
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cross-conjugation approach for high-performance diaryl-hemithioindigo photoswitches †
Max
Zitzmann
,
Frank
Hampel
and
Henry
Dube
 *
*
Friedrich-Alexander-Universität Erlangen-Nürnberg, Department of Chemistry and Pharmacy, Nikolaus-Fiebiger-Str. 10, 91058, Erlangen, Germany. E-mail: henry.dube@fau.de
First published on 1st May 2023
Abstract
Diaryl-hemithioindigos (diaryl-HTIs) are derivatives of a novel class of highly functionalized indigoid chromophores. In this work a systematic study of the electronic effects on their photoswitching reveals the design principles for achieving an excellent property profile. Two key elements need to be invoked for perfect diaryl-HTI performance, first introduction of strong electron donors and second establishment of cross-conjugation. The resulting photoswitches combine high thermal stability, large extinction coefficients, red-light responsiveness, pronounced photochromism, and strong isomer accumulation in the photostationary states with precise geometry changes. By using the inherent basicity of their strong electron donor moiety, diaryl-HTIs can be rendered into very potent tools for molecular logic applications. We demonstrate a variety of binary logic setups as well as sophisticated three- and four-input keypad locks for sequential logic operations. Three distinct states and up to four different stimuli are invoked for this multi-level molecular information processing. Diaryl-HTIs have thus entered the stage as very capable and promising photoswitch motives for anyone interested in reversible visible- and red-light as well as multi-stimuli responsive molecular behavior.
Introduction
Molecular photoswitches are versatile tools for introducing light-responsiveness into nano-scale systems.1–3 Of particular interest are variants that can be operated with visible to red light irradiation while maximizing geometric or electronic changes.4,5 In this regard double bond isomerization is oftentimes used for large geometrical changes and electrocyclization reactions for strong electronic changes. In a few photoswitch motives, both aspects can be combined, for example, in spiropyrans,6–8 Stenhouse dyes,9,10 or the recently discovered hemithioindigo (HTI)-TOND motive.11 Photoinduced double bond isomerization has been utilized for a long time with, e.g., stilbene12–14 and azobenzene chromophores,15 however additional substitutions are needed to shift their absorptions to the visible part of the spectrum.16–22 Indigoid chromophores have gained traction as intrinsically visible light responsive photoswitches for double bond isomerization reactions.23–26 From the different variants such as indigo,27–29 indirubin,30 or thioindigo,31 hemiindigo,32,33 and especially hemithioindigo25 (HTI, see also ref. 34–36 for some recent HTI-photoswitch examples) stand out as especially versatile core structures. HTI-based photoswitches consist of a central isomerizable double bond, which connects a benzothiophenone (a half- or hemithioindigo) fragment to one aromatic residue typically termed the stilbene fragment (Fig. 1a). Since one hydrogen atom remains at the double bond it is threefold substituted in the core chromophore structure. HTIs were employed early on in the photopharmacological or chemical biology context24,37–40 and found their way into supramolecular chemistry,41–44 molecular machine building,45–53 or molecular logic systems.54 However, particularly steep progress has been made since facile fourfold double-bond substitution became synthetically available.55,56 In particular, the combination of sp3-based (alkyl or ether) and aryl-based groups at the stilbene fragment side enabled the discovery of new types of thermal- and photoreactions,11 evidencing the long-debated hula-twist mechanism,57 generating new types of molecular motors,47,48 or establishing the first photogearing process.52 | ||
| Fig. 1 Diaryl-HTI as a highly functionalized photoswitch motive in comparison to the parent threefold double-bond substituted HTI. (a) The core chromophore of HTI contains a threefold substituted double bond. (b) Diaryl-HTIs contain a fourfold substituted double-bond with two geminal aromatic residues at the stilbene fragment side. Cross-conjugation involving donor/acceptor pairs offers very potent photoswitching properties in combination with well-controlled geometry changes and overall higher degree of functionalization. | ||
A different possibility is the introduction of two geminal aromatic residues at the stilbene fragment side. This substitution has already shown unique potential for multi-state switching and discovery of yet another unknown photoreaction, the dual single bond rotation (DSBR).58 In the eight-fold sequential switching, photoisomerization and thermal isomerization steps alternate while reversible photoswitching remains to be explored. In this work, we scrutinize the feasibility of reversible photoswitching for diaryl-HTI chromophores using a cross-conjugation approach to optimize the whole property profile. With this approach we achieve strongly red-shifted absorptions enabling red-light responsiveness, pronounced photochromism, high isomer enrichment in the photostationary states (PSS), good quantum yields for both switching processes, and high thermal bistability. These very favorable properties render diaryl-HTIs into particularly promising new photoswitch scaffolds for advanced applications, and we demonstrate a number of molecular logic operations as an example. To this end, the intrinsic basicity of the cross-conjugated and electron-rich aniline group is used to invoke acid/base responsiveness. Thus, another level of chemical stimuli is added and multi-stimuli switching allows the construction of diaryl-HTI based molecular logic operators.
Results and discussion
We investigated 10 different derivatives of diaryl-HTI photoswitches 1–10 bearing combinations of donor, acceptor, or rather neutral substituents at the aromatic para-positions (for all molecular structures investigated see Fig. 2). A systematic approach is followed in which all possible combinations of donor and acceptor substituents are scrutinized. Two different thioindigo-fragments are invoked in this study, one unsubstituted one and a pyridine-variant with the heterocyclic nitrogen next to the sulfur atom. The latter was used to scrutinize the effect of electron deficiency at the thioindigo fragment on the photoswitching properties. | ||
| Fig. 2 Diaryl-HTIs 1–10 investigated in this study. (a) Schematic depiction of all structures, only one isomer is shown in each case for clarity; (b) structures of 1, Z-3, 5, E/Z-7, and Z-8 in the crystalline state. | ||
Synthesis of diaryl-HTIs follows an established protocol reported earlier by our group (see the Synthesis section of the ESI† for details).56 Starting from commercially available thiosalicylic acids 11 and 12 a substitution reaction with α-bromo-ketones gives the corresponding substituted thioethers 13–16. Basic ring closure leading to ketones 17–20 was followed by subsequent chlorination to yield the chloro-HTI precursor molecules 21–24. The latter can be cross-coupled with aryl-boronic acids to introduce the second aryl residue at the stilbene fragment side of the central double bond. Crystals suitable for X-ray structural analysis were obtained for diaryl-HTI derivatives 1, Z-3, 5, E/Z-7, and Z-8 and for precursor compound 15 (see the ESI† for details on the latter). For derivative 7, both the E and the Z isomer are co-crystallizing and thus allow both isomeric forms to be structurally characterized in detail. The structures in the crystalline state of Z-3 and Z-8 also allowed us to directly assign the corresponding solution spectra to the specific double bond configuration. For derivatives 2, 3, 4, 7, 8, and 9 the double bond configuration in solution was elucidated using NOE-cross-signals in the corresponding NMR spectra (see Fig. S41–S46 in the ESI†).
The thermal behavior of diaryl-HTI derivatives 2–4 and 7–9 was investigated first to establish the relative stability of Z and E isomers as well as the kinetics of thermal isomerization of the metastable states in the dark (see Fig. 3a and the Thermal double bond isomerization section in the ESI†). All derivatives exhibit a greater stability for the Z-configured isomer with the exception of diaryl-HTI 4, for which the E isomer is more stable. The observed stability preference can be well explained by the general trend in which the more electron rich aryl substituent of the diaryl fragment is favored in a trans-relationship to the electron accepting carbonyl function of the thioindigo fragment. This stabilization is most pronounced in the strongest push–pull systems, i.e., diaryl-HTIs 3 and 8, where additionally a matching favorable trans-relationship of the electron-poor aryl substituent of the diaryl fragment to the electron donating sulfur of the thioindigo fragment is present.
 | ||
| Fig. 3 (a) Experimentally determined ΔG‡ values for the thermal double bond isomerization of diaryl-HTIs 2, 3, 7 and 8 in toluene and DMSO solution. (b) Excerpts of the aromatic region of 1H-NMR spectra of HTI 2 after irradiation with different wavelengths (E isomer highlighted in red and Z isomer highlighted in blue). (c) Excerpts of the aromatic region of 1H-NMR spectra of HTI 3 after irradiation with different wavelengths (E isomer highlighted in red and Z isomer highlighted in blue). (d) Molar extinction coefficients of diaryl-HTI 7 in toluene in the PSS measured after irradiation with different wavelengths (best performance for enrichment of the Z isomer at 617 nm (dark blue line) and for the E isomer at 450 nm (light blue line)) and observed color change. (e) Molar extinction coefficients of diaryl-HTI 8 in toluene in the PSS measured after irradiation with different wavelengths (best performance for enrichment of the Z isomer at 617 nm (dark blue line) and for the E isomer at 470 nm (light blue line)) and observed color change. | ||
In general, all derivatives show sizable Gibbs free energies of activation ΔG‡ for the thermal double bond isomerizations, giving rise to corresponding half-lives of the metastable states in the several hours to years range at ambient temperatures (Fig. 3e). The kinetics vary by about four orders in magnitude in terms of rate or by 5.5 kcal mol−1 in terms of ΔG‡ (Table 1). The fastest isomerization is seen for diaryl-HTI 7, giving rise to several hours persistence of the metastable state in solution. The highest barrier and thus slowest isomerization is observed for diaryl-HTI 2 with up to several years persistence of the metastable state. No clear trend is seen with respect to the particular electronic nature of the diaryl substituents. However, noticeably smaller ΔG‡ values are obtained for pyridine-thioindigo variants 7, 8, and 9 as compared to 2, 3, and 4. Interestingly, solvent polarity dependence in this series is typically opposite to the parent HTI derivatives with higher ΔG‡ values observed in the more polar solvent DMSO. Scrutiny of the thermal behavior of diaryl-HTIs 2–4 and 7–9 thus establishes favorable slow kinetics for the spontaneous thermal double bond isomerization in the dark, guaranteeing full photocontrol over their switching on a convenient time scale of hours (pyridine-thioindigo derivatives 7, 8, and 9) to years (diaryl 2, 3, and 4).
| Diaryl-HTI | ΔGE→Z‡ [kcal mol−1] | ΔG [kcal mol−1] (at T) | t eq. 23 °C1/2 | Isomer % in PSS (at LED nm) | λ max [nm] and εmax [L mol−1 cm−1] of Z/E isomers' lowest energy absorption band | Δλmax [nm] and Δεmax [L mol−1 cm−1] of E and Z isomers | Φ Z to E/ΦE to Z |
|---|---|---|---|---|---|---|---|
| a In toluene-d8. b In DMSO-d6. c In benzene-d6. d Linearly extrapolated to 23 °C. | |||||||
| 1 | — | — | — | — | 509; 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200a 200a |
— | |
| 526; 16800b |
|||||||
| 2 | 25.7a | 0.2 (50 °C)a | 12 da | 76% E (450 nm)a | 473; 19300a |
45; 4200a | 3%a |
| 96% Z (617 nm)a | 518; 15100a |
||||||
| 28.3b | 0.3 (100 °C)b | 3 ab | 75% E (470 nm)b | 490; 22900b |
48; 8800b | 11%a | |
| 97% Z (617 nm)b | 538; 14100b |
||||||
| 3 | 23.2a | 1.0 (40 °C)a | 4 ha | 75% E (450 nm)a | 496; 21100a |
27; 7700a | 3%a |
| 97% Z (617 nm)a | 523; 13400a |
||||||
| 27.7b | 1.6 (90 °C)b | 1 ab | 77% E (470 nm)b | 493; 21800b |
46; 6000b | 3%a | |
| 96% Z (617 nm)b | 539; 15800b |
||||||
| 4 | 27.8b | 0.5 (90 °C)b | 1 ab | 49% Z (490 nm)c | 444; 6300c | 6; 580c | 11%c |
| 69% E (420 nm)c | 438; 6800c | ||||||
| 55% Z (490 nm)b | 447; 5600b | 7; 940b | 6%c | ||||
| 64% E (420 nm)b | 440; 6600b | ||||||
| 5 | — | — | — | — | 440; 5200a | — | |
| 440; 5600b | |||||||
| 6 | — | — | — | — | 517; 14800a |
— | |
| 486; 16800b |
|||||||
| 7 | 23.9a | 0.3 (40 °C)a | 14 ha | 75% E (450 nm)a | 480; 20800a |
45; 7800a | 6%a |
| 97% Z (617 nm)a | 525; 13000a |
||||||
| 22.8b | 0.3 (30 °C)b | 2 hb | 79% E (470 nm)b | 496; 21200b |
49; 6600b | 5%a | |
| 94% Z (617 nm)b | 545; 14600b |
||||||
| 8 | 24.0a | 0.9 (30 °C)a | 16 ha | 83% E (470 nm)a | 486; 21100a |
46; 7300a | 4%a |
| 98% Z (625 nm)a | 532; 13800a |
||||||
| 24.5b | 0.9 (40 °C)b | 38 hb | 78% E (470 nm)b | 499; 20400b |
56; 7600b | 4%a | |
| 96% Z (650 nm)b | 555; 12800b |
||||||
| 9 | 25.6b | 0.4 (60 °C)b | 16 db | 49% E (490 nm)a | 426; 4700a | 0; 1180a | 5%a |
| 71% Z (435 nm)a | 426; 5800a | ||||||
| 52% E (505 nm)b | 424; 5100b | 0; 1290b | 3%a | ||||
| 69% Z (420 nm)b | 424; 6400b | ||||||
| 10 | — | — | — | — | 426; 4900a | — | |
| 422; 4500b | |||||||
The photochemical and photophysical properties of all diaryl-HTIs in the series 1–10 were quantified subsequently (see Fig. 3b–e and 4 for selected examples and the UV/Vis spectroscopic measurements, NMR irradiation experiments, and Quantum yield determination sections in the ESI† for more details). For reasons of simplicity, we refer to hypsochromic and bathochromic species in the following instead of Z and E isomers, as the former allows working out more precisely the structure-related trends. Hypsochromic species possess a trans-relationship of the thioindigo-fragments' carbonyl to the most electron-rich aromatic residue of the diaryl fragment, while the bathochromic species possess a corresponding cis-relationship.
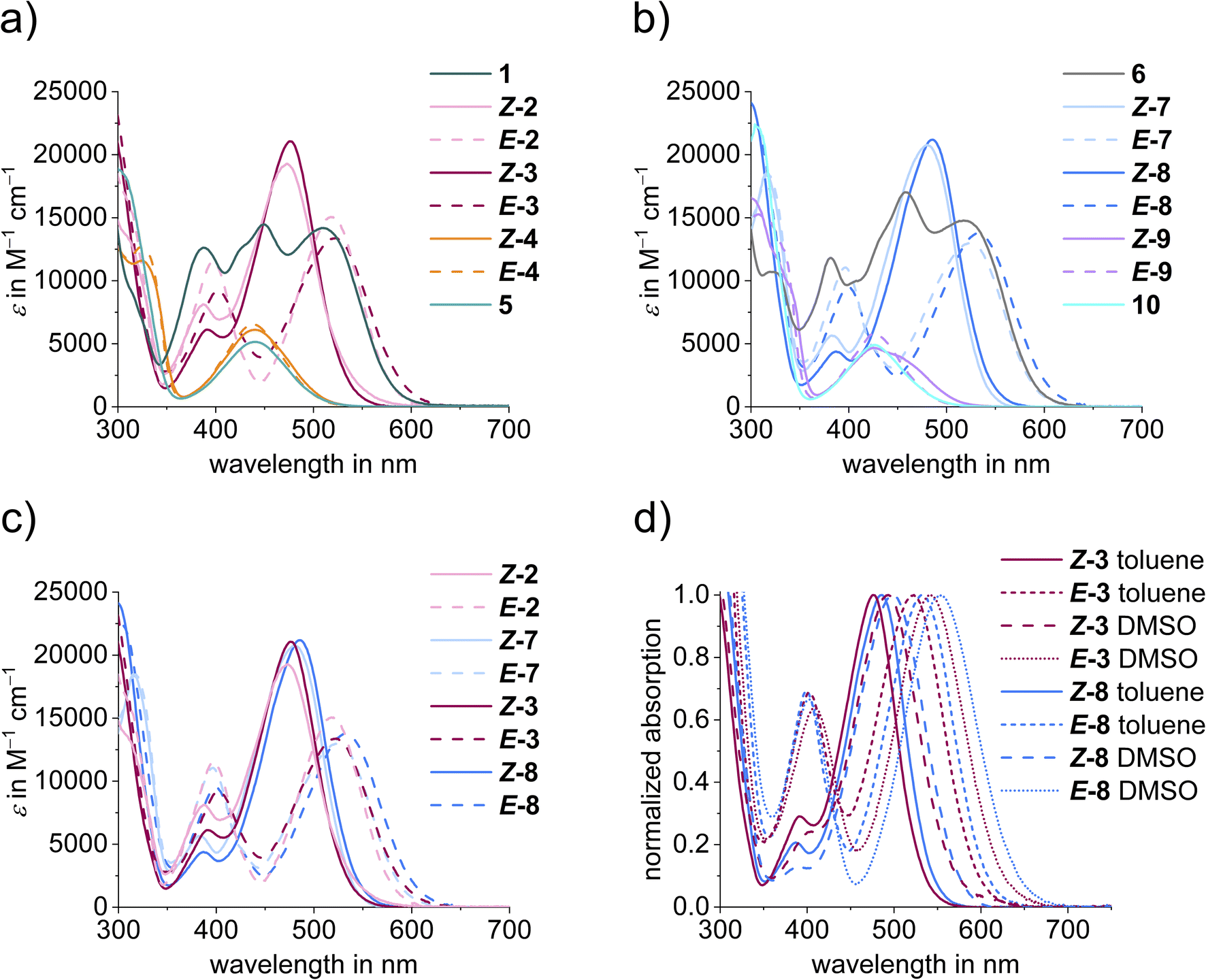 | ||
| Fig. 4 (a) Comparison of molar extinction coefficients of diaryl-HTIs 1–5 (solid line: Z isomers, dashed line: E isomers); (b) comparison of molar extinction coefficients of diaryl-HTIs 6–10 (solid line: Z isomers, dashed line: E isomers); (c) comparison of molar extinction coefficients of diaryl-HTIs 2 and 3 with their pyridine substituted counterparts 7 and 8 (solid line: Z isomers, dashed line: E isomers); (d) solvatochromism of diaryl-HTIs 3 and 8 in toluene and DMSO (solid line: Z isomers in toluene solution, dashed line: Z isomers in DMSO solution, dotted line: E isomers in toluene solution, short dotted line: E isomers in DMSO solution). | ||
Molar extinction coefficients show pronounced absorption in the visible region of the electromagnetic spectrum for all derivatives ranging from 5000 M−1 cm−1 to 22000 M−1 cm−1 (Fig. 4). Extinction coefficients are largest for dimethyl-aniline substituted variants. The combination of cross-conjugated donor–acceptor pairs gives rise to the most red-shifted absorptions, and non-symmetrically substituted diaryl-HTIs 3 and 8 exhibit absorptions beyond 650 nm for the bathochromic species. When comparing the influence of systematic electronic variations in the series, a consistent behavior is observed. The most hypsochromic absorption and rather low extinction coefficients are present in twofold acceptor substituted diaryl-HTIs 5 and 10, followed by slightly more redshifted derivatives 4 and 9 (see Fig. 4a and b). Substitution by dimethyl-aniline gives rise to significant redshifted absorptions and much stronger extinction coefficients as seen for diaryl-HTIs 2 and 7. The former is an accumulative effect as seen in the further redshifted absorptions of twofold dimethyl-aniline substituted diaryl-HTIs 1 and 6. Cross-conjugation of a strong donor–acceptor combination in diaryl-HTIs 3 and 8 leads to the largest achievable absorption redshift in the series, however not by a large margin (Fig. 4a–c). The effect of nitrogen-substitution at the thioindigo-fragment is also small as derivatives of the series 1–5 show essentially the same absorptions as their counterparts in the series 6–10 with the latter being a bit more redshifted (Fig. 4a–c).
Solvatochromism is absent for all derivatives lacking a dimethyl-aniline substituent, i.e., diaryl-HTIs 4, 5, 9, and 10 (Fig. 4d). The presence of a dimethyl-aniline substituent gives rise to pronounced solvatochromism with absorption red-shifts up to 20 nm in more polar DMSO as compared to toluene solutions. Again, a slightly larger solvatochromism and thus the largest effect overall is observed for cross-conjugated derivatives 3 and 8.
Strong photochromism is observed for diaryl-HTIs 2, 3, 7, and 8, with maxima differences between the hypsochromic and the bathochromic species reaching beyond 50 nm (Fig. 3c, d and 4c). As described above, the hypsochromic species bear the most electron rich aromatic residue of the diaryl fragment in a trans-relationship to the carbonyl function of the thioindigo fragment. Correspondingly, the bathochromic species shows a cis-relationship between these groups. This behavior is consistent with the parent HTIs, where the single aromatic substituent of the stilbene fragment serves the role of the most electron rich substituent. Because of their pronounced photochromism, photoswitching of diaryl-HTIs 2, 3, 7, and 8 is very effective. Isomer enrichment up to 83% of the bathochromic species as well as up to 98% of the hypsochromic species in the photostationary state (PSS) is possible using blue and red-light irradiation, respectively (see Fig. 3b–e, Table 1 and the ESI†). The polarity of the solvent (e.g., toluene versus DMSO) does not influence the proficient photoisomerization strongly, which establishes robust operation of diaryl-HTIs under different conditions.
Quantum yield measurements of diaryl-HTIs 2–4 and 7–9 established good photoisomerization efficiencies in both switching directions with values ranging from 3% up to 11% (see Table 1 and the Quantum yield determination section in the ESI†). Only small effects of nitrogen-substitution at the thioindigo-fragment are seen, which somewhat improve the Z to E photoisomerization and diminish the E to Z photoisomerization. All quantum yields measured are in the mid-range of measured quantum yields for HTI photoswitches and are sizable enough to allow for timely isomer conversions even under lower light conditions. It has to be mentioned at this point that fourfold double bond substitution in HTI photoswitches typically leads to significantly diminished quantum yields as compared to the parent threefold-double bond substituted variants.47,48,57,58 Additionally, quantum yields for Z to E and E to Z photoisomerizations are oftentimes very different, which hampers proper isomer accumulation in the PSS in many cases. For diaryl-HTI derivatives, both Z to E and E to Z photoisomerization reactions possess comparable quantum yields, which enable high isomer accumulation in the PSS with similar pacing because of their substantial photochromism.
A theoretical description of diaryl-HTIs 3 and 8 was undertaken to elucidate the character of the photoinduced electronic transition (see the Theoretical description section in the ESI†). In both cases this is essentially a π–π* transition at the central double bond as judged by the molecular orbitals shown in Fig. S39 and S40 in the ESI.† Theory also correctly predicts more stable Z isomers with absolute energy differences in good agreement with the experimental ΔGE–Z values obtained at elevated temperatures (theoretical values are ΔGE–Z(3) = 1.1 kcal mol−1 and ΔGE–Z(8) = 1.4 kcal mol−1 and experimental ones are ΔGE–Z(3) = 1.0 kcal mol−1 at 40 °C and ΔGE–Z(8) = 0.9 kcal mol−1 at 30 °C).
In order to demonstrate the value of the expanded structural space, functionality, and high-performance of diaryl-HTI photoswitches we explored an application that makes direct use of their defining characteristics. To this end we use the basicity of the electron-rich aniline moiety, which is needed for proper photoswitching, to introduce two more independent chemical stimuli, i.e., acid and base. Upon protonation of either isomer of diaryl-HTI 3 with trifluoroacetic acid (TFA) a third state, the protonated Z isomer Z-3H+, is obtained with a distinct absorption spectrum (Fig. 5a and b). At the same time photoisomerization of Z-3H+ is prohibited, which establishes acid-gating of the photoswitching property. Upon addition of triethylamine (TEA) unprotonated 3 is recovered and photoswitching is enabled again rendering the three-state/four-stimuli switching process between Z-3, E-3, and Z-3H+ fully reversible. Since the absorptions of the three states are significantly different, an advanced molecular logic system (see ref. 59–61 for examples of earlier molecular logic systems based on photoswitches) can be setup with diaryl-HTI 3. By thresholding absorption intensity at different wavelengths as output signals, a variety of binary logic gates such as AND, SAND, NOT, or Identity, as well as a binary multiplier can be constructed (see Fig. 5c–e, for specific examples and the Molecular logic application section of the ESI† for more details). In addition, application of diaryl-HTI 3 as a molecular sequential logic system is possible with a heightened level of selectivity. A three-input system (input A = TFA addition, input B = TEA addition, input C = 470 nm light) with one true input combination (one out of six possible, i.e. ABC) as well as a system with two true input combinations (two out of six, i.e. ACB and CAB) can be setup as shown in the Molecular logic application section of the ESI.† A more sophisticated four-input system can also be established with diaryl-HTI 3, which offers a heightened level of performance as the sequence of four different inputs (input A = TFA addition, input B = TEA addition, input C = 470 nm light, and input D = 617 nm light) is distinguished with three true input combinations (ABDC, ADBC, and DABC) out of 24 possible (Fig. 5f). With these applications diaryl-HTIs already show their potential as versatile multi-responsive molecular building blocks offering a range of optimal switching properties within a functionally expanded structure.
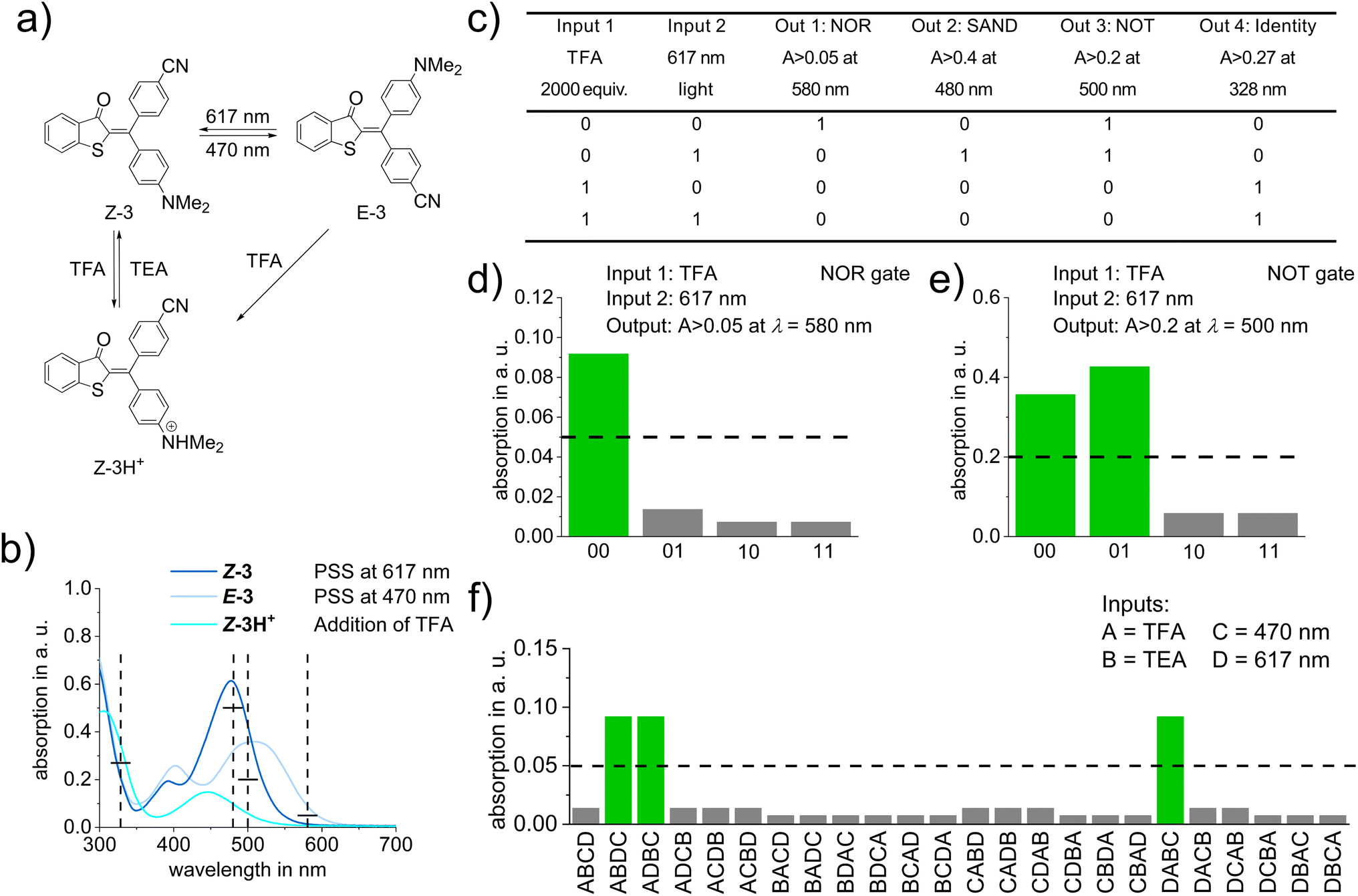 | ||
| Fig. 5 (a) Schematic representation of the multi-stimuli responsiveness of diaryl-HTI 3 towards 470 nm and 617 nm light as well as acid and base addition. Protonation with TFA (2000 equiv.) leads to the protonated Z isomer (Z-3H+) and subsequent deprotonation with TEA (2000 equiv.) to Z-3; (b) UV/Vis absorption spectra of Z-3, E-3, and Z-3H+ in toluene solution with indicated output readouts at 580 nm, 500 nm, 480 nm, and 328 nm (vertical dashed lines) and threshold limits (horizontal solid black lines); (c) truth table for four two-bit logic gates (NOR, SAND, NOT, and Identity) using TFA (2000 equiv.) and 617 nm light as orthogonal inputs; (d) experimental absorption outputs at 580 nm for a NOR gate (threshold level shown as horizonal dashed lines) starting from E-3; (e) experimental absorption outputs at 500 nm for a NOT gate (threshold level shown as horizonal dashed line) starting from E-3; (f) experimental absorption outputs at 580 nm for a keypad lock with three true outputs (threshold level shown as horizonal dashed line) starting from Z-3H+ and using four different inputs (TFA (input A), TEA (input B), 470 nm light (input C), and 617 nm light (input D)). | ||
Conclusion
In summary, a systematic study of diaryl-HTI photoswitching is presented optimizing the photochemical responses for these structurally expanded and visible-light responsive chromophores. It is revealed that the implementation of strong electron donation is key for achieving very good performance. Introduction of an additional pronounced cross-conjugation allows further improvement of the properties resulting in highly capable photoswitches responsive to blue and red-light irradiation. The introduction of a nitrogen atom within the thioindigo fragment does not lead to deterioration of this favorable behavior. As a result, the high thermal stability of metastable states across different solvent polarities is combined with efficient photoswitching, pronounced photochromism, and high isomer enrichment in the photostationary states. Acid and base can be used as further chemical stimuli to reversibly access a third protonated state of diaryl-HTI 3 with distinct spectral characteristics. Upon protonation photoisomerization capability is stalled, which establishes reversible acid gating of the photoresponse as well as multi-state and multi-stimuli responsiveness. The use of diaryl-HTIs in different molecular logic systems directly showcases the enabling effects of the diaryl-substitution for advanced switching applications. A variety of binary logic as well as sequential logic operations can be realized with these new chromophores, including expanded keypad locks that distinguish the sequences of up to four different inputs. Diaryl-HTIs are therefore very promising and highly functionalized multi-responsive photoswitches offering multi-addressability in combination with an outstanding property profile.Data availability
All relevant data have been included in ESI.† Crystal structural data have been deposited at the CCDC https://www.ccdc.cam.ac.uk. All original data underlying this work are available from the corresponding author upon request.Author contributions
M. Z. and H. D. conceived the study. M. Z. conducted the synthesis, full analysis including quantifying photochemical and photophysical properties, quantum chemical calculations, and molecular logic application experiments, F. H. provided X-ray analysis of the structures in the crystalline state, H. D. coordinated the study, helped in the analysis, and wrote the manuscript. All authors discussed and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
H. Dube thanks the Deutsche Forschungsgemeinschaft (DFG) for an Emmy Noether fellowship (DU 1414/1-2). This project has also received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (PHOTOMECH, grant agreement no. 101001794).References
-
B. L. Feringa and W. R. Browne, Molecular Switches, Wiley-VCH, Weinheim, 2011, vol. 1 Search PubMed
.
-
Z. L. Pianowski, Molecular Photoswitches. Chemistry, Properties, and Applications, Wiley-VCH, Weinheim, 2022, p. 1152 Search PubMed
- J. D. Harris, M. J. Moran and I. Aprahamian, New molecular switch architectures, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9414–9422 CrossRef CAS PubMed
- D. Bleger and S. Hecht, Visible-Light-Activated Molecular Switches, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef CAS PubMed
- L. N. Lameijer, S. Budzak, N. A. Simeth, M. J. Hansen, B. L. Feringa, D. Jacquemin and W. Szymanski, General Principles for the Design of Visible-Light-Responsive Photoswitches: Tetra-ortho-Chloro-Azobenzenes, Angew. Chem., Int. Ed., 2020, 59, 21663–21670 CrossRef CAS PubMed
- L. Kortekaas and W. R. Browne, The evolution of spiropyran: fundamentals and progress of an extraordinarily versatile photochrome, Chem. Soc. Rev., 2019, 48(12), 3406–3424 RSC
- B. S. Lukyanov and M. B. Lukyanova, Spiropyrans: Synthesis, Properties, and Application, Chem. Heterocycl. Compd., 2005, 41, 281–311 CrossRef CAS
- R. Klajn, Spiropyran-based dynamic materials, Chem. Soc. Rev., 2014, 43(1), 148–184 RSC
- M. M. Lerch, W. Szymański and B. L. Feringa, The (photo)chemistry of Stenhouse photoswitches: guiding principles and system design, Chem. Soc. Rev., 2018, 47(6), 1910–1937 RSC
- S. Helmy, S. Oh, F. A. Leibfarth, C. J. Hawker and J. Read de Alaniz, Design and synthesis of donor-acceptor Stenhouse adducts: a visible light photoswitch derived from furfural, J. Org. Chem., 2014, 79(23), 11316–11329 CrossRef CAS PubMed
- F. Kohl, A. Gerwien, F. Hampel, P. Mayer and H. Dube, Hemithioindigo-Based Trioxobicyclononadiene: 3D Multiswitching of Electronic and Geometric Properties, J. Am. Chem. Soc., 2022, 144(7), 2847–2852 CrossRef CAS PubMed
- D. H. Waldeck, Photoisomerization Dynamics of Stilbenes, Chem. Rev., 1991, 91(3), 415–436 CrossRef CAS
- D. Villaron and S. J. Wezenberg, Stiff-Stilbene Photoswitches: From Fundamental Studies to Emergent Applications, Angew. Chem., Int. Ed., 2020, 59(32), 13192–13202 CrossRef CAS PubMed
-
R. T. O'Neill and R. Boulatov, Stilbenes Revisited: Understanding the Mechanism of Mechanochemical Coupling, in Molecular Photoswitches, 2022, pp. 253–281 Search PubMed
- H. M. Bandara and S. C. Burdette, Photoisomerization in different classes of azobenzene, Chem. Soc. Rev., 2012, 41(5), 1809–1825 RSC
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Red-Shifting Azobenzene Photoswitches for in Vivo Use, Acc. Chem. Res., 2015, 48(10), 2662–2670 CrossRef CAS PubMed
- D. Bleger, J. Schwarz, A. M. Brouwer and S. Hecht, o-Fluoroazobenzenes as readily synthesized photoswitches offering nearly quantitative two-way isomerization with visible light, J. Am. Chem. Soc., 2012, 134(51), 20597–20600 CrossRef CAS PubMed
- J. Moreno, M. Gerecke, L. Grubert, S. A. Kovalenko and S. Hecht, Sensitized Two-NIR-Photon Z→E Isomerization of a Visible-Light-Addressable Bistable Azobenzene Derivative, Angew. Chem., Int. Ed., 2016, 55(4), 1544–1547 CrossRef CAS PubMed
- M. Dong, A. Babalhavaeji, C. V. Collins, K. Jarrah, O. Sadovski, Q. Dai and G. A. Woolley, Near-Infrared Photoswitching of Azobenzenes under Physiological Conditions, J. Am. Chem. Soc., 2017, 139(38), 13483–13486 CrossRef CAS PubMed
- P. Lentes, E. Stadler, F. Rohricht, A. Brahms, J. Grobner, F. D. Sonnichsen, G. Gescheidt and R. Herges, Nitrogen Bridged Diazocines: Photochromes Switching within the Near-Infrared Region with High Quantum
Yields in Organic Solvents and in Water, J. Am. Chem. Soc., 2019, 141(34), 13592–13600 CrossRef CAS PubMed
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, Highly Efficient Reversible Z-E Photoisomerization of a Bridged Azobenzene with Visible Light through Resolved S1(nπ*) Absorption Bands, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed
- Y. Yang, R. P. Hughes and I. Aprahamian, Near-infrared light activated azo-BF2 switches, J. Am. Chem. Soc., 2014, 136(38), 13190–13193 CrossRef CAS PubMed
- C. Petermayer and H. Dube, Indigoid Photoswitches: Visible Light Responsive Molecular Tools, Acc. Chem. Res., 2018, 51(5), 1153–1163 CrossRef CAS PubMed
- S. Kitzig, M. Thilemann, T. Cordes and K. Rück-Braun, Light-Switchable Peptides with a Hemithioindigo Unit: Peptide Design, Photochromism, and Optical Spectroscopy, ChemPhysChem, 2016, 17(9), 1252–1263 CrossRef CAS PubMed
- S. Wiedbrauk and H. Dube, Hemithioindigo—an emerging photoswitch, Tetrahedron Lett., 2015, 56(29), 4266–4274 CrossRef CAS
-
T. Bartelmann and H. Dube, Indigoid Photoswitches, in Molecular Photoswitches, 2022, pp. 283–302 Search PubMed
- J. Weinstein and G. M. Wyman, Spectroscopic Studies on Dyes. II. The Structure of N,N'-Dimethylindigo1, J. Am. Chem. Soc., 1956, 78(16), 4007–4010 CrossRef CAS
- C. Y. Huang, A. Bonasera, L. Hristov, Y. Garmshausen, B. M. Schmidt, D. Jacquemin and S. Hecht, N,N'-Disubstituted Indigos as Readily Available Red-Light Photoswitches with Tunable Thermal Half-Lives, J. Am. Chem. Soc., 2017, 139(42), 15205–15211 CrossRef CAS PubMed
- L. A. Huber, M. Peter and H. Dube, Photoisomerization of Mono-Arylated Indigo and Water-Induced Acceleration of Thermal cis to trans Isomerization, ChemPhotoChem, 2018, 2, 458–464 CrossRef CAS
- S. Thumser, L. Köttner, N. Hoffmann, P. Mayer and H. Dube, All-Red-Light Photoswitching of Indirubin Controlled by Supramolecular Interactions, J. Am. Chem. Soc., 2021, 143(43), 18251–18260 CrossRef CAS PubMed
- E. I. Stearns, Phototropic Dyes, J. Opt. Soc. Am., 1942, 32, 282–284 CrossRef CAS
- C. Petermayer, S. Thumser, F. Kink, P. Mayer and H. Dube, Hemiindigo: Highly Bistable Photoswitching at the Biooptical Window, J. Am. Chem. Soc., 2017, 139(42), 15060–15067 CrossRef CAS PubMed
- C. Petermayer and H. Dube, Circular Dichroism Photoswitching with a Twist: Axially Chiral Hemiindigo, J. Am. Chem. Soc., 2018, 140(42), 13558–13561 CrossRef CAS PubMed
- L. Köttner, M. Schildhauer, S. Wiedbrauk, P. Mayer and H. Dube, Oxidized Hemithioindigo Photoswitches-Influence of Oxidation State on (Photo)physical and Photochemical Properties, Chem.–Eur. J., 2020, 26(47), 10712–10718 CrossRef PubMed
- V. Josef, F. Hampel and H. Dube, Heterocyclic Hemithioindigos: Highly Advantageous Properties as Molecular Photoswitches, Angew. Chem., Int. Ed., 2022, 61(43), e202210855 CrossRef CAS PubMed
- M. Krell-Jørgensen, H. Zulfikri, M. G. Bonnevie, F. Simonsen Bro, A. O. Dohn and L. Laraia, Redshifted and Thermally Bistable One-Way Quantitative Hemithioindigo-derived Photoswitches Enabled by Isomer-Specific Excited State Intramolecular Proton Transfer, Chem. Commun., 2023, 59, 563–566 RSC
- T. Lougheed, V. Borisenko, T. Hennig, K. Rück-Braun and G. A. Woolley, Photomodulation of ionic current through hemithioindigo-modified gramicidin channels, Org. Biomol. Chem., 2004, 2(19), 2798–2801 RSC
- S. Herre, W. Steinle and K. Rück-Braun, Synthesis of photoswitchable hemithioindigo-based ω-amino acids and application in Boc-based peptide assembly, Synthesis, 2005, 3297–3300 CAS
- S. Herre, T. Schadendorf, I. Ivanov, C. Herrberger, W. Steinle, K. Rück-Braun, R. Preissner and H. Kuhn, Photoactivation of an inhibitor of the 12/15-lipoxygenase pathway, ChemBioChem, 2006, 7(7), 1089–1095 CrossRef CAS PubMed
- A. Sailer, J. C. M. Meiring, C. Heise, L. N. Pettersson, A. Akhmanova, J. Thorn-Seshold and O. Thorn-Seshold, Pyrrole Hemithioindigo Antimitotics with Near-Quantitative Bidirectional Photoswitching that Photocontrol Cellular Microtubule Dynamics with Single-Cell Precision, Angew. Chem., Int. Ed., 2021, 60(44), 23695–23704 CrossRef CAS PubMed
- K. Tanaka, K. Kohayakawa, S. Iwata and T. Irie, Application of 2-Pyridyl-Substituted Hemithioindigo as a Molecular Switch in Hydrogen-Bonded Porphyrins, J. Org. Chem., 2008, 73, 3768–3774 CrossRef CAS PubMed
- H. Dube and J. Rebek Jr, Selective guest exchange in encapsulation complexes using light of different wavelenghts, Angew. Chem., Int. Ed., 2012, 51(13), 3207–3210 CrossRef CAS PubMed
- G. Moncelsi, L. Escobar, H. Dube and P. Ballester, 2-(4'-Pyridyl-N-oxide)-Substituted Hemithioindigos as Photoresponsive Guests for a Super Aryl-Extended Calix[4]pyrrole Receptor, Chem.–Asian J., 2018, 13(12), 1632–1639 CrossRef CAS PubMed
- T. Bartelmann, F. Gnannt, M. Zitzmann, P. Mayer and H. Dube, Sulfoxide hemithioindigo tweezers – visible light addressable capture and release, Chem. Sci., 2021, 12, 3651–3659 RSC
- M. Guentner, M. Schildhauer, S. Thumser, P. Mayer, D. Stephenson, P. J. Mayer and H. Dube, Sunlight-powered kHz rotation of a hemithioindigo-based molecular motor, Nat. Commun., 2015, 6(1), 8406 CrossRef CAS PubMed
- R. Wilcken, M. Schildhauer, F. Rott, L. A. Huber, M. Guentner, S. Thumser, K. Hoffmann, S. Oesterling, R. de Vivie-Riedle, E. Riedle and H. Dube, Complete Mechanism of Hemithioindigo Motor Rotation, J. Am. Chem. Soc., 2018, 140(15), 5311–5318 CrossRef CAS PubMed
- A. Gerwien, P. Mayer and H. Dube, Photon-Only Molecular Motor with Reverse Temperature-Dependent Efficiency, J. Am. Chem. Soc., 2018, 140(48), 16442–16445 CrossRef CAS PubMed
- A. Gerwien, P. Mayer and H. Dube, Green light powered molecular state motor enabling eight-shaped unidirectional rotation, Nat. Commun., 2019, 10(1), 4449 CrossRef PubMed
- E. Uhl, S. Thumser, P. Mayer and H. Dube, Transmission of Unidirectional Molecular Motor Rotation to a Remote Biaryl Axis, Angew. Chem., Int. Ed., 2018, 57, 11064–11068 CrossRef CAS PubMed
- E. Uhl, P. Mayer and H. Dube, Active and Unidirectional Acceleration of Biaryl Rotation by a Molecular Motor, Angew. Chem., Int. Ed., 2020, 59(14), 5730–5737 CrossRef CAS PubMed
- N. N. Bach, V. Josef, H. Maid and H. Dube, Active Mechanical Threading by a Molecular Motor, Angew. Chem., Int. Ed., 2022, 61(19), e202201882 CAS
- A. Gerwien, F. Gnannt, P. Mayer and H. Dube, Photogearing as a concept for translation of precise motions at the nanoscale, Nat. Chem., 2022, 14, 670–676 CrossRef CAS PubMed
- K. Grill and H. Dube, Supramolecular Relay-Control of Organocatalysis with a Hemithioindigo-Based Molecular Motor, J. Am. Chem. Soc., 2020, 142(45), 19300–19307 CrossRef CAS PubMed
- F. Kink, M. P. Collado, S. Wiedbrauk, P. Mayer and H. Dube, Bistable Photoswitching of Hemithioindigo with Green and Red Light: Entry Point to Advanced Molecular Digital Information Processing, Chem.–Eur. J., 2017, 23(26), 6237–6243 CrossRef CAS PubMed
- L. A. Huber, K. Hoffmann, S. Thumser, N. Böcher, P. Mayer and H. Dube, Direct Observation of Hemithioindigo-Motor Unidirectionality, Angew. Chem., Int. Ed., 2017, 56(46), 14536–14539 CrossRef CAS PubMed
- A. Gerwien, T. Reinhardt, P. Mayer and H. Dube, Synthesis of Double-Bond Substituted Hemithioindigo Photoswitches, Org. Lett., 2018, 20(1), 232–235 CrossRef CAS PubMed
- A. Gerwien, M. Schildhauer, S. Thumser, P. Mayer and H. Dube, Direct evidence for hula twist and single-bond rotation photoproducts, Nat. Commun., 2018, 9(1), 2510 CrossRef PubMed
- A. Gerwien, B. Jehle, M. Irmler, P. Mayer and H. Dube, An Eight-State Molecular Sequential Switch Featuring a Dual Single-Bond Rotation Photoreaction, J. Am. Chem. Soc., 2022, 144(7), 3029–3038 CrossRef CAS PubMed
- J. Andreasson and U. Pischel, Molecules with a sense of logic: a progress report, Chem. Soc. Rev., 2015, 44, 1053–1069 RSC
- D. Gust, T. A. Moore and A. L. Moore, Molecular switches controlled by light, Chem. Commun., 2006, 1169–1178 RSC
- S. Erbas-Cakmak, S. Kolemen, A. C. Sedgwick, T. Gunnlaugsson, T. D. James, J. Yoon and E. U. Akkaya, Molecular logic gates: the past, present and future, Chem. Soc. Rev., 2018, 47, 2228–2248 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2227014–2227018 and 2227451. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06939c |
| This journal is © The Royal Society of Chemistry 2023 |