 1a with steric encumbrance by the three-membered bridge, as well as the cycloaddition process dictated by Woodward-Hoffmann rules, four isomers of tricyclononene would generate, in which the cyclopropane ring and the unsaturated bridge locate at syn-position, and the benzoyl and ester group from the dienophile adopt anti-position. Based on the absolute configuration of product 3aa determined by X-ray crystallography analysis, it is proposed that the transition state TS-(i) is the favorable one to yield the adduct 6-endo-3aa from(7S)-1a using the Re–Re-facial approach. The ability of the antipode to afford ent-6-endo-3aa from (7R)-1avia TS-(ii) is disfavored. The other two transition states (iii) and (iv) to form 6-exo-3aa were nearly inhibited in the presence of the optimal chiral catalyst.
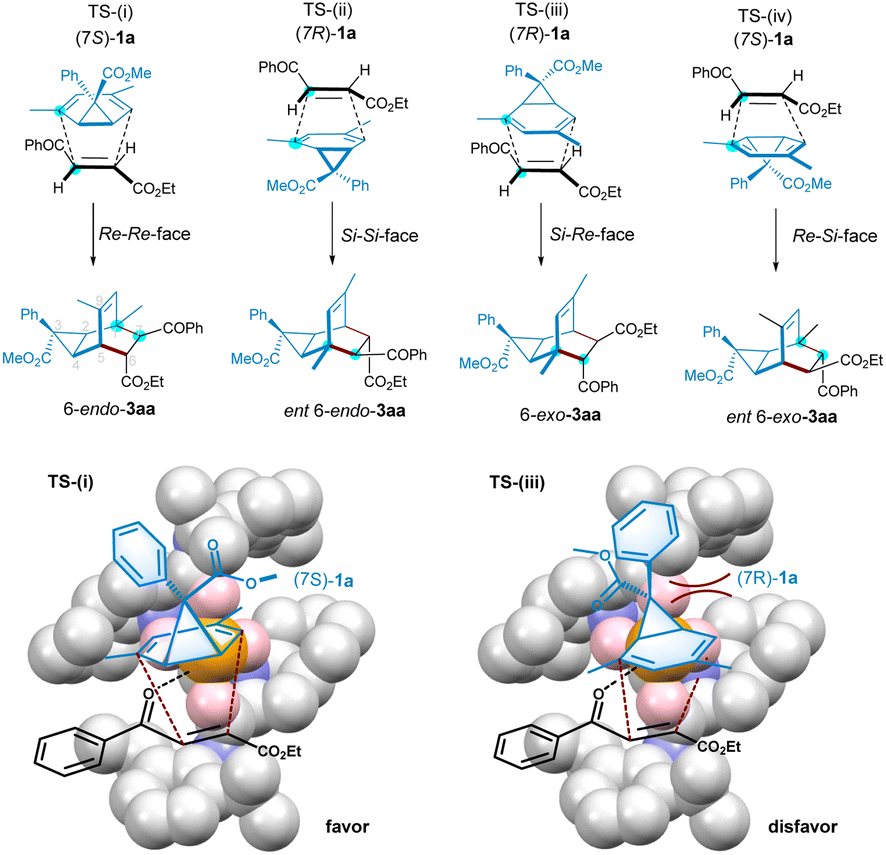 | ||
| Fig. 2 Possible transition states for the Diels–Alder reaction. | ||
Based on the X-ray crystal structure of Co(OTf)2/L3-PePr2 complex,18,26 and the activation manner of enone in cycloaddition, we rationalized the favorable enantioselective version of the Diels–Alder reaction and the kinetic resolution of norcaradiene (Fig. 2, below). As shown in the full view of TS-(i), the coordination of 4-oxo-4-arylbutenoate to the cobalt(II) center decreases the LUMO energy of the dieneophile, benefiting the cycloaddition. The Si-face of the enone is well shielded by the downward amide unit of the ligand. Thus, norcaradiene prefers to approach from the upper opening area. It is easy to see how to distinguish between the two enantiomers of norcaradiene, as shown in TS-(i) and TS-(iii). (7S)-1a could undergo [4 + 2]-cycloaddition with less steric hindrance, but the reaction of its enantiomer seems to be unfavorable owing to the steric hindrance of the inward cyclopropane-unit. Consequently, the facial selective addition in connection with the kinetic resolution yields the (1R, 2R, 3S, 4R, 5S, 6R, 7S)-3aa as the major adduct.
Conclusions
In summary, we have developed an enantioselective Diels–Alder reaction of norcaradienes with (E)-4-oxo-4-phenylbutenoates and β-nitroenones. The reactions catalyzed by a N,N′-dioxide/Co(II) complex use racemic norcaradienes prepared via visible-light-mediated dearomative cyclopropanation of m-xylene and α-diazoesters as the dienes and proceed via kinetic asymmetric transformation. Under otherwise identical reaction conditions, a series of complicated bridged tricyclo[3.2.2.0]nonene systems bearing seven contiguous stereocenters were accomplished under mild reaction conditions. Mechanism experiments also revealed the origin of the enantioselectivity and resolution of the isomers of norcaradienes. Further exploration of the catalyst system for other asymmetric reactions is under investigation.Data availability
Further details of experimental procedure, 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra and X-ray crystallographic data for 1a, 3aa, 5ab and Co(OTf)2/L3-PePr2 complex are available in the ESI.†Author contributions
S. Y. W. performed the experiments and prepared the ESI† and paper. Y. Q. Z. analyzed the X-ray diffraction crystal data and completed the theoretical calculation. W. L. X. and Z. G. L. repeated some experiments. X. M. F. and X. H. L. supervised the project. X. M. F. and X. H. L. helped with modifying the paper and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (no. 22188101), Sichuan Science and Technology Program (no. 2021YJ0561) and Sichuan University (2020SCUNL204) for financial support.Notes and references
- For selected reviews on biological activity of bicycle [2.2.2] octane, see: (a) L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113, 3516–3604 CrossRef CAS PubMed; (b) T. P. Stockdale and C. M. Williams, Chem. Soc. Rev., 2015, 44, 7737–7763 RSC; (c) K. Gao, J. L. Hu and H. F. Ding, Acc. Chem. Res., 2021, 54, 875–889 CrossRef CAS PubMed.
- (a) P. Yan, C. X. Zhong, J. Zhang, Y. Liu, H. Y. Fang and P. Lu, Angew. Chem., Int. Ed., 2021, 60, 4609–4613 CrossRef CAS PubMed; (b) A. Leverrier, K. Awang, F. Guéritte and M. Litaudon, Phytochemistry, 2011, 72, 1443–1452 CrossRef CAS PubMed.
- (a) J. S. Orman and G. M. Keating, Drugs, 2009, 69, 577–607 CrossRef CAS PubMed; (b) N. D. Campbell and A. M. Lovell, Ann. N. Y. Acad. Sci., 2012, 1248, 124–139 CrossRef CAS PubMed.
- D. L. Hughes, Org. Process Res. Dev., 2019, 23, 1298–1307 CrossRef CAS.
- For selected reviews, see: (a) F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 3364–3366 CrossRef CAS PubMed; (b) P. Tian, H.-Q. Dong and G.-Q. Lin, ACS Catal., 2012, 2, 95–119 CrossRef CAS; (c) Y. H. Huang and T. Hayash, Chem. Rev., 2022, 122, 14346–14404 CrossRef CAS PubMed . For selected examples, see:; (d) T. Hayashi, K. Ueyama, N. Tokunaga and K. Yoshida, J. Am. Chem. Soc., 2003, 125, 11508–11509 CrossRef CAS PubMed; (e) C. Fischer, C. Defieber, T. Suzuki and E. M. Carreira, J. Am. Chem. Soc., 2004, 126, 1628–1629 CrossRef CAS PubMed.
- For selected reviews, see: (a) B. K. Liebov and W. D. Harman, Chem. Rev., 2017, 117, 13721–13755 CrossRef CAS PubMed . For selected examples, see:; (b) A. C. Kinsman and M. A. Kerr, Org. Lett., 2000, 2, 3517–3520 CrossRef CAS PubMed; (c) M. D. Chordia, P. L. Smith, S. H. Meiere, M. Sabat and W. D. Harman, J. Am. Chem. Soc., 2001, 123, 10756–10757 CrossRef CAS PubMed; (d) S.-Y. Luo, Y.-J. Jang, J.-Y. Liu, C.-S. Chu, C.-C. Liao and S.-C. Hung, Angew. Chem., Int. Ed., 2008, 47, 8082–8085 CrossRef CAS PubMed; (e) I. Nakamura, K. Masukawa, Y. Ishida and M. Terada, Org. Lett., 2021, 23, 4127–4132 CrossRef CAS PubMed.
- For selected examples, see: (a) A. Bogdanova and V. V. Popik, Org. Lett., 2001, 3, 1885–1888 CrossRef CAS PubMed; (b) H. M. Mbuvi and L. K. Woo, J. Porphyrins Phthalocyanines, 2009, 13, 136–152 CrossRef CAS; (c) M. E. Jung, C. A. Roberts, F. Perez, H. V. Pham, L. F. Zou and K. N. Houk, Org. Lett., 2016, 18, 32–35 CrossRef CAS PubMed; (d) H. Sato, K. Fukaya, B. S. Poudel and M. J. Krische, Angew. Chem., Int. Ed., 2017, 56, 14667–14671 CrossRef CAS PubMed; (e) M. Hatano, T. Sakamoto, T. Mizuno, Y. Goto and K. Ishihara, J. Am. Chem. Soc., 2018, 140, 16253–16263 CrossRef CAS PubMed.
- For selected reviews, see: (a) S. E. Reisman, R. R. Nani and S. Levin, Synlett, 2011, 17, 2437–2442 CrossRef; (b) J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895–8918 CrossRef CAS. For selected examples, see: (c) R. Beniazza, V. Desvergnes and Y. Landais, Org. Lett., 2008, 10, 4195–4198 CrossRef CAS PubMed; (d) R. Beniazza, V. Desvergnes, E. Girard, B. Kauffmann, M. Berlande and Y. Landais, Chem.–Eur. J., 2012, 18, 11976–11986 CrossRef CAS PubMed; (e) R. R. Nani and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7304–7311 CrossRef CAS PubMed; (f) W. D. Mackay and J. S. Johnson, Org. Lett., 2016, 18, 536–539 CrossRef CAS PubMed; (g) Y. J. Guo, T. V. Nguyen and R. M. Koenigs, Org. Lett., 2019, 21, 8814–8818 CrossRef CAS PubMed; (h) S. Zhao, X.-X. Chen, N. Gao, M. C. Qian and X. Chen, J. Org. Chem., 2021, 86, 7131–7140 CrossRef CAS PubMed.
- For selected reviews, see: (a) W.-T. Wu, L. M. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570–1580 RSC; (b) Y.-Z. Cheng, Z. Feng, X. Zhang and S.-L. You, Chem. Soc. Rev., 2022, 51, 2145–2170 RSC . For selected examples, see:; (c) A. J. Anciaux, A. Demonceau, A. F. Noels, A. J. Hubert, R. Warin and P. Teyssie, J. Org. Chem., 1981, 46, 873–876 CrossRef CAS; (d) S. O'Neill, S. O'Keeffe, F. Harrington and A. R. Maguire, Synlett, 2009, 2009, 2312–2314 CrossRef; (e) X. F. Xu, X. B. Wang, P. Y. Zavalij and M. P. Doyle, Org. Lett., 2015, 17, 790–793 CrossRef CAS PubMed; (f) A. K. Clarke, W. P. Unsworth and R. J. K. Taylor, Tetrahedron, 2018, 74, 5374–5384 CrossRef CAS; (g) D. C. Crowley, D. Lynch and A. R. Maguire, J. Org. Chem., 2018, 83, 3794–3805 CrossRef CAS PubMed.
- (a) G. Maier, Angew. Chem., Int. Ed. Engl., 1967, 6, 402–413 CrossRef CAS; (b) Z. F. Chen, H. J. Jiao, J. I. Wu, R. Herges, S. B. Zhang and P. V. R. Schleyer, J. Phys. Chem. A, 2008, 112, 10586–10594 CrossRef CAS PubMed; (c) O. A. McNamara and A. R. Maguire, Tetrahedron, 2011, 67, 9–40 CrossRef CAS; (d) L. M. Bateman, O. A. McNamara, N. R. Buckley, P. O'Leary, F. Harrington, N. Kelly, S. O'Keeffe, A. Stack, S. O'Neill, D. G. McCarthy and A. R. Maguire, Org. Biomol. Chem., 2015, 13, 11026–11038 RSC; (e) M. H. Palmer, R. A. Aitken, M. Coreno, M. de Simone, C. Grazioli, S. V. Hoffmann and N. C. Jones, J. Chem. Phys., 2020, 152, 144301–144315 CrossRef CAS PubMed.
- (a) S. M. Wilkerson-Hill, B. E. Haines, D. G. Musaev and H. M. L. Davies, J. Org. Chem., 2018, 83, 7939–7949 CrossRef CAS PubMed; (b) S. H. Park, S.-G. Wang and N. Cramer, ACS Catal., 2019, 9, 10226–10231 CrossRef CAS; (c) J. T. Fu, N. Wurzer, V. Lehner, O. Reiser and H. M. L. Davies, Org. Lett., 2019, 21, 6102–6106 CrossRef CAS PubMed; (d) D. Antoniak and M. Barbasiewicz, Org. Lett., 2019, 21, 9320–9325 CrossRef CAS PubMed.
- K. L. Smith, C. L. Padgett, W. D. Mackay and J. S. Johnson, J. Am. Chem. Soc., 2020, 142, 6449–6455 CrossRef CAS PubMed.
- E. T. Crawford, K. L. Smith and J. S. Johnson, Org. Lett., 2022, 24, 1791–1795 CrossRef CAS PubMed.
- T. Ito, S. Harada, H. Homma, H. Takenaka, S. Hirose and T. Nemoto, J. Am. Chem. Soc., 2021, 143, 604–611 CrossRef CAS PubMed.
- (a) A. R. Maguire, P. O'Leary, F. Harrington, S. E. Lawrence and A. J. Blake, J. Org. Chem., 2001, 66, 7166–7177 CrossRef CAS PubMed; (b) R. Mose, G. Preegel, J. Larsen, S. Jakobsen, E. H. Iversen and K. A. Jørgensen, Nat. Chem., 2017, 9, 487–492 CrossRef CAS PubMed.
- For reviews on chiral N,N′-dioxides: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298–302 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621–2631 CrossRef CAS PubMed; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791–797 CrossRef CAS; (e) Z. Wang, X. H. Liu and X. M. Feng, Aldrichimica Acta, 2020, 53, 3–10 CAS; (f) W. D. Cao, X. H. Liu and X. M. Feng, Chin. Sci. Bull., 2020, 65, 2941–2951 CrossRef; (g) M. Y. Wang and W. Li, Chin. J. Chem., 2021, 39, 969–984 CrossRef CAS; (h) S. X. Dong, X. H. Liu and X. M. Feng, Acc. Chem. Res., 2022, 55, 415–428 CrossRef CAS PubMed . For selected examples on cycloaddition using chiral N,N′-dioxides:; (i) Y. H. Zhou, Y. Lu, X. Y. Hu, H. J. Mei, L. L. Lin, X. H. Liu and X. M. Feng, Chem. Commun., 2017, 53, 2060–2063 RSC; (j) D. Zhang, Z. S. Su, Q. W. He, Z. K. Wu, Y. Q. Zhou, C. J. Pan, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2020, 142, 15975–15985 CrossRef CAS PubMed; (k) B. Shen, Q. W. He, S. X. Dong, X. H. Liu and X. M. Feng, Chem. Sci., 2020, 11, 3862–3867 RSC; (l) Y. Luo, H. Zhang, S. Y. Wang, Y. Q. Zhou, S. X. Dong and X. M. Feng, Org. Lett., 2020, 22, 2645–2650 CrossRef CAS PubMed; (m) P. Zhao, Z. G. Li, J. He, X. H. Liu and X. M. Feng, Sci. China: Chem., 2021, 64, 1355–1360 CrossRef CAS; (n) L. Z. Hou, Y. Q. Zhou, H. Yu, T. Y. Zhan, W. D. Cao and X. M. Feng, J. Am. Chem. Soc., 2022, 144, 22140–22149 CrossRef CAS PubMed; (o) T. Y. Zhan, L. K. Yang, Q. Y. Chen, R. Weng, X. H. Liu and X. M. Feng, CCS Chem., 2022 DOI:10.31635/ccschem.022.202202405.
- H. K.-W. Hui and H. Shechter, Tetrahedron Lett., 1982, 23, 5115–5118 CrossRef CAS.
- CCDC: 2070991 (1a), CCDC: 2155946 (3aa), CCDC: 2212551 (5ab), and CCDC: 2212550 (Co(OTf)2/L3-PePr2) contains the supplementary crystallographic date for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- (a) E. Ciganek, J. Am. Chem. Soc., 1971, 93, 2207–2215 CrossRef CAS; (b) M. Balci, H. Fischer and H. Günther, Angew. Chem., Int. Ed. Engl., 1980, 19, 301–302 CrossRef; (c) V. N. G. Lindsay, D. Fiset, P. J. Gritsch, S. Azzi and A. B. Charette, J. Am. Chem. Soc., 2013, 135, 1463–1470 CrossRef CAS PubMed.
- (a) A. R. Choudhury, M. S. Manna and S. Mukherjee, Chem. Sci., 2017, 8, 6686–6690 RSC; (b) C. L. He, X. X. Tang, X. He, Y. Q. Zhou, X. H. Liu and X. M. Feng, Chin. Chem. Lett., 2023, 34, 107487–107492 CrossRef CAS.
- (a) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (c) L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- SambVca 2.0: a web application for analyzing catalytic pockets, https://www.molnac.unisa.it/OMtools/sambvca2.0/ Search PubMed.
- For selected examples, see: (a) W. Zhang, P. M. Waddell, M. A. Tiedemann, C. E. Padilla, J. J. Mei, L. Y. Chen and B. P. Carrow, J. Am. Chem. Soc., 2018, 140, 8841–8850 CrossRef CAS PubMed; (b) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed; (c) Q. F. Tan, H. Yu, Y. Luo, F. Z. Chang, X. H. Liu, Y. Q. Zhou and X. M. Feng, Chem. Commun., 2021, 57, 3018–3021 RSC.
- (a) J. Xu, Z. W. Zhong, M. Y. Jiang, Y. Q. Zhou, X. H. Liu and X. M. Feng, CCS Chem., 2021, 3, 1894–1902 CrossRef CAS; (b) G. H. Pan, C. L. He, M. Chen, Q. Xiong, W. D. Cao and X. M. Feng, CCS Chem., 2022, 4, 2000–2008 CrossRef CAS; (c) F. Q. Zhang, B.-T. Ren, Y. B. Liu and X. M. Feng, Org. Chem. Front., 2022, 9, 3956–3960 RSC; (d) F. Q. Zhang, B.-T. Ren, Y. B. Liu and X. M. Feng, Chem. Sci., 2022, 13, 5562–5567 RSC; (e) Z. D. Tan, S. B. Zhu, Y. B. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202203374 CAS; (f) W. Wang, F. Q. Zhang, Y. B. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202208837 CAS; (g) Y. Xu, H. K. Wang, Z. Yang, Y. Q. Zhou, Y. B. Liu and X. M. Feng, Chem, 2022, 8, 2011–2022 CrossRef CAS; (h) W. L. Xiao, L. C. Ning, S. Xin, S. X. Dong, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202211596 CAS; (i) Y. Wang, A. Y. Yihuo, L. F. Wang, S. X. Dong and X. M. Feng, Sci. China: Chem., 2022, 65, 546–553 CrossRef CAS; (j) W. Yang, Z. D. Yang, L. Chen, Y. C. Lu, C. F. Zhang, Z. S. Su, X. H. Liu and X. M. Feng, Chin. Chem. Lett., 2022, 32 DOI:10.1016/j.cclet.2022.107791.
- (a) X. X. Tang, Z. S. Su, Q. C. Lin, L. L. Lin, S. X. Dong and X. M. Feng, Chin. J. Chem., 2022, 40, 1793–1798 CrossRef CAS; (b) F. C. Zhang, X. P. Sang, Y. Q. Zhou, W. D. Cao and X. M. Feng, Org. Lett., 2022, 24, 1513–1517 CrossRef CAS PubMed; (c) Y. Liu, Y. S. Chen, A. Y. Yihuo, Y. Q. Zhou, X. H. Liu, L. L. Lin and X. M. Feng, ACS Catal., 2022, 12, 1784–1790 CrossRef CAS; (d) H. K. Wang, Y. Xu, F. Q. Zhang, Y. B. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202115715 CAS.
- (a) C. R. Xu, H. F. Zheng, B. W. Hu, X. H. Liu, L. L. Lin and X. M. Feng, Chem. Commun., 2017, 53, 9741–9744 RSC; (b) B. W. Hu, X. Y. Zhang, Y. H. Mo, J. Z. Li, L. L. Lin, X. H. Liu and X. M. Feng, Org. Lett., 2020, 22, 1034–1039 CrossRef CAS PubMed; (c) X. B. Lin, Z. Tan, W. K. Yang, W. Yang, X. H. Liu and X. M. Feng, CCS Chem., 2021, 3, 1423–1433 CrossRef CAS; (d) W. Yang, M. P. Pu, X. B. Lin, M. Chen, Y. J. Song, X. H. Liu, Y. D. Wu and X. M. Feng, J. Am. Chem. Soc., 2021, 143, 9648–9656 CrossRef CAS PubMed; (e) X. B. Lin, M. P. Pu, X. P. Sang, S. Y. Li, X. H. Liu, Y. D. Wu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202201151 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra (PDF). X-ray crystallographic data for 1a, 3aa, 5ab and Co(OTf)2/L3-PePr2 complex. CCDC 2070991, 2155946, 2212551 and 2212550. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06490a |
| This journal is © The Royal Society of Chemistry 2023 |