
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into structure–property relationships in ionic liquids using cyclic perfluoroalkylsulfonylimides†
Younes K. J.
Bejaoui
a,
Frederik
Philippi
 b,
Hans-Georg
Stammler
c,
Krzysztof
Radacki
a,
Ludwig
Zapf
a,
Nils
Schopper
a,
Kateryna
Goloviznina
d,
Kristina A. M.
Maibom
a,
Roland
Graf
a,
Jan A. P.
Sprenger
a,
Rüdiger
Bertermann
a,
Holger
Braunschweig
a,
Tom
Welton
b,
Nikolai V.
Ignat'ev
ae and
Maik
Finze
*a
b,
Hans-Georg
Stammler
c,
Krzysztof
Radacki
a,
Ludwig
Zapf
a,
Nils
Schopper
a,
Kateryna
Goloviznina
d,
Kristina A. M.
Maibom
a,
Roland
Graf
a,
Jan A. P.
Sprenger
a,
Rüdiger
Bertermann
a,
Holger
Braunschweig
a,
Tom
Welton
b,
Nikolai V.
Ignat'ev
ae and
Maik
Finze
*a
aJulius-Maximilians-Universität Würzburg, Institut für Anorganische Chemie, Institut für Nachhaltige Chemie & Katalyse mit Bor (ICB), Am Hubland, 97074 Würzburg, Germany. E-mail: maik.finze@uni-wuerzburg.de
bImperial College London, Department of Chemistry, Molecular Sciences Research Hub, White City Campus, London W12 0BZ, UK
cUniversität Bielefeld, Fakultät für Chemie, Lehrstuhl für Anorganische Chemie und Strukturchemie (ACS), Centre for Molecular Materials (CM2), Universitätsstr. 25, D-33615 Bielefeld, Germany
dSorbonne Université, CNRS, Physicochimie des Électrolytes et Nanosystèmes Interfaciaux, F-75005 Paris, France
eConsultant, Merck KGaA, 64293 Darmstadt, Germany
First published on 30th January 2023
Abstract
Room temperature ionic liquids of cyclic sulfonimide anions ncPFSI (ring size: n = 4–6) with the cations [EMIm]+ (1-ethyl-3-methylimidazolium), [BMIm]+ (1-butyl-3-methylimidazolium) and [BMPL]+ (BMPL = 1-butyl-1-methylpyrrolidinium) have been synthesized. Their solid-state structures have been elucidated by single-crystal X-ray diffraction and their physicochemical properties (thermal behaviour and stability, dynamic viscosity and specific conductivity) have been assessed. In addition, the ion diffusion was studied by pulsed field gradient stimulated echo (PFGSTE) NMR spectroscopy. The decisive influence of the ring size of the cyclic sulfonimide anions on the physicochemical properties of the ILs has been revealed. All ILs show different properties compared to those of the non-cyclic TFSI anion. While these differences are especially distinct for ILs with the very rigid 6cPFSI anion, the 5-membered ring anion 5cPFSI was found to result in ILs with relatively similar properties. The difference between the properties of the TFSI anion and the cyclic sulfonimide anions has been rationalized by the rigidity (conformational lock) of the cyclic sulfonimide anions. The comparison of selected IL properties was augmented by MD simulations. These highlight the importance of π+–π+ interactions between pairs of [EMIm]+ cations in the liquid phase. The π+–π+ interactions are evident for the solid state from the molecular structures of the [EMIm]+-ILs with the three cyclic imide anions determined by single-crystal X-ray diffraction.
Introduction
Ionic liquids (ILs) are salts with low melting temperatures, often at or below room temperature that have attracted wide interest in academia and industry during the last decades.1–3 The reason for this broad interest lies in their often favourable and tunable properties compared to alternative, non-ionic materials, i.e. traditional organic solvents. Examples of appealing properties of ILs are low dynamic viscosities, high specific conductivities, large liquid ranges, and negligible vapor pressure. The highly tuneable and versatile nature of ILs is mostly a consequence of the plethora of different combinations of anions and cations. Although major research activities have been devoted to the elucidation of nanostructures and structure–property-relationships in ILs since the early 1990s, a general concept is still missing.4–7 However, a more profound understanding of these structure–property relationships is necessary to guide scientists in designing tailor-made ILs.Due to the complex nature of ion interactions in ILs, many intertwining factors affecting the physicochemical properties have been identified.6–10 Thus, the study of the influence of a specific single factor is difficult. However, the importance of ion volume11–16 and mass17 as well as charge delocalization (weakly coordinating nature)18,19 of anion and cation has been studied.20 A further properties determining factor is the conformational flexibility of the ions as a higher number of conformers increases the entropy of the liquid.4,21–24 So, the introduction of flexible alkyl chains is a common strategy for the design of low-melting ILs, e.g. in 1-alkyl-3-methylimidazolium ions.10,25,26 However, a longer alkyl chain results in higher ion mass and size as well as increased dispersion interactions, which all have the opposite effect. Another example that shows the importance of conformational flexibility is the comparison of ILs with the 1-ethyl-3-methylimidazolium cation ([EMIm]+) and analogous ILs with the related 1-ethyl-2,3-dimethylimidazolium cation ([EMMIm]+). Although the [EMMIm]+ cation suppresses hydrogen bonding at the 2-position, its ILs exhibit significantly higher viscosities. This counterintuitive behaviour is rationalized by a loss in entropy due to (i) a relatively high internal rotational barrier of the ethyl group in the [EMMIm]+ cation,10,27 and (ii) a lower number of easily accessible ion–ion conformers in the [EMMIm]+-IL as a consequence of the loss of the H-bond.28–30 A third example is ILs with ammonium cations and their isostructural phosphonium analogues. Phosphonium-ILs exhibit typically significantly lower viscosities than their ammonium derivatives, which is in part attributed to the higher conformational flexibility at the larger central P atom.31
Studies on the influence of conformational flexibility of anions are rare.9,21 Most likely, this is because a targeted modification of anions is less developed compared to the derivatization of cations. However, a few examples have been described in the literature. For example, a recent molecular dynamics simulation on [BMIm][PF6] (BMIm = 1-butyl-3-methylimidazolium) and [BMIm][(C2F5)3PF3] ([BMIm]FAP) showed that the higher flexibility of the tris(pentafluoroethyl)trifluorophosphate (FAP) anion is one reason among others for the low dynamic viscosity of [BMIm][(C2F5)3PF3] compared to its [PF6]− counterpart.32
The bis(trifluoromethylsulfonyl)imide anion [(N(SO2CF3)2]−, TFSI)33 represents one of the most widely studied anions in IL research.4,34–37 It leads to low melting, low viscous and thermally robust ILs with many organic cations.4,38 In addition, the TFSI anion is chemically and electrochemically very robust. As a consequence of this combination of beneficial properties, TFSI-ILs are among the most widely studied ILs in battery technology.3,34,39–44 The TFSI anion exists in a trans and a cis conformation. According to experimental and theoretical studies, the trans conformer is slightly more stable than the cis form (Fig. 1, 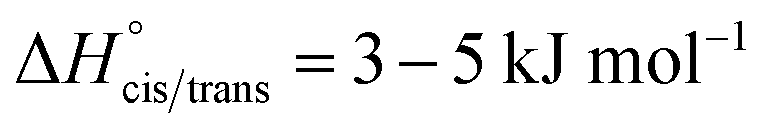 ) and the transition states for the interconversion of both conformers are of low energy (7–24 kJ mol−1).4 The high degree of conformational flexibility translates into a high ion mobility in and low viscosity of TFSI-ILs.9,45,46
) and the transition states for the interconversion of both conformers are of low energy (7–24 kJ mol−1).4 The high degree of conformational flexibility translates into a high ion mobility in and low viscosity of TFSI-ILs.9,45,46
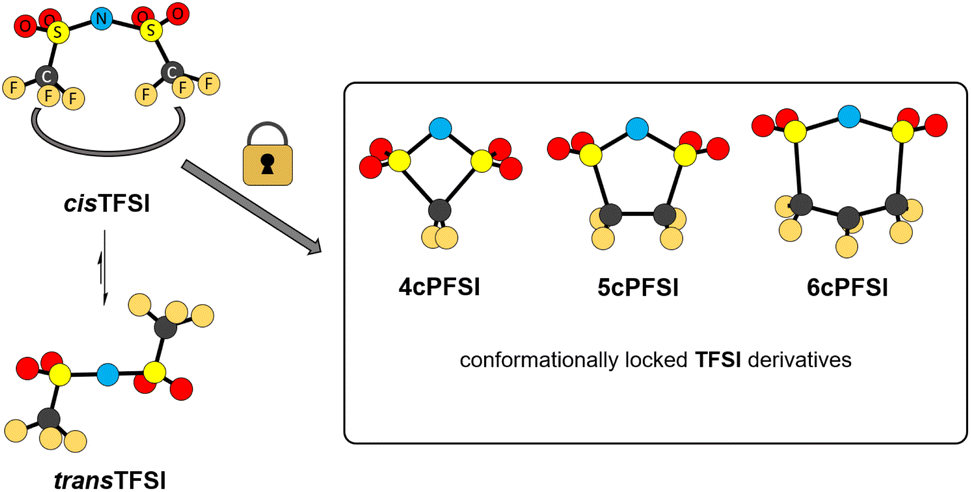 | ||
| Fig. 1 Conformational interconversion of the TFSI anion and conformationally locked cyclo-perfluoroalkanebis(sulfonyl)imide anions. | ||
In contrast to the TFSI anion, the related cyclo-hexafluoropropane-1,3-bis(sulfonyl)imide anion (6cPFSI, Fig. 1)47 is locked in the cis configuration. The reduced conformational flexibility of the 6cPFSI anion compared to the TFSI anion is, for example, reflected by higher melting points of [BMIm]6cPFSI (28 °C) compared to [BMIm]TFSI (−4 °C)48 and of [nBuPPh3]6cPFSI (119 °C) compared to [nBuPPh3]TFSI (89 °C).49 Similarly, the higher dynamic viscosity of [BMIm]6cPFSI (239 mPa s at 20 °C)48 than of [BMIm]TFSI (62.3 mPa s at 20 °C)48 has been attributed to the more rigid structure of the cyclic anion.4 The dynamic viscosity of the [BMIm]+-IL of the bis(pentafluoroethylsulfonyl)imide anion ([N(SO2C2F5)2]−) is much lower (140 mPa s at 20 °C)48 than the viscosity of [BMIm]6cPFSI. Thus, the higher molecular mass of the 6cPFSI anion compared to the TFSI anion is not the reason for the lower dynamic viscosity of [BMIm]TFSI. Further salts of the 6cPFSI anion with large organic cations have been described, which comprise comparably high melting points of more than 100 °C.50–54
In this study, ILs of the rigid cyclo-perfluoroalkanebis(sulfonyl)imide anions depicted in Fig. 1 with ring sizes of four ([cyclo-{CF2(SO2)2N}]−, 4cPFSI),55 five ([cyclo-{(CF2SO2)2N}]−, 5cPFSI)55 and six ([cyclo-{CF2(CF2SO2)2N}]−, 6cPFSI)47 with the widely applied countercations [EMIm]+, [BMIm]+ and [BMPL]+ (BMPL = 1-butyl-1-methylpyrrolidinium) are presented and selected physicochemical properties are described.56 Selected properties are compared to those of the respective TFSI-ILs. The structural features of the perfluoroalkanebis(sulfonyl)imide-ILs were elucidated by single-crystal X-ray diffraction (SC-XRD). We complement the experimental results with MD simulations which we performed for four prototypical ionic liquids.
Results and discussion
Synthesis
In contrast to salts of the 6cPFSI anion, salts of the cyclic imides 5cPFSI and 4cPFSI are not commercially available. Thus, the ammonium salts [NH4]5cPFSI and [NH4]4cPFSI were synthesized according to literature procedures. The first step is the electrochemical fluorination (ECF) according to the Simons process to give the perfluorinated disulfonylfluorides FSO2CF2SO2F and FSO2(CF2)2SO2F,57 which are reacted with ammonia to give [NH4]4cPFSI and [NH4]5cPFSI,55,56 respectively (Scheme 1). Treatment of the ammonium salts with aqueous KOH provides access to the corresponding potassium salts of the anions 5cPFSI and 4cPFSI.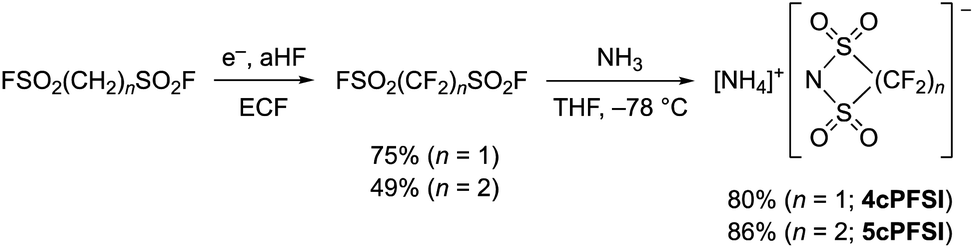 | ||
| Scheme 1 Electrochemical fluorination (ECF) of FSO2(CH2)nSO2F (n = 1, 2)57 and cyclization to result in [NH4]4cPFSI and [NH4]5cPFSI.55,56 | ||
These ammonium and potassium salts were used for metathesis in water to give the corresponding IL with [EMIm]+, [BMIm]+ and [BMPL]+ as countercations (Fig. 2). The corresponding 6cPFSI salts were obtained similarly using an aqueous solution of cyclo-hexafluoropropane-1,3-bis(sulfonyl)imide47 (Fig. 2). All ILs were obtained as colourless to pale yellow liquids from the reaction mixtures except for [EMIm]6cPFSI, which precipitated as colourless solid from the aqueous mixture and was isolated by filtration. The RTILs were typically phase separated from the aqueous phase.
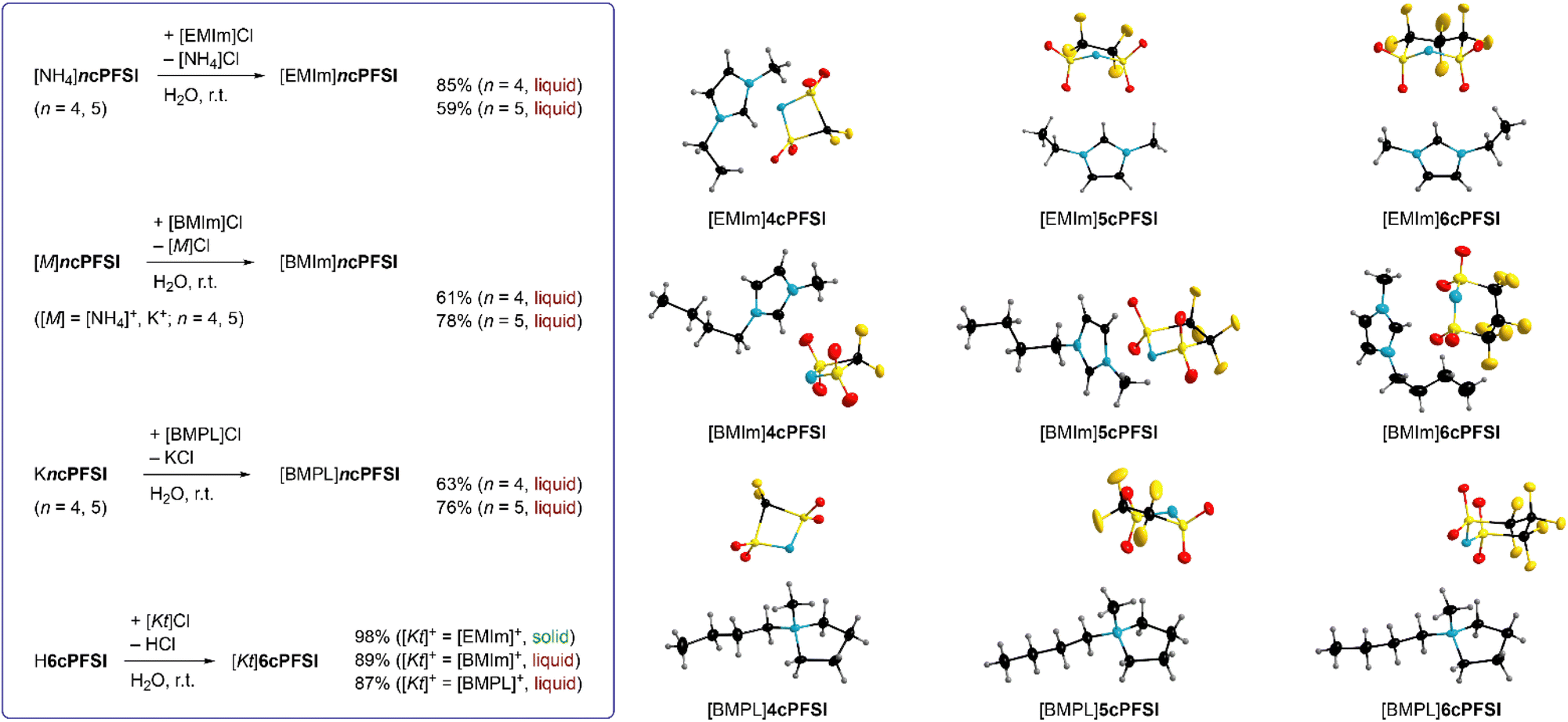 | ||
| Fig. 2 Synthesis of cyclo-perfluoroalkylbis(sulfonyl)imide-ILs via metathesis in water and ion pairs of the ILs in the crystal structure (thermal ellipsoids are set at 50% ([BMPL]5cPFSI: 20%) probability, H-atoms are depicted with arbitrary radii and any disorder omitted for clarity). | ||
Alternatively, extraction with CH2Cl2 can be used. The ILs were washed several times with double distilled water and subsequently dried under fine vacuum. The purity of the ILs was ensured by elemental analysis, NMR spectroscopy, Karl Fischer titration (<50 ppm H2O) and cyclic voltammetry (vide infra). Most likely, the repeated washing of the liquid salts (59–89%) is the reason for the relatively low yields compared to solid [EMIm]6cPFSI (98%).
The absence of chloride ions in the washing phase of the ILs with the cyclic imide anions 5cPFSI and 6cPFSI was confirmed by addition of a concentrated aqueous AgNO3 solution. For the 4cPFSI-ILs the silver nitrate test was not feasible. All three 4cPFSI-ILs studied herein revealed some solubility in water and they formed salts after addition of aqueous silver nitrate of the complex anion [Ag(4cPFSI)2]− that crystallized from the aqueous mother liquors (Fig. 3). Single crystals of [EMIm][Ag(4cPFSI)2], [BMIm][Ag(4cPFSI)2] and [BMPL][Ag(4cPFSI)2] were studied by SC-XRD (Fig. 3). So far, we have not optimized the syntheses of the complex salts and thus, no yields are given.
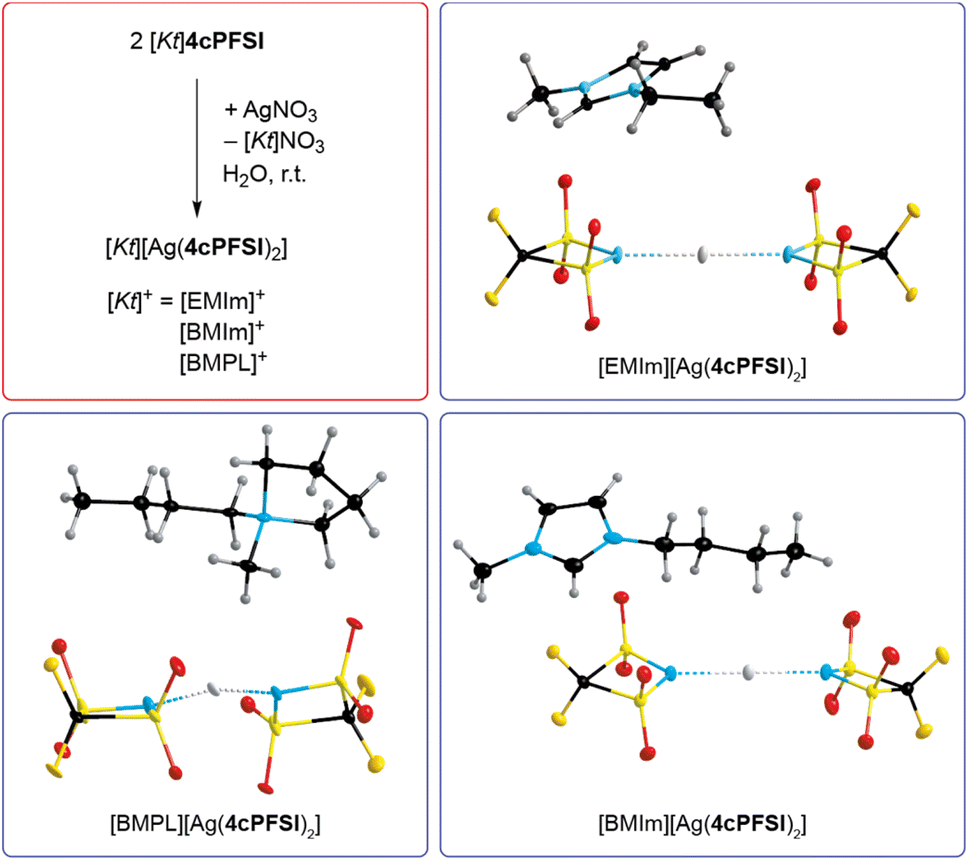 | ||
| Fig. 3 Synthesis of [Kt][Ag(4cPFSI)2] ([Kt]+ = [EMIm]+, [BMIm]+ and [BMPL]+) starting from the wash phase of the respective IL after addition of aqueous silver nitrate. Ion pairs of [Kt][Ag(4cPFSI)2] in the crystal structure (thermal ellipsoids are set at 25% ([Kt]+ = [EMIm]+) or 50% probability ([Kt]+ = [BMIm]+ and [BMPL]+), H atoms are shown with arbitrary radii, disorder of the cation in [EMIm][Ag(4cPFSI)2] and of both ions in [BMPL][Ag(4cPFSI)2] is omitted for clarity). | ||
The Ag atom of the [Ag(4cPFSI)2]− anion in the [EMIm]+ salt is located on an inversion center and hence, the N⋯Ag⋯N unit is linear (Fig. 3, Table S6 in the ESI†). In the [BMIm]+ salt the N⋯Ag⋯N unit is almost linear (178.7°) whereas in the [BMPL]+ salt it is slightly bent (166.7°). The Ag⋯N distances of the anions are similar in all three structures with 214.46(12) pm in [EMIm][Ag(4cPFSI)2], 215.2(6) and 214.4(6) pm in [BMIm][Ag(4cPFSI)2] and 213.7(3) and 213.8(3) pm in [BMPL][Ag(4cPFSI)2]. The related [Ag(TFSI)2]− anion is unknown but the complex salt [Ag(NCCH3)4]2[Ag(TFSI)3] with the dianion [Ag(TFSI)3]2− has been structurally characterized.58 Attempted crystallization of salts of the monoanionic silver(I) complex [Ag(TFSI)2]− remained unsuccessful, so far. For example, crystals of 3∞{AgTFSI} were obtained from a mixture of [EMIm]TFSI and AgTFSI in dichloromethane.59 Other bis(sulfonyl)imide anions such as [N(SO2F)2]− and [N(SO2CH3)2]− led to linear, two-coordinate argentate complexes, e.g. in [Ag(NCCH3)2][Ag{N(SO2F)2}2]60 and [Ag(NCCH3)4][Ag{N(SO2CH3)2}2].61
Crystal structures
The crystal structures of all ILs with the three cyclic imide anions 4cPFSI, 5cPFSI and 6cPFSI in combination with the countercations [EMIm]+, [BMIm]+ and [BMPL]+ were determined (Fig. 2). In addition, the crystal structure of [BMIm]cisTFSI was redetermined.62 The single-crystal X-ray diffraction studies provide a detailed insight into the arrangement and interactions of the ions in the solid state. Ion–ion interactions in the solid are known to often correlate to ion–ion interactions in the corresponding liquid phase,63–68i.e. in recent years, theories of quasi- and pseudo-lattices have been established, which view the liquid phase as a collapsed crystal.69–72 Crystals of salts with melting points below room temperature (vide infra) were grown by manual in situ cryocrystallization. Selected bond parameters of the cyclic imide anions 4cPFSI, 5cPFSI and 6cPFSI as well as the TFSI anion are collected in Table 1.| IL | Sym.c | d(N–S)d | d(S–C)d |
d(S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)d O)d |
∡(S–N–S) | ∡(N–S–C) | φ(S–N–S–C)e | |
|---|---|---|---|---|---|---|---|---|
| a [BMPL]5cPFSI was not considered due to disorder of the anion. b Bond lengths in Å and angles in °. c Symmetry of the anion; values in brackets indicate close higher symmetry. d Mean values where applicable. e Dihedral angle. f The disordered anion was not considered. g Two independent formula units in the unit cell. | ||||||||
| [EMIm]4cPFSI | C 1 (C2v) | 1.605(2) | 1.855(8) | 1.439(2) | 101.22(8) | 87.37(7)/87.46(8) | 1.64(9)/−1.64(9) | |
| [BMIm]4cPFSIf | C 1 (Cs) | 1.602(2) | 1.851(2) | 1.435(2) | 101.10(9) | 87.55(8)/87.13(8) | 4.83(9)/−4.81(9) | |
| [BMPL]4cPFSI | C 1 (Cs) | 1.611(2) | 1.859(3) | 1.437(2) | 100.97(11) | 86.83(10)/87.31(10) | 8.12(11)/−8.09(11) | |
| PBE0/def2-TZVPP | C 2v | 1.599 | 1.856 | 1.440 | 101.8 | 87.1 | 0 | |
| [EMIm]5cPFSI | C 1 | 1.587(2) | 1.853(2) | 1.433(3) | 112.9(1) | 96.13(8)/98.81(8) | 45.8(1)/−34.3(1) | |
| [BMIm]5cPFSI | Anion 1g | C 1 | 1.588(3) | 1.856(3) | 1.429(2) | 114.03(15) | 97.03(12)/99.41(12) | 25.9(2)/−41.7(2) |
| [BMIm]5cPFSI | Anion 2g | C 1 | 1.591(2) | 1.854(3) | 1.429(2) | 113.00(15) | 96.89(12)/99.02(12) | 33.8(2)/−44.7(2) |
| PBE0/def2-TZVPP | C 1 | 1.584 | 1.866 | 1.436 | 115.1 | 96.3/98.7 | 29.5/−42.7 | |
| [EMIm]6cPFSI | Anion 1g | C 1 (Cs) | 1.583(2) | 1.837(3) | 1.430(2) | 120.29(10) | 101.09(9)/101.77(9) | 64.21(13)/−64.47(13) |
| [EMIm]6cPFSI | Anion 2g | C 1 (Cs) | 1.582(2) | 1.842(3) | 1.430(2) | 120.33(10) | 101.44(9)/101.45(9) | 63.70(13)/−65.03(12) |
| [BMIm]6cPFSI | Anion 1g | C 1 (Cs) | 1.584(7) | 1.841(9) | 1.425(6) | 119.7(4) | 100.9(4)/102.0(4) | 64.2(5)/−65.2(5) |
| [BMIm]6cPFSI | Anion 2g | C 1 (Cs) | 1.585(5) | 1.839(9) | 1.425(6) | 119.9(3) | 100.9(3)/101.9(3) | 63.9(5)/−64.6(5) |
| [BMPL]6cPFSI | C 1 (Cs) | 158.6(3) | 184.5(4) | 142.9(4) | 119.7(2) | 100.66(14)/100.71(14) | 66.5(2)/−66.2(2) | |
| PBE0/def2-TZVPP | C s | 1.579 | 1.856 | 1.433 | 122.2 | 101.0/101.0 | 64.1/−64.1 | |
| [BMIm]cisTFSI | Anion 1g | C 1 | 1.575(5) | 1.836(7) | 1.430(5) | 125.9(3) | 100.3(3)/106.7(3) | 117.6(4)/−82.5(4) |
| [BMIm]cisTFSI | Anion 2g | C 1 | 1.578(6) | 1.842(9) | 1.429(5) | 126.9(4) | 101.8(3)/104.5(4) | 116.2(5)/−99.8(5) |
| PBE0/def2-TZVPP | C 1 | 1.571 | 1.853 | 1.434 | 127.9 | 98.7/104.6 | 88.5/−131.9 | |
| [BMPL]transTFSI64 | C 1 | 1.583(3) | 1.839(4) | 1.431(3) | 124.6(2) | 104.8(2)/104.4(2) | 85.5(3)/89.3(3) | |
| PBE0/def2-TZVPP | C 2 | 1.574 | 1.853 | 1.433 | 127.2 | 102.2 | 94.0 | |
The symmetry of the 4cPFSI anion in its [EMIm]+ salt is close to C2v, which nicely agrees with the calculated structure (Table 1). In the [BMIm]+ and [BMPL]+ salt, the N atom is significantly bent out of the S–C–S plane, which leads to a reduced Cs symmetry. A similar deviation from C2v symmetry of the 4cPFSI anion was reported for [NH4]4cPFSI73 and K4cPFSI.55 The 5cPFSI anion adopts a half-chair conformation (C1 symmetry) in the three crystal structures and in Rb5cPFSI that was studied, earlier.55 The 6cPFSI anion reveals an almost ideal chair conformation close to Cs symmetry in the three crystal structures. The same conformation was reported for salts of the 6cPFSI anion with inorganic and organic cations, previously.49,53,54,74–76 So, the 5cPFSI anion has the lowest symmetry of the three related imide anions.
The S–N–S angle increases with increasing ring size from ca. 101° in 4cPFSI to approximately 114° in 5cPFSI and to ca. 120° in 6cPFSI (Table 1). The latter angle is still slightly smaller than the S–N–S angle in the cis- and trans-conformer of the parent TFSI anion with ca. 125–127° (Table 1). The significantly smaller S–N–S angle, especially in 4cPFSI, reflects a different electronic situation at nitrogen, which is also indicated by the ready formation of the argentate complex [Ag(4cPFSI)2]− (Fig. 3). Similar to the S–N–S angle, the N–S–C angles increase with increasing ring size. The distances d(N–S), d(S–C) and d(SO) decrease in the row 4cPFSI, 5cPFSI and 6cPFSI. However, the differences are small and thus, in most cases not significant but in line with calculated bond distances. The bonding parameters of the six-membered ring derivative 6cPFSI are closest to those of the cisTFSI anion (Table 1).
The van der Waals volume of the imide anions was assessed using the program packages Olex2 (ref. 77) and Platon78–80 (Table 2). Both methods gave very similar volumes. The van der Waals volume increases in the order 4cPFSI, 5cPFSI and 6cPFSI. The volumes of the cis- and trans-conformers of the non-cyclic TFSI anion do not differ. This result agrees with earlier studies that resulted in very similar volumes of both conformers, as well.81,82 The volume of the TFSI anion is a little smaller than the volume of 6cPFSI but significantly larger than Vm(vdW)− of 5cPFSI. The van der Waals volume linearly increases with the molecular mass (Fig. S41 in the ESI†).
|
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Anion | M anion [g mol−1] | Cation | T [°C] | s.g.b | Z | Z′ | V cell [Å3] | V m [Å3] | P(Platon)d [%] | V m(vdW)(Platon)e [Å3] | V m(vdW) − (Platon)f [Å3] | V m(vdW) − (Olex2)g [Å3] | V m(vdW) + (Olex2)g [Å3] |
| a Calculated at the PBE0/def2-TZVPP level of theory. b s.g. = space group. c V m = Vcell × Z−1. d P = packing index.78,79 e van der Waals volume of one formula unit obtained with the Platon program package.78–80 f V m(vdW) −(Platon) = Vm(vdW)(Platon) − Vm(vdW)+; Vm(vdW)+([EMIm]+) = 110 ± 1.4 Å3, Vm(vdW)+([BMIm]+) = 142 ± 1.5 Å3, Vm(vdW)+([BMPL]+) = 158 ± 7 Å3, 171 ± 0.7 Å3.83 g van der Waals volume calculated from the crystallographic data using the Olex2 program.77 | |||||||||||||
| 4cPFSI | 192.13 | [EMIm]+ | 100 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
2 | 1 | 603.04(3) | 302 | 70.9 | 214 | 104 | 103 | 104 |
| [BMIm]+ | 100 | Pbca | 16 | 2 | 5725.43(8) | 358 | 68.1 | 244 | 102 | 103 | 133 | ||
| [BMPL]+ | 100 | P212121 | 4 | 1 | 1484.43(2) | 371 | 69.9 | 259 | 101 | 104 | 144 | ||
| 5cPFSI | 242.14 | [EMIm]+ | 100 |
P |
2 | 1 | 692.05(7) | 346 | 69.0 | 239 | 129 | 128 | 104 |
| [BMIm]+ | 100 |
P |
4 | 2 | 1568.45(3) | 392 | 68.9 | 270 | 128 | 128 | 133 | ||
| [BMPL]+ | 100 | P21/n | 4 | 1 | 1663.88(6) | 416 | 67.8 | 282 | 124 | 128 | 143 | ||
| 6cPFSI | 292.15 | [EMIm]+ | 100 |
P |
4 | 2 | 1542.43(6) | 386 | 69.4 | 268 | 158 | 154 | 104 |
| [BMIm]+ | 100 | Pca21 | 8 | 2 | 3507.12(10) | 438 | 67.4 | 295 | 153 | 153 | 133 | ||
| [BMPL]+ | 100 |
P |
2 | 1 | 882.98(2) | 441 | 70.5 | 311 | 153 | 154 | 144 | ||
| cisTFSI | 280.14 | [BMIm]+ | 100 | P21/n | 8 | 2 | 3471.91(5) | 434 | 66.7 | 289 | 147 | 148 | 134 |
| cisTFSI | [HexPyrr]+ | 125 |
P |
4 | 2 | 1966.0(6) | 492 | 64.7 | 318 | 147 | 148 | 159 | |
| transTFSI | [BMPL]+ | 130 | P212121 | 4 | 1 | 1811.1(4) | 453 | 67.4 | 305 | 147 | 148 | 145 | |

Hirshfeld surface analyses,84–88 which is a well-established tool for the interpretation of intermolecular interactions in crystal structures in general, and of organic salts and ionic liquids in particular,49,54,89,90 have been performed to study the noncovalent interionic interactions in the crystals. The program CrystalExplorer17 was used for the analysis.91,92
The [EMIm]+ salts of the three cyclic anions 4cPFSI, 5cPFSI and 6cPFSI crystallize in the triclinic space group P (Table S5 in the ESI†). In contrast, [EMIm]cisTFSI crystallizes in the non-centrosymmetric orthorhombic space group Pca21.63 In all four structures, the anions and cations are interlinked via SO⋯H, N⋯H and F⋯H hydrogen bonds,30,93 as exemplified by the Hirshfeld surfaces in Fig. 4 and evident from the Hirshfeld surface contributions (Table S7 in the ESI†) and the fingerprint plots84–88,92 depicted in Fig. S44–S48 in the ESI.† The Hirshfeld analyses show that the SO⋯H interactions are the shortest cation–anion interactions in all crystals, which parallels earlier observations on crystal structures of salts of imide anions, e.g.TFSI,49,63,906cPFSI,49 and [N(SO2CH3)2]− ([NMes2]−).90 The H-bonded motifs are complex and include bi- and trifurcated interactions. The most pronounced H-bonds are found for the H atoms of the central imidazolium ring, which are the most acidic ones. However, further H-bonds are present for the alkyl chains.
 | ||
| Fig. 4 Environments of the cyclic imide anions 4cPFSI, 5cPFSI and 6cPFSI in the crystal structures of their [EMIm]+ salts. dnorm mapped onto the Hirshfeld surfaces of the respective central imide anion (thermal ellipsoids set at 50% probability, H-atoms are shown with arbitrary radii). | ||
In the crystals of the [EMIm]+ salts of the three cyclic anions 4cPFSI, 5cPFSI and 6cPFSI the imidazolium rings of two neighbouring [EMIm]+ cations reveal antiparallel anion-templated π+–π+ stacking as depicted in Fig. 5. The π+–π+ stacking is evident from the pattern of red and blue triangles in the same region of the shape index placed onto the Hirshfeld surface of the [EMIm]+ cation.84 It is also obvious from the plots of the surrounding of the anions 4cPFSI, 5cPFSI and 6cPFSI by the cations in Fig. 4. In all three salts the imidazolium planes are parallel to each other as a result of crystallographic symmetry. The centroid–centroid distances (dπ) are similar for the three analogues and they range from 3.6874(19) to 4.0454(19) Å. The antiparallel aligned [EMIm]+ cations are shifted from an ideal stacked position and hence, the distances between the planes (dplane) of 3.243(4) to 3.447(3) Å are shorter than dπ (Fig. 4). The occurrence of π+–π+ stacking between imidazolium rings has been observed for different imidazolium cations by SC-XRD.94–98 In most cases, small anions that efficiently compensate for the positive charge are present and the imidazolium cations are typically arranged in an antiparallel fashion. Furthermore, imidazolium cations with small, sterically non-demanding substituents, especially hydrogen and methyl, tend to form anion-templated π+–π+ dimers. So, π+–π+ stacking of [EMIm]+ cations has been rarely observed in the solid state. One of the rare examples is the crystal structure of [EMIm]Cl.95 The shortest distances in the crystal structure of [EMIm]Cl are 3.791(17) Å for the ring–ring distance (dπ) and 3.60(3) Å for dplane. Both values are similar to those found for the related salt [EMMIm]Cl98 and for [EMIm]ncPFSI (n = 4–6) in the present study.
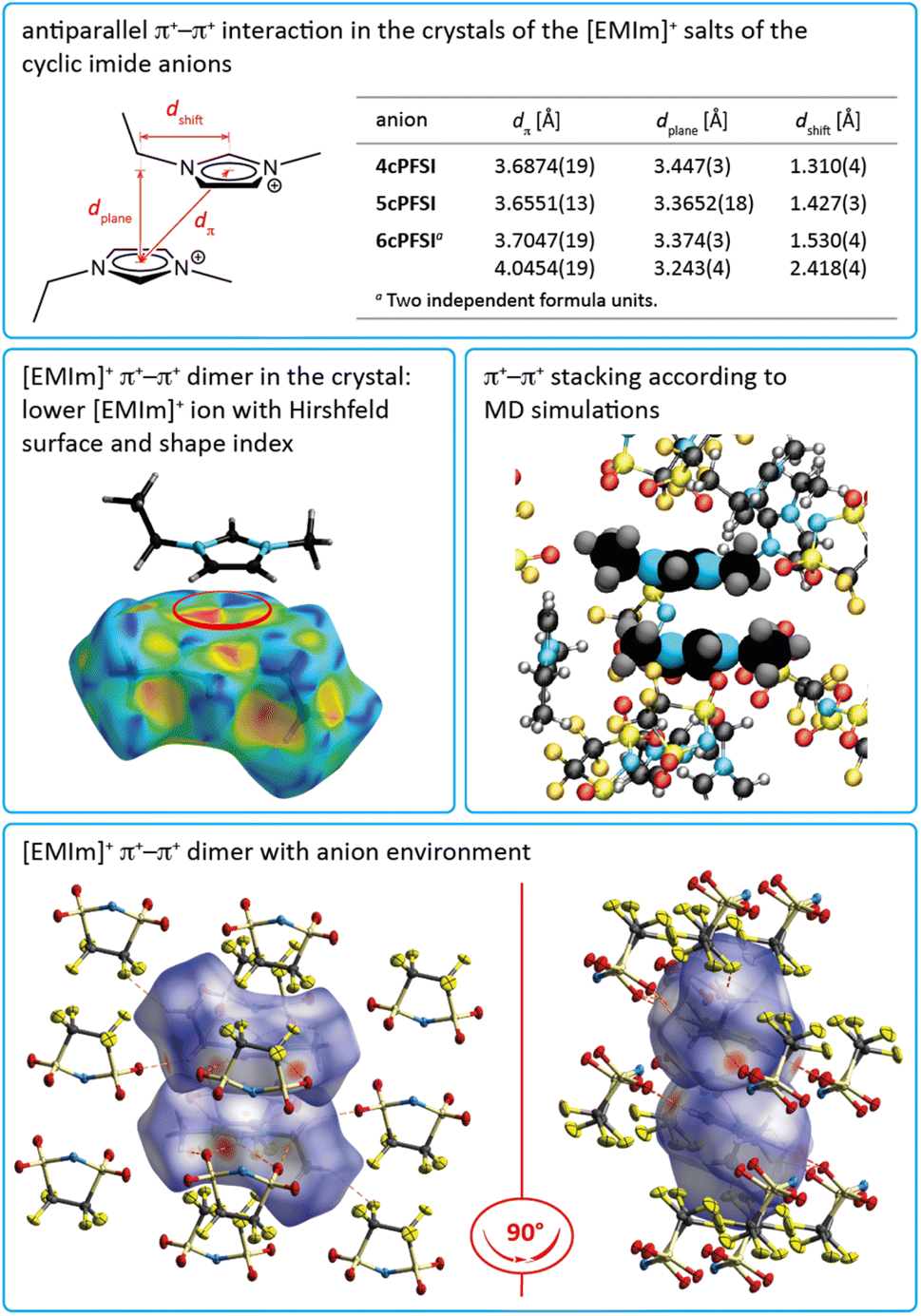 | ||
| Fig. 5 Antiparallel displaced π+–π+ stacking of [EMIm]+ cations in the crystal of [EMIm]+ salts with ncPFSI (n = 4–6) counterions (top), Hirshfeld surface with shape index of an [EMIm]+ cation in [EMIm]5cPFSI (middle, left), sketch of two [EMIm]+ cations showing the π+–π+ stacking according to MD simulations (middle, right) and two different views of a π+–π+ [EMIm]+ dimer with Hirshfeld surfaces and surrounding 5cPFSI anions in the crystal of [EMIm]5cPFSI (bottom). | ||
The nature of π+–π+ stacking, in general, and for imidazolium cations, in particular, has been the subject of theoretical studies.94,99–105 In these studies it has been pointed out that smaller anions that form comparably strong hydrogen bonds foster the occurrence of π+–π+ stacking while efficient anion–π+ interactions that tend to increase with increasing anion size compete with cation–cation π+–π+ stacking.99–102 Although the binding energy between the imidazolium rings is rather small, π+–π+ interactions are predicted to significantly influence the physicochemical properties of imidazolium ILs, e.g. viscosity102 and interactions with surfaces of electrodes.103
In contrast to the structures of the three cyclic imide ILs [EMIm]ncPFSI (n = 4–6), in the crystal structure of [EMIm]cisTFSI no π+–π+ stacking is found.63 Thus, the occurrence of π+–π+ stacking interactions in the [EMIm]+ salts of the three cyclic imide anions and the absence of such interactions in the crystal of the cisTFSI salt points towards significant differences of the properties of the closely related anions and their salts (vide infra). However, although the missing π+–π+ stacking interactions in [EMIm]cisTFSI highlight a significant difference, the plots of the three-dimensional packing diagrams in Fig. 6 of [EMIm]5cPFSI and [EMIm]cisTFSI show the similarity of the packing, in general. The anions and cations form stacks in the crystals and the anion–cation arrangement can be distinguished into more ionic and more non-ionic domains.
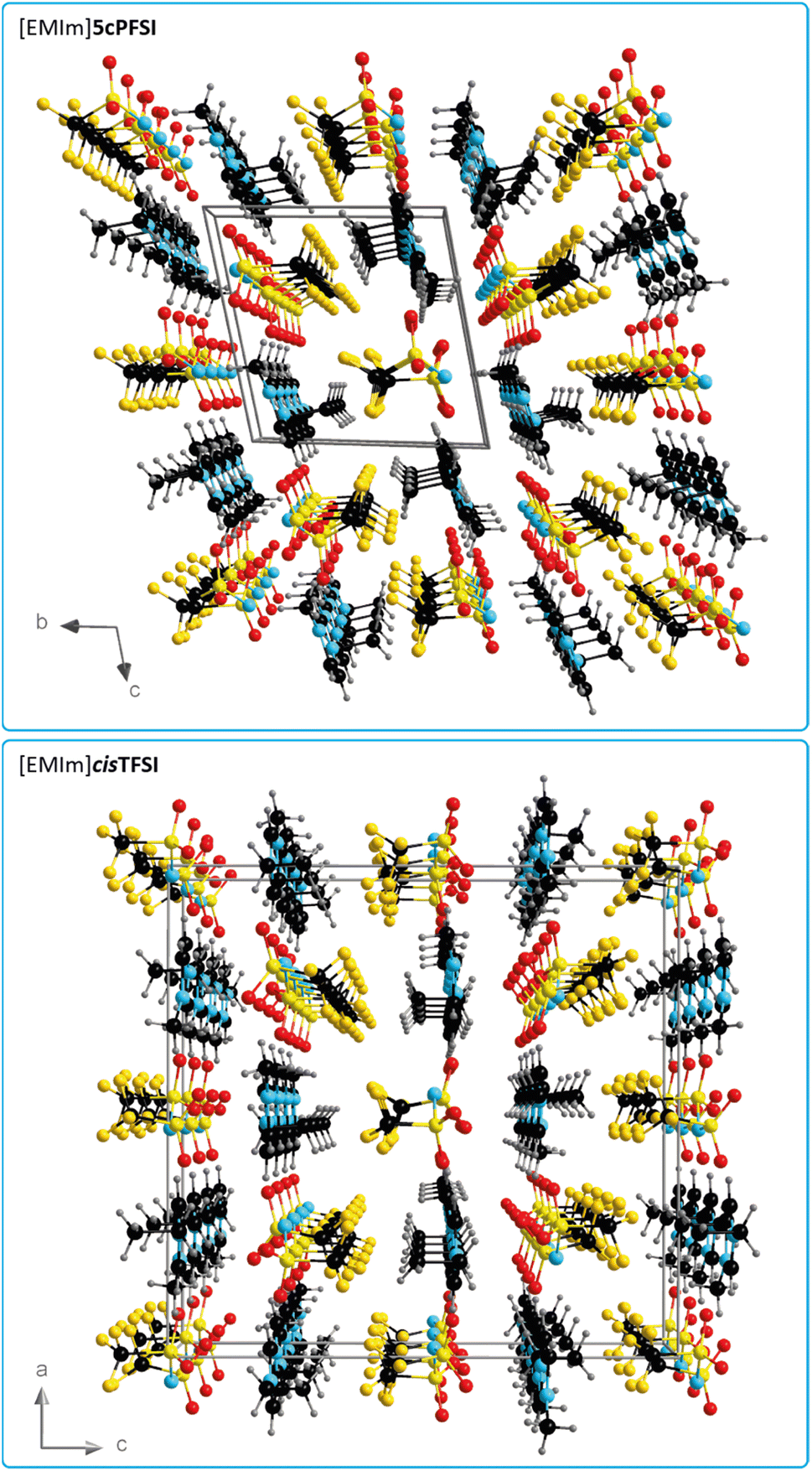 | ||
| Fig. 6 Three-dimensional packing diagram with unit cell of [EMIm]5cPFSI (top) and [EMIm]cisTFSI (bottom)63 (all atoms are depicted with arbitrary radii). | ||
The MD simulations demonstrate that the π+–π+ stacking observed in the experimental crystal structure is also present in the bulk liquid [EMIm]5cPFSI (Fig. 5). Specifically, a comparable degree of stacking was observed for the two ionic liquids [EMIm]TFSI and [EMIm]5cPFSI (Fig. S21–S23 in the ESI†).
The crystal structures of [BMIm]ncPFSI (n = 4–6) and [BMIm]cisTFSI show similar interactions and distances between anions and cations, and anions and anions, as their [EMIm]+ counterparts as exemplified for [BMIm]5cPFSI in Fig. 7. However, in contrast to the [EMIm]+ salts of 4cPFSI, 5cPFSI and 6cPFSI, the respective [BMIm]+ salts do not show any π+–π+ interactions in the crystal. Most likely, the sterically more demanding n-butyl groups, which are aligned next to each other (Fig. 7), prevent the stacking between the imidazolium rings. This observation is in line with earlier experimental observations, e.g. for picrate ILs π+–π+ stacking was found for the [MMIm]+ salt but not for the related [PMIm]+ salt (MMIm = 1,3-dimethylimidazolium; PMIm = 1-propyl-3-methylimidazolium).96
 | ||
| Fig. 7 Excerpt of the crystal structure of [BMIm]5cPFSI showing the packing of cations and anions (top) and surrounding of [BMIm]+ by anions with C–H⋯O and C–H⋯N hydrogen bonding illustrated by dnorm mapped onto the Hirshfeld surface of [BMIm]+ (bottom; thermal ellipsoids are set at 50% probability, H-atoms are depicted with arbitrary radii). | ||
The interactions between the fluorinated imide anions are based on weak F⋯F contacts. These interactions are evident from the Hirshfeld surface contributions in Table S7 in the ESI† and illustrated in the 2D-fingerprint plots of the anions in Fig. S44, S46 and S48 in the ESI.† In addition, they are shown for the structures of the [EMIm]+-ILs in Fig. 4 in the anion representations (bottom figures). The orientations of the anions 5cPFSI and the cisTFSI in Fig. 6 show the close proximity of the fluorine atoms of different anions, which is indicative for weak F⋯F interactions. These F⋯F interactions range in [EMIm]5cPFSI from 2.85 to 3.57 Å. The importance of F⋯F interactions decreases with decreasing number of fluorine atoms per cyclic imide anion as shown by the Hirshfeld surface contributions in Table S7 in the ESI† and the 2D fingerprint plots in Fig. S44, S46 and S48 in the ESI.† For [BMPL]5cPFSI and [BMPL]4cPFSI no F⋯F interactions are present, at all.
Pronounced anion–anion close contacts were also observed in the MD simulations of the bulk ionic liquid [EMIm]5cPFSI. Fig. 8 shows the atom-resolved spatial distribution function for the anion surrounded by other anions. Fluorine–fluorine interactions dominate in these first shell contacts, see Fig. 8 for a snapshot from the simulation. We identified four prevalent mutual orientations, see ESI† for more details.
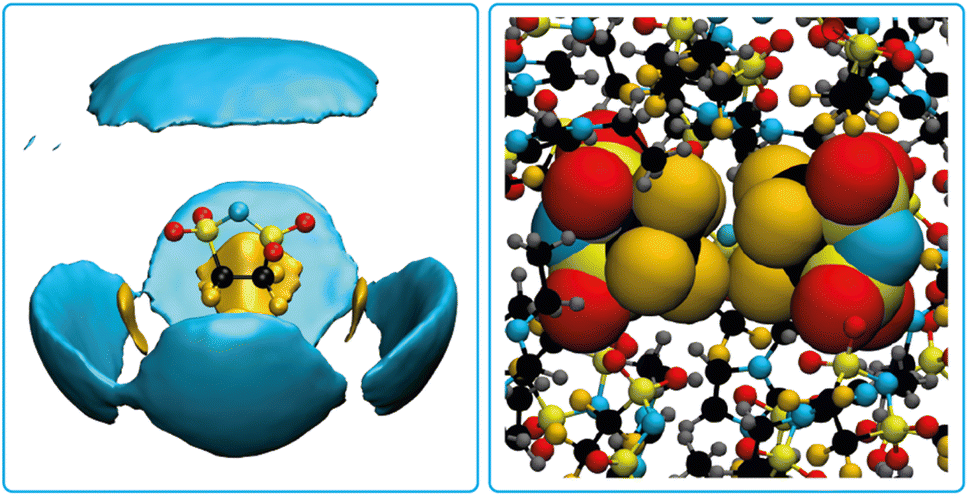 | ||
| Fig. 8 Atom-resolved spatial distribution function for the 5cPFSI anion with surrounding anions (left) and a snapshot from the MD simulation showing a pair of 5cPFSI anions (right). | ||
The TFSI anion was found to easily adopt the cis or the trans conformation in crystals of imidazolium salts, i.e. [EMIm]cisTFSI,63 [EMIm]transTFSI62 and [BMIm]cisTFSI.62 Most likely, the occurrence of the cis conformation is due to comparably strong H-bonds between the TFSI anion and the relatively acidic H atoms of the central imidazolium rings that force the TFSI anion into the enthalpically less favoured conformer. In general, the H-bonded motifs with bi- and trifurcated H-bonds of the cisTFSI anion in its [EMIm]+ and [BMIm]+ salts are very similar to those found in the respective crystal structures of [EMIm]6cPFSI and [BMIm]6cPFSI. For the [BMPL]+ cation weaker H-bonds are only possible. So, it is not surprising that only the transTFSI anion was observed in the crystal of [BMPL]TFSI,64 so far.
Thermal properties
The melting point and liquid range of an IL are obvious key parameters to assess its performance and potential for various applications. The thermal parameters depend on the ion mass and size, ion symmetry, the surface electron density and the ion size ratio among others. The correlation between ion rigidity/flexibility and low melting points has been rationalized through the entropy of the liquid phase.22 A higher number of available conformers in the liquid phase typically results in an increase of entropy leading to lower melting temperatures of the IL.23 The influence of conformational flexibility on the thermal properties is reflected by the phase transitions of the ILs presented in this study (Table 3). Hence, it is not surprising that the TFSI anion, which readily switches between the cis and trans conformation, leads to ILs with very low melting points (Table 3). The respective ILs with the related cyclic imide anion 5cPFSI have similar low melting points. In contrast, the melting points of the ILs of the imide anions 4cPFSI and 6cPFSI are significantly higher, with [EMIm]6cPFSI having a melting point as high as +67 °C. For the small cyclic 4cPFSI anion, the relatively high electron density as evident from the ESP plot in Table 2 results in relatively strong cation–anion interactions. However, the argument of a high electron density does not hold for the 6-membered ring imide anion 6cPFSI (ESP plot in Table 2). Therefore, the explanation for the high melting points of the 6cPFSI-ILs must be different and presumably the rigidity of the 6cPFSI anion, which favours the chair conformation, is the reason. In contrast, switching between the half-chair and the envelope conformation of the 5cPFSI anion is accompanied by almost no change in energy (Fig. 9). Despite the aforementioned strong cation–anion interactions between [EMIm]+ and the 4cPFSI anion, this small 4-membered cyclic imide anion is rigid (Fig. S37 in the ESI†), which might contribute to the comparably high melting point of 27 °C. Most of the ILs studied herein form supercooled liquids with glass transition temperatures below −50 °C (Table 3). The only exceptions are [EMIm]6cPFSI, [BMIm]6cPFSI and [BMPL]4cPFSI that crystallize upon cooling. So, again the rigid anion 6cPFSI shows the strongest tendency for crystallization.| IL | T mp [°C] | T g [°C] | T dec [°C] |
|---|---|---|---|
| a Temperatures are onset values (DSC). b T mp = melting point. c T g = glass transition temperature. d T dec = decomposition temperature. e Melting point was determined from the in situ cryocrystallization experiments performed for the SC-XRD study (vide infra). The melting point was not observed in the DSC curve. f n.o. = not observed. | |||
| [EMIm]4cPFSI | 27 | −77 | 285 |
| [EMIm]5cPFSI | −18e | −91 | 423 |
| [EMIm]6cPFSI | 67 | n.o.f | 412 |
| [EMIm]TFSI | −18 | −98 | 394 |
| [BMIm]4cPFSI | 16 | −76 | 262 |
| [BMIm]5cPFSI | 2 | −52 | 405 |
| [BMIm]6cPFSI | 28 | n.o. | 409 |
| [BMIm]TFSI | −4 | −87 | 404 |
| [BMPL]4cPFSI | 26 | n.o. | 251 |
| [BMPL]5cPFSI | −19 | −97 | 383 |
| [BMPL]6cPFSI | 20 | −73 | 405 |
| [BMPL]TFSI | −21 | −88 | 400 |
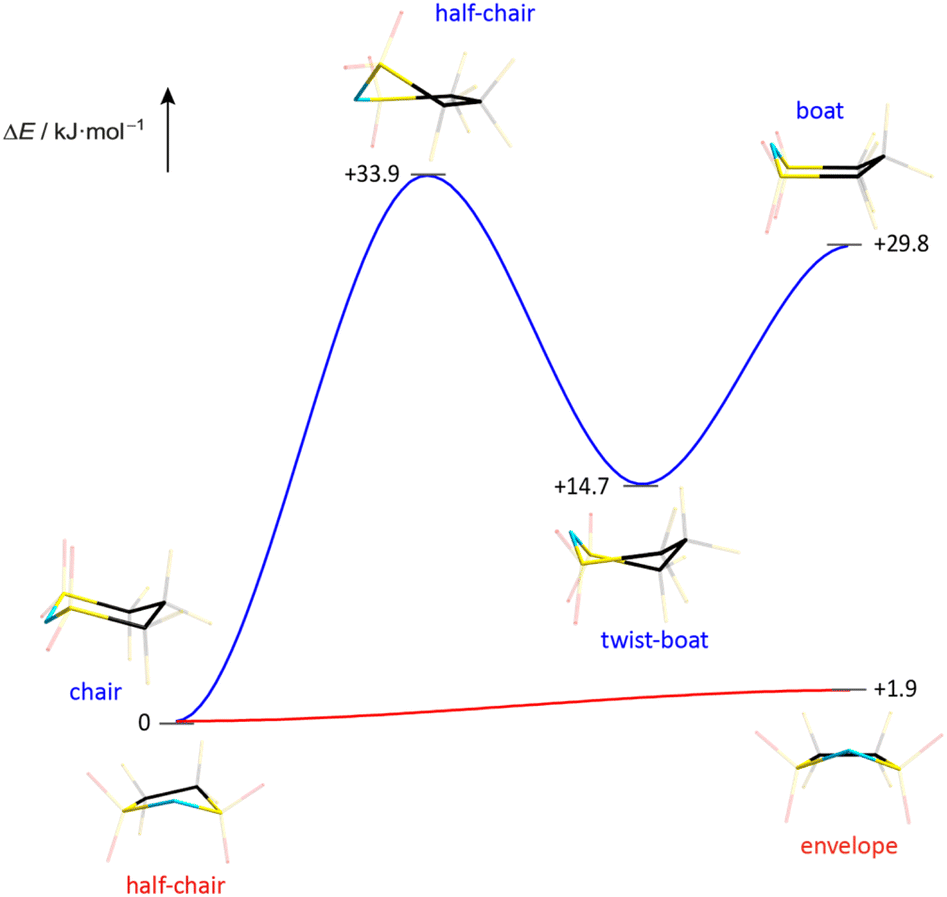 | ||
| Fig. 9 Energy profile for the conformational change of the cyclic imide anions 5cPFSI and 6cPFSI (PBE0/def2-TZVPP). | ||
The thermal stabilities of the ILs with the cyclic imide anions 5cPFSI and 6cPFSI with all three countercations [EMIm]+, [BMIm]+ and [BMPL]+ are in the range of 383–423 °C and thus, similar to the stabilities of the TFSI salts. Although all 4cPFSI-ILs exhibit significantly lower decomposition temperatures than the other imide-ILs studied herein, their thermal stabilities are surprisingly high with decomposition temperatures of more than 250 °C (Table 3). Probably, the comparably low decomposition temperature is related to the ring strain of the small cyclic imide anion 4cPFSI, which is also the reason for its relatively high sensitivity to strong Brønsted acids.106
Behaviour against elemental lithium
A freshly cut piece of elemental lithium was added to a small sample of the neat ionic liquid [Kt]ncPFSI ([Kt]+ = [BMIm]+, [BMPL]+; n = 4–6) under an Ar atmosphere and stored at 30 °C for 24 hours. The lithium surfaces became brownish and the ionic liquids changed their colour to yellow or pale brownish. In some cases, formation of a few gas bubbles was observed. However, the 1H, 19F and 13C NMR spectra revealed no significant changes of the ILs after storage in the presence of lithium at 30 °C and most of the 7Li NMR spectra revealed the presence of a minor amount of a Li species (−1.0 to −2.5 ppm). The six ILs were heated to 70 °C for further 24 hours in the presence of lithium but no significant changes were detected neither visually nor by multinuclear NMR spectroscopy. Thus, the ILs based on the three cyclic imide anions 4cPFSI, 5cPFSI and 6cPFSI are stable against lithium after initial passivation. Furthermore, we have no signs for any reaction of the three anions with elemental lithium.Electrochemical properties
Since ionic liquids consist purely of ions this substance class of compounds is of utmost interest for various electrochemical applications, e.g. batteries and supercapacitors (supercaps),2,3,43,44,107–116 and for chemical synthesis where electrochemical stability is of relevance.117–120 Thus, their electrochemical stability is a decisive parameter and the understanding of factors that determine their key physicochemical properties is of utmost importance.The [EMIm]+-, [BMIm]+- and [BMPL]+-ILs with the cyclic imide anions ncPFSI (n = 4–6) possess large electrochemical windows of 4.2 to 5.3 V in acetonitrile (0.1 mol L−1) that are similar to those of the related TFSI-ILs (Fig. 10 and Table 4). The smallest anion 4cPFSI has the lowest oxidative stability for all three types of ILs. The ILs with the larger imide anions 5cPFSI, 6cPFSI and TFSI have very similar anodic limits (Ea) and do not show a clear trend with respect to oxidative stability. The reductive stability of the ILs in CH3CN is determined by the cation and thus, no dependence on the counteranion is evident.
 | ||
| Fig. 10 Cyclic voltammograms of imide ionic liquids with the cations [EMIm]+, [BMIm]+ and [BMPL]+ (solid line: neat IL; dashed line: solution of the IL in CH3CN (0.1 mol L−1) at 20 °C, scan rate: 50 mV s−1, glassy carbon working electrode). | ||
| IL | Neat IL | Solution in CH3CN (0.1 mol L−1) | ||||
|---|---|---|---|---|---|---|
| E c [V] | E a [V] | ΔE [V] | E c [V] | E a [V] | ΔE [V] | |
| a Cathodic and anodic limits Ec and Ea; electrochemical window ΔE = Ea − Ec. b Supercooled melt. c Solid at 20 °C. | ||||||
| [EMIm]4cPFSI | −2.4b | 2.8b | 5.2b | −2.4 | 1.8 | 4.2 |
| [EMIm]5cPFSI | −2.4 | 2.8 | 5.2 | −2.5 | 2.2 | 4.7 |
| [EMIm]6cPFSI | —c | —c | —c | −2.4 | 2.1 | 4.5 |
| [EMIm]TFSI | −2.4 | 2.3 | 4.7 | −2.4 | 2.1 | 4.5 |
| [BMIm]4cPFSI | −2.4 | 2.9 | 5.4 | −2.4 | 1.8 | 4.2 |
| [BMIm]5cPFSI | −2.3 | 2.3 | 4.6 | −2.5 | 2.5 | 5.0 |
| [BMIm]6cPFSI | −2.1 | 2.5 | 4.6 | −2.5 | 2.2 | 4.7 |
| [BMIm]TFSI | −2.1 | 2.6 | 4.7 | −2.5 | 2.4 | 4.9 |
| [BMPL]4cPFSI | —c | —c | —c | −3.0 | 2.0 | 5.0 |
| [BMPL]5cPFSI | −2.1 | 2.5 | 4.6 | −3.0 | 2.3 | 5.3 |
| [BMPL]6cPFSI | −2.2 | 2.8 | 5.0 | −3.0 | 2.3 | 5.3 |
| [BMPL]TFSI | −2.1 | 2.3 | 4.4 | −3.1 | 2.5 | 5.6 |
The neat ILs reveal similarly large electrochemical windows as their solutions in CH3CN (Fig. 10 and Table 4). Noticeably, [EMIm]4cPFSI and [BMIm]4cPFSI are found to provide larger electrochemical windows than the respective TFSI-ILs, which is in stark contrast to the behaviour of the solutions in acetonitrile. This difference may be a consequence of relatively strong ion–ion interactions in the neat ILs due to the small size and thus high electron density; see ESP plots in Table 2 and compare to the easy formation of silver(I) complexes (Fig. 3). The cyclic voltammograms of the neat RTILs in Fig. 10 show a correlation between the amperage of reduction and oxidation with the viscosity and diffusivity of the ions (vide infra). A higher dynamic viscosity results in a smaller current for the respective redox events.
Viscosities and conductivities
The viscosity is a key parameter for the application of ionic liquids in materials science and a low viscosity that leads to a high conductivity is preferable, in general.21,121–123 The viscosity of an ionic liquid is determined by a number of in part mutually influencing factors,21,69,124 including ion volume11–16 and mass,17 ion–ion interactions (Coulomb interaction, hydrogen bond, etc.)15,18,19,125 and conformational flexibility.4,22–24For all three different types of ILs with the cations, [EMIm]+, [BMIm]+ and [BMPL]+, the respective TFSI-IL possesses the lowest dynamic viscosity (Table 5 and Fig. 11). [EMIm]6cPFSI melts at 67 °C and does not form supercooled melts, which prevented its study by viscometry. The ILs with the [BMPL]+ cation possess the highest dynamic viscosities, respectively, while η of the BMIm-ILs are in between.
| IL | η [mPa s] | ρ [g· cm−3] | c IL [mol L−1] | σ [mS cm−1] | D + [10−11 m2 s−1] | D − [10−11 m2 s−1] | Λ NMR [cm2 S mol−1] | Λ imp [cm2 S mol−1] | I |
|---|---|---|---|---|---|---|---|---|---|
| a Dynamic viscosity η; density ρ; concentration cIL; diffusion coefficients of cations (D+) and anions (D−); specific conductivity σ measured via impedance spectroscopy; molar conductivities calcd by ΛNMR = (D+ + D−)NAe2k−1T−1 and Λimp = σMρ−1; ionicity I = ΛimpΛNMR−1. b Solid at room temperature; η and ρ measured on a supercooled sample. | |||||||||
| [EMIm]4cPFSI | 50.4 | 1.47 | 4.85 | 4.37 | 3.0 | 2.4 | 2.06 | 0.90 | 0.44 |
| [EMIm]5cPFSI | 53.0 | 1.53 | 4.33 | 2.84 | 3.0 | 1.9 | 1.87 | 0.66 | 0.35 |
| [EMIm]TFSI | 37.8 | 1.52 | 3.88 | 7.22 | 4.4 | 2.5 | 2.64 | 1.86 | 0.70 |
| [BMIm]4cPFSI | 93.8 | 1.38 | 4.16 | 2.00 | 1.4 | 1.2 | 0.99 | 0.48 | 0.48 |
| [BMIm]5cPFSI | 85.0 | 1.44 | 3.78 | 1.84 | 1.6 | 1.2 | 1.07 | 0.49 | 0.46 |
| [BMIm]6cPFSIb | 250.3 | 1.49 | 3.45 | — | 0.6 | 0.4 | 0.41 | — | — |
| [BMIm]TFSI | 62.0 | 1.44 | 3.43 | 3.11 | 2.2 | 1.6 | 1.96 | 0.91 | 0.46 |
| [BMPL]4cPFSIb | 129.6 | 1.33 | 3.97 | — | — | — | — | — | — |
| [BMPL]5cPFSI | 143.5 | 1.39 | 3.62 | 1.13 | 1.0 | 0.8 | 0.69 | 0.27 | 0.40 |
| [BMPL]6cPFSI | 469.2 | 1.50 | 3.45 | 0.34 | 0.3 | 0.2 | 0.21 | 0.10 | 0.48 |
| [BMPL]TFSI | 98.8 | 1.40 | 3.31 | 1.36 | 1.3 | 1.1 | 0.92 | 0.41 | 0.45 |
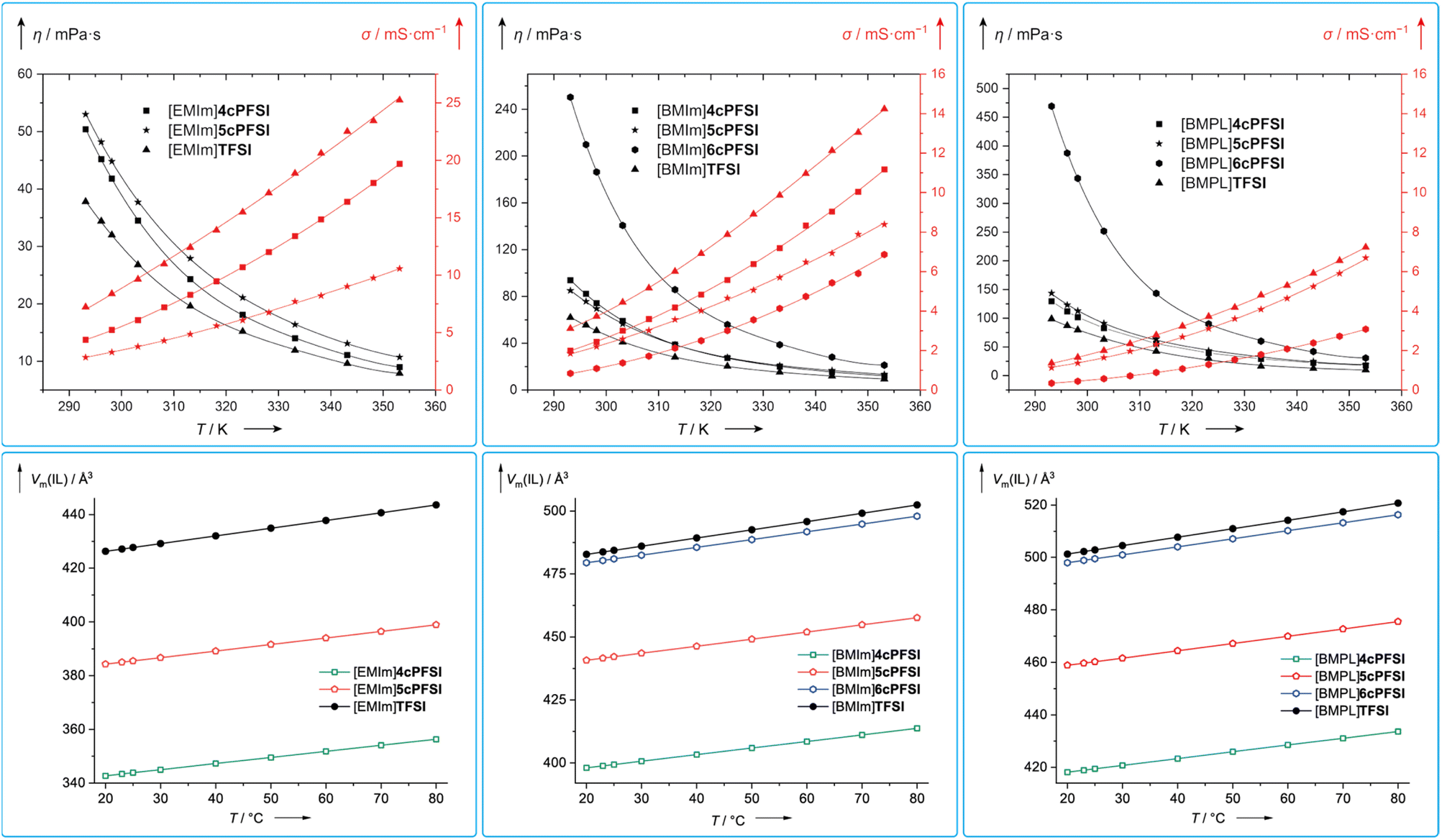 | ||
| Fig. 11 Temperature dependence of the dynamic viscosities (black) and specific conductivities (red) of the neat RTILs investigated in this study (top) and their molecular volumina (Vm(IL)) at different temperatures calculated from the densities of the neat ILs (ρ; Table S3 in the ESI†) according to Vm(IL) = (ρ × MIL−1 × NA)−1 (MIL = molecular mass of the IL; NA = Avogadro constant; bottom). | ||
The dynamic viscosities of the ILs with the cyclic imide anions 4cPFSI and 5cPFSI are similar for the three different countercations in the full temperature range investigated (20–80 °C; Fig. 11). For [EMIm]+ and [BMPL]+ the ILs with the 4cPFSI anion are less viscous than those with the slightly larger and heavier 5cPFSI anion whereas with the [BMIm]+ cation the opposite behaviour is found (Table 5 and Fig. 11). The ILs with the non-cyclic TFSI anion show the lowest viscosities with all three different cations and at 20 °C, η is approximately 1.3–1.5 times higher for the ILs with 4cPFSI and 5cPFSI compared to the TFSI-ILs (Table 5). ILs with the 6cPFSI anion reveal significantly higher viscosities than the related ILs with the smaller cyclic imide anions and the TFSI anion (Table 5 and Fig. 11). The large, almost equal dynamic viscosities of the ILs with the anions 4cPFSI and 5cPFSI and the strong increase for the analogous ILs with the 6cPFSI anion cannot be rationalized by differences in mass or volume as these should result in a consistent trend for η for these anions. Since the delocalization of the negative charge is most effective in 6cPFSI and least effective in 4cPFSI, one would expect the highest fluidity for the 6cPFSI-ILs, which is not the case. However, this might be one reason for the very similar dynamic viscosities of 4cPFSI- and 5cPFSI-ILs. So, the most reasonable explanation for the high dynamic viscosity of ILs with the 6cPFSI anion is the rigidity of this cyclic imide anion (vide supra, Fig. 9). The low dynamic viscosities of the TFSI-ILs nicely agree with this argument because the TFSI anion is the least rigid, most flexible one in the series studied, herein.21,23 If ion mass and size (Table 2) were the most relevant factors determining the dynamic viscosity, the TFSI-ILs would have properties in between those of the respective ILs with the anions 4cPFSI and 5cPFSI.
Similar to ILs with cyclic bis(perfluoroalkyl)imide anions, the dynamic viscosity of ILs with cyclic alkylammonium cations depends on the ring size. Typically, larger rings result in higher viscosities,126,127e.g. [BMPL]TFSI (85 mPa s, 298.15 K), [BMPip]TFSI (182 mPa s, 298.15 K; BMPip = N-butyl-N-methylpiperidinium) and [BMAzp]TFSI (315 mPa s, 298.15 K; BMAzp = N-butyl-N-methylazepanium).127 A related trend was reported for the two dicyanamide-ILs [BMPL][N(CN)2]128,129 (33.7–40.13 mPa s, 298.15 K)130–132 and [BMPip][N(CN)2] (119.7 mPa s, 298.15 K).132 But the dicyanamide-IL with the smaller N-butyl-N-methylazetidinium cation [BMAze]+ reveals a higher viscosity (86.7 mPa s, 298.15 K) than the respective [BMPL]+-IL.132,133 Thus, the [BMPL]+ cation with the five-membered ring leads to the IL with the lowest viscosity. The general low dynamic viscosity of [BMPL]+-ILs may be related to a high conformational flexibility of the five-membered ring of the [BMPL]+ cation.134 The high conformational flexibility of the [BMPL]+ cation compared to the smaller [BMAze]+ and the larger [BMPip]+ cation also mirrors the well documented high relative entropy of the all carbon ring in cyclopentane versus cyclobutane and cyclohexane.135,136
The conductivity is another crucial bulk property of ILs. Typically, the specific conductivity increases with decreasing viscosity and generally, it correlates with the dynamic viscosity and the concentration of charge carriers.137 The ILs studied in this contribution follow this trend, in general (Table 5 and Fig. 11). So, the TFSI-ILs have higher specific conductivities than their counterparts with the cyclic imide anions.
Assuming that the molecular volume of an IL (Vm(IL)), i.e. the concentration (cIL), correlates with its molecular mass (MIL), one would expect an increase in Vm(IL) in the order 4cPFSI < 5cPFSI < TFSI < 6cPFSI for a series of ILs with the same cation. The ILs with the cyclic imide anions strictly follow this trend (Fig. 11). In case of the ILs with [BMIm]6cPFSI and [BMIm]TFSI as well as [BMPL]6cPFSI and [BMPL]TFSI the reverse behaviour is found since the TFSI-IL possesses a larger molecular volume than the respective 6cPFSI-IL as calculated from the densities (ρ) of the neat ILs. These observations cannot be rationalized by the anion size as the lighter TFSI anion has a smaller van der Waals volume (Vm(vdW)−) compared to the heavier 6cPFSI anion (Table 2). Thus, the TFSI-ILs are comparably poorly packed, revealing a relatively large free volume, which nicely agrees with the low dynamic viscosities and high specific conductivities.
Ion diffusivity
Since the macroscopic properties viscosity and conductivity are determined by the diffusion of the ions,10,138,139 the diffusion in the neat ionic liquids was studied by pulsed field gradient stimulated echo (PFGSTE) NMR spectroscopy in the temperature range of 20–80 °C (Fig. 12 and S38–S40 in the ESI†). The diffusion coefficients nicely display the dynamic viscosities and the specific conductivities (vide supra).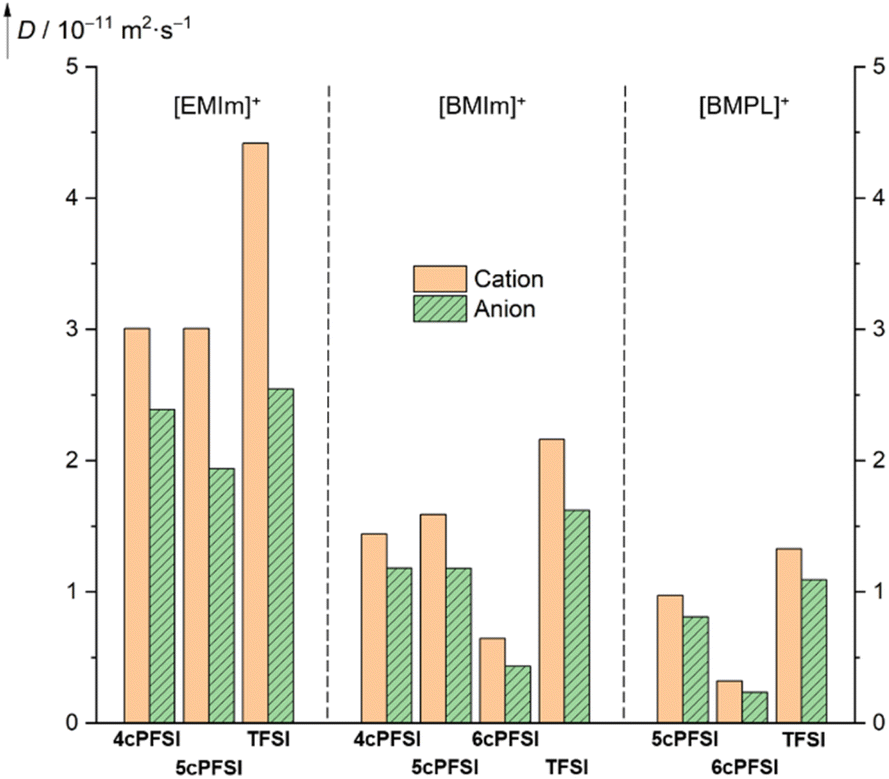 | ||
| Fig. 12 Translational self diffusion coefficients of the ions of neat RTILs determined at 20 °C by PFGSTE NMR spectroscopy. | ||
Within the series of three [EMIm]+-ILs with the anions 4cPFSI, 5cPFSI and TFSI, the TFSI anion possesses the highest diffusion coefficient (2.545 × 10−11 m2 s−1; Fig. 12). Its conformationally locked counterpart 5cPFSI reveals a significantly lower (24%) diffusivity. The diffusion coefficient of the 4cPFSI anion is in between those of the anions TFSI and 5cPFSI. However, the difference in the diffusion coefficients of the three related imide anions in the [EMIm]+-ILs is rather small. Interestingly, the [EMIm]+ cations in [EMIm]4cPFSI and [EMIm]5cPFSI exhibit almost identical diffusion coefficients, while the coefficient of [EMIm]+ cation in neat [EMIm]TFSI is 1.47 times higher. A possible explanation arises from X-ray diffraction study. The crystal structures of the [EMIm]+-ILs with cyclic anions exhibit antiparallel displaced π+–π+-stacking (vide infra, Fig. 5). This greater tendency for cation–cation interactions decreases the ion mobility, which is reflected in the corresponding diffusion coefficients. These results experimentally indicate that in the liquid phase of the cyclic imide-ILs the cation–cation association by π+–π+-stacking evident in the crystalline state is, at least in part, retained. To establish such interactions between two cations a sufficient Coulomb stabilization provided by the counterions is needed.
The diffusion properties of the [BMIm]+- and [BMPL]+-RTILs reflect the effect of conformational lock of the cyclic imide anions more strongly than their [EMIm]+-counterparts. The ion diffusion of the cyclic [BMIm]+- and [BMPL]+-ILs is significantly smaller with all cyclic imide ions compared to [BMIm]TFSI (Fig. 12). This strongly suggests that the reduced diffusion observed, is significantly affected by the conformational lock of the anions and not solely by other accompanying factors such as size, mass, degree of fluorination or electronic structure, since these properties differ for the three cyclic imide anions and those of the TFSI anion are in between.
It has been demonstrated for anions with conformational constraints that the physicochemical properties of their ILs reveal a successive reduced influence of these constraints with increasing temperature.9 With an increasing temperature conformational barriers are overcome and the interconversion between ion conformers is more easily achieved resulting in an increased conformational flexibility.9 Obviously the strict conformational lock present in the cyclic imide anions prevents such a behaviour, which explains that the temperature-dependent diffusion coefficients of the cyclic imide ILs do not show an analogous behaviour (Fig. S38–S40 in the ESI†).
Conclusions
Room temperature ionic liquids (RTILs) with 4- to 6-membered cyclic sulfonimide anions 4cPFSI, 5cPFSI and 6cPFSI have been synthesized, which are closely related to the well-established ILs with the TFSI anion. The conformational lock of the cyclic sulfonimide anions is well reflected by the physicochemical properties of their ILs. Thus, the TFSI-ILs with the three cations [EMIm]+, [BMIm]+ and [BMPL]+ possess lower melting points, lower viscosities and higher specific conductivities than their counterparts with the cyclic sulfonimide anions. The properties of the 5cPFSI-ILs are closest to those of the respective TFSI-ILs, in general, while those of the analogous 6cPFSI-ILs are significantly different. This is remarkable as the molecular mass and volume of the 5cPFSI and the 6cPFSI anion are similar (5cPFSI) and very similar (6cPFSI) to the molecular mass and size of the TFSI anion. So, the differences in melting point, viscosity and conductivity are mostly attributed to the conformational flexibility of the anions. The 6cPFSI anion has comparably high barriers for the interconversion between its conformers and the 5cPFSI anion reveals a higher conformational flexibility. In stark contrast, the TFSI anion easily switches between its cis-conformation, which resembles the cyclic sulfonimide anions, and the trans-conformation.SC-XRD studies on the [EMIm]+ salts of the four imide anions TFSI and ncPFSI (n = 4–6) show that the salts with the cyclic imide anions reveal distinctive π+–π+ interactions between two [EMIm]+ cations whereas for crystalline [EMIm]TFSI no π+–π+ stacking has been observed. The π+–π+ interactions translate into properties of the liquid salts as demonstrated by their ion diffusion coefficients and in agreement with results of MD simulations. This observation is one of the very rare experimental examples of π+–π+ interactions for imidazolium cations with alkyl chains longer than methyl.
In summary, the present study shows that minor modifications of anionic IL building blocks lead to significant changes of the macroscopic properties as exemplified by a study on the ring size of cyclic anions.
Data availability
Experimental and computational data is deposited in the ESI† as well as on the Github pages given in the methodology.Author contributions
Younes K. J. Bejaoui: conceptualisation, synthesis, spectroscopy, SC-XRD (data collection and structure solution) and interpretation, DFT calculations, writing (original draft, review and editing). Frederik Philippi: conceptualisation, MD simulations, writing (review). Hans-Georg Stammler: SC-XRD (data collection and structure solution). Krzysztof Radacki: SC-XRD (data collection and structure solution). Ludwig Zapf: cyclic voltammetry, impedance spectroscopy and SC-XRD (data collection and structure solution). Nils Schopper: cyclic voltammetry and impedance spectroscopy. Kateryna Goloviznina: force field development. Kristina A. M. Maibom: SC-XRD (data collection and structure solution). Roland Graf: SC-XRD (data collection and structure solution). Jan A. P. Sprenger: viscometry and thermal analysis. Rüdiger Bertermann: NMR studies (PFGSTE and spectra on neat ILs). Holger Braunschweig: writing (editing). Tom Welton: conceptualisation, writing (review and editing). Nikolai V. Ignat'ev: conceptualisation, writing (original manuscript, review and editing). Maik Finze: supervision, conceptualisation, writing (original manuscript, review and editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of Würzburg for generous support and Laura Wolz for performing the PFGSTE NMR experiments (all University of Würzburg).Notes and references
- M. Kar, O. Tutusaus, D. R. MacFarlane and R. Mohtadi, Energy Environ. Sci., 2019, 12, 566–571 RSC.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. Zheng, S. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- F. Philippi and T. Welton, Phys. Chem. Chem. Phys., 2021, 23, 6993–7021 RSC.
- Y.-L. Wang, B. Li, S. Sarman, F. Mocci, Z.-Y. Lu, J. Yuan, A. Laaksonen and M. D. Fayer, Chem. Rev., 2020, 120, 5798–5877 CrossRef CAS PubMed.
- K. Dong, X. Liu, H. Dong, X. Zhang and S. Zhang, Chem. Rev., 2017, 117, 6636–6695 CrossRef CAS PubMed.
- W. Silva, M. Zanatta, A. S. Ferreira, M. C. Corvo and E. J. Cabrita, Int. J. Mol. Sci., 2020, 21, 7745 CrossRef CAS PubMed.
- H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan and H. Watanabe, J. Phys. Chem. B, 2004, 108, 16593–16600 CrossRef CAS.
- F. Philippi, D. Pugh, D. Rauber, T. Welton and P. A. Hunt, Chem. Sci., 2020, 11, 6405–6422 RSC.
- S. Tsuzuki, ChemPhysChem, 2012, 13, 1664–1670 CrossRef CAS PubMed.
- U. P. R. M. Preiss, J. M. Slattery and I. Krossing, Ind. Eng. Chem. Res., 2009, 48, 2290–2296 CrossRef CAS.
- Y. Marcus, J. Mol. Liq., 2015, 209, 289–293 CrossRef CAS.
- Y. V. Nelyubina, A. S. Shaplov, E. I. Lozinskaya, M. I. Buzin and Y. S. Vygodskii, J. Am. Chem. Soc., 2016, 138, 10076–10079 CrossRef CAS PubMed.
- J. M. Slattery, C. Daguenet, P. J. Dyson, T. J. S. Schubert and I. Krossing, Angew. Chem., 2007, 119, 5480–5484 ( Angew. Chem., Int. Ed. , 2007 , 46 , 5384–5388 ) CrossRef.
- A. Rupp, N. Roznyatovskaya, H. Scherer, W. Beichel, P. Klose, C. Sturm, A. Hoffmann, J. Tübke, T. Koslowski and I. Krossing, Chem.–Eur. J., 2014, 20, 9794–9804 CrossRef CAS PubMed.
- W. Beichel, Y. Yu, G. Dlubek, R. Krause-Rehberg, J. Pionteck, D. Pfefferkorn, S. Bulut, D. Bejan, C. Friedrich and I. Krossing, Phys. Chem. Chem. Phys., 2013, 15, 8821–8830 RSC.
- P. Barthen, W. Frank and N. Ignatiev, Ionics, 2015, 21, 149–159 CrossRef CAS.
- W. Beichel, N. Trapp, C. Hauf, O. Kohler, G. Eickerling, W. Scherer and I. Krossing, Angew. Chem., 2014, 126, 3207–3210 ( Angew. Chem., Int. Ed. , 2014 , 53 , 3143–3146 ) CrossRef.
- C. Hardacre, J. D. Holbrey, M. Nieuwenhuyzen and T. G. A. Youngs, Acc. Chem. Res., 2007, 40, 1146–1155 CrossRef CAS PubMed.
- I. Krossing and J. M. Slattery, Z. Phys. Chem., 2006, 220, 1343–1359 CrossRef CAS.
- F. Philippi, D. Rauber, O. Palumbo, K. Goloviznina, J. McDaniel, D. Pugh, S. N. Suarez, C. C. Fraenza, A. Padua, C. W. M. Kay and T. Welton, Chem. Sci., 2022, 13, 9176–9190 RSC.
- T. Endo, K. Sunada, H. Sumida and Y. Kimura, Chem. Sci., 2022, 13, 7560–7565 RSC.
- I. Krossing, J. M. Slattery, C. Daguenet, P. J. Dyson, A. Oleinikova and H. Weingärtner, J. Am. Chem. Soc., 2006, 128, 13427–13434 CrossRef CAS PubMed.
- P. M. Dean, J. M. Pringle and D. MacFarlane, Phys. Chem. Chem. Phys., 2010, 12, 9144–9153 RSC.
- H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan and H. Watanabe, J. Phys. Chem. B, 2005, 109, 6103–6110 CrossRef CAS PubMed.
- S. Tsuzuki, H. Matsumoto, W. Shinoda and K. Mikami, Phys. Chem. Chem. Phys., 2011, 13, 5987–5993 RSC.
- T. Endo, M. Imanari, H. Seki and K. Nishikawa, J. Phys. Chem. A, 2011, 115, 2999–3005 CrossRef CAS PubMed.
- P. A. Hunt, J. Phys. Chem. B, 2007, 111, 4844–4853 CrossRef CAS PubMed.
- S. Zahn, G. Bruns, J. Thar and B. Kirchner, Phys. Chem. Chem. Phys., 2008, 10, 6921–6924 RSC.
- P. A. Hunt, C. R. Ashworth and R. P. Matthews, Chem. Soc. Rev., 2015, 44, 1257–1288 RSC.
- L. K. Scarbath-Evers, P. A. Hunt, B. Kirchner, D. R. MacFarlane and S. Zahn, Phys. Chem. Chem. Phys., 2015, 17, 20205–20216 RSC.
- M. H. Kowsari and S. Ebrahimi, Phys. Chem. Chem. Phys., 2018, 20, 13379–13393 RSC.
- J. Foropoulos and D. D. DesMarteau, Inorg. Chem., 1984, 23, 3720–3723 CrossRef CAS.
- K. Liu, Z. Wang, L.-X. Shi, S. Jungsuttiwong and S. Yuan, J. Energy Chem., 2021, 59, 320–333 CrossRef CAS.
- T. Rüther, A. I. Bhatt, A. S. Best, K. R. Harris and A. F. Hollenkamp, Batteries Supercaps, 2020, 3, 793–827 CrossRef.
- C. Lian, H. Liu, C. Li and J. Wu, AIChE J., 2019, 65, 804–810 CrossRef CAS.
- B. A. Shainyan and L. L. Tolstikova, Chem. Rev., 2013, 113, 699–733 CrossRef CAS PubMed.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef PubMed.
- A. Mauger, C. M. Julie, A. Paolella, M. Armand and K. Zaghib, Mater. Sci. Eng., R, 2018, 134, 1–21 CrossRef.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- G. A. Giffin, J. Mater. Chem. A, 2016, 4, 13378–13389 RSC.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154–2175 CrossRef CAS PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis Jr, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- S. N. Suarez, A. Rúa, D. Cuffari, K. Pilar, J. L. Hatcher, S. Ramati and J. F. Wishart, J. Phys. Chem. B, 2015, 119, 14756–14765 CrossRef CAS PubMed.
- O. Borodin, W. Gorecki, G. D. Smith and M. Armand, J. Phys. Chem. B, 2010, 114, 6786–6798 CrossRef CAS PubMed.
- R. Koshar, EP0057327, Minnesota Mining & Manufacturing Company, 1982.
- S. Kakinuma, T. Ishida and H. Shirota, J. Phys. Chem. B, 2017, 121, 250–264 CrossRef CAS PubMed.
- B. O'Rourke, C. Lauderback, L. I. Teodoro, M. Grimm, M. Zeller, A. Mirjafari, G. L. Guillet and P. C. Hillesheim, ACS Omega, 2021, 6, 32285–32296 CrossRef PubMed.
- S. Yamaguchi, H. Yamada, Y. Takeoka, M. Rikukawa and M. Yoshizawa-Fujita, New J. Chem., 2019, 43, 4008–4012 RSC.
- S. Kakinuma and H. Shirota, J. Phys. Chem. B, 2019, 123, 1307–1323 CrossRef CAS PubMed.
- M. Moriya, T. Watanabe, W. Sakamoto and T. Yogo, RSC Adv., 2012, 2, 8502–8507 RSC.
- M. Moriya, T. Watanabe, S. Nabeno, W. Sakamoto and T. Yogo, Chem. Lett., 2014, 43, 108–110 CrossRef CAS.
- J. Traver, E. Chenard, M. Zeller, G. L. Guillet, W. E. Lynch and P. C. Hillesheim, J. Mol. Struct., 2021, 1232, 130046 CrossRef CAS.
- R. Jüschke, G. Henkel and P. Sartori, Z. Naturforsch., B: Chem. Sci., 1997, 52, 359–366 CrossRef.
- L. Pohl, V. Hilarius, P. Sartori and R. Jüschke, WO97/31909, Merck Patent GmbH, 1997.
- R. Jüschke, D. Velayutham and P. Sartori, J. Fluorine Chem., 1997, 83, 145–149 CrossRef.
- S. Schaltin, N. R. Brooks, L. Stappers, K. Van Hecke, L. Van Meervelt, K. Binnemans and J. Fransaer, Phys. Chem. Chem. Phys., 2012, 14, 1706–1715 RSC.
- M. Stricker, B. Oelkers, C. P. Rosenau and J. Sundermeyer, Chem.–Eur. J., 2013, 19, 1042–1057 CrossRef CAS PubMed.
- Y. Tang and B. Yu, Eur. J. Inorg. Chem., 2020, 107–118 CrossRef CAS.
- A. Blaschette, P. G. Jones, T. Hamann, M. Nävcke, D. Schomburg, H. K. Cammenga, M. Epple and I. Steppuhn, Z. Anorg. Allg. Chem., 1993, 619, 912–922 CrossRef CAS.
- Y. U. Paulechka, G. J. Kabo, A. V. Blokhin, A. S. Shaplov, E. I. Lozinskaya, D. G. Golovanov, K. A. Lyssenko, A. A. Korlyukov and Y. S. Vygodskii, J. Phys. Chem. B, 2009, 113, 9538–9546 CrossRef CAS PubMed.
- A. R. Choudhury, N. Winterton, A. Steiner, A. I. Cooper and K. A. Johnson, CrystEngComm, 2006, 8, 742–745 RSC.
- A. R. Choudhury, N. Winterton, A. Steiner, A. I. Cooper and K. A. Johnson, J. Am. Chem. Soc., 2005, 127, 16792–16793 CrossRef CAS PubMed.
- H. Li, L. Su, X. Cheng, K. Yang and G. Yang, J. Phys. Chem. B, 2014, 118, 8684–8690 CrossRef CAS PubMed.
- L. Su, M. Li, X.-L. Zhu, Z. Wang, Z.-N. Chen, F. Li, Q. Zhou and S. Hong, J. Phys. Chem. B, 2010, 114, 5061–5065 CrossRef CAS PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- A.-V. Mudring, Aust. J. Chem., 2010, 63, 544–564 CrossRef CAS.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- C. M. Burba, J. Janzen, E. D. Butson and G. L. Coltrain, J. Phys. Chem. B, 2013, 117, 8814–8820 CrossRef CAS PubMed.
- L. M. Varela, J. Carrete, M. Turmine, E. Rilo and O. Cabeza, J. Phys. Chem. B, 2009, 113, 12500–12505 CrossRef CAS PubMed.
- S. Bouguerra, I. B. Malham, P. L'etellier, A. Mayaffre and M. Turmine, J. Chem. Thermodyn., 2008, 40, 146–154 CrossRef CAS.
- M. Finze and G. J. Reiss, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, E68, o1992–o1993 CrossRef PubMed.
- T. Mochida, Y. Funasako, T. Inagaki, M.-L. Li, K. Asahara and D. Kuwahara, Chem.–Eur. J., 2013, 19, 6257–6264 CrossRef CAS PubMed.
- T. Mochida, M. Ishida, T. Tominaga, K. Takahashi, T. Sakurai and H. Ohta, Phys. Chem. Chem. Phys., 2018, 20, 3019–3028 RSC.
- D. D. DesMarteau, S. S. Zuberi, W. T. Pennington and B. B. Randolph, Eur. J. Solid State Inorg. Chem., 1992, 29, 777–789 CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9–18 CrossRef CAS PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2010 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. B: Struct. Sci., 2009, D65, 148–155 Search PubMed.
- F. Capitani, S. Gatto, P. Postorino, O. Palumbo, F. Trequattrini, M. Deutsch, J.-B. Brubach, P. Roy and A. Paolone, J. Phys. Chem. B, 2016, 120, 1312–1318 CrossRef CAS PubMed.
- F. Capitani, F. Trequattrini, O. Palumbo, A. Paolone and P. Postorino, J. Phys. Chem. B, 2016, 120, 2921–2928 CrossRef CAS PubMed.
- W. Beichel, P. Eiden and I. Krossing, ChemPhysChem, 2013, 14, 3221–3226 CrossRef CAS PubMed.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- M. A. Spackman, J. J. McKinnon and D. Jayatilaka, CrystEngComm, 2008, 10, 377–388 CAS.
- B. Burgenmeister, K. Sonnenberg, S. Riedel and I. Krossing, Chem.–Eur. J., 2017, 23, 11312–11322 CrossRef CAS PubMed.
- P. M. Dean, J. M. Pringle, C. M. Forsyth, J. L. Scott and D. R. MacFarlane, New J. Chem., 2008, 32, 2121–2126 RSC.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017 Search PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- T. Steiner, Angew. Chem., 2002, 114, 50–80 ( Angew. Chem., Int. Ed. , 2002 , 41 , 48–76 ) CrossRef.
- I. Geronimo, N. J. Singh and K. S. Kim, Phys. Chem. Chem. Phys., 2011, 13, 11841–11845 RSC.
- C. J. Dymek, D. A. Grossie, A. V. Fratini and W. W. Adams, J. Mol. Struct., 1989, 213, 25–34 CrossRef CAS.
- S. K. Panja, N. Dwivedi, H. Noothalapati, S. Shigeto, A. K. Sikder, A. Saha, S. S. Sunkari and S. Saha, Phys. Chem. Chem. Phys., 2015, 17, 18167–18177 RSC.
- J. S. Wilkes and M. J. Zaworotko, Supramol. Chem., 1993, 1, 191–193 CrossRef CAS.
- I. de Pedro, A. García-Saiz, J. Dupont, P. Migowski, O. Vallcorba, J. Junquera, J. Rius and J. Rodríguez Fernández, Cryst. Growth Des., 2015, 15, 5207–5212 CrossRef CAS.
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2014, 16, 3238–3253 RSC.
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2015, 17, 14437–14453 RSC.
- W. Gao, Y. Tian and X. Xuan, J. Mol. Graphics Modell., 2015, 60, 118–123 CrossRef CAS PubMed.
- H. Weber and B. Kirchner, J. Phys. Chem. B, 2016, 120, 2471–2483 CrossRef CAS PubMed.
- F. Tang, T. Ohto, T. Hasegawa, M. Bonn and Y. Nagata, Phys. Chem. Chem. Phys., 2017, 19, 2850–2856 RSC.
- Y.-L. Wang, A. Laaksonen and M. D. Fayer, J. Phys. Chem. B, 2017, 121, 7173–7179 CrossRef CAS PubMed.
- Y.-L. Wang, J. Phys. Chem. B, 2018, 122, 6570–6585 CrossRef CAS PubMed.
- Y. K. J. Bejaoui, N. V. Ignat'ev and M. Finze, manuscript in preparation.
- Electrochemical Aspects of Ionic Liquids, ed. H. Ohno, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2nd edn, 2011 Search PubMed.
- M. Salanne, Top. Curr. Chem., 2017, 375, 63 CrossRef PubMed.
- A. Vioux and B. Coasne, Adv. Energy Mater., 2017, 7, 1700883 CrossRef.
- Q. Yang, Z. Zhang, X.-G. Sun, Y.-S. Hu, H. Xing and S. Dai, Chem. Soc. Rev., 2018, 47, 2020–2064 RSC.
- A. Lahiri, N. Borisenko and F. Endres, Top. Curr. Chem., 2018, 376, 55–83 CrossRef PubMed.
- A. Balducci, Top. Curr. Chem., 2017, 375, 1–27 CrossRef CAS PubMed.
- L. Yu and G. Z. Chen, Front. Chem., 2019, 7, 272 CrossRef CAS PubMed.
- A. Basile, M. Hilder, F. Makhlooghiazad, C. Pozo-Gonzalo, D. R. MacFarlane, P. C. Howlett and C. M. Forsyth, Adv. Energy Mater., 2018, 8, 1703491 CrossRef.
- M. Forsyth, L. Porcarelli, X. Wang, N. Goujon and D. Mecerreyes, Acc. Chem. Res., 2019, 52, 686–694 CrossRef CAS PubMed.
- M. Kar, N. V. Plechkova, K. R. Seddon, J. M. Pringle and D. R. MacFarlane, Aust. J. Chem., 2019, 72, 3–10 CrossRef CAS.
- C. Dai, J. Zhang, C. Huang and Z. Lei, Chem. Rev., 2017, 117, 6929–6983 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- Y. Chen and T. Mu, Green Chem., 2019, 21, 2544–2574 RSC.
- B. Mandal, S. Ghosh and B. Basu, Top. Curr. Chem., 2019, 377, 30 CrossRef PubMed.
- K. Paduszyński and U. Domańska, J. Chem. Inf. Model., 2014, 54, 1311–1324 CrossRef PubMed.
- S. Jiang, Y. Hu, Y. Wang and X. Wang, J. Phys. Chem. Ref. Data, 2019, 48, 033101 CrossRef.
- N. V. Ignat'ev, M. Finze, J. A. P. Sprenger, C. Kerpen, E. Bernhardt and H. Willner, J. Fluorine Chem., 2015, 177, 46–54 CrossRef.
- A. Shakeel, H. Mahmood, U. Farooq, Z. Ullah, S. Yasin, T. Iqbal, C. Chassagne and M. Moniruzzaman, ACS Sustainable Chem. Eng., 2019, 7, 13586–13626 CrossRef CAS.
- F. Philippi, D. Rauber, K. L. Eliasen, N. Bouscharain, K. Niss, C. W. M. Kay and T. Welton, Chem. Sci., 2022, 13, 2735–2743 RSC.
- T. Yim, Y. J. Lee, H.-J. Kim, J. Mun, S. Kim, S. M. Oh and Y. G. Kim, Bull. Korean Chem. Soc., 2007, 28, 1567–1572 CrossRef CAS.
- T. Belhocine, S. A. Forsyth, H. Q. N. Gunaratne, M. Nieuwenhuyzen, P. Nockemann, A. V. Puga, K. R. Seddon, G. Srinivasan and K. Whiston, Green Chem., 2011, 13, 3137–3155 RSC.
- D. R. MacFarlane, J. Golding, S. Forsyth, M. Forsyth and G. B. Deacon, Chem. Commun., 2001, 1430–1431 RSC.
- D. R. MacFarlane, S. A. Forsyth, J. Golding and G. B. Deacon, Green Chem., 2002, 4, 444–448 RSC.
- N. Zec, M. Bešter-Rogač, M. Vraneš and S. Gadžurić, J. Chem. Thermodyn., 2015, 91, 327–335 CrossRef CAS.
- G. McHale, C. Hardcare, R. Ge, N. Doy, R. W. K. Allen, J. M. MacInnes, M. R. Bown and M. I. Newton, Anal. Chem., 2008, 80, 5806–5811 CrossRef CAS PubMed.
- Y. Jin, Y. Shi, W. Zhang, X. Qi, H. Xia and Q. Zhang, J. Mol. Liq., 2021, 336, 116572 CrossRef CAS.
- B. Zheng, Y. Zhang, Z. Zhang, L.-S. Long, S. Chen and S. Zhang, ChemistrySelect, 2018, 3, 284–288 CrossRef CAS.
- J. N. C. Lopes, K. Shimizu, A. A. H. Pádua, Y. Umebayashi, S. Fukuda, K. Fujii and S.-i. Ishiguro, J. Phys. Chem. B, 2008, 112, 1465–1472 CrossRef PubMed.
- J. E. Kilpatrick, K. S. Pitzer and R. Spitzer, J. Am. Chem. Soc., 1947, 69, 2483–2488 CrossRef CAS.
- L. Chan, G. M. Morris and G. R. Hutchinson, J. Chem. Theory Comput., 2021, 17, 2099–2106 CrossRef CAS PubMed.
- L. A. Bischoff, M. Drisch, C. Kerpen, P. T. Hennig, J. Landmann, J. A. P. Sprenger, R. Bertermann, M. Grüne, Q. Yuan, J. Warneke, X.-B. Wang, N. V. Ignat'ev and M. Finze, Chem.–Eur. J., 2019, 25, 3560–3574 CrossRef CAS PubMed.
- V. Mazan and M. Boltoeva, J. Mol. Liq., 2017, 240, 74–79 CrossRef CAS.
- J. R. Sangoro, C. Iacob, S. Naumov, R. Valiullin, H. Rexhausen, J. Hunger, R. Buchner, V. Strehmel, J. Kärger and F. Kremer, Soft Matter, 2011, 7, 1678–1681 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2155593, 2222987–2222997 and 2236907. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06758g |
| This journal is © The Royal Society of Chemistry 2023 |