
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of functionalized 2,3-diaminopropionates and their potential for directed monobactam biosynthesis†
Michael S.
Lichstrahl‡
 a,
Lukas
Kahlert‡
a,
Rongfeng
Li‡
a,
Trevor A.
Zandi
bcd,
Jerry
Yang
a and
Craig. A.
Townsend
*a
a,
Lukas
Kahlert‡
a,
Rongfeng
Li‡
a,
Trevor A.
Zandi
bcd,
Jerry
Yang
a and
Craig. A.
Townsend
*a
aDepartment of Chemistry, The Johns Hopkins University, 3400 N Charles St, Baltimore, MD, USA. E-mail: ctownsend@jhu.edu
bNovartis Institutes for Biomedical Research, Cambridge, MA, USA
cChemical Biology and Therapeutics Science Program, Broad Institute, Cambridge, MA, USA
dT. C. Jenkins Department of Biophysics, Johns Hopkins University, Baltimore, MD, USA
First published on 20th March 2023
Abstract
The N-sulfonated monobactams harbor considerable potential to combat emerging bacterial infections that are problematic to treat due to their metallo-β-lactamase mediated resistance against conventional β-lactam antibiotics. Herein, we report a divergent synthesis of C3-substituted 2,3-diaminopropionates featuring an array of small functional groups and examine their potential as alternative precursors during monobactam biosynthesis in a mutant strain (ΔsulG) of Pseudomonas acidophila that is deficient in the supply of this native precursor. In vitro assays revealed high diastereoselectivity, as well as a substrate tolerance by the terminal adenylation domain of the non-ribosomal peptide synthetase (NRPS) SulM toward the majority of synthetic analogs. Chemical complementation of this mutant yielded a fluorinated, bioactive monobactam through fermentation as confirmed by a combination of spectrometric data and microbiological assays. This study demonstrates site-specific functionalization of a clinically important natural product and sets in place a platform for further strain improvements and engineered NRPS-biosynthesis of non-native congeners.
Introduction
Non-proteinogenic α,β-diamino acids are valuable building blocks for both natural product biosynthesis and the development of synthetic drugs.1–3L-2,3-Diaminopropionate (L-Dap, 1) as well as its C3-functionalized derivatives are the structural backbone of the azetidinone warhead of monobactams, a structurally distinct class of β-lactam antibiotics. The growing emergence of Ambler class B metallo-β-lactamases (MBLs), for which no FDA approved inhibitor is yet available, has brought renewed attention to monobactams because of their intrinsic stability to MBLs and their promise to overcome otherwise resistant bacterial infections where traditional β-lactam antibiotics have failed.4,5The isolation of the first monobactam sulfazecin (2) from Pseudomonas acidophila6 and subsequent discovery of several structurally related congeners from natural sources inspired and ultimately resulted in the development of aztreonam7 (3), currently the only FDA approved monobactam for clinical use.
Our group recently elucidated the biosynthesis of sulfazecin combining in vivo gene inactivation and in vitro enzyme studies.8,9 The final module of the NRPS SulM incorporates L-Dap, which itself is derived from the primary metabolite L-phosphoserine through the successive action of two enzymes, SulG and SulH, onto the upstream D-Glu-D-Ala dipeptide precursor (Scheme 1A). Following a distinct enzymatic mechanism, this linear tripeptide is N-sulfonated in trans by the sulfotransferase SulN, a step that is essential for the unusual thioesterase-mediated cyclization to yield the sulfonated β-lactam core. Hydroxylation and O-methylation catalyzed by SulO and SulP, respectively, furnish the final structure of sulfazecin 2.8,9
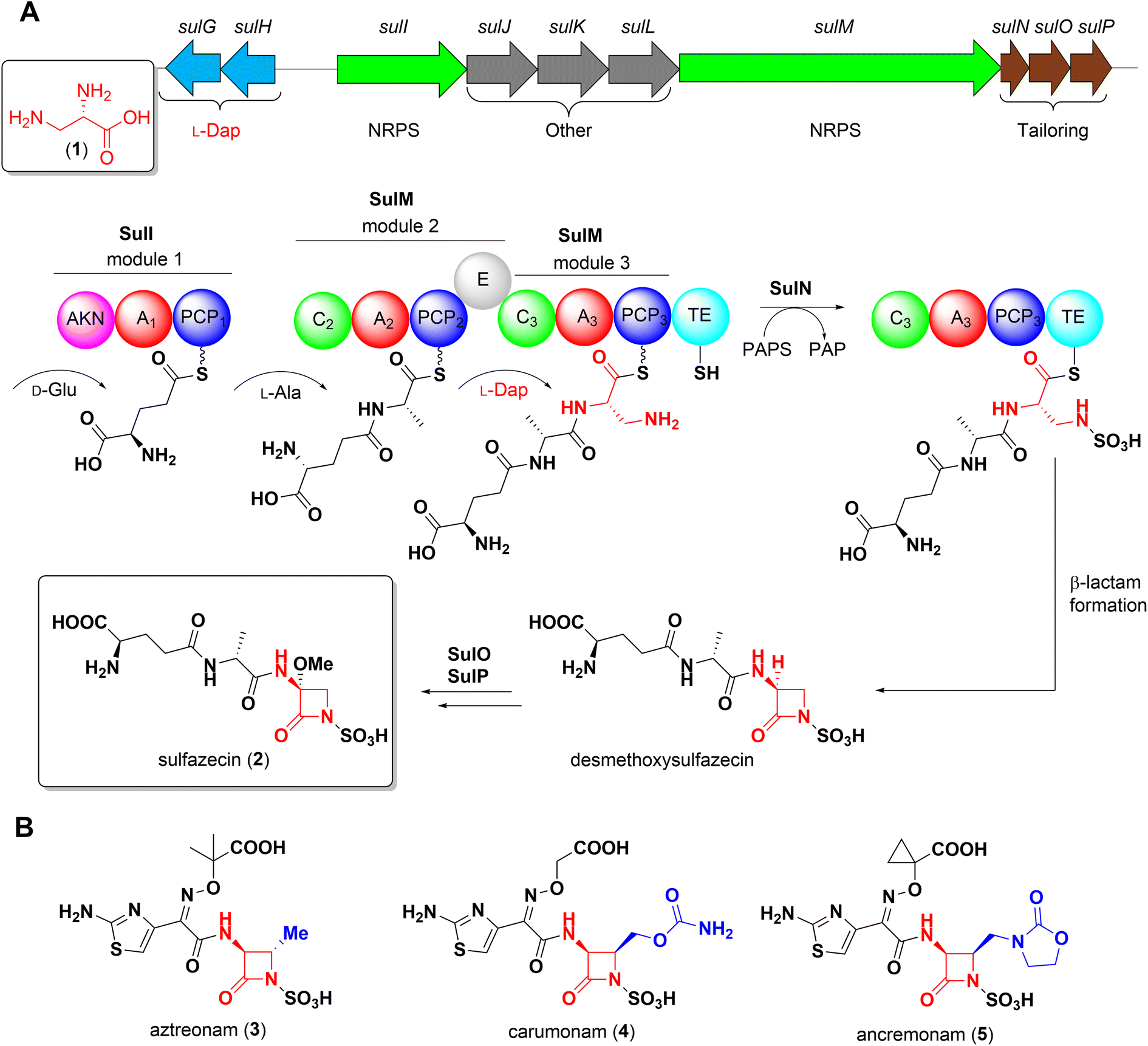 | ||
| Scheme 1 Biosynthesis of sulfazecin 2. (A) Biosynthetic gene cluster (top) and biosynthesis of sulfazecin 2. The L-Dap 1 derived subunit is shown in red. (B) Representative synthetic monobactams. Additional functionalization is highlighted in blue. AKN: predicted adenylylsulfate kinase of unknown function, A: adenylation domain, PCP: peptidyl-carrier-protein, C: condensation domain, E: epimerase domain, TE: thioesterase domain. | ||
Owing to the lack of a traceable chromophore and the formally charged monobactam core that prevents facile extraction with organic solvents from the aqueous fermentation media, multi-step isolation processes are necessary to obtain these natural products from their native producers.10,11 Therefore, synthetic procedures have historically been utilized to access derivatives of this potent pharmacophore for clinical development.12
Unlike naturally-occurring 2, a prominent feature of synthetic monobactams is a substitution at C4 of the azetidinone moiety, as exemplified by carumonam 4 and ancremonam 5 (Scheme 1B).13,14 This structural modification has been shown to improve both stability against serine β-lactamases and antibiotic activity.15,16 However, unlike the majority of naturally occurring monobactams, the C3-methoxy substituent is absent in the synthetic compounds. This functionalization is similarly observed in the cephamycin class of β-lactam natural products and semi-synthetic drugs such as cefoxitin where it has been demonstrated to improve the stability of these cephalosporin derivatives to serine β-lactamases.17
In a previous key experiment disruption of sulG abolished sulfazecin production, but could be fully restored by exogenous supplementation of L-Dap 1 to the fermentation medium.8 Such strains that lack supply of native building blocks have the potential to expand structural product diversity by means of mutasynthesis.18 This approach of mutasynthesis has been developed for the production of manifold valuable compounds such as the antiparasitic ivermectin,19 and enabled structural modification associated with enhanced bioactivity of established drugs including vancomycin, neomycin, and rapamycin.20–22 Contemporary approaches in the field of NRPS-based mutasynthesis, however, are usually limited to substituted aromatic non-proteinogenic building blocks such as hydroxyphenylglycines, β-hydroxytyrosine or salicylates, where synthetic analogs are readily available.20,23–26
In this study we aimed to extend this concept through supplementation of non-native derivatives of L-Dap to P. acidophila ΔsulG. Here, these synthetic analogs both functionalize an unreactive sp3-carbon and introduce an additional stereocenter into the molecule. Incorporation of any analog would allow the biosynthetic production of non-native, potentially bioactive monobactams or relay compounds for semi-synthesis that are substituted at the azetidinone C4 position, just like their purely synthetic counterparts. These molecules would further bear the clinically relevant C3-methoxy moiety, which is not present in synthetic monobactams and provide a biosynthetic route to chemical space that is challenging to cleanly access by synthetic means.27 Moreover, access to these densely functionalized diamino acid building blocks affords great utility in traditional synthetic and medicinal chemistry for the construction of bioactive compounds.2
Results and discussion
During NRPS-mediated biosynthesis the adenylation (A) domain selects its amino acid substrate in a highly chemo- and stereoselective manner before activating it for downstream processing.28–30 Since introduction of any substituent at the β-carbon of L-Dap introduces diastereomers, we reasoned that A3, the adenylation domain in SulM that activates L-Dap during sulfazecin biosynthesis, would be biased towards one or the other. We initially prepared the known compounds (2S,3R)-6 and (2S,3S)-methyl-Dap 7, the structurally simplest derivatives bearing an additional methyl group, by adapting established methodology (Scheme S1†). To probe the chemo- and stereoselectivity of A3 toward the methyl-analogs 6 and 7, we performed two complementary colorimetric adenylation domain activity assays in vitro: The first one detects inorganic pyrophosphate (PPi) that is released during the ATP-driven amino acid activation,31,32 whereas the second assay relies on direct detection of the aminoacyl-adenylate through capture with hydroxylamine and complexation of the resulting hydroxamate species with Fe(III) (Fig. 1A).33,34 Both assays consistently revealed that A3 exhibits a strong preference for the (2S,3R)-diastereomer 6 with only marginal activation of the (2S,3S)-diastereomer 7 (Fig. 1B). Interestingly, while L-serine is not activated, as shown previously,8 both D-Dap and the L-2,4-diaminobutyric acid (L-Dab) 8 control, a non-native substrate that would preclude downstream β-lactam formation, showed lower, but clear activation.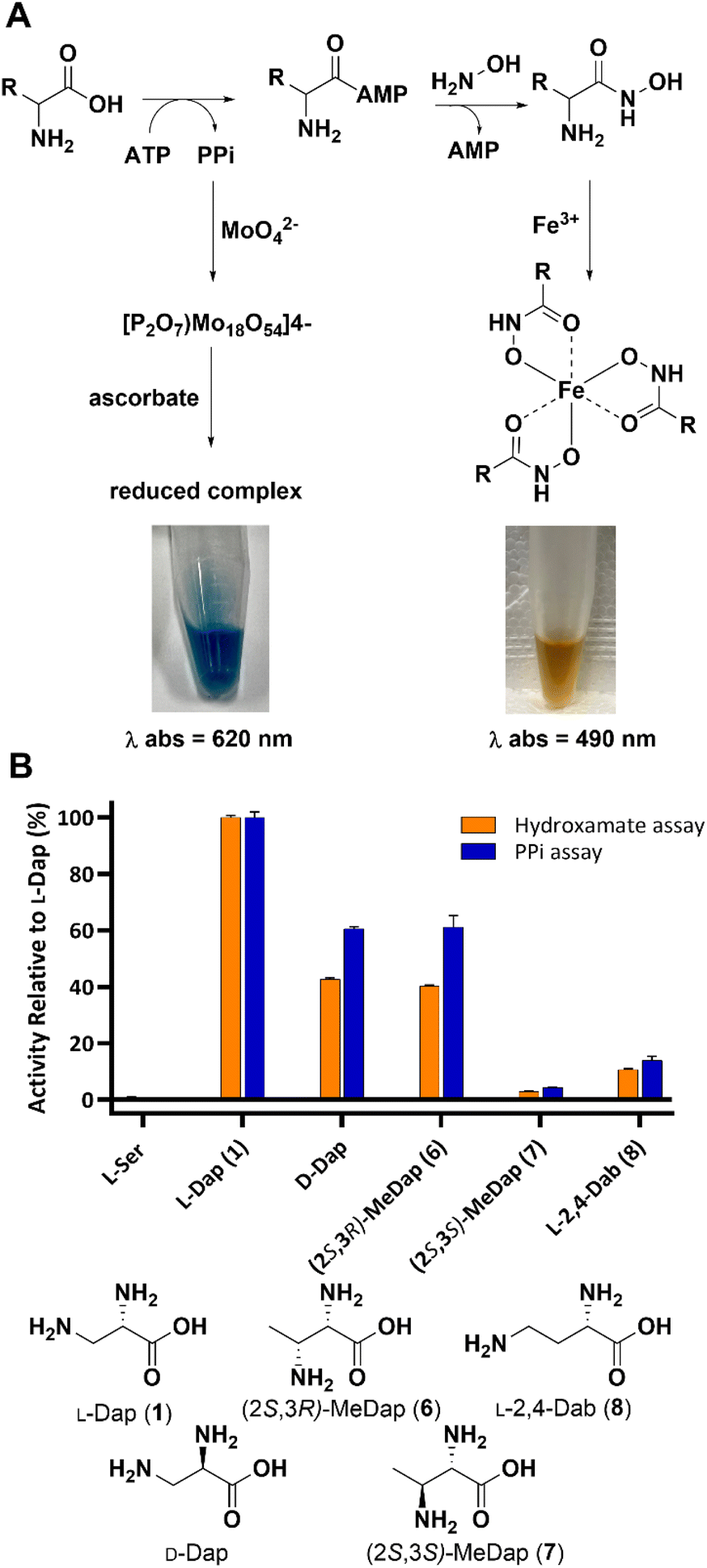 | ||
| Fig. 1 (A) Principle of A-domain in vitro assays. (B) Substrate preference of A3 towards Dap isomers based on in vitro assays. | ||
Encouraged by these initial results we continued to construct an extended library of functionalized L-Dap derivatives bearing the preferred threo stereochemistry. It is worth noting that the absolute configuration at C4 of the azetidinone core in the anticipated final monobactam products would correlate with that in penicillins, cephalosporins, carbapenems and clavulanic acid where this stereocenter is essential to potent antibiotic activity.35 Notably, the erythro (4S) stereochemistry is present in aztreonam 3, which simplified its commercial production from readily available L-threonine. Newer synthetic monobactams, however, and the 4R-diasteomer of aztreonam itself have been demonstrated to have greater β-lactamase stability and antibiotic activity than the 4S-diastereomer.15 The selection of the sulfazecin biosynthetic machinery for the more potent diastereomer therefore constitutes an important intrinsic advantage.
Hydroxymethyl-Dap 9 was initially targeted to focus construction of our library, believing the hydroxyl moiety would be a useful handle for further functionalization (Scheme 2A). Evaluating the literature, we identified butyrolactone 11 as a key synthetic intermediate that could be prepared by the procedure of Hanessian et al. from L-aspartate.36 Unexpectedly, upon repeating the reported procedure with minor modifications we isolated a product that we believed to be 11, but that did not agree with the reported spectral characterization. Single crystal X-ray crystallographic analysis, however, confirmed the identity of this product with the anticipated anti stereochemistry (Fig. S1 and Table S1†).
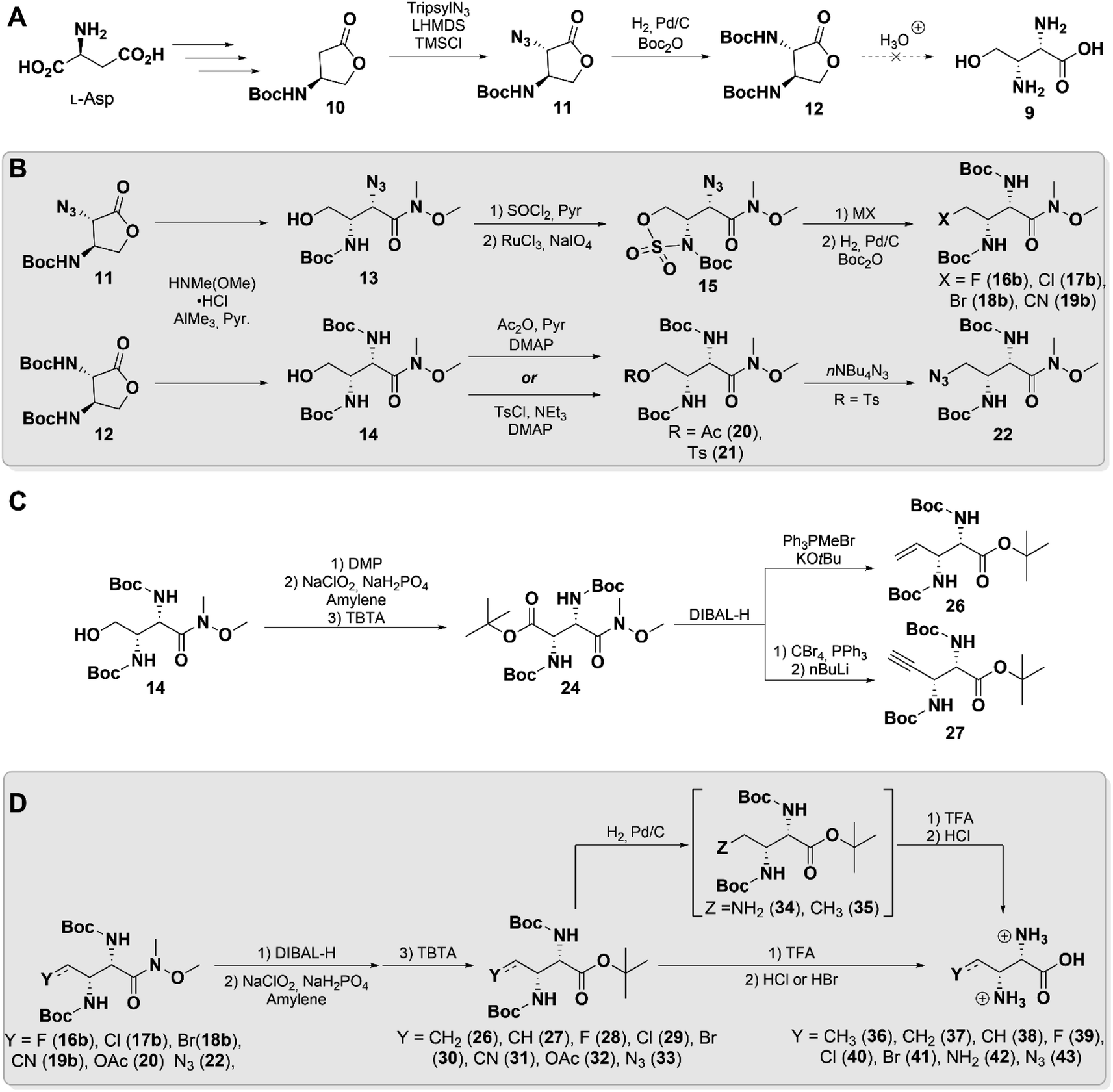 | ||
| Scheme 2 Synthesis of functionalized 2,3-diaminopropionates. (A) Diastereoselective synthesis of diamino-butyrolactone 12. (B) C4 functionalization by nucleophilic substitution. (C) Preparation of alkenyl/alkynyl analogs by head-to-tail inversion. (D) Amide cleavage and global deprotection to afford final 2,3-diaminopropionate analogues. | ||
With butyrolactone 11 in hand, we proceeded to 12 through a one-pot hydrogenation and Boc-protection. A single-step acid deprotection and hydrolysis of 12 was attempted to obtain 9, but appreciable re-lactonization was observed upon isolation. Lactonization is a well characterized phenomenon in related systems such as homoserine,37 which we believe was made even more thermodynamically favorable by the threo stereochemistry in 9. Therefore, we elected to pursue other derivatives that would not be subject to competing re-lactonization.
Literature precedence for similar systems showed that a Weinreb amide effectively suppresses this interfering reaction and permits isolation of the γ-hydroxy compound.38 Accordingly, both lactones 11 and 12 were subjected to an AlMe3-mediated amidation,39 which cleanly generated 13 and 14 without the need for further purification (Scheme 2B). This factor proved to be particularly important, as both compounds were found to nonetheless undergo re-lactonization on silica gel as well as upon prolonged standing at room temperature. Extended reaction telescoping and careful selection of reaction conditions enabled desired downstream transformations to be carried out while minimizing re-lactonization of intermediates (see ESI† Synthetic Procedures).
From 13 and 14, our syntheses diverged slightly to access various derivatives. Weinreb amide 13 was efficiently converted to its cyclic sulfamidate 15 in 65% yield over 3-steps from 11, simultaneously “capping” and activating the δ-hydroxy group. Compound 15 proved to be an excellent common intermediate for the generation of derivatives through facile nucleophilic ring opening by halides (16a–18a, see ESI†) and cyanide (19a, see ESI†), followed by hydrolysis of the N-sulfate (Scheme 2B).40 The α-azide in these products was then reduced and Boc-protected to yield 16b–19b in moderate yields, likely due to steric congestion. To access additional derivatives, Boc-protected diamino lactone 12 was employed as a precursor instead. Acetylation of its Weinreb amide 14 yielded acetoxymethyl 20, which we hoped could serve as a “masked” hydroxyl group. Sulfonylation of 14 generated tosylate 21, which was displaced to provide azidomethyl 22, which was not accessible in our hands from 13 (Scheme 2B).
Next, we attempted to oxidize 14 to the aldehyde (23, see ESI†) and further convert it to the olefin using Wittig chemistry. However, the latter reaction only yielded a small amount of the anticipated product, presumably due to competing reaction of the phosphonium ylide at the Weinreb amide. We therefore took advantage of its molecular symmetry to perform a head-to-tail inversion by oxidizing and protecting 14 as the di-amino succinate 24. Its Weinreb amide was selectively reduced to the aldehyde 25 (see ESI†) and subjected to Wittig olefination or Corey–Fuchs reaction to yield vinyl 26 and alkynyl 27, respectively (Scheme 2C).
For the remaining Weinreb amides basic hydrolysis proved very difficult, requiring extended reaction times and often leading to racemization, as well as destruction of labile functionalities at C4. Instead, the amide was readily reduced to the aldehyde and subsequently re-oxidized, followed by protection of the free carboxyl to simplify final purification. This protocol allowed preparation of the globally-protected Dap derivatives 26–33 in ample yields without detectable racemization. At this stage azidomethyl 33 was also converted to the tri-amine 34, and vinyl 26 was reduced to ethyl 35. This series of compounds, encompassing diverse chemical functionalities of small size, was then subjected to a final global deprotection and anion exchange to afford amino acids 36–43 (Scheme 2D). All of these compounds exhibited spectral signatures consistent with the threo stereochemistry of 2S,3R-methyl-Dap (6), by displaying H2–H3 vicinal coupling constants of ca. 3–4 Hz.41 Unfortunately, in the case of cyanomethyl 31 and acetoxymethyl 32, this final reaction led to decomposition, excluding them from further experiments. Bromide 41 was obtained with trace lactone, likely due to favorable 5-exo-tet cyclization.
With our extended substrate library in hand we employed the operationally simple hydroxamate assay to once again probe the promiscuity of SulM A3in vitro, as this domain represents the entry point of these substrate analogs into the monobactam biosynthetic machinery. In vitro assays revealed appreciable activation for the fluoromethyl-Dap 39, although less than observed for the smaller (2S,3R)-methyl-Dap 6. Except for the rigid alkyne 38, all synthetic L-Dap analogs exhibited low but measurable activity (Fig. 2A).
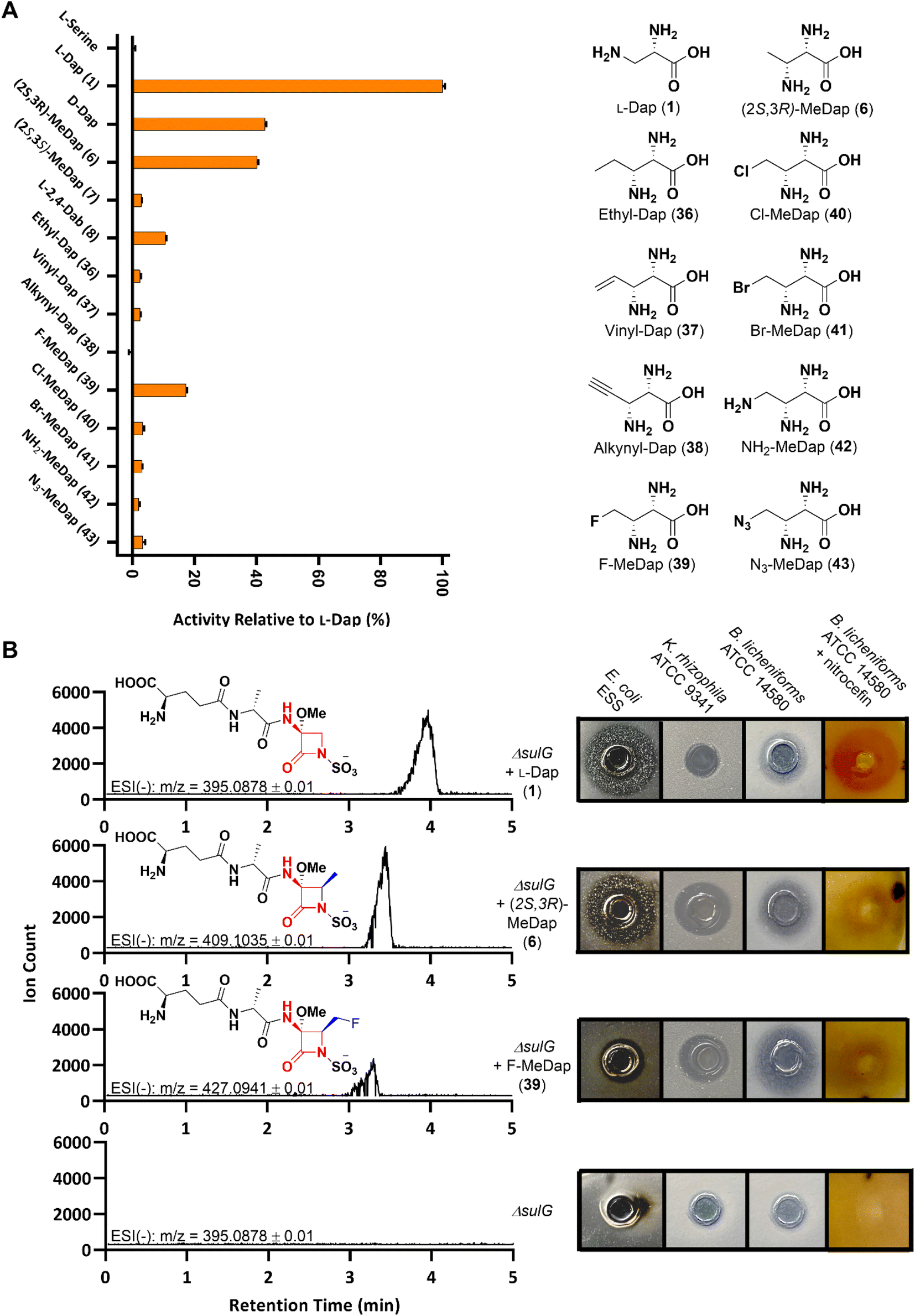 | ||
| Fig. 2 (A) Hydroxamate-based in vitro assays with A3 against the synthetic L-Dap library. (B) UPLC-HRMS analysis and bioassays of concentrated P. acidophila ΔsulG cell-free supernatant (semi-purified for UPLC-MS) supplemented with selected L-Dap derivatives. | ||
To obtain a more detailed understanding how the additional substituents affect the reactivity of A3, we applied the commonly used continuous 6-methyl-7-thioguanosine (MesG)/hydroxylamine coupled assay42,43 to record kinetic profiles of A3 for its native substrate 1 and the two analogues 6 and 39, which show the highest relative activity in end-point assays. While the size of the substituent did not have a large effect on kcat, it did notably increase the KM by approx. factor 10 for 6 (0.09 mM for 1vs. 1.08 mM for 6) and 200 for 39 (0.09 mM for 1vs. 20.7 mM for 39), in line with the observed relative activities (Fig. S2.†) Although the measured kcat/KM of 78.4 mM−1 min−1 is low for the native substrate 1, this value might not necessarily reflect the actual catalytic efficiency of A3 under in vivo conditions, as the enzyme might favour a more acidic environment that is not compatible the MesG-hydroxylamine continuous assay.44 Furthermore, it has been shown that the turnover number can be much higher in the presence of a native acceptor molecule (here the holo-PCP3) instead of hydroxylamine.42,45
Finally, we set out to examine whether any synthetic analog could substitute for L-Dap throughout the entire sulfazecin biosynthetic pathway in vivo, resulting in the production of non-native monobactams. We therefore selected the most promising candidates (2S,3R)-methyl-Dap 6 and fluoromethyl-Dap 39, as well as chloromethyl-Dap 40 and vinyl-Dap 37 because of their potential as chemical handles for semi-synthetic modification, and performed a chemical complementation of the P. acidophila ΔsulG mutant that is deficient in L-Dap 1 biosynthesis and, therefore, sulfazecin production by itself.8
Initial bioassays on super-sensitive E. coli ESS46 plates showed antibacterial activity for the cell-free supernatant (CFS) of the ΔsulG culture supplemented with 6, whereas the inhibition zone was smaller compared to that of the positive control culture with added L-Dap 1 (Fig. S3†). Both samples also showed β-lactamase-induction activity (1 > 6) by nitrocefin assay,47 further corroborating the microbiological growth inhibition data for β-lactam production (Fig. S3†). No positive signals from either bioassay were obtained for the CFS of the ΔsulG cultures supplemented with any other synthetic analog. We suspected that either lower production or an altered antibacterial spectrum might be the reason other potential monobactam analogs eluded detection. Therefore, semi-purified fractions of each CFS were analyzed by Ultra Performance Liquid Chromatography-High Resolution Mass Spectrometry (UPLC-HRMS) as a more sensitive detection method. Indeed, a signal corresponding to the anticipated monobactam product was obtained not just for the cultures supplemented with 1 and 6, but also for the culture supplemented with fluoromethyl 39, the only other analog that showed distinct activation by A3 besides 6 (Fig. 2B). Moreover, a slightly shorter retention time (39 < 6 < 1) with increasing product hydrophobicity met expectations for the chromatographic parameters used.
Analysis of the tandem MS/MS (MSe) spectra (Fig. S4–S7†) confirmed the presence of imine fragments consistent with the highly diagnostic, formal [2 + 2] cycloreversion, a fragmentation pattern common to other β-lactams48 including aztreonam (Fig. S8†). These fragments not only bear the charged sulfamate species for detection in ESI(−) but also contain the corresponding substitution at the C4 position. The observation of these characteristic fragments leaves little doubt as to the assigned structures.
Reassured by these spectrometric data, we repeated our previous chemical complementation and bioassays using 7× concentrated CFS of ΔsulG cultures supplemented with either 1, 6 or 39. In fact, nitrocefin-positive and antibacterial activities were observed for all concentrated samples except that for the non-supplemented ΔsulG mutant negative control (Fig. 2B). The methylated sulfazecin analog (termed MM42842) obtained by fermentation in the presence of (2S,3R)-methyl-Dap 6, wherein the methyl- and methoxy-substituents are trans, has been previously isolated from P. cocovenenans.27 Our supplementation studies suggest that P. cocovenenans might directly employ 6, rather than L-Dap 1, as the native precursor during the biosynthesis of MM42842. In contrast to sulfazecin 2 its antibacterial activity has been described as mainly directed against Gram-positive bacteria, in line with our observed activity against Bacillus licheniformis ATTC 14580 and Kocuria rhizophila ATCC 9341.27
A novel fluorinated monobactam, fluoromethyl-sulfazecin, was identified when the ΔsulG mutant was fermented in the presence of 39. In line with the methylated analog it showed pronounced antibacterial activities against the two Gram-positive strains tested, but weak activity against E. coli ESS (Fig. 2B). While these data suggest that C4-substitutions alter the bioactivity profiles compared to sulfazecin 2, extended studies will be required to assess their antibacterial spectrum in greater detail. Nevertheless, these initial bioassay results and spectrometric data support the generation of bioactive monobactam derivatives. This outcome furthermore shows that despite the unfavourable kinetic performance of A3in vitro, under native conditions the biosynthetic machinery can successfully utilize 6 and 39 as alternative building blocks.
Currently, our ability to isolate these new products in sufficient quantities for downstream applications is met by numerous challenges. The low titre of sulfazecin 1 in wild type P. acidophila combined with the decreased affinity of the native biosynthetic machinery towards our unnatural substrates and an extensive purification protocol10,49 necessitate fermentations on a larger scale. While our synthetic route (cf.Scheme 2) was intended to generate substantial product diversity from a common precursor, it was not designed to yield large quantities of the corresponding functionalized 2,3-diaminopropionates.
It has been long recognized that introduction of fluorine into drugs can have beneficial effects on pharmacokinetic properties due to improved metabolic stability, enhanced membrane permeation and increased target binding affinity.50,51 The incorporation of even a single fluorine into natural products either by chemical synthesis51 or mutasynthesis26 has been demonstrated to significantly improve their potency. Due to its extreme scarcity among natural products, there is high interest in manipulating biosynthetic pathways to enable the site-selective incorporation of fluorine to yield compounds with potentially improved bioactivity that remain largely untapped.52 Recent successes have utilized fluorinated extender units in engineered polyketide synthases (PKS) to generate previously unknown macrolide products bearing aliphatic C–F bonds.53,54 In NRPS biosynthesis, fluorinated amino acids have been successfully incorporated into peptidyl natural products, however these examples consist almost exclusively of halogen-substituted aromatic α-amino acids.25,55,56 Owing to the much simpler installation of C-sp2–F bonds, these biosynthetic precursors are either commercially available or prepared in few synthetic steps. For instance, in order to isolate a fluorinated analog of balhimycin, Süssmuth et al. applied a three-step synthetic sequence to access racemic fluorinated beta-hydroxy tyrosine, which was required in multi-gram quantities to isolate the mutasynthetic product.24 Here, although unable to obtain such quantities, we have installed fluorine at an aliphatic position to prepare fluoromethyl-Dap 39 as a single stereoisomer. Exploiting the inherent substrate flexibility of the entire native late-stage sulfazecin biosynthetic machinery, we were able to selectively produce a bioactive fluorinated monobactam using conventional fermentation methods. To the best of our knowledge, we report the first example for the generation of non-native monobactams through precursor-directed fermentation.
Conclusions
The monobactam class of antibiotics is unique among the β-lactams not only for their unusual structure and biosynthetic rationale but also due to their inherent stability against the clinical threat of Ambler class B MBLs. Here, a divergent synthesis was developed to prepare densely functionalized 2,3-diaminopropionates as biosynthetic replacements for the essential L-Dap 1 motif in sulfazecin 2. Combining in vitro assays, and in vivo chemical complementation coupled with spectrometric and microbiological data, we demonstrate that these substituted analogs illustrate the potential to be incorporated into bioactive monobactam products.Adenylation domain activity end-point assays of our L-Dap library roughly correlate with spectrometric data of monobactam products obtained from fermentation experiments with selected substrates. These observations imply that A3 is flux-limiting in the efficient production of non-native sulfazecin analogs due to its decreasing activation of larger substrates (cf.Fig. 2A). Enzyme engineering is a powerful approach to overcome this hurdle by altering the active site to accommodate modified substrates, although rational optimization of A-domains to-date has been met with limited success.57–59 Here the terminal NRPS module of sulfazecin biosynthesis presents unique advantages we intend to exploit – the pronounced discrimination of A3 against cellularly ubiquitous canonical α-amino acids8 and strict selection of α,β-diamino acids is a fortunate circumstance for reprogrammed biosynthesis. Future work will aim to determine the structural basis for this α,β-diamino selectivity and to streamline A3 towards the effective activation of dedicated non-native L-Dap analogs.
Nevertheless, adenylation is only the first step in a short sequence of terminal biosynthetic transformations that include condensation of the non-native precursor with the upstream dipeptide, followed by in trans N-sulfonation and TE-catalyzed β-lactam formation with concurrent release from the NRPS. A two-step methoxylation, which is difficult to install stereoselectively using conventional synthetic methods, then furnishes the structure of the sulfazecin-like product.9 Studies to probe and optimize the substrate tolerance of participating enzymes are currently ongoing, with the aim to improve the scope and efficiency of our biosynthetic platform in vivo. Additionally, establishment of a more scalable synthetic or biosynthetic route to selected precursor amino acids will enable large-scale fermentation and isolation of monobactams for further characterization. Although it was recognized from the outset that mutasynthesis of monobactams presents a difficult target due to their naturally low production titre, the present work establishes the feasibility and lays the foundation to manipulate the biosynthetic machinery to produce customized monobactams by fermentation.
Data availability
ESI† contains everything needed to repeat any experiment.Author contributions
MSL, LK, RFL and CAT conceptualized experiments, analysed data, and wrote the manuscript. MSL, LK and JY carried out chemical synthesis. LK, RFL and MSL performed in vitro assays. RFL performed fermentation experiments and bioassays. TAZ and MSL developed and carried out UPLC-HRMS analyses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by National Institutes of Health Research Grant R01 AI121072. LK acknowledges financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 492438365. TAZ was supported by T32 GM135131. We would like to thank Dr M. A. Siegler for the collection of X-ray crystallographic data. We would also like to thank Dr I.P. Mortimer and Dr J. Catazaro for their help with ESI-MS and NMR experiments, respectively.Notes and references
- A. Viso, R. Fernández de la Pradilla, A. García and A. Flores, Chem. Rev., 2005, 105, 3167–3196 CrossRef CAS PubMed.
- A. Viso, R. F. De La Pradilla, M. Tortosa, A. García and A. Flores, Chem. Rev., 2011, 111, 3167–3196 CrossRef PubMed.
- C. Müller, S. Nolden, P. Gebhardt, E. Heinzelmann, C. Lange, O. Puk, K. Welzel, W. Wohlleben and D. Schwartz, Antimicrob. Agents Chemother., 2007, 51, 1028–1037 CrossRef PubMed.
- K. Bush and P. A. Bradford, Clin. Microbiol. Rev., 2020, 33(2) DOI:10.1128/cmr.00047-19.
- P. Hinchliffe, D. M. Moreno, M. A. Rossi, M. F. Mojica, V. Martinez, V. Villamil, B. Spellberg, G. L. Drusano, C. Banchio, G. Mahler, R. A. Bonomo, A. J. Vila and J. Spencer, ACS Infect. Dis., 2021, 7, 2697–2706 CrossRef CAS PubMed.
- R. B. Sykes, C. M. Cimarusti, D. P. Bonner, K. Bush, D. M. Floyd, N. H. Georgopapadakou, W. H. Koster, W. C. Liu, W. L. Parker, P. A. Principe, M. L. Rathnum, W. A. Slusarchyk, W. H. Trejo and J. S. Wells, Nature, 1981, 291, 489–491 CrossRef CAS PubMed.
- R. B. Sykes, D. P. Bonner, K. Bush and N. H. Georgopapadakou, Antimicrob. Agents Chemother., 1982, 21, 85–92 CrossRef CAS PubMed.
- R. Li, R. A. Oliver and C. A. Townsend, Cell Chem. Biol., 2017, 24, 24–34 CrossRef CAS PubMed.
- R. A. Oliver, R. Li and C. A. Townsend, Nat. Chem. Biol., 2018, 14, 5–7 CrossRef CAS PubMed.
- M. Asai, K. Haibara, M. Muroi, K. Kintaka and T. Kishi, J. Antibiot., 1981, 34, 621–627 CrossRef CAS PubMed.
- W. L. Parker, W. H. Koster, C. M. Cimarusti, D. M. Floyd, W. C. Liu and M. L. Rathnum, J. Antibiot., 1982, 35, 189–195 CrossRef CAS PubMed.
- C. M. Cimarusti, D. P. Bonner, H. Breuer, H. W. Chang, A. W. Fritz, D. M. Floyd, T. P. Kissick, W. H. Koster, D. Kronenthal, F. Massa, R. H. Mueller, J. Pluscec, W. A. Slusarchyk, R. B. Sykes, M. Taylor and E. R. Weaver, Tetrahedron, 1983, 39, 2577–2589 CrossRef CAS.
- A. Imada, M. Kondo, K. Okonogi, K. Yukishige and M. Kuno, Antimicrob. Agents Chemother., 1985, 27, 821–827 CrossRef CAS PubMed.
- M. Osborn, N. Stachulski, H. Sun, J. Blais, V. Venishetty, M. Raccuglia, M. Kankam, R. Colvin and F. Segal, Antimicrob. Agents Chemother., 2019, 63(7), e02592–18 CrossRef PubMed.
- R. B. Sykes and D. P. Bonner, Rev. Infect. Dis., 1985, 7(Suppl 4), S579–S593 CrossRef CAS PubMed.
- F. Reck, A. Bermingham, J. Blais, V. Capka, T. Cariaga, A. Casarez, R. Colvin, C. R. Dean, A. Fekete, W. Gong, E. Growcott, H. Guo, A. K. Jones, C. Li, F. Li, X. Lin, M. Lindvall, S. Lopez, D. McKenney, L. Metzger, H. E. Moser, R. Prathapam, D. Rasper, P. Rudewicz, V. Sethuraman, X. Shen, J. Shaul, R. L. Simmons, K. Tashiro, D. Tang, M. Tjandra, N. Turner, T. Uehara, C. Vitt, S. Whitebread, A. Yifru, X. Zang and Q. Zhu, Bioorg. Med. Chem. Lett., 2018, 28, 748–755 CrossRef CAS PubMed.
- H. R. Onishi, D. R. Daoust, S. B. Zimmerman, D. Hendlin and E. O. Stapley, Antimicrob. Agents Chemother., 1974, 5, 38–48 CrossRef CAS PubMed.
- K. L. Rinehart, Pure Appl. Chem., 1977, 49, 1361–1384 CAS.
- C. D. Denoya, R. W. Fedechko, E. W. Hafner, H. A. I. McArthur, M. R. Morgenstern, D. D. Skinner, K. Stutzman-Engwall, R. G. Wax and W. C. Wernau, J. Bacteriol., 1995, 177, 3504–3511 CrossRef CAS PubMed.
- S. Weist, C. Kittel, D. Bischoff, B. Bister, V. Pfeifer, G. J. Nicholson, W. Wohlleben and R. D. Süssmuth, J. Am. Chem. Soc., 2004, 126, 5942–5943 CrossRef CAS PubMed.
- W. T. Shier, K. L. Rinehart and D. Gottlieb, Proc. Natl. Acad. Sci. U. S. A., 1969, 63, 198–204 CrossRef CAS PubMed.
- M. A. Gregory, H. Petkovic, R. E. Lill, S. J. Moss, B. Wilkinson, S. Gaisser, P. F. Leadlay and R. M. Sheridan, Angew. Chem., Int. Ed., 2005, 44, 4757–4760 CrossRef CAS PubMed.
- Z. Hojati, C. Milne, B. Harvey, L. Gordon, M. Borg, F. Flett, B. Wilkinson, P. J. Sidebottom, B. A. M. Rudd, M. A. Hayes, C. P. Smith and J. Micklefield, Chem. Biol., 2002, 9, 1175–1187 CrossRef CAS PubMed.
- S. Weist, B. Bister, O. Puk, D. Bischoff, S. Pelzer, G. J. Nicholson, W. Wohlleben, G. Jung and R. D. Süssmuth, Angew. Chem., Int. Ed., 2002, 41, 3383–3385 CrossRef CAS PubMed.
- Y. Shi, Z. Jiang, X. Lei, N. Zhang, Q. Cai, Q. Li, L. Wang, S. Si, Y. Xie and B. Hong, Microb. Cell Fact., 2016, 15, 77 CrossRef PubMed.
- F. Xie, S. Dai, Y. Zhao, P. Huang, S. Yu, B. Ren, Q. Wang, Z. Ji, G. Alterovitz, Q. Zhang, J. Zhang, X. Chen, L. Jiang, F. Song, H. Liu, F. M. Ausubel, X. Liu, H. Dai and L. Zhang, Cell Chem. Biol., 2020, 27, 1532–1543.e6 CrossRef CAS PubMed.
- S. J. Box, A. G. Brown, M. L. Gilpin, M. N. Gwynn and S. R. Spear, J. Antibiot., 1988, 41, 7–12 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 CrossRef CAS PubMed.
- B. R. M. Villiers and F. Hollfelder, ChemBioChem, 2009, 10, 671–682 CrossRef CAS PubMed.
- H. Katano, H. Watanabe, M. Takakuwa, C. Maruyama and Y. Hamano, Anal. Sci., 2013, 29, 1095–1098 CrossRef CAS PubMed.
- C. Maruyama, H. Niikura, M. Takakuwa, H. Katano and Y. Hamano, in Methods in Molecular Biology, Humana Press, New York, NY, 2016, vol. 1401, pp. 77–84 Search PubMed.
- N. Kadi and G. L. Challis, Methods Enzymol., 2009, 458, 431–457 CAS.
- R. Hara, R. Suzuki and K. Kino, Anal. Biochem., 2015, 477, 89–91 CrossRef CAS PubMed.
- C. A. Townsend, Curr. Opin. Chem. Biol., 2016, 35, 97–108 CrossRef CAS PubMed.
- S. Hanessian, B. Vanasse, H. Yang and M. Alpegiani, Can. J. Chem., 1993, 71, 1407–1411 CrossRef CAS.
- M. D. Armstrong, J. Am. Chem. Soc., 1949, 71, 3399–3402 CrossRef CAS.
- C. Pearson, K. L. Rinehart and M. Sugano, Tetrahedron Lett., 1999, 40, 411–414 CrossRef CAS.
- B. T. Kelley and M. M. Joullié, Org. Lett., 2010, 12, 4244–4247 CrossRef CAS PubMed.
- S. Bolek and J. Ignatowska, J. Fluorine Chem., 2019, 217, 13–21 CrossRef CAS.
- H. Han, J. Yoon and K. D. Janda, J. Org. Chem., 1998, 63, 2045–2048 CrossRef CAS.
- D. J. Wilson and C. C. Aldrich, Anal. Biochem., 2010, 404, 56–63 CrossRef CAS PubMed.
- B. P. Duckworth, D. J. Wilson and C. C. Aldrich, in Methods in Molecular Biology, Humana Press Inc., 2016, vol. 1401, pp. 53–61 Search PubMed.
- M. R. Webb, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 4884–4887 CrossRef CAS PubMed.
- D. E. Ehmann, C. A. Shaw-Reid, H. C. Losey and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2509–2514 CrossRef CAS PubMed.
- H. Aoki, H. I. Sakai, M. Kohsaka, T. Konomi, J. Hosoda, E. Iguchi, H. Imanaka and Y. Kubochi, J. Antibiot., 1976, 29, 492–500 CrossRef CAS PubMed.
- C. H. O'Callaghan, A. Morris, S. M. Kirby and A. H. Shingler, Antimicrob. Agents Chemother., 1972, 1, 283–288 CrossRef PubMed.
- L. A. Mitscher, H. D. H. Showalter, K. Shirahata and R. L. Foltz, J. Antibiot., 1975, 28, 668–675 CrossRef CAS PubMed.
- A. Imada, K. Kitano and M. Asai, US Pat., US4229436A, 1980 Search PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071–1081 CrossRef.
- M. C. Walker and M. C. Y. Chang, Chem. Soc. Rev., 2014, 43, 6527–6536 RSC.
- S. Sirirungruang, O. Ad, T. M. Privalsky, S. Ramesh, J. L. Sax, H. Dong, E. E. K. Baidoo, B. Amer, C. Khosla and M. C. Y. Chang, Nat. Chem. Biol., 2022, 18, 886–893 CrossRef CAS PubMed.
- A. Rittner, M. Joppe, J. J. Schmidt, L. M. Mayer, S. Reiners, E. Heid, D. Herzberg, D. H. Sherman and M. Grininger, Nat. Chem., 2022, 1–7 Search PubMed.
- C. Wang, C. Lambert, M. Hauser, A. Deuschmann, C. Zeilinger, K. Rottner, T. E. B. Stradal, M. Stadler, E. J. Skellam and R. J. Cox, Chem. - Eur. J., 2020, 26, 13578–13583 CrossRef CAS PubMed.
- K. M. J. De Mattos-Shipley, C. Greco, D. M. Heard, G. Hough, N. P. Mulholland, J. L. Vincent, J. Micklefield, T. J. Simpson, C. L. Willis, R. J. Cox and A. M. Bailey, Chem. Sci., 2018, 9, 4109–4117 RSC.
- H. Kries, R. Wachtel, A. Pabst, B. Wanner, D. Niquille and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 10105–10108 CrossRef CAS PubMed.
- S. Meyer, J. C. Kehr, A. Mainz, D. Dehm, D. Petras, R. D. Süssmuth and E. Dittmann, Cell Chem. Biol., 2016, 23, 462–471 CrossRef CAS PubMed.
- D. L. Niquille, D. A. Hansen, T. Mori, D. Fercher, H. Kries and D. Hilvert, Nat. Chem., 2017, 10, 282–287 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplemental figures and schemes, materials and methods, experimental details, characterization data and NMR spectra of new compounds. Check CIF file for single crystal X-ray structure of 11. CCDC 2207290. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06893a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |