
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSARS-CoV-2 spike proteins react with Au and Si, are electrically conductive and denature at 3 × 108 V m−1: a surface bonding and a single-protein circuit study†
Essam M.
Dief
and
Nadim
Darwish
 *
*
School of Molecular and Life Sciences, Curtin University, Bentley, WA 6102, Australia. E-mail: nadim.darwish@curtin.edu.au
First published on 17th February 2023
Abstract
Developing means to characterise SARS-CoV-2 and its new variants is critical for future outbreaks. SARS-CoV-2 spike proteins have peripheral disulfide bonds (S–S), which are common in all spike proteins of SARS-CoV-2 variants, in other types of coronaviruses (e.g., SARS-CoV and MERS-CoV) and are likely to be present in future coronaviruses. Here, we demonstrate that S–S bonds in the spike S1 protein of SARS-CoV-2 react with gold (Au) and silicon (Si) electrodes. Bonding to Si is induced by a spontaneous electrochemical reaction that involves oxidation of Si–H and the reduction of the S–S bonds. The reaction of the spike protein with Au enabled single-molecule protein circuits, by connecting the spike S1 protein between two Au nano-electrodes using the scanning tunnelling microscopy-break junction (STM-BJ) technique. The conductance of a single spike S1 protein was surprisingly high and ranged between two states of 3 × 10−4G0 and 4 × 10−6G0 (1G0 = 77.5 μS). The two conductance states are governed by the S–S bonds reaction with Au which controls the orientation of the protein in the circuit, and via which different electron pathways are created. The 3 × 10−4G0 level is attributed to a single SARS-CoV-2 protein connecting to the two STM Au nano-electrodes from the receptor binding domain (RBD) subunit and the S1/S2 cleavage site. A lower 4 × 10−6G0 conductance is attributed to the spike protein connecting to the STM electrodes from the RBD subunit and the N-terminal domain (NTD). These conductance signals are only observed at electric fields equal to or lower than 7.5 × 107 V m−1. At an electric field of 1.5 × 108 V m−1, the original conductance magnitude decreases accompanied by a lower junction yield, suggesting a change in the structure of the spike protein in the electrified junction. Above an electric field of 3 × 108 V m−1, the conducting channels are blocked and this is attributed to the spike protein denaturing in the nano-gap. These findings open new venues for developing coronavirus-capturing materials and offer an electrical method for analysing, detecting and potentially electrically deactivating coronaviruses and their future variants.
1. Introduction
In 2019, the emergence of a novel coronavirus, severe acute respiratory syndrome coronavirus (SARS-CoV-2), with the subsequent pandemic of COVID-19 infections, has highlighted the need for ways to reduce transmission of coronaviruses. Individuals acquire infection with coronaviruses primarily through direct contact with virus-positive individuals via aerosols.1 It is also debated that coronaviruses can be transmitted by touching the surfaces that an infected person has coughed or sneezed on.2,3 In 2020, it was reported that the viability of the SARS-CoV-2 virus in aerosol droplets varied on different surfaces. While the virus remained viable for three days on plastic, cardboard, and stainless steel, it was only viable for three hours on copper.4 Although the study highlighted the effect of surfaces on the lifespan of the viral particles, the reason of its low viability on copper compared to other surfaces remains unknown.The structures of the spike proteins (S1 and S2) of most of the coronaviruses, including SARS-CoV-2 (Fig. 1a(i)), revealed that these proteins possess multiple disulfide (S–S) bonds. For example, the spike proteins (S1 and S2) of SARS-CoV-2 contain 14 S–S bonds in well-defined regions, with 10 S–S bridges in the S1 subunit. (Fig. 1a(ii)),5–7 MERS-CoV contains 11 S–S bonds, and HCoV-229E spike protein contains 13 S–S bonds.8 The SARS-CoV-2 spike protein S1 is composed of three domains, the receptor binding domain (RBD) that contains 4 S–S bridges, the N-terminal domain (NTD) that contains 3 S–S bridges and the S1/S2 cleavage site that contains 3 S–S bridges (Fig. 1a(iii)). Such abundant S–S bridges indicate their important structural role in the formation and stabilisation of the proper spike architecture and are likely to be present in future types of coronaviruses and their variants. For instance, SARS-CoV-2 spike protein RBD has an extra S–S bond as compared to SARS-CoV spike protein RBD.9 In SARS-CoV-2, four pairs of S–S bridges (C336:C361, C379:C432, C391:C525, C480:C488) are found in the RBD (Fig. 1a(iv)). Among these four pairs, three are in the core, which help to stabilise the β sheet structure; the remaining S–S pair connects the loops in the distal end of the receptor-binding motif (RBM).10 These S–S bonds are essential for the SARS-CoV-2 spike protein structure and its ability to infect, by interacting with the angiotensin-converting enzyme 2 (ACE2) human cell surface receptor via a thiol–disulfide exchange process.10–15
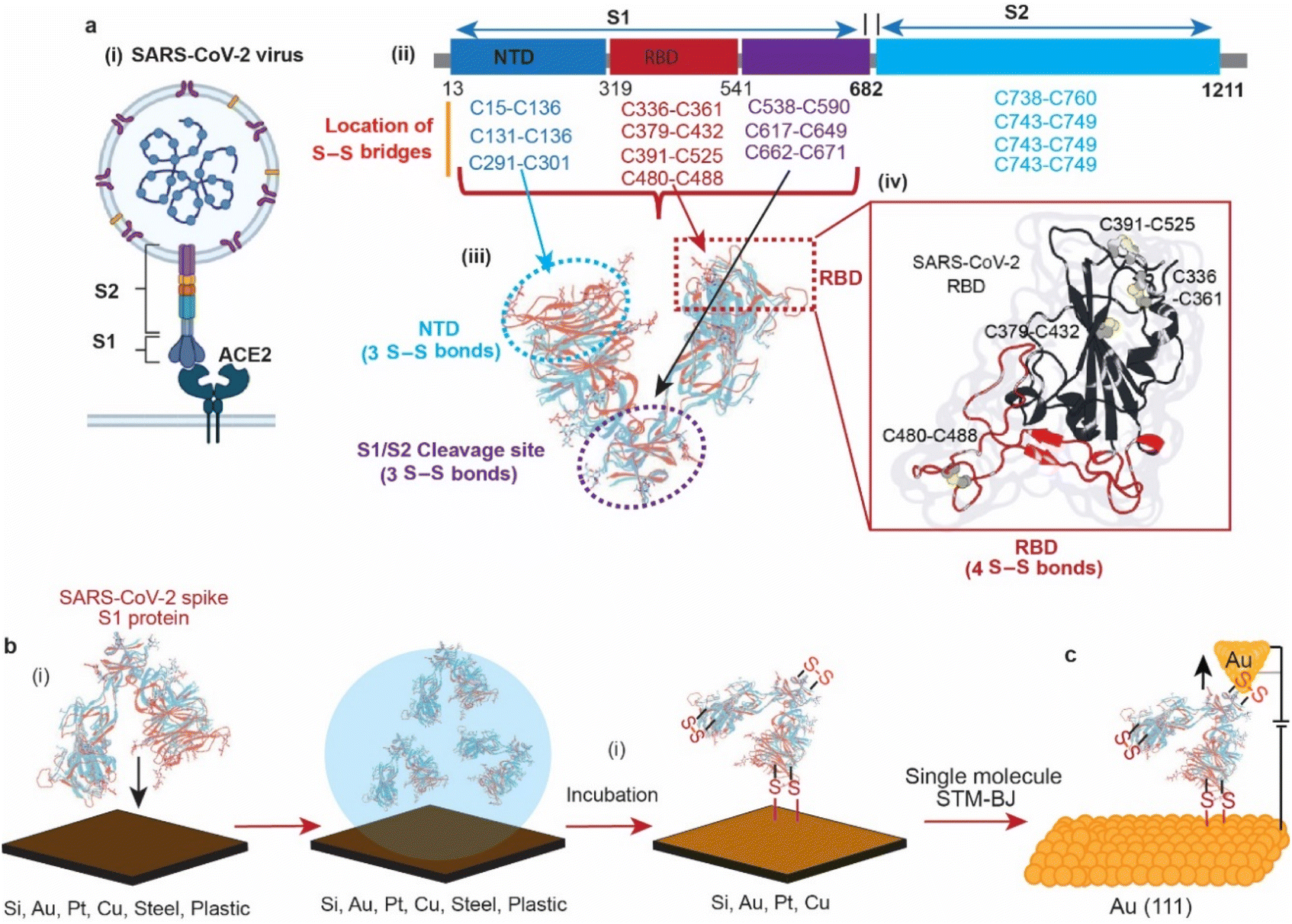 | ||
| Fig. 1 Structural properties of the SARS-CoV-2 spike S1 protein and its interaction with different surfaces. (a) Schematic representation of the SARS-CoV-2 virus (i), its spike protein (S1 + S2) amino acid sequence depicting the positions of the disulfide bridges in each domain (ii), with the structure of the S1 subunit containing 4 disulfide bridges at the RBD, 3 disulfide bridges at the NTD and 3 disulfide bridges at the S1/S2 cleavage site (iii). (b) Schematic describing the interaction of SARS-CoV-2 (2019-nCoV) spike protein with different surfaces. The surfaces were incubated in the spike protein solution in phosphate buffer saline (PBS), pH 7.4 before further analysis. (c) Schematic showing the wiring of the SAR-CoV-2 spike S1 protein between two gold nano-electrodes in a scanning tunnelling microscopy-break junction (STM-BJ) experiment. | ||
On surfaces, S–S residues have been reported to form covalent bonding to noble metals such as Au and have been widely used in many applications.16–27 While these metal–organic molecule contacts are known to occur, the reaction mechanism is still debated.16,28–34 In addition to metal surfaces, oxide-free silicon (Si–H), which is obtained by etching the native oxide away from the top layer of Si wafers, possesses a low reduction potential that allows for reducing chemical compounds and metal ions on its surface.35–38 Recently, it has been demonstrated that Si–H surfaces can spontaneously reduce the S–S bonds in disulfide-terminated organic molecules and in the protein azurin that contains a peripheral S–S bond, enabling connecting these molecules to the Si surface via covalent S–Si bonds.39–41
In this article, we use surface spectroscopy, electrochemical and single-molecule scanning tunnelling break junction techniques to (i) study the chemical reactivity of SARS-CoV-2 with surfaces of electrodes, (ii) electrically detect the spike protein and (iii) study the effect of electric fields on spike proteins at the single-molecule level (Fig. 1).
2. Results and discussion
2.1. Surfaces that covalently bond SARS-CoV-2 spike S1 protein
AFM imaging revealed that the spike S1 protein remains connected to some surfaces, particularly Si and Au, despite significant washing. For example, a freshly etched Si (111)–H surface incubated in the spike S1 protein solution showed the presence of the protein, despite excessive washing of the surfaces (Fig. 2a). In addition, incubating an Au electrode in the spike S1 protein for the same time showed a fully covered surface (Fig. 2c). After setting up a background threshold on the topography height images (see Fig. S1, ESI† for details), particle analysis for the protein spots on the Au surface shows a histogram with an average height size of ∼15 nm (Fig. 2a and b). On Si, the average height distribution of the protein on the surface is ∼8 nm. The variations in the size of the protein on Si and on Au can be attributed to the nature of the reaction between the S–S bonds on the protein and the surfaces. While on Si the kinetics of the reaction is faster than that on Au, it is reasonable to expect that the protein aggregates less on the Si than on Au.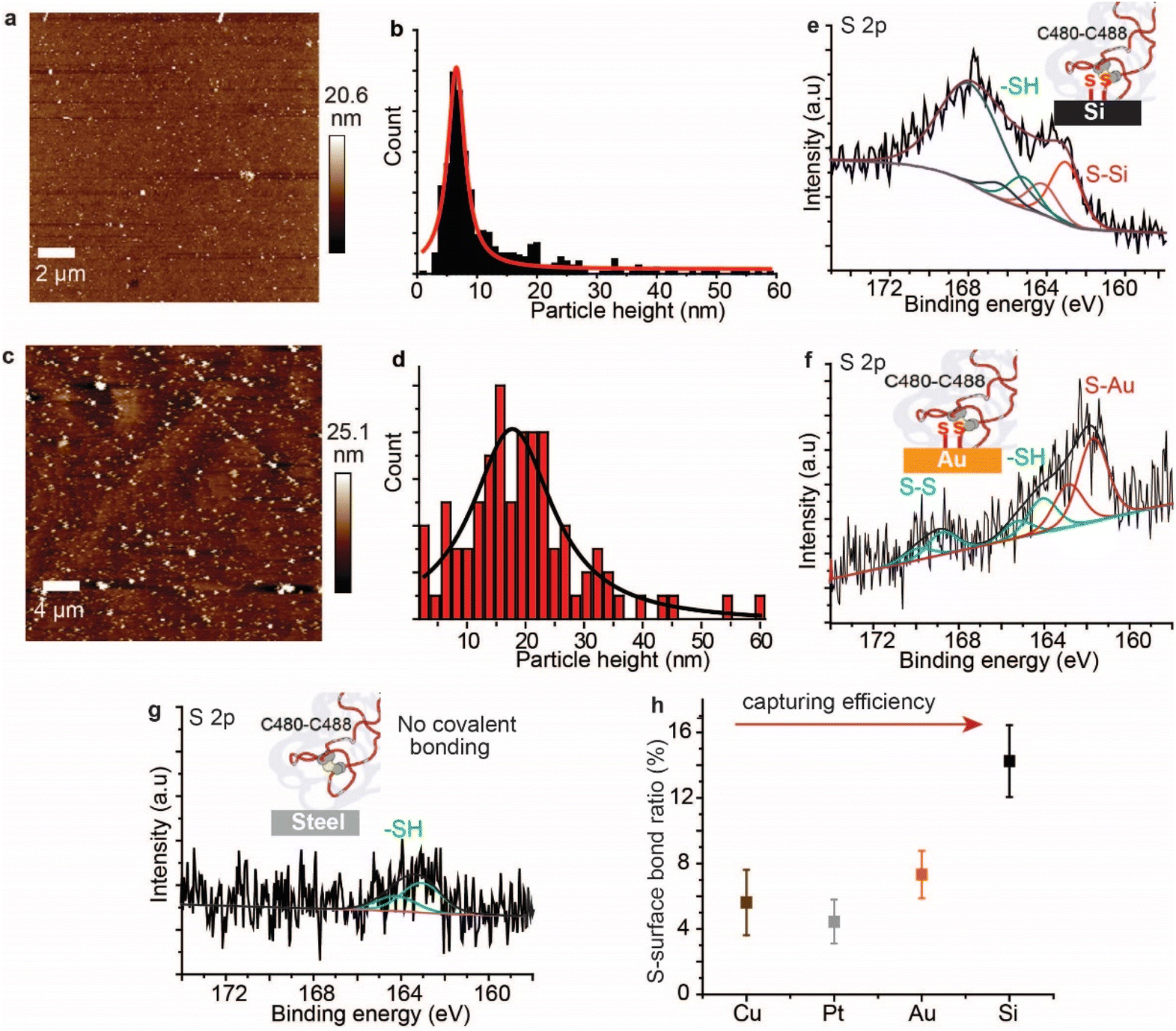 | ||
| Fig. 2 Surfaces that covalently bond SARS-CoV-2 spike S1 protein. (a) AFM topography image of a Si (111)–H surface after incubation in a solution of the spike protein for 12 h, and then washed thoroughly, showing the surface covered with the spike protein. (b) A histogram representing the particle height distribution for the SARS-CoV-2 spike S1 protein shown in (a), fitted to a Lorentzian distribution. (c) AFM topography image of an Au (111) surface after incubation in the spike protein for 12 h, and then washed thoroughly, showing the surface covered with the protein. (d) A histogram representing the particle height distribution for the SARS-CoV-2 spike S1 protein shown in (c), fitted to a Lorentzian distribution. (e) The S 2p envelope for the spike protein on the Si–H surface, showing two spin–orbit splits. The spin–orbit split centred at 163.1 eV (red) is assigned to Si–S while the spin–orbit split at 165.3 eV is attributed to non-bound disulfides, respectively. (f) The S 2p envelope for the spike protein on the Au surface with two spin–orbit splits with the spin–orbit split centred at 162.3 eV (red) assigned to Au–S bonding, while that at 164.1 eV is attributed to –SH and the doublet at ∼168.8 eV is ascribed to unreacted S–S bonding. (g) S 2p envelope for the spike protein on stainless steel after the washing step, showing only one spin–orbit split at 163.7 eV that is ascribed to thiols (–SH). (h) The ratio of the S–surface covalent bonding as a function of the surface type, showing that Si can capture more spike protein as compared to metal surfaces. | ||
Control experiments in which Au (111) surfaces were kept in the PBS buffer followed by washing showed flat terraces with some nodules induced by flame annealing that are characteristic of unmodified freshly annealed Au (111) surfaces (ESI Fig. S1b†). It is noteworthy that for the microscopy experiments, Si and Au surfaces were chosen because they expose an atomically flat surface suitable for AFM imaging.42,43 In a control experiment, the spike protein was found to be washed away from mica surfaces which do not form covalent bonding with the protein (Fig. S2, ESI†).
To investigate the nature of the chemical bonding between the spike S1 protein and each surface, XPS analysis was carried out. The high-resolution S 2p signal for the spike protein on the Si–H surface shows two spin–orbit splits centred at 163.1 eV and 165.3 eV, corresponding to the covalently bonded sulfur (S–Si) and the unbound thiols (–SH), respectively (Fig. 1e, see also ESI Fig. S3†). In addition, the emission peak at ∼168 eV is attributed to the Si plasmon loss, due to the strong interaction between Si surface electrons and the emitted photoelectrons. The emission from Si plasmon loss is relatively large that the S–S bonding could be present in the case of the spike protein interaction with Si, but its signal is shadowed underneath the Si plasmon loss spectra. An emission from the Si plasmon loss was also observed from a bare Si–H surface and from the interaction of the Si with a model disulfide molecule, alpha lipoic acid (ALA) (Fig. S4†).
In a control experiment using the protein azurin, which has only one S–S at one of the very peripheral ends and a copper metal centre at the opposite terminal,44 it formed S–Si bonds with Si surfaces (ESI Fig. S5†). The high-resolution S 2p of the spike S1 protein on Au showed a doublet centred at 161.9 eV and a doublet centred at 164.2 eV corresponding to the S–Au and the –SH, respectively, and the doublet at ∼168.7 eV is ascribed to S–S bonds that are buried inside the protein (Fig. 2f).45 The high-resolution N 1s showed a central emission at 400.1 eV attributed to the –NH2 groups and a small emission of a lower binding energy (398.2 eV) that is attributed to NH2 groups that are bound to the Au substrate (ESI Fig. S6†). Similarly, XPS of the spike S1 protein on Pt (ESI Fig. S7†) and Cu (ESI Fig. S8†) surfaces showed covalent bonding between the protein S–S bridges and these metals. In contrast to metal surfaces and Si, plastic and stainless-steel surfaces that are incubated in the spike S1 protein, followed by washing, showed no covalent bonding between the protein S–S bonds and these surfaces. The S 2p emission from the spike S1 protein on steel was shifted to higher binding energy and is fitted into one spin–orbit split centred at ∼163.7 eV that is ascribed to –SH bonding (Fig. 2g, see also ESI Fig. S9†).
The spike S1 protein on plastic showed two spin–orbit splits in the S 2p signal, at 163.6 eV and 167.9 eV, which are ascribed to the –SH and unbroken S–S bonds, respectively, suggesting that the disulfide bonds in the protein are present in the –SH or the S–S form, but with no covalent bonding to the surface (ESI Fig. S10†). Control experiments in which the surfaces were exposed to the spike protein solution, and then dried without washing showed a higher content of the unreacted S–S moieties on metals (ESI Fig. S11†) and on plastic surfaces (ESI Fig. S12†). Detailed quantitative analysis of the ratio of the covalently bound sulfur in the XPS of the spike protein on different surfaces showed that the capability of each surface to break and react with the S–S bonds varies (Fig. 2h). While Si showed the highest ratio of S–Si/Si bonding, the metal substrates (Au, Pt and Cu) showed two-fold lower S–metal/metal bonding. This difference can be attributed to a different reaction mechanism on the different surfaces.
2.2. Disulfide bond cleavage of the spike S1 protein
The variations in the ratios of the bound sulfur shown in the XPS of the spike protein on different surfaces are attributed to the difference in the rate of the reaction by which each surface breaks and reacts with the S–S bonds. For instance, Au and Pt have high reduction potentials (standard reduction potential of +1.48 V and +1.20 V for Au and Pt, respectively) and therefore they are not likely capable of reducing the disulfide bonds by a spontaneous electrochemical reaction. In contrast, because of their low reduction potential (standard reduction potential of −0.516 V for the closely related SiH4),46,47 Si–H surfaces have been reported to spontaneously reduce metals such as Au, and Pd.48 The reduction is attributed to the electrochemical reaction between the metal ions in solution and the Si, resulting in the reduction of metals and partial oxidation of Si. Si–H has been also reported to spontaneously reduce S–S bonds in organic molecules.49,50Here, we used cyclic voltammetry (CV) to determine the half-wave potential (E1/2) for the reduction of the S–S bonds in SARS-CoV-2 and it was benchmarked against a model disulfide-terminated molecule, alpha lipoic acid (ALA). Fig. 3a shows a CV for the spike protein on a glassy carbon electrode (GCE), in PBS buffer (pH 7.4), with a main oxidation wave at +0.59 V and a reduction wave at −0.74 V (vs. Ag/AgCl), and an E1/2 of −0.15 V. We hypothesise that these waves are for the reduction/oxidation of the most easily accessible S–S bonds in SARS-COV-2 spike protein S1. Another smaller oxidation peak appears at a higher potential (+1.3 V vs. Ag/AgCl) and can be attributed to the re-oxidation to disulfide bridges in an environment different to that observed at +0.59 V. The position of these signals agrees with what has been previously observed for the reduction/oxidation of cystine/cysteine containing compounds and proteins.51,52
 | ||
| Fig. 3 Schematic describing the suggested reaction mechanisms for the reaction of the disulfide bonds with Si–H and Au. (a) Cyclic voltammograms (CV) of the SARS-CoV-2 spike protein (100 μg mL−1) in PBS buffer at pH 7.4 on Si–H showing a reduction wave at −0.74 V vs. Ag/AgCl. (b) CV for a model disulfide molecule (alpha lipoic acid) in organic electrolyte on a glassy carbon electrode with an E1/2 of −0.06 V vs. Ag/AgCl. The E1/2 value is close to 0 V (vs. Ag/AgCl) in both the protein and the cyclic disulfide molecule (1 mM), suggesting that Si–H which has a lower reduction potential that can spontaneously reduce the S–S bonds. (c) Comparison of the S–metal reaction rate on Si and Au surfaces at different times. (d) Disulfide bond reduction mechanism via an electron transfer process from the Si surface that reduces the disulfide bond in the spike protein forming a thiyl radical and a thiolate anion. Covalent bonds are then formed between the Si and S, leaving an active surface radical that undergoes a radical chain reaction. (e) Schematic illustrating the suggested mechanism of S–S reaction with metal electrodes (Au, Pt and Cu). The S–S containing protein is first physically adsorbed on the surface followed by a homolytic cleavage forming two thiyl radicals which then react to form covalent bonds to the metal surface. | ||
In a control experiment, ALA, that has only one exposed S–S bond, in dry DMF on a GCE electrode showed an oxidation wave at +1.09 V and a reduction wave at −1.15 V, with an E1/2 of −0.06 V vs. Ag/AgCl (Fig. 3b), which is comparable to what has been observed for the spike protein on GCE.
ALA was chosen as the control molecule because its S–S bond is strained within the ring making it more reactive and it is exposed to the electrolyte, mimicking the situation in the spike protein where the S–S bonds are peripheral and are strained due to their role in holding the tertiary structure of the protein by bridging the protein beta sheets.
Since the reduction potential of the S–S bonds is more positive than that of Si–H, it is reasonable to suggest that Si–H surfaces reduce the S–S bond by a spontaneous electrochemical reaction and consequently covalently bond the spike protein to the surface (Fig. 3d). Such a mechanism would involve the oxidation of the Si–H surfaces at the nanoscale by water vapour from the ambient environment, generating electrons that break the disulfide bonds in the spike protein forming a thiyl radical S˙ and S− anion (i). S˙ can then react with Si (111)–H to abstract a surface hydrogen (ii), forming a thiol RSH physisorbed to a Si-surface radical site that forms a covalent bond between the S and the Si atoms – releasing H2, and resulting in a radical on the neighboring Si atom (ii). The Si radical reacts with the thiolate anion forming the second Si–S covalent contact (iv), leaving a radical that leads to a chain reaction on the Si surface.
On metal surfaces, the mechanism is suggested to be different to that on Si, with the electrochemical reduction scenario not possible due to the high reduction potential of the Au, but with the same outcome of Au–S covalent bonding (Fig. 3e). Therefore, the reaction of the spike protein with Au is expected to occur via physical adsorption of S–S bonds to the surface (Fig. 3e(i)), followed by a homolytic cleavage forming thiyl radicals that react with the metal surface (Fig. 3e(ii)). This hypothesis is supported by electrochemical studies for the adsorption/desorption process of cyclic disulfides on Au electrodes.28
The difference in the reaction rates of the spike protein on Si and Au agrees with the electrochemical characterisation of a control monolayer formed from a ferrocene-terminated model disulfide molecule, where the molecular coverage achieved on Si was three-fold higher than that achieved on Au when the incubation time was fixed to 1 h for both surfaces (ESI Fig. S13†).
2.3. Reaction of SARS-CoV-2 spike S1 with Au nanoparticles
To test whether SARS-CoV-2 spike protein reacts with Au other than that of flat surfaces, we tested the reaction with gold nanoparticles (AuNPs). AuNPs were exposed to the spike protein for 12 h under ambient conditions (Fig. 4a), and then UV-vis spectra (Fig. 4b) were recorded for the bare (unmodified) AuNPs (black trace) and for the AuNPs after their reaction with SARS-CoV-2 spike protein (red trace). The absorbance spectrum of the AuNP localized surface plasmon resonance (LSPR) is shifted from 519 nm to 525 nm upon connection to the spike protein – a shift which is consistent with LSPR shifts of 20 nm for AuNPs functionalised with organic alkanethiols.53,54 Similar shifts were also observed for the reaction of spike S1 protein with 50 nm AuNPs (Fig. 4c).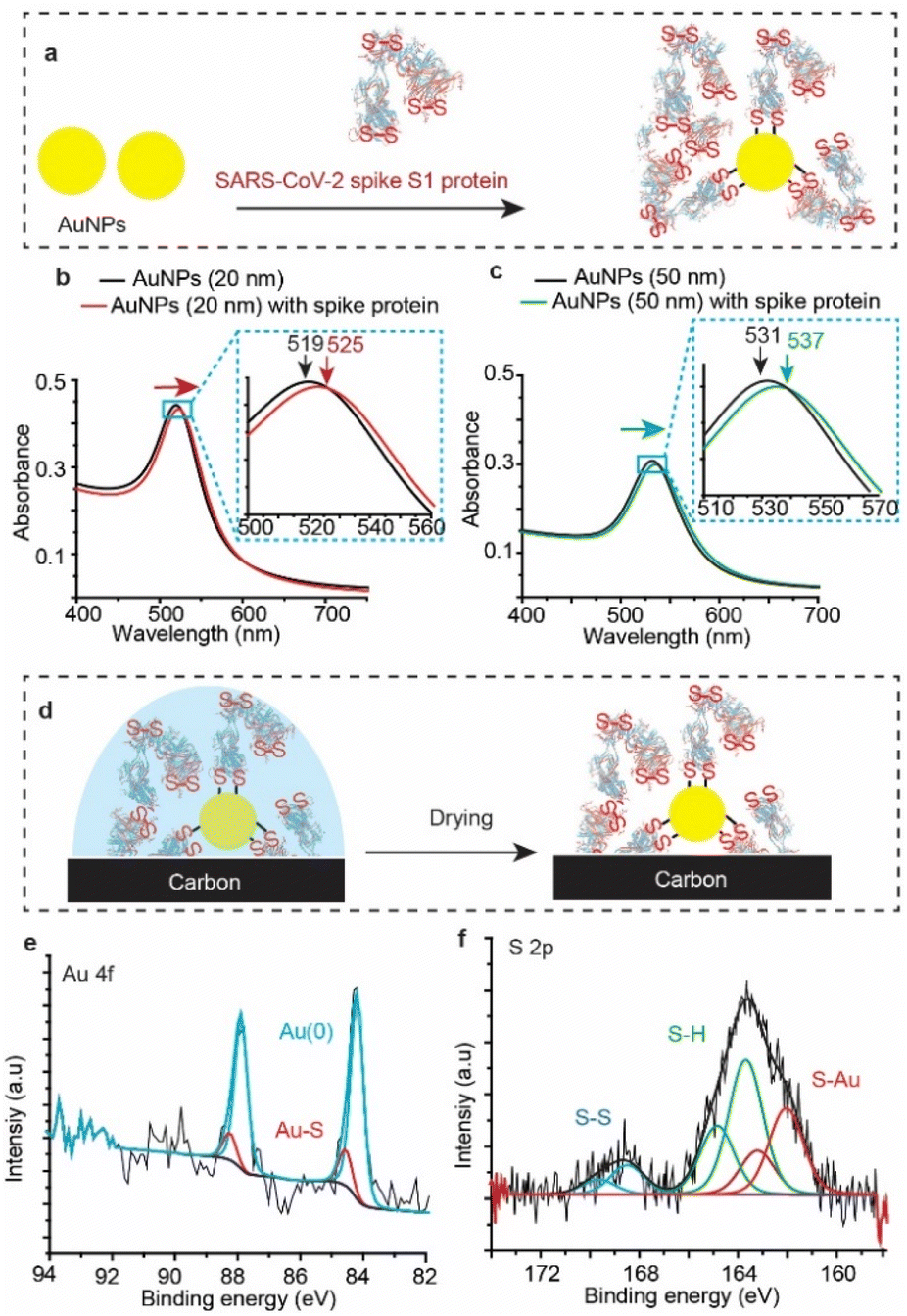 | ||
| Fig. 4 SARS-CoV-2 spike S1 protein reaction with AuNPs. (a) Schematic describing the functionalisation process by exposing a solution of AuNPs to the spike protein (100 μg mL−1). (b) UV-vis spectra of 20 nm AuNP solution before functionalisation (black trace) and the 20 nm AuNP solution incubated with the spike protein solution for 12 h (red trace). (c) UV-vis spectra of 50 nm AuNP solution before functionalisation (black trace) and the 50 nm AuNP solution incubated with the spike protein solution for 12 h (green trace). (d) Schematic describing the XPS analysis of the spike protein functionalised AuNPs, where 100 μL of the nanoparticles (6 × 1011 particle per mL) exposed to 100 μL of the spike protein (100 μg mL−1), for 12 h, were deposited on a carbon surface and allowed to dry before the XPS analysis. (e and f) Au 4f (e) and the S 2p (f) emissions from the spike protein-functionalised AuNPs on the carbon surface. | ||
XPS analysis (Fig. 4d) showed an Au 4f signal with a main spin–orbit split with two emission peaks at 84.3 eV and 88.0 eV corresponding to the metallic gold Au 4f7/2 Au(0) and Au 4f5/2 Au(0), respectively (Fig. 4e). The emissions at 85.4 eV and 89.1 eV correspond to the Au 4f7/2 and the Au 4f5/2 of the gold bound to sulfur (Au–S), demonstrating covalent bonding between the protein S–S bonds and the AuNPs. Similar to the spike protein on flat Au surfaces, the S 2p emission showed two spin–orbit splits centred at ∼162.0 eV and 163.8 eV corresponding to the bound sulfur (Au–S) and the free thiols (–SH), respectively (Fig. 4f). The spin–orbit split centred at 168.5 eV is ascribed to the unbound disulfides (S–S), similar to those observed on unwashed flat surfaces (Fig. 2). Interestingly, the ratio of the sulfur![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :gold (S/Au) was found to be 19%, significantly higher than an S/Au ratio of 4.8% observed on flat Au surfaces. This difference suggests that the high surface-to-volume ratio of spherical nanoparticles enables capturing more of the spike protein from the solution, compared to that with flat Au surfaces. In addition, UV-vis absorption spectra of AuNPs incubated in the spike S1 protein solution for different times show that the localized surface plasmon peak shifts to a higher wavelength (ESI Fig. S14†), suggesting that the surface coverage increases with increasing incubation time.
:gold (S/Au) was found to be 19%, significantly higher than an S/Au ratio of 4.8% observed on flat Au surfaces. This difference suggests that the high surface-to-volume ratio of spherical nanoparticles enables capturing more of the spike protein from the solution, compared to that with flat Au surfaces. In addition, UV-vis absorption spectra of AuNPs incubated in the spike S1 protein solution for different times show that the localized surface plasmon peak shifts to a higher wavelength (ESI Fig. S14†), suggesting that the surface coverage increases with increasing incubation time.
2.4. Single SARS-CoV-2 spike protein circuits
It has been demonstrated above that the SARS-CoV-2 spike S1 protein spontaneously reacts with Au, via the S–S bonds located at the extremities of the three protein domains, RBD, NTD and the S1/S2 cleavage site (Fig. 5a). We proceeded to test the capability of this reaction in enabling wiring a single protein in nano-circuits. The spontaneous reaction of the terminal S–S bonds enabled wiring the SARS-CoV-2 spike S1 protein between two gold electrodes using the scanning tunnelling microscopy break junction (STM-BJ) technique. In the STM-BJ approach, the Au STM tip is pushed in and out at a rate of 0.5 Å ms−1 from the surface in the presence of a 100 μg mL−1 solution of SARS-CoV-2 spike S1 protein in PBS (pH 7.4). The STM Au tip was coated with Apiezon wax to reduce leakage current in the aqueous environment.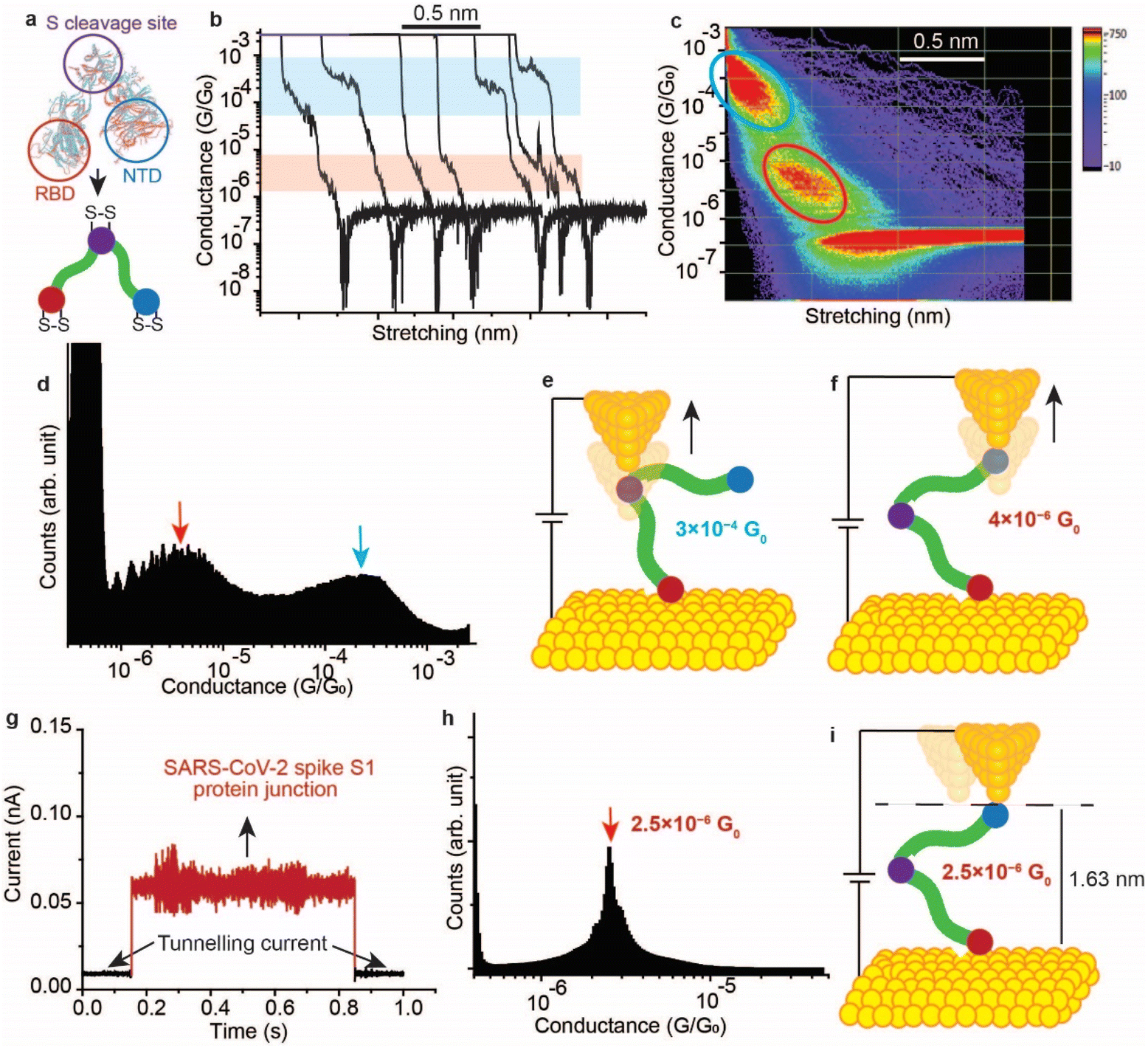 | ||
| Fig. 5 Single molecule circuits of SARS-CoV-2 spike S1 protein enabled by the reaction of S–S bonds with Au in a scanning tunnelling microscopy-break junction (STM-BJ). (a) An illustration of the SARS-CoV-2 spike S1 protein showing the S–S bond location at the peripheral of each protein domain. (b) Representative conductance versus distance traces of the STM-BJ operated in current–distance mode with bias set to 50 mV, for the Au–S–spike S1 protein–S–Au circuit. (c) Two-dimensional conductance versus distance histograms formed by accumulating 4000 individual traces without selection. The blue circle highlights one conductance state at 3 × 10−4G0 and the red circle highlights the second conductance state at 4 × 10−6G0. (d) One-dimensional histogram of the Au–S–spike S1 protein–S–Au circuit formed by accumulating 4000 traces with no selection. (e and f) Schematics representing the possible electron pathways in the Au–S–spike S1 protein–S–Au circuits. (g) Representative blink for a single spike S1 protein bonded between two Au nano-electrodes in an STM-BJ operating in the blinking (current–time) mode, measured at a bias voltage of 300 mV. (h) Blinking histogram of the Au–S–spike S1 protein–S–Au junction with an average conductance of 2.5 × 10−6G0. The blinking histogram was formed by accumulating 100 blinks. (i) Schematic representing the electron pathway in the Au–S–spike S1 protein–S–Au circuit in blinking mode where the protein is likely wired between the NTD and RBD sites which has the most peripheral S–S bonds and which can form spontaneous bonding with the Au tip and surface. | ||
Fig. 5b shows typical current versus distance traces for an Au–S–spike protein–S–Au circuit, with two conductance signals appearing at 4 × 10−6G0 and 3 × 10−4G0 with G0 = 2e2/ℏ = 77.5 μS. Fig. 5c and d show 2D and 1D histograms, respectively, formed of 4000 current vs. distance traces without applying any selection procedures (see the Experimental method section55–63). The two conductance signals are attributed to wiring the SARS-CoV-2 spike S1 protein in different configurations. Control STMBJ experiments in the PBS buffer solution but in the absence of SARS-CoV-2 protein showed no conductance peaks even at small biases (Fig. S15†). The higher conductance signal, 3 × 10−4G0, is attributed to the case where the S–S bonds in the RBD are connected to one of the junction electrodes and the other is connected to the S1/S2 cleavage site (Fig. 5e). This assignment is supported by the fact that the S1/S2 cleavage site contains a peripheral S–S bond connecting the amino acids at C662 and C671 (see Fig. 1b). On the other hand, the lower conductance 4 × 10−6G0 signal is attributed to a second configuration in which the spike protein is connected to one of the junction electrodes through the RBD and to the other STM electrode through the NTD S–S bonds (Fig. 5f). The NTD site also contains a very terminal S–S bond at positions C15 and C156 (see Fig. 1a). The two measured conductance states show two different plateaus stretching lengths of 0.4 nm and 0.6 nm corresponding to the higher (3 × 10−4G0) and the lower (4 × 10−6G0) conductance states, respectively. It should be noted here that these distances are not calibrated for the Au–Au snap-back distance (typically ∼0.5 nm).64 This is because a narrow-range linear current-amplifier was used which does not ensure the formation of Au–Au junctions during the pushing cycles. Hence, the plateau stretching length is estimated from the piezo retraction speed from a point close to the surface but not necessarily the Au–Au contact (see Experimental method section). The use of this approach can potentially explain the discrepancy between the molecular stretching observed and the dimension of the protein which is ∼7 nm from the cleavage site to either the RBD or the NTD end. Since S–S bonds are present at the terminal sites in all of the three subunits in the spike S1 protein, there is a third possibility of the protein circuit forming in which both the RBD and the NTD are both bound to the surface and the tip is connected to the S1/S2 cleavage site (ESI Fig. S16†), but this scenario is less likely to occur and signals corresponding to this configuration are not observed experimentally. This is consistent with the fact that the position of the first S–S bonds in the NTD is very exposed, at the amino acid residue C13 (the spike S1 protein is terminated by the amino acid residue C12). In contrast, most terminal S–S bonds in the S1/S2 cleavage site are located at the amino acid residue C671 which is not exposed. The terminal amino acid residue in the S1/S2 domain is C682 and does not contain S–S bonds. The position of the S–S bonds in the S1/S2 cleavage site is therefore more buried inside the protein compared to the NTD subunit. The RBD has 4 S–S bonds with an S–S bond at C480–C488 being the most exposed in the whole protein. Therefore, the probability of the surface binding to the S–S bond of the NTD and the RBD is higher than binding to that of the S1/S2 cleavage site.
We next measured the SARS-CoV-2 single molecule conductivity using the blinking approach of STM (current vs. time approach).65,66 In the blinking approach, the STM tip is fixed at a specific distance from the surface in the presence of a dilute concentration of the SARS-CoV-2 protein (10 μg mL−1). The blinking approach differs from STMBJ in that no physical contact takes place between the STM tip and the Au (111) surface. Once a specific tunnelling current is reached (which determines the distance separating the electrodes), the STM feedback system is disabled. Occasionally, a SARS-COV-2 spike protein bridges between the Au tip and the surface, and this event is accompanied by a sudden jump in the current (blink) above the tunnelling current background. In the absence of the protein in the circuit, the current decay value obtained (β) was ∼9 nm−1, and the electrode–electrode gap distance can be evaluated using the equation G = G0e(−βd) where d is the distance separating the two electrodes and G0 = 2e2/ℏ is the quantum of conductance (see Experimental method section). Fig. 5g shows a typical current vs. time “blink” for the Au–spike S1 protein–Au junction when the separation of the electrodes is ∼1.63 nm. The average conductance occurs at 2.5 × 10−6G0 (Fig. 5h), a value that is close to the lower conductance signal obtained using the current–distance STM-BJ approach (Fig. 5f) suggesting that only one connection pathway is enabled using the spontaneous junction approach of the blinking method. We hypothesise here that the fixed spacing between the junction electrodes enables only one specific configuration of the spike protein in which the protein connects to the Au electrodes from sites with most peripheral disulfide bonds. These sites are likely to be the NTD and the RBD because the RBD contains the most exposed S–S bond in the protein at the amino acid location 480–488, and the first S–S bond in the NTD is located at the third amino acid in the domain (amino acids 15–136, Fig. 1a).
In contrast to the blinking approach, the current–distance approach of STM-BJ (also referred to as tapping mode) the tip moves in and out of the surface. Therefore, the STM tip can penetrate deeper in the protein enabling contact with sites (e.g., S1/S2 cleavage site) other than that naturally occurring connection from the RBD and NTD. The penetration capability of the current–distance “tapping” approach explains the multiple conductance signals obtained (Fig. 5c and d).
2.5. Effect of electric fields on single SARS-CoV-2 spike protein circuits
The effect of the magnitude of the electric field on the structure of the SAR-CoV-2 spike S1 protein, in the electrified nano-gap, was studied by measuring the conductance, using STM-BJ experiments, at different bias-voltages. The electric field was estimated by dividing the bias magnitude by the stretching length of the protein junction during the pulling cycle. This is a simple approximation of the magnitude of the electric field, and it does not take into account the shape of the tip and the likelihood that the electric field can reach very high magnitudes when the tip is very close to the surface. At small biases of 50 mV (5 × 107 V m−1) and 75 mV (7.5 × 107 V m−1), the two conductance signals at 4 × 10−6G0 and 3 × 10−4G0 remain clearly visible (Fig. 6a). At 150 mV however, the two signals significantly change in magnitude (the two well-separated signals at 4 × 10−6G0 and 3 × 10−4G0 observed at low biases become closely connected in the range of 8 × 10−6G0 and 2 × 10−5G0). and the yield of the junctions significantly decreases (Fig. 6b). At 350 mV (∼3.5 × 108 V m−1), the two conductance signals almost disappear despite the histograms being composed of a similar number of curves (3500 curves) to that used to build the histograms at low biases. Only small signals that do not form clear conductance peaks remain visible in the range of 10−5 to 10−7G0 (Fig. 6c). | ||
| Fig. 6 Effect of applied electric field on the conductance of the Au-SARS-CoV-2 spike S1 protein–Au circuit obtained by STMBJ. (a–c) One-dimensional histograms of the Au–S–spike S1 protein–S–Au circuit at three different applied bias voltages, 75 mV (a), 150 mV (b) and 350 mV (c), formed by accumulating 3500 traces with no selection. (d–f) Two-dimensional histograms of the Au–S–spike S1 protein–S–Au circuit at the same three different applied bias voltages, 75 mV (d), 150 mV (e) and 350 mV (f). | ||
At a bias-voltage of 500 mV with an estimated electric field of 5 × 108 V m−1, no traces of any conductance signals remain (Fig. 7a and c). Applying a bias-voltage of 50 mV directly after 500 mV, the original signals at 4 × 10−6G0 and 3 × 10−4G0 are restored although with broader conductance distribution and a lower yield of the conductance plateaus (Fig. 7b and d). These results can be interpreted by the structure of SARS-CoV-2 spike S1 protein being stable under electric fields equal to or below 7.5 × 107 V m−1. At electric fields equal to or higher than 1.5 × 108 V m−1, we propose that the structure of the SARS-CoV-2 spike S1 protein changes in a way that prevents the protein from properly aligning in the junction which disrupts the efficient electron pathways that are naturally present in SARS-CoV-2 spike protein at low electric fields. The absence of the original conductance signals above 3.5 × 108 V m−1 can be possibly ascribed to SARS-CoV-2 protein denaturation, which prevents a proper orientation and connection to the junction electrodes. Another possibility is that at high electric fields, the protein is still intact in the junction but in an unfavourable orientation for efficient electron transfer. These propositions are inferred from observing less junction yield (smaller conductance peaks in the histograms) with applied voltage and changes in the conductance to lower values with applied bias. It should be noted here that in STMBJ of any molecule, clearer conductance peaks in the histograms and higher junction yield are typically obtained at low biases. However, in typical molecules, a change in the magnitude of the measured conductance with a change in the bias-voltage does not often occur. In addition, a significant change in the yield of the junctions to an extent of complete disappearance from the conductance histograms usually happens at very high biases (typically >1.5 V for molecules of ∼1 nm, e.g., experiencing an electric field of at least 1.5 × 109 V m−1).67–69 Therefore, the change in the magnitude of the conductance and the yield of the junctions observed here for the SARS-CoV-2 S1 protein cannot be simply related to the better environment of measurements at lower biases but is more likely due to a specific sensitivity of the SARS-CoV-2 spike S1 protein to electric fields. It is also worth noting that the effect of electric fields on the junction yield was only previously studied on small molecules with contact groups different from the S1 protein. Hence, further study of the effect of electric fields on the structure of the S1 protein, specifically their ability to expose S–S bonds to the junction electrodes, is needed.
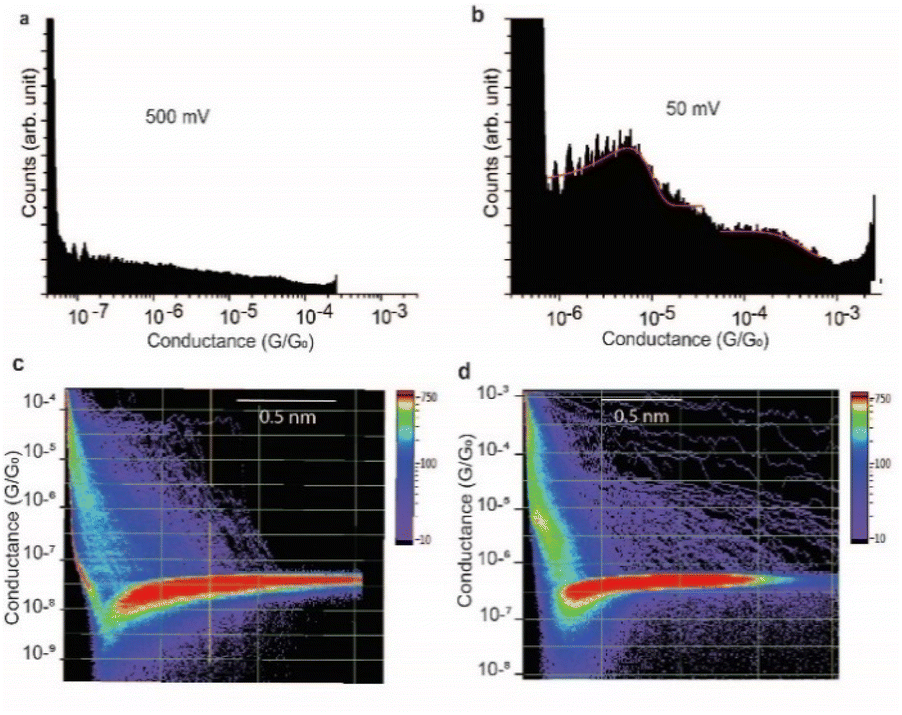 | ||
| Fig. 7 Effect of switching the magnitude of the electric field on SARS-CoV2 S1 protein conductance. (a) One-dimensional histograms of the Au–S–spike S1 protein–S–Au circuit at an applied bias voltage of 500 mV. (b) One-dimensional histograms of the Au–S–spike S1 protein–S–Au circuit after switching the bias voltage back to 50 mV. (c) and (d) 2D histograms of the Au–S–spike S1 protein–S–Au circuits at 500 mV and 50 mV, respectively. | ||
It has been recently shown via atomistic simulations that electric fields of moderate strengths (107 to 108 V m−1) can dramatically change the conformation of the SARS-CoV-2 S1 protein.70 One vital predicted field-induced conformational change occurs at the level of the recognition loop (L3) of the RBD where two parallel beta sheets, that are responsible for a high affinity to ACE2, undergo a change into an unstructured coil, making the RBD have no binding possibilities to the ACE2 receptor. The effect of moderate fields on the spike protein can be understood in terms of the interactions of the electric field with the permanent dipoles located in the protein. The protein's dipole moments orient along the field direction in order to minimise the electrostatic energy. With the rearrangements of the dipoles, the protein can undergo a significant conformational change.
We propose that the sensitivity of SARS-CoV-2 S1 protein to the electric field is the reason behind our observation of only clear conductance signals at 50–75 mV at which we estimated the electric field to be 5–7.5 × 107 V m−1. At higher fields, the conductance signals change and become unrecognisable at 3.5 × 108 V m−1. This does not quantitatively match the theoretical prediction of a change in the structure of the protein at field magnitudes as low as 107 V m−1.70 The conductance signals at 5–7.5 × 107 V m−1 reappear after their disappearance at high electric fields. Fig. 7 shows histograms at 500 mV at which no traces of conductance signals were observed (5 × 108 V m−1) and then their reappearance at a bias of 50 mV (5 × 107 V m−1), immediately after. Since each push–pull cycle in STMBJ experiments is likely to occur on a different area of the bottom Au surface, it is likely that fresh un-denatured proteins diffuse to the junction gap at low biases immediately after applying high biases and the conductance signals are recovered. Changes detected in the conductance of SARS-CoV-2 spike protein demonstrated here can be potentially used for characterising, detecting, and potentially denaturing and i.e., deactivating coronaviruses. For example, such electric fields can be incorporated in air filtering systems with magnitudes below what causes ionisation of air.
3. Conclusions
In summary, we demonstrate that SARS-CoV-2 spike protein reacts and forms covalent bonds with specific metals and Si. Metal surfaces that have affinity to thiols/disulfides such as Au, Pt and Cu covalently bond to the spike protein via M–S bonding. Si surfaces also showed covalent Si–S bonding between the protein's S–S bonds and the surface, a process that is triggered by a spontaneous electrochemical reaction that involves partial oxidation of the Si surface and the reduction of disulfide bonds in the protein. The rate of the reaction is two-fold higher on Si than on metals which is attributed to a slow homolytic cleavage of the S–S bonds in the spike protein when in contact with metals unlike the electrochemically initiated reaction on Si. In contrast, common surfaces such as plastic and stainless steel showed no covalent bonding between the protein and the surface, and the protein remains only physically adsorbed on these surfaces. The spike protein was also shown to react efficiently with AuNPs with one order of magnitude higher S/Au ratio for AuNPs compared to flat Au surfaces. The capability of Si, Pt, Au and Cu to react with the spike protein can potentially be used to develop anti-coronavirus surfaces that are capable of irreversibly trapping the virus via strong covalent bonds. This covalent bonding potentially explains why SARS-CoV-2 survives a limited amount of time on copper compared to its viability on stainless steel and plastics.4 The reaction of S–S in SARS-CoV-2 with metals and Si is particularly relevant because all previous and likely future coronaviruses will possess peripheral disulfide bonds in their spike proteins.Another important implication of the reaction is that it enables efficient electrical coupling of the spike proteins to commonly used electrode materials such as Si and Au. Owing to the efficient electrical connection to Au,71–74 we demonstrate that the spike protein is highly conductive at the single molecule level, with two conductive channels associated with the different orientation of the protein in the electrified nano-gap. A 3 × 10−4G0 conductance is attributed to a single SARS-COV-2 spike protein S1 connecting to the two STM nano-electrodes from the RBD subunit and the S1/S2 cleavage site. Another conductance signal of 4 × 10−6G0 is attributed to the spike protein connecting to the STM electrodes from the RBD subunit and the NTD. Tuning to a specific conducting channel can be achieved by fixing the distances between the electrodes such that the conducting channel is dictated by the spike protein connection to the Au electrodes from sites that contain the most peripheral S–S bonds. Clear conductance signals are only observed at electric fields equal to or lower than 7.5 × 107 V m−1. At an electric field of 1.5 × 108 V m−1, the conductance signals change to a lower magnitude and exhibit a lower yield. This change in the electrical signals can be possibly interpreted by a change in the structure of the spike protein at that field magnitude. It is worth noting here that this field magnitude is higher than what is predicted to cause a change in the structure of the RBD and therefore the efficient conductivity observed at low field magnitudes could possibly be due to an already altered structure of the RBD and the entire S1 protein in the junction.
Above an electric field of 3.5 × 108 V m−1, no junctions with the protein were observed. The disappearance of the protein's current signature at such field magnitudes can be explained by (i) the protein denaturing in the electrified nano-gap which would consequently lead to the electron channels across the protein to be blocked and (ii) the unfavourable orientation of the protein in the junction at high electric fields, which can also lead to the blocking of the electron channels. Electric fields have been theorised and experimentally demonstrated to have a profound effect on the chemical structure and reactivity75–78 and this study indicates that similar effects need to be considered for SARS-CoV-2 spike proteins. Also needed is an accurate determination of the magnitude of the electric field in STMBJ experiments which largely depends on the structure of the STM tip and the distance between the two junction electrodes. These findings open new directions for electrically detecting, characterising, and potentially disabling coronaviruses and their future variants using external electric fields.
4. Experimental method
4.1. General chemicals and materials
All chemicals were used without further modifications and were of analytical grade, unless otherwise stated. The SARS-CoV-2 spike protein S1 subunit was purchased from Thermo Fisher Scientific, catalog number (RP-87679). The spike protein S1 subunit was lyophilized from sterile PBS buffer pH 7.4, 5% mannitol and 0.01% Tween-80. Silicon wafers were purchased from Siltronix, S.A.S. (Archamps, France). The silicon was p-type, boron highly doped, with resistivity ca. 0.007 Ω cm, and a wafer thickness of 500 ± 25 μm oriented ±0.5° away from the (111) plane. Au, Pt, Cu, and stainless-steel foils were purchased from Goodfellow, UK. The thickness of the foils was 0.125 mm with a purity of 99.999%. The PTFE substrate was purchased from McMaster-Carr, US. The Piranha solutions used in silicon cleaning were sulfuric acid (Puranal TM, 95–97%) and hydrogen peroxide (30 wt% in water) of semiconductor grades from Sigma-Aldrich. Ammonium fluoride (Puranal TM, 40 wt% in water) was used in the Si etching step and was bought from Sigma-Aldrich. (±)-α-Lipoic acid ≥ 98.0 was obtained from Sigma-Aldrich. The dichloromethane (DCM), acetonitrile (ACN), isopropyl alcohol, and ethanol solvents were purified by distillation before use. Tetrabutylammonium hexafluorophosphate 98%, used in electrochemical reduction experiments, was purchased from Sigma-Aldrich. Milli-Q water (>18 MΩ cm) and was used for all the cleaning procedures and the electrolyte preparation. 20 and 50 nm AuNPs, citrate coated, were purchased from Sigma-Aldrich and were used as received.4.2. Preparation of the spike protein solution
The lyophilized powder of the SARS-CoV-2 spike protein (103 kDa) was dissolved in 1 mL of sterile water to form a stock solution of 100 μg mL−1, split into portions to avoid defreezing and refreezing and stored at −20 °C.4.3. Surface interaction with the spike protein
Metal surfaces including Pt, Au, Cu, stainless steel, and the PTFE plastic surfaces were cleaned with acetone, isopropanol, and water sequentially under sonication for 5 minutes and then dried out with a stream of argon gas, before further use. The Si–H surface was prepared by cutting a Si wafer into pieces (1 cm × 1 cm), and then cleaned with DCM, isopropanol, and water before treating the surface with a Piranha solution of sulfuric acid:hydrogen peroxide (3:1, v/v) for 30 min at 100 °C. The Piranha treated wafers were then washed thoroughly with Milli-Q water before etching in ammonium fluoride solution (40%) in the presence of traces of sodium sulfite salt as a radical scavenger, for 13 minutes. The etched Si–H surfaces were then washed with Milli-Q water and DCM and then dried out with a stream of argon gas before further use. The clean surfaces were then incubated in the SARS-CoV-2 spike protein solution (by adding 200 μL of the spike protein (100 μg mL−1) on the surface) for 12 h (or 1 h), then washed in Milli-Q water and dried with argon before further analysis. For the azurin control experiment, 200 μL of the azurin solution (500 μg mL−1) was deposited on the Si surface, and then left for 12 h before washing the surface and drying with a stream of Ar gas. For kinetics experiments, the same procedures were followed except for incubating the surfaces in the spike protein for different periods of time.
4.4. Cyclic voltammetry measurements
All electrochemical measurements were performed with a CHI650 (CH Instruments, USA) electrochemical workstation, using a three-electrode system cell, a Pt wire as the counter electrode and Ag/AgCl as the reference electrode. Cyclic voltammetry measurements were carried out in 1 M NaClO4 solution. For the protein CV experiments, 400 μL of the protein stock was mixed with 2 mL PBS buffer pH 7.4 (final concentration 20 μg mL−1), and the voltammograms were recorded against an aqueous Ag/AgCl reference electrode. For the CV experiments with the cyclic disulfide molecule, alpha lipoic acid, the voltammograms were recorded for a solution of the alpha lipoic acid (1 mM) in an organic electrolyte of ammonium tertbutyl hexafluorophosphate (0.5 M) in dry DMF electrolyte, against a leakless Ag/AgCl reference electrode.4.5. Atomic force microscopy imaging
AFM topography images were acquired using a Bruker Dimension microscope, operating in tapping mode. All images were recorded using silicon nitride cantilevers (TESPA from Bruker, with a spring constant of 20 N m−1) under ambient conditions.4.6. X-ray photoelectron spectroscopy (XPS) analysis
XPS measurements were performed on a Kratos Axis Ultra DLD spectrometer, using a monochromatic Al-Kα (1486.6 eV) irradiation source operating at 150 W. The spectra of C 1s (277–300 eV) and S 2p (163–164 eV) were taken in normal emission at or below 7 × 10−9 torr. Data files were processed using CasaXPS© software, and the reported XPS energies are binding energies expressed in eV. After background subtraction (Shirley), spectra were fitted with Voigt functions. To correct for energy shifts caused by adventitious charging, all peak energies were corrected with a rigid shift to bring the C 1s emission to 284.7 eV.4.7. UV-vis spectrometry
1 mL of 20 nm (or 50 nm) size AuNPs at a stock concentration of 6 × 1011 particles per mL were mixed with 100 μL of the spike S1 protein (100 μg mL−1), and then the mixture was rested for 12 h. The absorbance was scanned in the range of 200–800 nm, at a step size of 0.2 nm, and recorded on a Shimadzu 2401 PC UV-vis spectrophotometer.4.8. STM break junction measurements
Scanning tunnelling microscopy-break junction (STM-BJ) experiments were carried out using a PicoSPM I microscope head that is controlled by a “Picoscan 2500” controller from Agilent Technologies. Data acquisition was carried out using a NI-DAQmx/BNC-2110 National Instruments and analysed using a home-built LABVIEW code. The STMBJ was first calibrated by recording direct Au–Au quantum point contacts (Fig. S17, ESI†) at 1G0 (77.5 μS). Single-protein conductivity measurements were performed in a solution of the spike S1 protein in PBS (pH 7.4). The STM Au tip was coated with Apiezon wax to reduce leakage current in the aqueous environment. The leakage current was typically 2–3 pA. Details about the STM-BJ technique are published elsewhere.55,57,58,60–62,79–82 ∼4000 current–distance curves were collected for the Au–spike protein–Au circuit and were accumulated in 1D and 2D conductance histograms without selection. In the blinking (current–time) mode, to ensure mechanical stability as much as possible, the set-point tunnelling current was first set low such that the tip is placed away from the surface with applying a waiting period of 1 h. The set-point tunnelling current was then set to the desired value and the tip is brought closer to the surface. Another waiting period of 1 h is then applied, after which the i–t blinks were collected. Also in the (current–time) mode, the junction separation was estimated from the set-point current and the applied bias voltage. First, the current decay values (β) were obtained in the absence of proteins and were found to be ∼9 nm−1. The electrode–electrode gap distance was then evaluated using the equation G = G0e(−βd) where d is the distance separating the two electrodes and G0 = 2e2/ℏ is the quantum of conductance; e is the electron charge and ℏ is the Planck's constant. For a tunnelling current of 0.01 nA and a bias-voltage of 300 mV, the distance between the Au electrodes in the blinking (current–time) approach was estimated to be 1.63 nm.In order to facilitate estimation of the molecular stretching during the pulling cycle, we used a code to shift all the current–distance decays on the x-axis to have as a starting point the current dropping directly from current saturation such that they are all equivalent in time. The time scale was then changed to applied voltage knowing the V s−1 ramp, and then to piezo's retraction via the particular piezo's sensitivity (nm V−1) used. The current versus piezo's retraction was then plotted without taking into account the Au–Au snap distance.
The STMBJ setup utilises a linear current amplification. Therefore, the current–distance plots are collected in the linear scale but plotted on the log scale (y-axis) for clarity. This leads to the appearance of pronounced dips near the noise level as seen in Fig. 5 and 6.
Data availability
Additional experimental details and data are provided in the ESI,† including XPS, AFM, UV-vis spectra, electrochemical measurements, and additional STMBJ data. STM experimental data are available from N. D. upon request. Single-molecule acquisition and treatment codes are available from N. D. upon request.Author contributions
Essam Dief: investigation, formal analysis, writing – original draft. Nadim Darwish: conceptualization, supervision, investigation, formal analysis, methodology, writing – original draft, resources.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
N. D. acknowledges the support by the Western Australian Future Health Research and Innovation Fund and the Australian Research Council Discovery Project DP190100735.Notes and references
- O. World Health, Coronavirus disease 2019 (COVID-19): situation report, 51, World Health Organization, Geneva, 2020 Search PubMed.
- G. Kampf, D. Todt, S. Pfaender and E. Steinmann, J. Hosp. Infect., 2020, 104, 246–251 CrossRef CAS PubMed.
- P. Y. Chia, K. K. Coleman, Y. K. Tan, S. W. X. Ong, M. Gum, S. K. Lau, X. F. Lim, A. S. Lim, S. Sutjipto, P. H. Lee, T. T. Son, B. E. Young, D. K. Milton, G. C. Gray, S. Schuster, T. Barkham, P. P. De, S. Vasoo, M. Chan, B. S. P. Ang, B. H. Tan, Y. S. Leo, O. T. Ng, M. S. Y. Wong and K. Marimuthu, Nat. Commun., 2020, 11, 2800 CrossRef CAS PubMed.
- N. van Doremalen, T. Bushmaker, D. H. Morris, M. G. Holbrook, A. Gamble, B. N. Williamson, A. Tamin, J. L. Harcourt, N. J. Thornburg, S. I. Gerber, J. O. Lloyd-Smith, E. de Wit and V. J. Munster, N. Engl. J. Med., 2020, 382, 1564–1567 CrossRef PubMed.
- A. C. Walls, Y.-J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 181, 281–292 CrossRef CAS PubMed.
- F. Li, W. Li, M. Farzan and C. H. Stephen, Science, 2005, 309, 1864–1868 CrossRef CAS PubMed.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C. L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS PubMed.
- Z. Li, A. C. Tomlinson, A. H. Wong, D. Zhou, M. Desforges, P. J. Talbot, S. Benlekbir, J. L. Rubinstein and J. M. Rini, eLife, 2019, 8, e51230 CrossRef CAS PubMed.
- S. Suhail, J. Zajac, C. Fossum, H. Lowater, C. McCracken, N. Severson, B. Laatsch, A. Narkiewicz-Jodko, B. Johnson, J. Liebau, S. Bhattacharyya and S. Hati, Protein J., 2020, 39, 644–656 CrossRef PubMed.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang, L. Zhang and X. Wang, Nature, 2020, 581, 215–220 CrossRef CAS PubMed.
- M. Manček-Keber, I. Hafner-Bratkovič, D. Lainšček, M. Benčina, T. Govednik, S. Orehek, T. Plaper, V. Jazbec, V. Bergant, V. Grass, A. Pichlmair and R. Jerala, FASEB J., 2021, 35, e21651 CrossRef PubMed.
- Y. Shi, A. Zeida, C. E. Edwards, M. L. Mallory, S. Sastre, M. R. Machado, R. J. Pickles, L. Fu, K. Liu, J. Yang, R. S. Baric, R. C. Boucher, R. Radi and K. S. Carroll, Proc. Natl. Acad. Sci. U. S. A., 2022, 119 CAS.
- C. Watanabe, Y. Okiyama, S. Tanaka, K. Fukuzawa and T. Honma, Chem. Sci., 2021, 12, 4722–4739 RSC.
- K. Sargsyan, C.-C. Lin, T. Chen, C. Grauffel, Y.-P. Chen, W.-Z. Yang, H. S. Yuan and C. Lim, Chem. Sci., 2020, 11, 9904–9909 RSC.
- A. M. Harbison, C. A. Fogarty, T. K. Phung, A. Satheesan, B. L. Schulz and E. Fadda, Chem. Sci., 2022, 13, 386–395 RSC.
- J. R. Reimers, M. J. Ford, A. Halder, J. Ulstrup and N. S. Hush, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E1424 CrossRef CAS PubMed.
- E. Pensa, E. Cortés, G. Corthey, P. Carro, C. Vericat, M. H. Fonticelli, G. Benítez, A. A. Rubert and R. C. Salvarezza, Acc. Chem. Res., 2012, 45, 1183–1192 CrossRef CAS PubMed.
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805–1834 RSC.
- C. Vericat, M. E. Vela, G. Corthey, E. Pensa, E. Cortés, M. H. Fonticelli, F. Ibañez, G. E. Benitez, P. Carro and R. C. Salvarezza, RSC Adv., 2014, 4, 27730–27754 RSC.
- T. Wink, S. J. van Zuilen, A. Bult and W. P. van Bennekom, Analyst, 1997, 122, 43R–50R RSC.
- M. Frasconi, F. Mazzei and T. Ferri, Anal. Bioanal. Chem., 2010, 398, 1545–1564 CrossRef CAS PubMed.
- S. Dong and J. Li, Bioelectrochem. Bioenerg., 1997, 42, 7–13 CrossRef CAS.
- S. Y. Yeung, A. Mucha, R. Deshmukh, M. Boutrus, T. Arnebrant and B. Sellergren, ACS Cent. Sci., 2017, 3, 1198–1207 CrossRef CAS PubMed.
- M. C. Walkey, C. R. Peiris, S. Ciampi, A. C. Aragonès, R. B. Domínguez-Espíndola, D. Jago, T. Pulbrook, B. W. Skelton, A. N. Sobolev, I. Díez Pérez, M. J. Piggott, G. A. Koutsantonis and N. Darwish, ACS Appl. Mater. Interfaces, 2019, 11, 36886–36894 CrossRef CAS PubMed.
- N. Darwish, M. N. Paddon-Row and J. J. Gooding, Acc. Chem. Res., 2014, 47, 385–395 CrossRef CAS PubMed.
- N. Darwish, P. K. Eggers, S. Ciampi, Y. Tong, S. Ye, M. N. Paddon-Row and J. J. Gooding, J. Am. Chem. Soc., 2012, 134, 18401–18409 CrossRef CAS PubMed.
- E. M. Dief and N. Darwish, Curr. Opin. Electrochem., 2022, 101019, DOI:10.1016/j.coelec.2022.101019.
- R. Sahli, C. Fave, N. Raouafi, K. Boujlel, B. Schöllhorn and B. Limoges, Langmuir, 2013, 29, 5360–5368 CrossRef CAS PubMed.
- Y. Zhou, T. Nagaoka and G. Zhu, Biophys. Chem., 1999, 79, 55–62 CrossRef CAS PubMed.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS PubMed.
- F. Lisdat and I. Karube, Biosens. Bioelectron., 2002, 17, 1051–1057 CrossRef CAS PubMed.
- M. S. Inkpen, Z. F. Liu, H. Li, L. M. Campos, J. B. Neaton and L. Venkataraman, Nat. Chem., 2019, 11, 351–358 CrossRef CAS PubMed.
- J. Yan, R. Ouyang, P. S. Jensen, E. Ascic, D. Tanner, B. Mao, J. Zhang, C. Tang, N. S. Hush, J. Ulstrup and J. R. Reimers, J. Am. Chem. Soc., 2014, 136, 17087–17094 CrossRef CAS PubMed.
- Q. Chi, M. J. Ford, A. Halder, N. S. Hush, J. R. Reimers and J. Ulstrup, Curr. Opin. Electrochem., 2017, 1, 7–15 CrossRef CAS.
- M. J. Sailor, in Handbook of Porous Silicon, ed. L. Canham, Springer International Publishing, Cham, 2014, pp. 355–380, DOI:10.1007/978-3-319-05744-6_37.
- T. G. Truong, C. Mériadec, B. Fabre, J. F. Bergamini, O. de Sagazan, S. Ababou-Girard and G. Loget, Nanoscale, 2017, 9, 1799–1804 RSC.
- H. Sugimoto, M. Fujii and K. Imakita, Nanoscale, 2016, 8, 10956–10962 RSC.
- I. Choi, Y. Shin, J. Song, S. Hong, Y. Park, D. Kim, T. Kang and L. P. Lee, ACS Nano, 2016, 10, 7639–7645 CrossRef CAS PubMed.
- C. R. Peiris, S. Ciampi, E. M. Dief, J. Zhang, P. J. Canfield, A. P. Le Brun, D. S. Kosov, J. R. Reimers and N. Darwish, Chem. Sci., 2020, 11, 5246–5256 RSC.
- E. M. Dief, Y. B. Vogel, C. R. Peiris, A. P. Le Brun, V. R. Gonçales, S. Ciampi, J. R. Reimers and N. Darwish, Langmuir, 2020, 36, 14999–15009 CrossRef CAS PubMed.
- J. R. Reimers, J. Yang, N. Darwish and D. S. Kosov, Chem. Sci., 2021, 12, 15870–15881 RSC.
- C. Godon, J. M. Teulon, M. Odorico, C. Basset, M. Meillan, L. Vellutini, S. W. Chen and J. L. Pellequer, J. Struct. Biol., 2017, 197, 322–329 CrossRef CAS PubMed.
- E. Migliorini, M. Weidenhaupt and C. Picart, Biointerphases, 2018, 13, 06D303 CrossRef PubMed.
- N. Bonander, J. Leckner, H. Guo, B. G. Karlsson and L. Sjölin, Eur. J. Biochem., 2000, 267, 4511–4519 CAS.
- X. Zhang, L. Wang, Z. Chen, Y. Li, X. Luo and Y. Li, RSC Adv., 2019, 9, 13550–13560 RSC.
- G. Milazzo, S. Caroli and R. D. Braun, J. Electrochem. Soc., 1978, 125, 261C CrossRef.
- A. J. Bard, R. Parsons, J. Jordan and International Union of Pure and Applied Chemistry, ed. M. Dekker, Standard Potentials in Aqueous Solution, New York, 1st edn, 1985 Search PubMed.
- L. Zhao, N. Heinig and K. T. Leung, Langmuir, 2013, 29, 927–931 CrossRef CAS PubMed.
- E. M. Dief, Y. B. Vogel, C. R. Peiris, A. P. Le Brun, V. R. Gonçales, S. Ciampi, J. R. Reimers and N. Darwish, Langmuir, 2020, 36, 14999–15009 CrossRef CAS PubMed.
- M. Karimi, M. T. Ignasiak, B. Chan, A. K. Croft, L. Radom, C. H. Schiesser, D. I. Pattison and M. J. Davies, Sci. Rep., 2016, 6, 38572 CrossRef CAS PubMed.
- T. R. Ralph, M. L. Hitchman, J. P. Millington and F. C. Walsh, J. Electroanal. Chem., 1994, 375, 1–15 CrossRef CAS.
- M. T. Stankovich and A. J. Bard, J. Electroanal. Chem. Interfacial Electrochem., 1977, 85, 173–183 CrossRef CAS.
- X. Huang and M. A. El-Sayed, J. Adv. Res., 2010, 1, 13–28 CrossRef.
- M. Ouellette, F. Masse, M. Lefebvre-Demers, Q. Maestracci, P. Grenier, R. Millar, N. Bertrand, M. Prieto and É. Boisselier, Sci. Rep., 2018, 8, 14357 CrossRef PubMed.
- B. Xu and N. J. Tao, Science, 2003, 301, 1221 CrossRef CAS PubMed.
- W. Haiss, R. J. Nichols, H. van Zalinge, S. J. Higgins, D. Bethell and D. J. Schiffrin, Phys. Chem. Chem. Phys., 2004, 6, 4330–4337 RSC.
- S. V. Aradhya and L. Venkataraman, Nat. Nanotechnol., 2013, 8, 399–410 CrossRef CAS PubMed.
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Chem. Rev., 2016, 116, 4318–4440 CrossRef CAS PubMed.
- Y. Li, C. Yang and X. Guo, Acc. Chem. Res., 2020, 53, 159–169 CrossRef CAS PubMed.
- T. Albrecht, Nat. Commun., 2012, 3, 829 CrossRef CAS PubMed.
- M. S. Inkpen, M. Lemmer, N. Fitzpatrick, D. C. Milan, R. J. Nichols, N. J. Long and T. Albrecht, J. Am. Chem. Soc., 2015, 137, 9971–9981 CrossRef CAS PubMed.
- Y. Zang, A. Pinkard, Z.-F. Liu, J. B. Neaton, M. L. Steigerwald, X. Roy and L. Venkataraman, J. Am. Chem. Soc., 2017, 139, 14845–14848 CrossRef CAS PubMed.
- R. S. Klausen, J. R. Widawsky, T. A. Su, H. Li, Q. Chen, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Chem. Sci., 2014, 5, 1561–1564 RSC.
- W. Hong, D. Z. Manrique, P. Moreno-García, M. Gulcur, A. Mishchenko, C. J. Lambert, M. R. Bryce and T. Wandlowski, J. Am. Chem. Soc., 2012, 134, 2292–2304 CrossRef CAS PubMed.
- R. J. Nichols, W. Haiss, S. J. Higgins, E. Leary, S. Martin and D. Bethell, Phys. Chem. Chem. Phys., 2010, 12, 2801–2815 RSC.
- R. J. Nichols and S. J. Higgins, Nat. Mater., 2015, 14, 465–466 CrossRef CAS PubMed.
- H. Li, T. A. Su, V. Zhang, M. L. Steigerwald, C. Nuckolls and L. Venkataraman, J. Am. Chem. Soc., 2015, 137, 5028–5033 CrossRef CAS PubMed.
- A. C. Aragones, N. L. Haworth, N. Darwish, S. Ciampi, N. J. Bloomfield, G. G. Wallace, I. Diez-Perez and M. L. Coote, Nature, 2016, 531, 88–91 CrossRef CAS PubMed.
- L. Yuan, L. Jiang and C. A. Nijhuis, Adv. Funct. Mater., 2018, 28, 1801710 CrossRef.
- C. R. Arbeitman, P. Rojas, P. Ojeda-May and M. E. Garcia, Nat. Commun., 2021, 12, 5407 CrossRef CAS PubMed.
- T. Q. Ha, I. J. Planje, J. R. G. White, A. C. Aragonès and I. Díez-Pérez, Curr. Opin. Electrochem., 2021, 28, 100734 CrossRef CAS.
- M. P. Ruiz, A. C. Aragonès, N. Camarero, J. G. Vilhena, M. Ortega, L. A. Zotti, R. Pérez, J. C. Cuevas, P. Gorostiza and I. Díez-Pérez, J. Am. Chem. Soc., 2017, 139, 15337–15346 CrossRef CAS PubMed.
- B. Zhang, W. Song, P. Pang, H. Lai, Q. Chen, P. Zhang and S. Lindsay, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5886 CrossRef CAS PubMed.
- B. Zhang, H. Deng, S. Mukherjee, W. Song, X. Wang and S. Lindsay, ACS Nano, 2020, 14, 1360–1368 CrossRef CAS PubMed.
- S. Shaik, D. Mandal and R. Ramanan, Nat. Chem., 2016, 8, 1091–1098 CrossRef CAS PubMed.
- S. Ciampi, N. Darwish, H. M. Aitken, I. Diez-Perez and M. L. Coote, Chem. Soc. Rev., 2018, 47, 5146–5164 RSC.
- S. Shaik, R. Ramanan, D. Danovich and D. Mandal, Chem. Soc. Rev., 2018, 47, 5125–5145 RSC.
- H. Wang and H. Li, Chem. Sci., 2020, 11, 12512–12521 RSC.
- W. Haiss, R. J. Nichols, H. van Zalinge, S. J. Higgins, D. Bethell and D. J. Schiffrin, Phys. Chem. Chem. Phys., 2004, 6, 4330–4337 RSC.
- C. Gu, C. Jia and X. Guo, Small Methods, 2017, 1, 1700071 CrossRef.
- Y. Li, C. Yang and X. Guo, Acc. Chem. Res., 2020, 53, 159–169 CrossRef CAS PubMed.
- Y. Zang, Q. Zou, T. Fu, F. Ng, B. Fowler, J. Yang, H. Li, M. L. Steigerwald, C. Nuckolls and L. Venkataraman, Nat. Commun., 2019, 10, 4482 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental characterisation. See DOI: https://doi.org/10.1039/d2sc06492h |
| This journal is © The Royal Society of Chemistry 2023 |