
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcid-induced nitrite reduction of nonheme iron(II)-nitrite: mimicking biological Fe–NiR reactions†
Kulbir
 a,
Sandip
Das
a,
Tarali
Devi
c,
Somnath
Ghosh
a,
Subash
Chandra Sahoo
b and
Pankaj
Kumar
*a
a,
Sandip
Das
a,
Tarali
Devi
c,
Somnath
Ghosh
a,
Subash
Chandra Sahoo
b and
Pankaj
Kumar
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research (IISER), Tirupati 517507, India. E-mail: pankajatiisert@gmail.com; pankaj@iisertirupati.ac.in
bDepartment of Chemistry, Punjab University, Chandigarh, Punjab, India
cHumboldt-Universität zu Berlin, Institut für Chemie, Brook-Taylor-Straße 2, D-12489 Berlin, Germany
First published on 23rd February 2023
Abstract
Nitrite reductase (NiR) catalyzes nitrite (NO2−) to nitric oxide (NO) transformation in the presence of an acid (H+ ions/pH) and serves as a critical step in NO biosynthesis. In addition to the NiR enzyme, NO synthases (NOSs) participate in NO production. The chemistry involved in the catalytic reduction of NO2−, in the presence of H+, generates NO with a H2O molecule utilizing two H+ + one electron from cytochromes and is believed to be affected by the pH. Here, to understand the effect of H+ ions on NO2− reduction, we report the acid-induced NO2− reduction chemistry of a nonheme FeII-nitrito complex, [(12TMC)FeII(NO2−)]+ (FeII–NO2−, 2), with variable amounts of H+. FeII–NO2− upon reaction with one-equiv. of acid (H+) generates [(12TMC)Fe(NO)]2+, {FeNO}7 (3) with H2O2 rather than H2O. However, the amount of H2O2 decreases with increasing equivalents of H+ and entirely disappears when H+ reaches ≅ two-equiv. and shows H2O formation. Furthermore, we have spectroscopically characterized and followed the formation of H2O2 (H+ = one-equiv.) and H2O (H+ ≅ two-equiv.) and explained why bio-driven NiR reactions end with NO and H2O. Mechanistic investigations, using 15N-labeled-15NO2− and 2H-labeled-CF3SO3D (D+ source), revealed that the N atom in the {Fe14/15NO}7 is derived from the NO2− ligand and the H atom in H2O or H2O2 is derived from the H+ source, respectively.
Nitric oxide (NO), a critical biological component, participates in numerous bio-physiological processes such as neurotransmission, vascular regulation, inhibiting platelet aggregation, and immune response to multiple infections at nanomolar concentration.1 Also, NO is known to be involved in plant growth and development.2 NO meagerness may cause pathogenic effects such as atherosclerosis, diabetic hypertension, etc.3 However, at micromolar concentrations, NO is highly toxic and utilized for immune defense against harmful pathogens,4 in addition to its oxidized species, i.e., peroxynitrite (ONOO−)5 or/nitrogen dioxide (˙NO2).6 In contrast to the immune response towards pathogens, oxidized NO species also show various toxicological actions in biological systems.5b,7
Hence, sensible production of NO is required to maintain physiological homeostasis and is usually achieved by two metalloenzymes, i.e., NO synthases (NOSs)8 and/or nitrite reductases (NiRs).8a,9 NOS enzymes are heme-proteins that generate NO by catalyzing the conversion of L-arginine to L-citrulline under aerobic conditions.8b,c However, under ischemia and hypoxic conditions, the suppression of NOS activity results in the decrease of NO generation. Under such conditions, NO2− works as an active NO source in biological systems, generating NO in acid-induced NO2− reduction reactions.10 Sometimes, under abnormal conditions, biochemical dysfunction may cause NO overproduction by NiRs or NOSs. Under such conditions, NO dioxygenase (NOD) enzymes available in vivo convert excess NO to biologically benign nitrate (NO3−).11 NO3−, the product of the NOD reaction, serves as a critical component of NO2− generation and a precursor to the biological NO cycle.12 In humans, commensal bacteria in the oral cavity play a vital role in converting NO3− to NO2−.12a Bacteria reduce NO3− to NO2−via an OAT reaction mediated by molybdenum-based NR enzymes.13 The interconversion of NO3− to/NO2−/to NO (or vice versa) is the critical step of the denitrification process.14In vivo studies have proven that NO2− is a fundamental source of NO in mammalian or bacterial systems, an intermediate species of the biological nitrogen cycle (NO3− → NO2− → NO → N2O → N2).14 At the bio-physiological level, NO2− gets reduced to NO, primarily by globins15 or by acid-catalyzed NO2− reduction in the stomach16,17 or by Fe/Cu-NiR enzymes/cytochrome c oxidase (CcO)/xanthine oxidase,18 which reduces NO2− to NO in the presence of two-equiv. of H+ ions, i.e.,8a,9,15
| NO2− + e− + 2H+ → NO + H2O | (1) |
However, few functional mimicking models were developed and investigated in vivo/in vitro to explore the mechanistic insight of microbial NiR enzymatic chemistry. Brooks and coworkers proposed the NiR activity of mammalian hemoglobin (Hb) protein under anaerobic conditions, which converts NO2− to NO with the formation of metHb.19 E. T. Papish et al. explored the Cu-NiR chemistry and showed NO production with H2O as a side product via a CuI–NO+ ↔ CuII–NO intermediate in the reaction of Cu–NO2− with two-equiv. of H+.20 A heme-Fe/Cu assembly model has been developed to mimic the cytochrome c oxidase, illustrating the reversible conversion of NO2− to NO.21 For the first time, Murphy and coworkers explored the explained Cu–NO2− reduction reaction to release NO via the {CuNO} intermediate and characterized it structurally.9b,22 Patra and coworkers have mimicked NO2− reduction reactivity using CuII–NO2− with two-equiv. of H+ and one e−, leading to NO and H2O molecule formation.23 Lehnert and coworkers have explored electrocatalytic reduction using CuII–NO2− producing NO in aqueous media24 and compiled the electronic structure and reactivity of the biologically relevant coordination chemistry of iron and NO.15 In addition to acid-encouraged NO2− reduction to NO in different model systems. Various models have been explored for the reduction of metal-bound NO2− to NO via (i) oxygen atom transfer (OAT) caused by (R2S)25/thiol (RSH)}18a,26/triphenylphosphine (PPh3)27/vanadium chloride (VCl3)28 and (ii) photo-induced reactions29 of metal–bound nitrite (M–NO2−).
In addition to the developments on biomimetic synthetic modeling of M–NOs/or active sites associated with NiR and/or NOS.11d,30 Recently, Lehnert and coworkers have established the synthetic strategy for Fe–NOs,31 and Nam and coworkers have explored the photo-induced NiR reactivity of Fe–NO2− to generate Fe–NOs and also stabilize Co–NOs.32 Ford and coworkers continuously discover the NiR chemistry of various heme systems.33 Although NiR is the key source of NO in the biological system, such reactions are not investigated extensively to characterize the intermediates and transition states of NO2− reduction reactions. Hence, several research groups are working to understand the proper NO2− reduction reaction mechanism. There are only very few reports on acid-induced NO2− reduction reactions to mimic NiR enzymatic reactions and understand the mechanistic aspects.9b,29,34 In this investigation, we intend to characterize different intermediates of H+-induced NO2− reduction in FeII–NO2− and its reaction products and then explore its mechanistic aspects. This report will focus on how different amounts of acid (H+ ion) affect the reaction mechanism and regulate the side products in addition to NO.
Herein, we report the NO2− reduction chemistry of a nonheme FeII–NO2− complex, [(12TMC)FeII(NO2−)]+ (2), bearing the 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane (12TMC) ligand (Scheme 1, reaction I). Complex 2 reacts with one-equiv. of triflic acid (HSO3CF3, H+ source) and generates the corresponding nonheme Fe–nitrosyl complex, [(12TMC)Fe(NO)]2+({FeNO}, 3), and H2O2 (Scheme 1, reactions II & III) in CH3CN at 233 K. However, upon reaction with a base (OH−), 2 does not form 3. Mechanistic investigations using 15N-labeled-15 NO2− demonstrated explicitly that the N atom in the NO moiety of 3 is derived from the NO2− anion and H2O2 by the protonation of the O atom of the NO2− moiety. Conversely, an increased H+ concentration showed a significant fall in H2O2, which disappeared completely when the H+ ion quantity was ≅ two-equiv. with the simultaneous formation of a substantial amount of H2O (Scheme 1, reaction IV). To the extent of our knowledge, the present work reports the very first comparative study for the reaction of an FeII–NO2− complex with varying H+ concentrations and the evidence showing the formation of H2O2 (one-equiv. of H+) and H2O (≅ two-equiv. of H+), illustrating a new approach for NiR enzyme activity (Scheme 1).
Results and discussion
Preparation of the FeII-nitrito complex, [(12-TMC)FeII(NO2−)]+ (2)
The initial FeII–NO2− complex, [(12TMC)FeII(NO2−)]+ (2), was prepared by the addition of one equivalent of NaNO2 in the presence of a 15-crown-5 to FeII-complex, [(12TMC)FeII(NCCH3)]2+ (1), in CH3CN at 298 K (Scheme 1 & reaction I; also see the ESI and Experimental section (ES)). Complex 2 was further characterized by various spectroscopic measurements, including the determination of the single-crystal X-ray structure. A UV-vis absorption band (λmax = 325 nm and ε = 356 M−1 cm−1) was formed upon adding an equivalent amount of NaNO2 to the CH3CN solution of 1, which corresponds to 2 (Fig. 1a). A characteristic peak for metal-bound NO2− stretching at 1270 cm−1 was observed in the FT-IR spectrum of 2, which shifted to 1247 cm−1 when 2 was prepared using 15N-labeled-nitrite (15N16O2−) (inset: Fig. 1a andS1; ESI†).35 The electrospray ionization mass spectrum (ESI-MS) recorded for 2 showed a prominent ion peak at m/z 330.1, which shifted to m/z 331.1 when prepared with 15N-labeled Na15N16O2, and their mass and isotope distribution pattern corresponds to [(12TMC)Fe(NO2−)]+ (calc. m/z 330.1) and [(12TMC)Fe(15NO2−)]+ (calc. m/z 331.1), respectively (Fig. 1b and S2; ESI†). The 1H-NMR spectrum of 2 showed fairly clean paramagnetic proton signals for the protons of the 12TMC ligand (Fig. S3a†), suggesting a magnetically active Fe center. The spin-state of the Fe center in 2 was determined by calculating the magnetic moment of the FeII center by Evans' method and found to be 5.19 BM, suggesting a high spin FeII ion (S = 2) in complex 2 (ESI,† ES, Fig. S3b). The electrochemical measurement of 2 showed a reversible cyclic voltammogram (redox potential + 0.56 V vs. Ag/AgNO3−) (ESI, Fig. S4a†). In addition to the above spectral measurements, the structural details of 2 were obtained by its single-crystal X-ray structure determination (Fig. 2). The FeII center of 2 was found to have O, O/-chelated bi-dentate NO2− anions in a distorted octahedral geometry (ESI,† ES, Fig. S5 and Tables 1 & 2).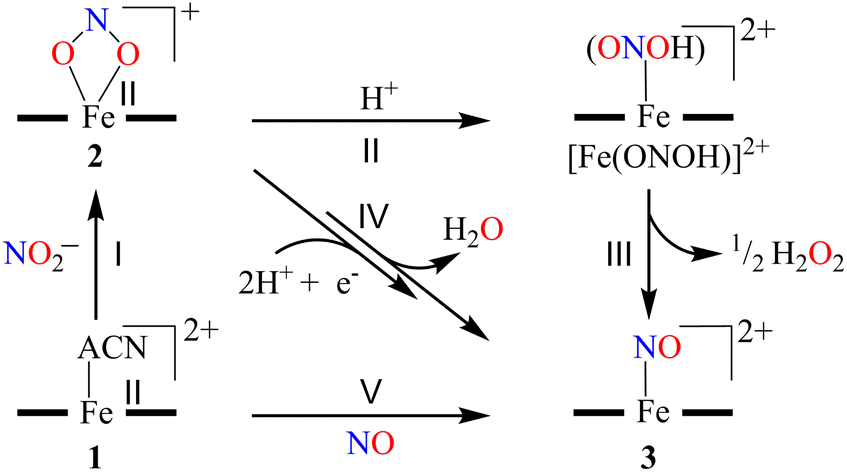 | ||
| Scheme 1 | ||
 | ||
| Fig. 1 (a) UV-visible spectra of 1 (0.50 mM, black line) and 2 (0.50 mM, red line) in CH3CN under Ar at 298 K. Inset: IR spectra of 2-14NO2− (red line) and 2-15NO2− (blue line) in KBr. (b) ESI-MS spectra of 2. The peak at 330.1 is assigned to [(12TMC)FeII(NO2−)]+ (calcd m/z 330.1). Inset: isotopic distribution pattern for 2-14NO2− (red line) and 2-15NO2− (blue line). | ||
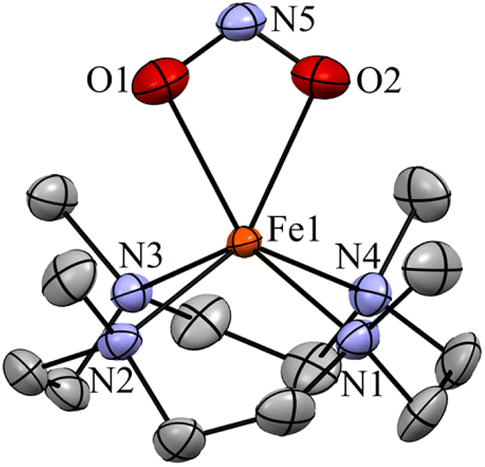 | ||
| Fig. 2 Displacement ellipsoid plot (15% probability) of 2 at 100 K. Disorder C atoms of TMC, anions and H atoms have been removed for clarity. | ||
The nitrite reduction reaction of the FeII–NO2− complex (2)
To further investigate the NO2− reduction chemistry of FeII–NO2− (2), we explored its reaction with different equivalents of acid (H+ ions). When 2 was reacted with H+, we observed a visible color change from yellow to green and a new absorption band (λmax = 350 nm and ε = 1450 M−1 cm−1), characteristic of a new species (3), formed over ∼2 minutes in CH3CN under Ar at 233 K (Fig. 3a; ESI,† ES, and Fig. S6).8a,9 Complex 2 was found to be very stable in CH3CN and at 298 K as it did not show any spectral variations in the absence of H+ (ESI,† Fig. S7a). Complex 2 was also found inert towards OH− as it does not indicate any change in UV-vis spectra when treated with tetrabutylammonium hydroxide (ESI, Fig. S7b†), suggesting that Fe–NO2− reacts only with H+. The amount of H+ required to reduce the NO2− moiety was determined by spectral titration, which confirmed the ratio-metric equivalent of 2 with H+ as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ESI,† Fig. S8). The compound 3, obtained in the reaction of 2 with H+ was determined to be an Fe–nitrosyl complex, {FeNO}7, based on various spectroscopic characterization techniques (vide infra). The other product of the NO2− reduction using one-equiv. of H+ was determined to be H2O2, in contrast to previous reports on biological NiR and NO2− reduction chemistry, via a proposed thermally unstable ONOH intermediate as reported in the literature (Scheme 1, reactions II & III).36 However, when reacted with more than one-equiv. of H+ (≅ two) 2 generated 3, but the amount of H2O2 decreased gradually with increasing H+. This suggests the decomposition of H2O2 or utterly new chemistry in the presence of more than one-equiv. of H+; the new product was confirmed to be H2O by using various spectral measurements (Scheme 1, reaction IV). To the best of our knowledge, this work reports the first-ever study where the side products of NO2− reduction are regulated by different amounts of H+, which opens a new pathway of acid-induced NO2− reduction chemistry to the scientific community.
:1 (ESI,† Fig. S8). The compound 3, obtained in the reaction of 2 with H+ was determined to be an Fe–nitrosyl complex, {FeNO}7, based on various spectroscopic characterization techniques (vide infra). The other product of the NO2− reduction using one-equiv. of H+ was determined to be H2O2, in contrast to previous reports on biological NiR and NO2− reduction chemistry, via a proposed thermally unstable ONOH intermediate as reported in the literature (Scheme 1, reactions II & III).36 However, when reacted with more than one-equiv. of H+ (≅ two) 2 generated 3, but the amount of H2O2 decreased gradually with increasing H+. This suggests the decomposition of H2O2 or utterly new chemistry in the presence of more than one-equiv. of H+; the new product was confirmed to be H2O by using various spectral measurements (Scheme 1, reaction IV). To the best of our knowledge, this work reports the first-ever study where the side products of NO2− reduction are regulated by different amounts of H+, which opens a new pathway of acid-induced NO2− reduction chemistry to the scientific community.
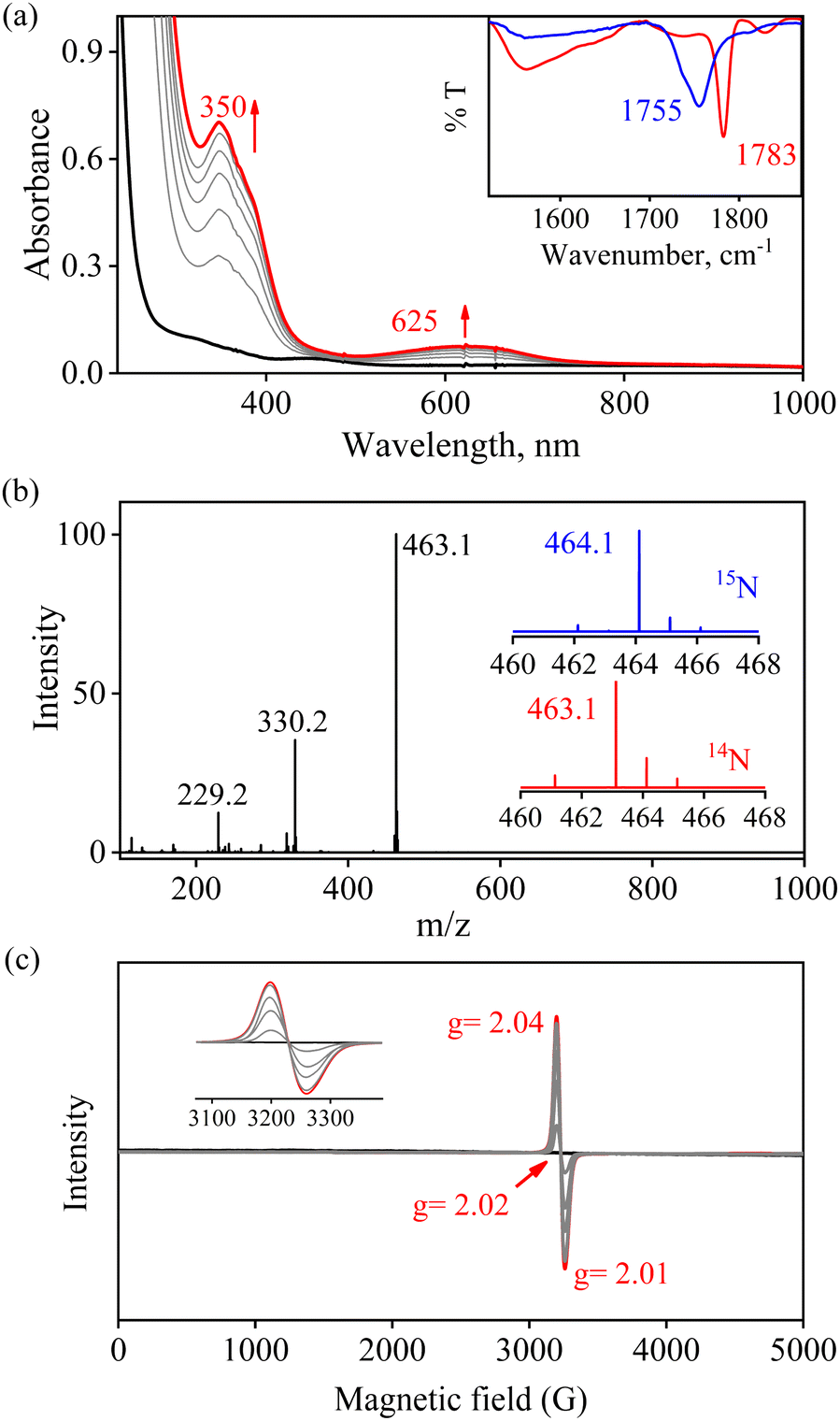 | ||
| Fig. 3 (a) UV-visible spectral changes of 2 (0.50 mM, black line) upon addition of H+ (one-equiv.) in CH3CN at 233 K. Black line (2) changed to a red line (3) upon addition of H+. Inset: IR spectra 3-14NO (red line) and 3-15NO (blue line) in KBr. (b) ESI-MS spectra of 3. The peak at 463.1 is assigned to [(12TMC)FeII(NO)(OTf)]+ (calcd m/z 463.1). Inset: isotopic distribution pattern for 3-14NO (red line) and 3-15NO (blue line). (c) Time-dependent EPR spectra of the generation of 3 (red line) in the reaction of 2 and H+ (one-equiv.) in CH3CN at 77 K. | ||
We have performed various spectral measurements to track the products of H+ (or D+)-induced reduction of Fe-bound 14/15NO2− in 2. The FT-IR spectrum of 3 showed a characteristic peak for Fe-bound nitrosyl stretching at 1783 cm−1 ({Fe14NO}7), which shifted to 1755 cm−1 ({Fe15NO}7) when 3 was prepared by the reaction of 15N-labeled-nitrite (2-15NO2−) with one-equiv. of H+ (inset, Fig. 3a and S9; ESI†). This shifting in NO stretching frequency (Δ = 28 cm−1) indicates that the N atom in NO moiety is derived from the 14/15NO2− ligand of 2. Similarly, the ESI-MS spectrum of 3 showed a prominent peak at m/z 463.1, [(12TMC)Fe(NO)(OTf)]+ (calc. m/z 463.1), which shifted to 464.1, [(12TMC)Fe(15NO)(OTf)]+ (calc. m/z 464.1), when FeII–15NO2− was reacted with H+ (Fig. 3b and S10; ESI†), specifying clearly that NO in 3 is derived from the NO2− moiety. The 1H-NMR spectrum of 3 showed shifting in the 1H-signals of the 12TMC ligand framework suggesting a paramagnetic system (ESI, Fig. S11a†).32a,b We determined the spin-state of the Fe centre in 3 by calculating its magnetic moment using Evans' method and found it to be 2.3 BM, suggesting a low-spin Fe center in 3 (S = 1/2) for the complex 3 (ESI, ES, and Fig. S11b†).37 Additionally, time-dependent EPR measurements were followed for the generation of 3 in the reaction mixture of 2 + H+. EPR measurements (77 K), performed at different time intervals, showed the formation of a new species (g = 2.04) (Fig. 3c), which is characteristic of the EPR signal of isolated species {FeNO}7 (Scheme 1, reaction V), confirming the formation of low-spin 3 in the above reaction (ESI, Fig. S11c†). The electrochemical measurement of 3 showed a reversible cyclic voltammogram (redox potential + 0.36 V vs. Ag/AgNO3−) (ESI, Fig. S4b†). Additionally, we have determined the binding constants Kb(FeII–NO2−) and Kb{(Fe(NO)}7 using the Benesi–Hildebrand equation28,38 for the generation of 2 and 3 in the reaction of [(12TMC)FeII(CH3CN)]2+ with NO2− and NO. The values were Kb(FeII–NO2−) = 4.7 × 102 M−1 & Kb{(FeNO} = 8.4 × 102 M−1 (ESI,† ES, and Fig. S12), which also supports the forward reaction. In addition, the yield for the formation of 3 was calculated by comparing the UV-vis absorption spectra of 3 formed in the reaction of 2 with one-equiv. of H+ with the authentic Fe–nitrosyl complex ({FeNO}7), prepared in a separate reaction of [(12TMC)FeII(CH3CN)]2+ + NO, and was found to be 95%. However, the yield decreased to 85% when the reaction was carried out using two-equiv of H+ (ESI, ES, and Fig. S13†). Furthermore, the structural details of 3, obtained in the reaction of 2 and H+, were obtained by its single-crystal X-ray structure determination (ESI, ES & Fig. 4). The NO moiety showed the coordination via the N atom to the Fe center of 3 with the a angle of 168° (ESI, ES Fig. S14;† & Tables T1 and T2). This arrangement suggests a neutral ˙NO moiety with an FeII center37,39 (further supported by BVS calculation from the crystal parameters of 3, ESI,† ES) and can be formulated as [(12TMC)FeII(NO)]2+; however, in this manuscript, we used Enemark–Feltham notation for 3 ({FeNO}7).
 | ||
| Fig. 4 Displacement ellipsoid plot (50% probability) of 3 at 100 K. Disordered C atoms of TMC, anions and H atoms have been removed for clarity. | ||
Mechanistic investigation of NO2− reduction
In vivo and in vitro biomimetic studies on acid-induced NO2− reduction reactions produce NO with H2O (as explained in the biological NiR chemistry)34a,40 or in some cases ˙OH (H2O2)36 or a metal hydroxide41 and should be accomplished via the proposed ONOH intermediate, as reported by Murphy et al.,9b and Rose et al.40g for the biological Cu-NiR chemistry. Similarly, Fujii and coworkers also proposed a Cu(ONOH) intermediate in the acid-induced biomimetic NO2− reduction on the CuI center.40h Meanwhile, Shigeta et al.40i & Chen and coworkers40j theoretically established the presence of a Cu(ONOH) intermediate in acid-induced NO2− reduction. The present work elucidated how varying equivalents of H+ ions determine the side products of FeII-bound NO2− reduction chemistry in addition to NO and should be accomplished by a similar proposed ONOH intermediate. In this regard, we proposed the reaction sequences, where the preliminary step of the NO2− reduction reaction consists of an electrophilic addition of H+ to the NO2− anion of 2 and generating the suggested [Fe–ONOH]2+ intermediate species (Scheme 2, reaction I), as proposed previously.36,41 The presumed [Fe–(ONOH)]2+ intermediate is believed to produce {FeNO}7via the homolytic cleavage of the ON–OH moiety, as reported in NO2− reduction on the FeII center and Cu-NiR,36,41 and ˙OH (1/2 H2O2)42 (Scheme 2, reaction II). In contrast, NO2− reduction in the presence of ≈ two-equiv. or more H+ produced 3 with H2O as a side product in a multiple-step reaction (Scheme 2, reaction III). This reaction is believed to occur via the reduction of the NO2− anion of 2 in the presence of two H+, as reported in biological NiR9b,40g and biomimetic NO2− reduction40h–j,43 reactions. The H2O molecule may be generated either by (a) step-wise protonation of NO2− species of 2 as observed in biology,40a,b (b) acidic decomposition of H2O2,44 or (c) by auto-decomposition of H2O2.45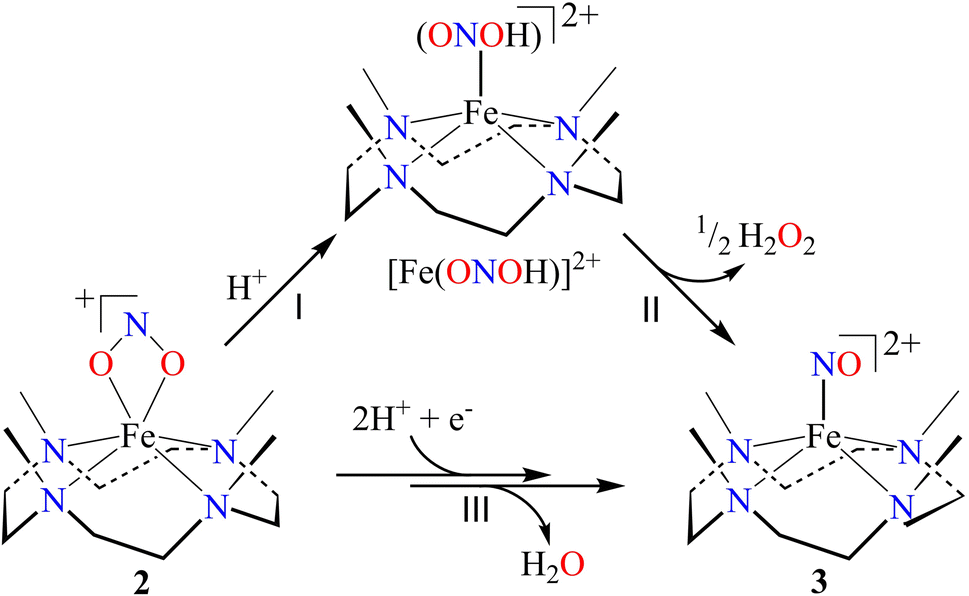 | ||
| Scheme 2 | ||
To validate our proposed H+-induced NO2− reduction chemistry mechanism, we have reacted 2 with different equivalents of H+ and characterized all the products formed in the reaction mixture. In both the acid-induced reactions, we observed the formation of 3. However, the side product of NO2− reduction changed to H2O instead of H2O2 when the H+ amount was ≥ two-equiv. (ESI,† ES). H2O2/& H2O formed in the NO2− reduction reaction was followed/characterized and quantified using 1H-NMR spectroscopic measurements. A characteristic signal for H2O2 (8.66 ppm, ESI,† Fig. S15a)46 was observed in the 1H-NMR spectrum of 2 with one-equiv. of H+ in CD3CN. Our proposal of H2O2 formation in one-equiv. of H+ induced NO2− reduction was authenticated by comparing this spectrum with those of the authentic samples: (i) H2O2 plus 3 (8.66 ppm; ESI,† Fig. S15b) and (ii) H2O2 only (8.66 ppm; ESI,† Fig. S15c).46 The amount of H2O2 in the above reaction was confirmed to be more than 50% (defining ½ equivalent of H2O2 relative to 2 as 100% yield) from 1H-NMR spectral measurements and using benzene as the internal standard (ESI,† ES, and Fig. S15a).46 Time-based 1H-NMR spectral measurements for the above reaction showed the gradual formation of H2O2 (8.66 ppm), which starts decreasing after reaching its maxima, suggesting the decomposition of H2O2 to H2O (Fig. 5a).44,45 In addition to 1H-NMR, iodometric titration likewise confirmed H2O2 formation in the reaction of 2 with one-equiv. of H+ which was determined to be ∼65% (ESI, ES, and Fig. S16a†) (defining 1/2 equivalent of H2O2 relative to 2 as 100% yield). However, no H2O2 was observed in iodometric titration when the reaction was carried out in the presence of two-equiv. of H+ (ESI, ES, and Fig. S16b†).47 Furthermore, we have also established H2O formation in the NO2− reduction reaction in the presence of two-equiv. of H+ by 1H-NMR spectroscopic measurements (Fig. 5b and S15d, ESI†). To establish that H+ is only responsible for forming 1H-NMR signals at 2.2 ppm, we have explored the same reaction using CF3SO3D (D+ source). Surprisingly, when the source was D+, we did not observe the formation of the H2O peak (at 2.2 ppm); this clearly suggests that H+ ions are responsible for the H2O formation in the two-equiv. of H+ induced NO2− reduction (ESI, ES, and Fig. S17†). In addition, time-dependent 1H-NMR spectral measurements for the reaction of 2 with two-equiv. of H+ showed an increment in the peak of H2O protons and a kind of first time-base measurement, further supporting our proposal of H2O formation (Fig. 5b). However, our efforts to quantify the amount of H2O formed in the reaction are futile; but they scientifically established the mechanistic aspect of acid-induced NO2− reduction in the presence of different equivalents of H+. These results are the only example where tracking H+-induced NO2− reduction products has confirmed that the variable amounts of H+ (pH/acidic conditions) generate NO with H2O2 (one-equiv. of H+) or H2O (≥two-equiv. of H+).
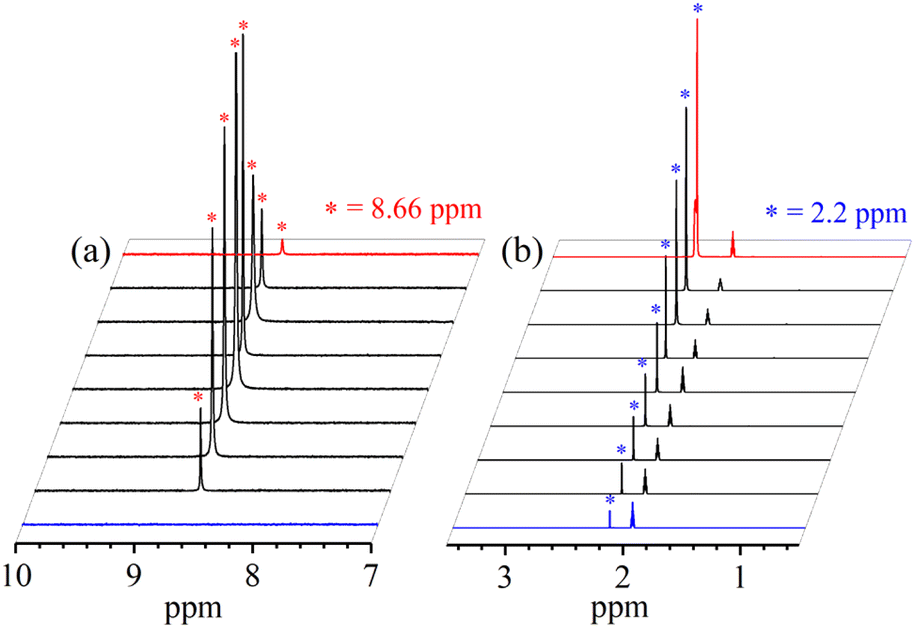 | ||
| Fig. 5 1H-NMR spectrum of (a) H2O2 formation and (b) H2O formation in the reaction of 2 with one-equiv. & two-equiv. of H+ in CD3CN recorded at different times, respectively. | ||
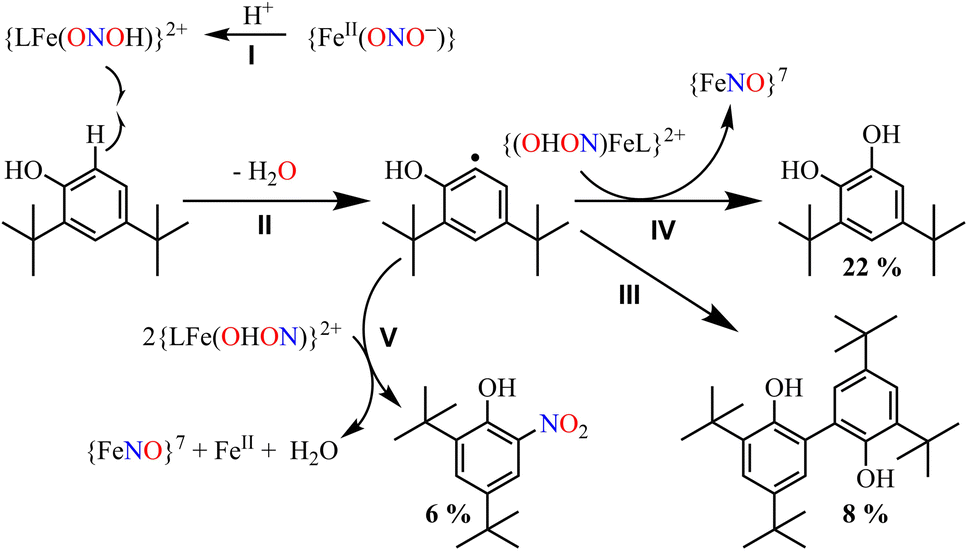
Furthermore, we attempted to characterize the proposed [Fe–ONOH]2+ intermediate to illustrate its conversion mechanism to 3. However, after several attempts, we failed to detect/stabilize the intermediate even at low temperature (193 K) in UV-vis & FT-IR spectroscopic measurements, suggesting a kinetically driven reaction.48 Since metal-nitrous acid intermediates are known to be highly unstable intermediates,34b,36,40h–j there are only a few reports about the metal-bound nitrous acid species.34b,40h,40j,49 However, to support our mechanistic proposal for the formation of an ˙OH radical (H2O2) via the homolytic cleavage of the N–O bond, we pursued the ˙OH radical trapping experiment using 2,4-di-tert-butylphenol (2,4-DTBP).50 In the one-equiv. of H+ induced NO2− reduction reaction, we have observed the formation of 3,5-Di-tert-butylcatechol (3,5-DTBC, ∼22%) and 2,4-DTBP-dimer (2,4-DTBP-D, ∼8%) with a minimal amount of nitro-2,4-DTBP (NO2-2,4-DTBP, ∼6%) (ESI, ES, and Fig. S18 & S19†). The generation of 3,5-DTBC51 in the above experiments undoubtedly confirmed the ˙OH formation via the N–O bond homolysis of the ON–OH moiety. Hence, the formation of 3,5-DTBC and other products confirms the reaction sequences (Scheme 3) and supports the presence of the [Fe-(ONOH)]2+ intermediate in the one-equiv. of H+ induced NO2− reduction reaction.
 | ||
| Scheme 3 | ||
The transformation of 2,4-DTBP in the presence of one-equiv. of H+ can be explained based on the radical coupling reaction.51 The sequences of the 2,4-DTBP conversion are believed to be (i) the generation of the phenoxyl radical and the release of Fe–NOs by the H-atom abstraction reaction of [Fe–ONOH]2+ from DTBP (Scheme 3, reaction I & II). After that, the phenoxyl radical either (ii) dimerizes to give 2,4-DTBP-D (Scheme 3, reaction III) or (iii) produces 3,5-DTBC upon radical coupling with another molecule of [Fe–ONOH]2+ and releases 3 (Scheme 3, reaction IV). In some cases, NO2-DTBP and 3 may generate in the presence of two molecules of [Fe–ONOH]2+ and a phenoxyl radical (Scheme 3, reaction V). Also, when the above radical trapping experiments were performed using CF3SO3D, we observed the generation ˙OD-driven products of DTBC (ESI,† ES, and Fig. S20). In addition, we reacted 2-16O14N18O− with one equiv. of H+ in the presence of 2,4-DTBP. Surprisingly, we observed the formation of 3,5-DTBC (18OH) with 16O14N18O-DTBP as a side product (ESI, Fig. S21†). These experiments support that one equiv. of H+ induced NO2− reduction in 2 generates 3 + H2O2via the homolytic cleavage of the N–O bond of the ON-OH intermediate.36
Conclusion
The mechanistic investigation of acid-induced NO2− reduction became an important research topic in modern-day chemistry as it deals with NO2− to NO transformation, an essential signaling molecule in biosystems.40b,52 The mechanistic aspects of NiR chemistry mediated by H+ are still challenging to the scientific community and yet to be resolved as two different side products have been proposed to form in vivo and in vitro studies.8a,9,16,25,30 Also, the pH/or H+ ion concentration effect is yet to be confirmed as it affects the reaction mechanism and the side products of NO2− reduction reactions.48 In this report, we have shown the reduction of NO2− in a nonheme FeII-nitrito complex, [(12TMC)FeII(NO2−)]+ (2), to an Fe–nitrosyl complex [(12TMC)Fe(NO)]2+, {FeNO}7 (3), in the presence of different equivalents of H+ (CF3SO3D, D+ ion source), a biomimetic functional model of NiR. The structural details of {FeNO}7 showed an axially coordinated NO moiety to the Fe center. In addition, 15N-labeled 15NO2− experiments confirm that the N atom of the NO moiety in 3 is derived from the NO2− anion of 2. Acid-induced NO2− reduction of 2 showed the formation of {FeNO}7 along with H2O2 or H2O as a side product when treated with different ratios of H+, one-equiv. or ≥two-equiv., respectively. Reports on acid-induced biomimetic NO2− reduction40h,j,43 and biological NiR reactions9b,40g,41 suggested a metal-ONOH intermediate before NO formation; hence, we believe that the H+-induced NO2− reduction on the FeII center in 2 should generate 3via the proposed [Fe–ONOH]2+ intermediate and follow the NiR chemistry. The N–O bond homolysis of the proposed ONOH intermediate was supported by the observation of 3,5-DTBC-16 OH(18 OH) in ˙OH radical trapping experiments using 2,4-DTBP51 in the reaction of 2-ON16O2−(16ON18O−) with one equiv. of H+. Also, the observation of DTBC(OD) in the presence of D+ further supports the acid-induced reduction of NO2−. In addition, a significant amount of H2O2 formation was also confirmed using 1H-NMR/or UV-vis iodometric titration along 3.46 However, the generation of the H2O molecule was believed to occur either (i) by NO2− reduction in the presence of two-equiv. of H+ and an electron9b,40c,40g,40h,40j or (ii) by the acid-induced decay of H2O2 or auto-decomposition of H2O2.44,45 The redox potential of 2 was higher than that of the enzymatic iron-site, making 2 more prone to reduction.53 At this time, we are not sure about the source of another electron; however, we are currently exploring various NO2− bound CuI/II & FeII/III complexes to understand the reaction sequences and track the electron source using the known electron donor species. These results provide entirely new reaction sequences for acid-induced NO2− reduction chemistry, a functional model of biomimetic NiR chemistry, and show how the H+ ion concentration determines H2O2 or H2O as a side product along with NO.Experimental Section
For the experimental details, see the ESI.†Data availability
All the required data is already provided in the ESI† and manuscript.Author contributions
PKK discovered /conceptualized the initial project. Kulbir carried out most of the experiments and gathered the data. PKK, SG & TD helped in interpreting the experimental results. SCS, Kulbir & SD worked on growing the crystals and recording the crystallographic data. Kulbir and SD write the first draft of the article. PKK & TD have corrected the manuscript, finalized the final draft, and guided during the revision. PKK followed and guided the whole project work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grants-in-aid (Grant No. CRG/2021/003371, & EEQ/2021/000109) from SERB-DST and AvH (ID: 1219648). Kulbir and S. D. thank IISER Tirupati for their fellowship.References
- (a) R. F. Furchgott, Angew. Chem., Int. Ed., 1999, 38, 1870–1880 CrossRef CAS; (b) L. J. Ignarro, Angew. Chem., Int. Ed., 1999, 38, 1882–1892 CrossRef CAS; (c) L. J. Ignarro, Nitric Oxide: Biology and Pathobiology, Academic press, 2000 Search PubMed; (d) G. B. Richter-Addo, P. Legzdins and J. Burstyn, Chem. Rev., 2002, 102, 857–860 CrossRef CAS PubMed; (e) I. M. Wasser, S. de Vries, P. Moënne-Loccoz, I. Schröder and K. D. Karlin, Chem. Rev., 2002, 102, 1201–1234 CrossRef CAS PubMed.
- R. B. S. Nabi, R. Tayade, A. Hussain, K. P. Kulkarni, Q. M. Imran, B. G. Mun and B. W. Yun, Environ. Exp. Bot., 2019, 161, 120–133 CrossRef CAS.
- F. Vargas, J. M. Moreno, R. Wangensteen, I. Rodriguez-Gomez and J. Garcia-Estan, Eur. J. Endocrinol., 2007, 156, 1–12 CAS.
- (a) H. T. Dong, S. Camarena, D. Sil, M. O. Lengel, J. Zhao, M. Y. Hu, E. E. Alp, C. Krebs and N. Lehnert, J. Am. Chem. Soc., 2022, 144, 16395–16409 CrossRef CAS PubMed; (b) D. J. Stuehr, S. S. Gross, I. Sakuma, R. Levi and C. F. Nathan, J. Exp. Med., 1989, 169, 1011–1020 CrossRef CAS PubMed.
- (a) R. E. Huie and S. Padmaja, Free Radical Res. Commun., 1993, 18, 195–199 CrossRef CAS PubMed; (b) P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS PubMed; (c) C. Prolo, M. N. Alvarez and R. Radi, Biofactors, 2014, 40, 215–225 CrossRef CAS PubMed.
- (a) W. C. Nottingham and J. R. Sutter, Int. J. Chem. Kinet., 1986, 18, 1289–1302 CrossRef CAS; (b) C. H. Lim, P. C. Dedon and W. M. Deen, Chem. Res. Toxicol., 2008, 21, 2134–2147 Search PubMed.
- (a) R. Radi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4003–4008 CrossRef CAS PubMed; (b) B. Kalyanaraman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11527–11528 CrossRef CAS PubMed; (c) P. C. Dedon and S. R. Tannenbaum, Arch. Biochem. Biophys., 2004, 423, 12–22 CrossRef CAS PubMed.
- (a) N. Lehnert, T. C. Berto, M. G. I. Galinato and L. E. Goodrich, in Handbook of Porphyrin Science, ed. K. Kadish, K. Smith and R. Guilard, World Scientific Publishing, Singapore, 2011, p. 1 Search PubMed; (b) T. B. McCall, N. K. Boughton-Smith, R. M. Palmer, B. J. Whittle and S. Moncada, Biochem. J., 1989, 261, 293–296 CrossRef CAS PubMed; (c) R. G. Knowles and S. Moncada, Biochem. J., 1994, 298(Pt 2), 249–258 CrossRef CAS PubMed.
- (a) B. A. Averill, Chem. Rev., 1996, 96, 2951–2964 CrossRef CAS PubMed; (b) E. I. Tocheva, F. I. Rosell, A. G. Mauk and M. E. Murphy, Science, 2004, 304, 867–870 CrossRef CAS PubMed.
- L. Ma, L. Hu, X. Feng and S. Wang, Aging Dis., 2018, 9, 938–945 CrossRef PubMed.
- (a) P. C. Ford and I. M. Lorkovic, Chem. Rev., 2002, 102, 993–1018 CrossRef CAS PubMed; (b) M. P. Schopfer, B. Mondal, D. H. Lee, A. A. Sarjeant and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 11304–11305 CrossRef CAS PubMed; (c) M. P. Doyle and J. W. Hoekstra, J. Inorg. Biochem., 1981, 14, 351–358 CrossRef CAS PubMed; (d) M. Yenuganti, S. Das, K. Kulbir, S. Ghosh, P. Bhardwaj, S. S. Pawar, S. C. Sahoo and P. Kumar, Inorg. Chem. Front., 2020, 7, 4872–4882 RSC.
- (a) E. Weitzberg and J. O. Lundberg, Nitric Oxide – Biol. Chem., 1998, 2, 1–7 CrossRef CAS PubMed; (b) J. O. Lundberg and M. Govoni, Free Radical Biol. Med., 2004, 37, 395–400 CrossRef CAS PubMed.
- (a) L. I. Hochstein and G. A. Tomlinson, Annu. Rev. Microbiol., 1988, 42, 231–261 CrossRef CAS PubMed; (b) W. H. Campbell, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1999, 50, 277–303 CrossRef CAS PubMed.
- P. Tavares, A. S. Pereira, J. J. Moura and I. Moura, J. Inorg. Biochem., 2006, 100, 2087–2100 CrossRef CAS PubMed.
- N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. S. Alfaro, Chem. Rev., 2021, 121, 14682–14905 CrossRef CAS PubMed.
- N. Benjamin, F. O'Driscoll, H. Dougall, C. Duncan, L. Smith, M. Golden and H. McKenzie, Nature, 1994, 368, 502 CrossRef CAS PubMed.
- J. O. Lundberg, E. Weitzberg, J. M. Lundberg and K. Alving, Gut, 1994, 35, 1543–1546 CrossRef CAS PubMed.
- (a) S. Kundu, W. Y. Kim, J. A. Bertke and T. H. Warren, J. Am. Chem. Soc., 2017, 139, 1045–1048 CrossRef CAS PubMed; (b) K. Cosby, K. S. Partovi, J. H. Crawford, R. P. Patel, C. D. Reiter, S. Martyr, B. K. Yang, M. A. Waclawiw, G. Zalos, X. Xu, K. T. Huang, H. Shields, D. B. Kim-Shapiro, A. N. Schechter, R. O. Cannon and M. T. Gladwin, Nat. Med., 2003, 9, 1498–1505 CrossRef CAS PubMed; (c) U. B. Hendgen-Cotta, M. W. Merx, S. Shiva, J. Schmitz, S. Becher, J. P. Klare, H.-J. Steinhoff, A. Goedecke, J. Schrader, M. T. Gladwin, M. Kelm and T. Rassaf, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10256–10261 CrossRef CAS PubMed.
- J. Brooks and D. Keilin, Proc. R. Soc. London, Ser. B, 1937, 123, 368–382 CAS.
- M. Kumar, N. A. Dixon, A. C. Merkle, M. Zeller, N. Lehnert and E. T. Papish, Inorg. Chem., 2012, 51, 7004–7006 CrossRef CAS PubMed.
- (a) S. Hematian, M. A. Siegler and K. D. Karlin, J. Am. Chem. Soc., 2012, 134, 18912–18915 CrossRef CAS PubMed; (b) S. Hematian, I. Kenkel, T. E. Shubina, M. Dürr, J. J. Liu, M. A. Siegler, I. Ivanovic-Burmazovic and K. D. Karlin, J. Am. Chem. Soc., 2015, 137, 6602–6615 CrossRef CAS PubMed.
- M. E. Murphy, S. Turley and E. T. Adman, J. Biol. Chem., 1997, 272, 28455–28460 CrossRef CAS PubMed.
- R. C. Maji, S. K. Barman, S. Roy, S. K. Chatterjee, F. L. Bowles, M. M. Olmstead and A. K. Patra, Inorg. Chem., 2013, 52, 11084–11095 CrossRef CAS PubMed.
- A. P. Hunt, A. E. Batka, M. Hosseinzadeh, J. D. Gregory, H. K. Haque, H. Ren, M. E. Meyerhoff and N. Lehnert, ACS Catal., 2019, 9, 7746–7758 CrossRef CAS PubMed.
- T. S. Kurtikyan, A. A. Hovhannisyan, A. V. Iretskii and P. C. Ford, Inorg. Chem., 2009, 48, 11236–11241 CrossRef CAS PubMed.
- (a) B. C. Sanders, S. M. Hassan and T. C. Harrop, J. Am. Chem. Soc., 2014, 136, 10230–10233 CrossRef CAS PubMed; (b) S. Zhang, M. M. Melzer, S. N. Sen, N. Celebi-Olcum and T. H. Warren, Nat. Chem., 2016, 8, 663–669 CrossRef CAS PubMed.
- (a) L. Cheng, D. R. Powell, M. A. Khan and G. B. Richter-Addo, Chem. Commun., 2000, 2301–2302, 10.1039/B006775J; (b) A. K. Patra, R. K. Afshar, J. M. Rowland, M. M. Olmstead and P. K. Mascharak, Angew. Chem., Int. Ed. Engl., 2003, 42, 4517–4521 CrossRef CAS PubMed.
- K. Kulbir, S. Das, T. Devi, M. Goswami, M. Yenuganti, P. Bhardwaj, S. Ghosh, S. C. Sahoo and P. Kumar, Chem. Sci., 2021, 12, 10605–10612 RSC.
- S. Hong, J. J. Yan, D. G. Karmalkar, K. D. Sutherlin, J. Kim, Y. M. Lee, Y. Goo, P. K. Mascharak, B. Hedman, K. O. Hodgson, K. D. Karlin, E. I. Solomon and W. Nam, Chem. Sci., 2018, 9, 6952–6960 RSC.
- (a) J. Heinecke and P. C. Ford, Coord. Chem. Rev., 2010, 254, 235–247 CrossRef CAS; (b) S. Das, K. Kulbir, S. Ghosh, S. Chandra Sahoo and P. Kumar, Chem. Sci., 2020, 11, 5037–5042 RSC; (c) S. Das, K. Kulbir, S. Ray, T. Devi, S. Ghosh, S. S. Harmalkar, S. N. Dhuri, P. Mondal and P. Kumar, Chem. Sci., 2022, 13, 1706–1714 RSC.
- (a) A. P. Hunt and N. Lehnert, Acc. Chem. Res., 2015, 48, 2117–2125 CrossRef CAS PubMed; (b) A. L. Speelman, B. Zhang, C. Krebs and N. Lehnert, Angew. Chem., Int. Ed., 2016, 55, 6685–6688 CrossRef CAS PubMed.
- (a) P. Kumar, Y. M. Lee, Y. J. Park, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2015, 137, 4284–4287 CrossRef CAS PubMed; (b) P. Kumar, Y. M. Lee, L. Hu, J. Chen, Y. J. Park, J. Yao, H. Chen, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2016, 138, 7753–7762 CrossRef CAS PubMed; (c) S. Hong, J. J. Yan, D. G. Karmalkar, K. D. Sutherlin, J. Kim, Y. M. Lee, Y. Goo, P. K. Mascharak, B. Hedman, K. O. Hodgson, K. D. Karlin, E. I. Solomon and W. Nam, Chem. Sci., 2018, 9, 6952–6960 RSC.
- (a) J. L. Heinecke, C. Khin, J. C. Pereira, S. A. Suarez, A. V. Iretskii, F. Doctorovich and P. C. Ford, J. Am. Chem. Soc., 2013, 135, 4007–4017 CrossRef CAS PubMed; (b) T. S. Kurtikyan, A. A. Hovhannisyan and P. C. Ford, Inorg. Chem., 2016, 55, 9517–9520 CrossRef CAS PubMed.
- (a) B. A. Averill, Chem. Rev., 1996, 96, 2951–2964 CrossRef CAS PubMed; (b) M. A. Puthiyaveetil Yoosaf, S. Ghosh, Y. Narayan, M. Yadav, S. C. Sahoo and P. Kumar, Dalton Trans., 2019, 48, 13916–13920 RSC.
- C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 12023–12031 CrossRef CAS PubMed.
- W. M. Ching, P. P. Chen and C. H. Hung, Dalton Trans., 2017, 46, 15087–15094 RSC.
- C. H. Hsieh, S. Ding, O. F. Erdem, D. J. Crouthers, T. Liu, C. C. McCrory, W. Lubitz, C. V. Popescu, J. H. Reibenspies, M. B. Hall and M. Y. Darensbourg, Nat. Commun., 2014, 5, 3684 CrossRef CAS PubMed.
- (a) S. Goswami, D. Sen, N. K. Das, H. K. Fun and C. K. Quah, Chem. Commun., 2011, 47, 9101–9103 RSC; (b) J. Chen, H. Yoon, Y. M. Lee, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Chem. Sci., 2015, 6, 3624–3632 RSC; (c) Y. M. Lee, M. Yoo, H. Yoon, X. X. Li, W. Nam and S. Fukuzumi, Chem. Commun., 2017, 53, 9352–9355 RSC.
- (a) P. C. Ford, J. C. M. Pereira and K. M. Miranda, in Nitrosyl Complexes in Inorganic Chemistry, Biochemistry and Medicine II, ed. D. M. P. Mingos, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 12–44, DOI: DOI:10.1007/430_2013_117; (b) G. R. Wyllie and W. R. Scheidt, Chem. Rev., 2002, 102, 1067–1090 CrossRef CAS PubMed; (c) D. M. P. Mingos, in Nitrosyl Complexes in Inorganic Chemistry, Biochemistry and Medicine I, ed. D. M. P. Mingos, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 1–44, DOI: DOI:10.1007/430_2013_116.
- (a) Y. Li, M. Hodak and J. Bernholc, Biochemistry, 2015, 54, 1233–1242 CrossRef CAS PubMed; (b) O. Einsle, A. Messerschmidt, R. Huber, P. M. Kroneck and F. Neese, J. Am. Chem. Soc., 2002, 124, 11737–11745 CrossRef CAS PubMed; (c) S. Basu, N. A. Azarova, M. D. Font, S. B. King, N. Hogg, M. T. Gladwin, S. Shiva and D. B. Kim-Shapiro, J. Biol. Chem., 2008, 283, 32590–32597 CrossRef CAS PubMed; (d) S. Besson, C. Carneiro, J. J. Moura, I. Moura and G. Fauque, Anaerobe, 1995, 1, 219–226 CrossRef CAS PubMed; (e) M. J. Boulanger, M. Kukimoto, M. Nishiyama, S. Horinouchi and M. E. Murphy, J. Biol. Chem., 2000, 275, 23957–23964 CrossRef CAS PubMed; (f) J. Brooks and D. Keilin, Proc. R. Soc. London, Ser. B, 1937, 123, 368–382 CrossRef CAS; (g) S. L. Rose, S. V. Antonyuk, D. Sasaki, K. Yamashita, K. Hirata, G. Ueno, H. Ago, R. R. Eady, T. Tosha, M. Yamamoto and S. S. Hasnain, Sci. Adv., 2021, 7, eabd8523 CrossRef CAS PubMed; (h) M. Kujime and H. Fujii, Angew. Chem., Int. Ed. Engl., 2006, 45, 1089–1092 CrossRef CAS PubMed; (i) S. Maekawa, T. Matsui, K. Hirao and Y. Shigeta, J. Phys. Chem. B, 2015, 119, 5392–5403 CrossRef CAS PubMed; (j) S. C. Hsu, Y. L. Chang, W. J. Chuang, H. Y. Chen, I. J. Lin, M. Y. Chiang, C. L. Kao and H. Y. Chen, Inorg. Chem., 2012, 51, 9297–9308 CrossRef CAS PubMed.
- M. Lintuluoto and J. M. Lintuluoto, Metallomics, 2018, 10, 565–578 CrossRef CAS PubMed.
- (a) B. Chen, Y. Xia, R. He, H. Sang, W. Zhang, J. Li, L. Chen, P. Wang, S. Guo, Y. Yin, L. Hu, M. Song, Y. Liang, Y. Wang, G. Jiang and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2209056119 CrossRef CAS PubMed; (b) P. Ulanski and C. von Sonntag, J. Chem. Soc., Perkin Trans. 2, 1999, 165–168, 10.1039/a808543i.
- J. A. Halfen, S. Mahapatra, E. C. Wilkinson, A. J. Gengenbach, V. G. Young, L. Que and W. B. Tolman, J. Am. Chem. Soc., 1996, 118, 763–776 CrossRef CAS.
- (a) B. Mlasi, D. Glasser and D. Hildebrandt, Ind. Eng. Chem. Res., 2015, 54, 5589–5597 CrossRef CAS; (b) F. M. Fomin and K. S. Zaitseva, Russ. J. Phys. Chem. A, 2014, 88, 466–470 CrossRef CAS.
- P. Pędziwiatr, F. Mikołajczyk, D. Zawadzki, K. Mikołajczyk and A. Bedka, Acta Innov., 2018, 45–52, DOI:10.32933/ActaInnovations.26.5.
- N. A. Stephenson and A. T. Bell, Anal. Bioanal. Chem., 2005, 381, 1289–1293 CrossRef CAS PubMed.
- K. Mase, K. Ohkubo, Z. Xue, H. Yamada and S. Fukuzumi, Chem. Sci., 2015, 6, 6496–6504 RSC.
- (a) Z. H. L. Abraham, B. E. Smith, B. D. Howes, D. J. Lowe and R. R. Eady, Biochem. J., 1997, 324, 511–516 CrossRef CAS PubMed; (b) Z. Huang, S. Shiva, D. B. Kim-Shapiro, R. P. Patel, L. A. Ringwood, C. E. Irby, K. T. Huang, C. Ho, N. Hogg, A. N. Schechter and M. T. Gladwin, J. Clin. Invest., 2005, 115, 2099–2107 CrossRef CAS PubMed; (c) C. A. Clark, C. P. Reddy, H. Xu, K. N. Heck, G. H. Luo, T. P. Senftle and M. S. Wong, ACS Catal., 2020, 10, 494–509 CrossRef CAS; (d) H.-Y. Hu, N. Goto and K. Fujie, Water Res., 2001, 35, 2789–2793 CrossRef CAS PubMed.
- (a) A. Samouilov, P. Kuppusamy and J. L. Zweier, Arch. Biochem. Biophys., 1998, 357, 1–7 CrossRef CAS PubMed; (b) K. Tsuchiya, Y. Kanematsu, M. Yoshizumi, H. Ohnishi, K. Kirima, Y. Izawa, M. Shikishima, T. Ishida, S. Kondo, S. Kagami, Y. Takiguchi and T. Tamaki, Am. J. Physiol. Heart Circ., 2005, 288, H2163–H2170 CrossRef CAS PubMed.
- A. Dubey, V. Rives and S. Kannan, J. Mol. Catal. A: Chem., 2002, 181, 151–160 CrossRef CAS.
- (a) A. Arnold, R. Metzinger and C. Limberg, Chemistry, 2015, 21, 1198–1207 CrossRef CAS PubMed; (b) C. Citek, C. T. Lyons, E. C. Wasinger and T. D. Stack, Nat. Chem., 2012, 4, 317–322 CrossRef CAS PubMed; (c) P. T. Kaye, K. W. Wellington and G. M. Watkins, Arkivoc, 2010, 2009, 301–313 Search PubMed.
- S. Suzuki, K. Kataoka and K. Yamaguchi, Acc. Chem. Res., 2000, 33, 728–735 CrossRef CAS PubMed.
- C. E. Immoos, J. Chou, M. Bayachou, E. Blair, J. Greaves and P. J. Farmer, J. Am. Chem. Soc., 2004, 126, 4934–4942 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2181978 and 2181979. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06704h |
| This journal is © The Royal Society of Chemistry 2023 |