
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective copper-catalyzed hydrophosphination of alkenyl isoquinolines†‡
Qingjing
Yang
a,
Jian
Zhou
ab and
Jun (Joelle)
Wang
 *b
*b
aDepartment of Chemistry, Southern University of Science and Technology (SUSTech), Shenzhen, 518055, China
bDepartment of Chemistry, Hong Kong Baptist University, Kowloon, Hong Kong, China. E-mail: junwang@hkbu.edu.hk
First published on 21st March 2023
Abstract
An enantioselective hydrophosphination of alkenyl isoquinolines is developed by using a copper-chiral diphosphine ligand catalyst. It provides a direct and atom-efficient approach to prepare a variety of chiral phosphines with an isoquinoline unit in good yields and high enantioselectivities. In addition, these chiral phosphine products are useful bidentate P,N-ligands which showed potential application in asymmetric catalysis.
Chiral organophosphorus compounds play a significant role in bioactive molecules, agrochemistry, and functional materials.1 In addition, they are also broadly applied in transition metal catalysis and organocatalysis as chiral ligands and chiral catalysts.2 Compared with conventional methods using a stoichiometric amount of chiral starting materials or chiral reagents,3 asymmetric catalytic approaches have attracted increasing attention in the construction of chiral phosphines. Among them, catalytic asymmetric hydrophosphination is one of the most direct and atom-economical ways for preparation of optically active phosphines.
Pd-Catalyzed asymmetric hydrophosphination of α,β-unsaturated compounds (including α,β-unsaturated aldehydes, ketones, esters, pyrrole amides, sulfonic esters, and sulfonamides) with secondary phosphines has emerged as a versatile method for the construction of chiral phosphine compounds (Scheme 1a).4 Other transition metal complexes such as chiral Ni,5 Pt,6 Cu7 and Mn8 catalysts and organocatalysts9 were also applied in the asymmetric hydrophosphination of electron-deficient alkenes, recently. However, examples where heteroarenes were employed in alkene activation have not appeared due to the relatively poor reactivity of alkenylheteroarenes. In 2021, Terada reported hydrophosphinylation of β-unsubstituted alkenylheteroarene N-oxides with SPO (secondary phosphine oxide) catalyzed by their chiral bis(guanidino)iminophosphorane organosuperbase (Scheme 1b).10 The subsequent reduction of the phosphine oxide product could give quinoline phosphine. Inspired by Yin's recent elegant work on the Cu/TANIAPhos-catalyzed enantioselective hydrophosphination of α,β-unsaturated amides in which the olefin moiety is sluggishly activated by the adjacent carboxamide group (Scheme 1c),7a we envisioned that the long-time unsolved asymmetric hydrophosphination of intrinsic low electrophilic alkenyl-heteroarenes might be realized with a suitable Cu/chiral phosphine catalyst because of its relative stability in the presence of excess HPPh2. Furthermore, the chiral tertiary phosphine products bearing a N-heteroaromatic ring themselves are potentially useful chiral bidentate P,N-ligands in asymmetric catalysis.
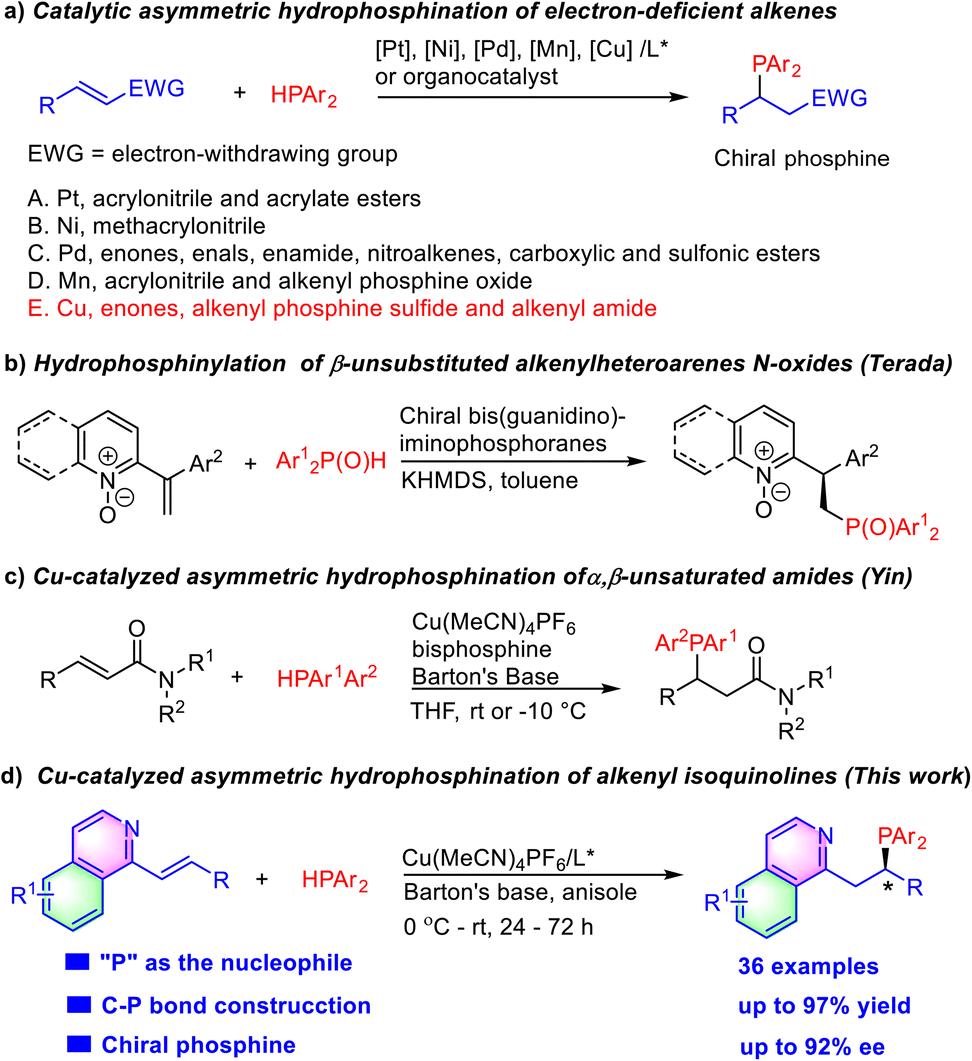 | ||
| Scheme 1 Examples of asymmetric hydrophosphination of electron-deficient alkenes and asymmetric hydrophosphination to alkenyl azaarenes. | ||
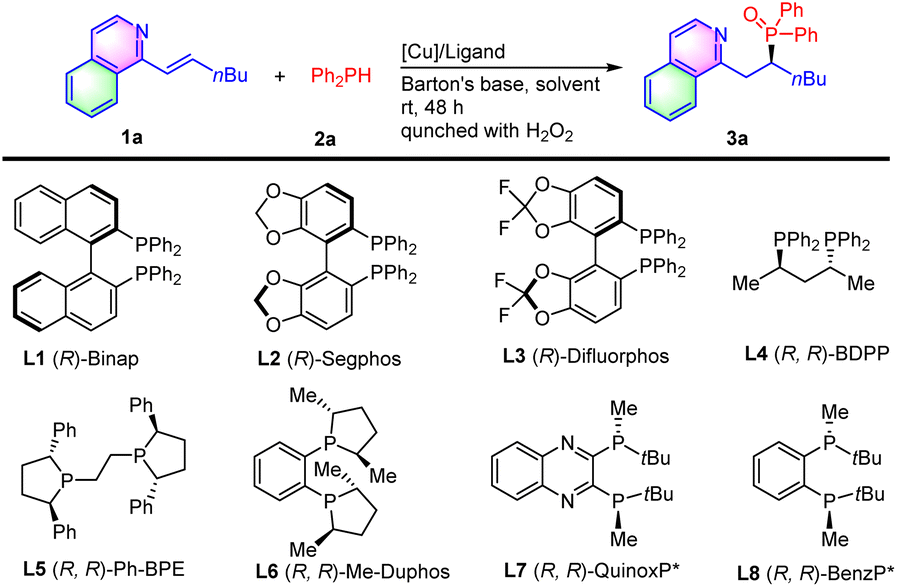
Based on these design considerations and our prior experience in the construction of chiral organophosphorus compounds,11 we began our study by examining the hydrophosphination of alkenyl isoquinoline 1a with diphenyl phosphine 2a in the presence of a copper complex and Barton's base in toluene at room temperature (Table 1). (R)-Binap (L1) gave the bench-stable secondary phosphine oxide 3a in 85% yield by the in situ oxidation of the labile secondary phosphine product with hydrogen peroxide, avoiding their isolation and purification while maintaining their structural diversity. However, the enantioselectivity of 3a was very poor (entry 1). Other biphosphine ligands-(R)-Segphos (L2), (R)-difluorophos (L3), (R, R)-BDPP (L4), (R, R)-Ph-BPE (L5) and (R, R)-Me-Duphos (L6) resulted in low yields and enantio-control (entries 2–6). Catalyzed by (R, R)-QuinoxP* (L7), 68% ee was obtained with 77% yield (entry 7). To our delight, the reaction with (R, R)-BenzP* (L8) provided the hydrophosphination product 3a in 88% yield with 83% ee (entry 8). Therefore, (R, R)-BenzP* (L8) was chosen for further optimization. Other Cu precursors, such as Cu(MeCN)4BF4, Cu(OAc)2, Cu(ClO4)2·6H2O, and Cu(OTf)2, were employed, indicating that Cu(MeCN)4PF6 was the best choice (entry 8 vs. entries 9–12). Next, we examined the effect of a solvent on the reaction. It was showed that the reactions in THF, 1,4-dioxane, DME, and anisole provided product 3a with similar high yields, but the reaction in anisole gave the highest ee (entries 13–16). Other bases, including DABCO, K2CO3, and KOAc, were then tested, and led to a slight decrease of the reaction yields (entries 17–19). On increasing the molar ratio of Barton's base/1a from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1.5/1, the ee of product 3a was enhanced to 87% (entry 20). Finally, product 3a was obtained in 94% yield with 90% ee by lowering the temperature to 0 °C (entry 21).
:1 to 1.5/1, the ee of product 3a was enhanced to 87% (entry 20). Finally, product 3a was obtained in 94% yield with 90% ee by lowering the temperature to 0 °C (entry 21).
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Cu] | Ligand | Solvent | Yieldb (%) | eec (%) |
| a Reaction conditions: [Cu] (10 mol%), ligand (11 mol%), 1a (0.2 mmol), 2a (0.3 mmol), Barton's base (1.0 eq.), solvent (1.0 mL). b Isolated yields. c The ee values were determined by chiral HPLC. d DABCO. e K2CO3. f KOAc. g 1.5 eq. Barton’s base. h 2.0 eq. Barton’s base. i 0 °C. | |||||
| 1 | Cu(MeCN)4PF6 | L1 | Toluene | 85 | 3 |
| 2 | Cu(MeCN)4PF6 | L2 | Toluene | 51 | 32 |
| 3 | Cu(MeCN)4PF6 | L3 | Toluene | 52 | 20 |
| 4 | Cu(MeCN)4PF6 | L4 | Toluene | 44 | 40 |
| 5 | Cu(MeCN)4PF6 | L5 | Toluene | 30 | −35 |
| 6 | Cu(MeCN)4PF6 | L6 | Toluene | 11 | 7 |
| 7 | Cu(MeCN)4PF6 | L7 | Toluene | 77 | 68 |
| 8 | Cu(MeCN)4PF6 | L8 | Toluene | 88 | 83 |
| 9 | Cu(MeCN)4BF4 | L8 | Toluene | 36 | 83 |
| 10 | Cu(OAc)2 | L8 | Toluene | 48 | 86 |
| 11 | Cu(ClO4)2·6H2O | L8 | Toluene | 74 | 66 |
| 12 | Cu(OTf)2 | L8 | Toluene | 41 | 73 |
| 13 | Cu(MeCN)4PF6 | L8 | THF | 86 | 84 |
| 14 | Cu(MeCN)4PF6 | L8 | 1,4-Dioxane | 91 | 83 |
| 15 | Cu(MeCN)4PF6 | L8 | DME | 93 | 82 |
| 16 | Cu(MeCN)4PF6 | L8 | Anisole | 92 | 85 |
| 17d | Cu(MeCN)4PF6 | L8 | Anisole | 20 | 73 |
| 18e | Cu(MeCN)4PF6 | L8 | Anisole | 35 | 70 |
| 19f | Cu(MeCN)4PF6 | L8 | Anisole | 24 | 68 |
| 20g | Cu(MeCN)4PF6 | L8 | Anisole | 90 | 87 |
| 21h | Cu(MeCN)4PF6 | L8 | Anisole | 91 | 86 |
| 22g,i | Cu(MeCN)4PF6 | L8 | Anisole | 94 | 90 |
With the optimal conditions, the substrate scope of substituted alkenyl isoquinolines was then investigated (Table 2). As for β-alkyl substituted alkenyl isoquinolines, both linear alkyl (3b and 3c), benzyl (3d) and cyclopropanyl (3e) reacted efficiently with diphenyl phosphine 2a to afford the corresponding products in 80–96% yields with 80–86% ee. The absolute configuration of product 3e was confirmed as (R) by X-ray crystal structure analysis.12 Interestingly, there was only 1,4-addition when the conjugate divinyl isoquinoline 1f was investigated, and the hydrophosphination product 3f was obtained in 70% yield with 86% ee. Subsequently, substrates with aryl groups were also evaluated. The 1-styrylisoquinoline 1g reacted smoothly under the reaction conditions, providing the product 3g in 87% yield and 92% ee. A wide range of alkenyl isoquinolines with an electron-donating group (3h and 3i) or an electron-withdrawing group (3j, 3k, 3l, 3m, and 3n) at the para-position of the phenyl ring were tolerated. In general, the reactions afforded products in moderate to excellent yields with moderate to high enantioselectivity. A good outcome was also achieved upon introducing a methyl group at the meta-position of the aryl group of the substrate (3p). The reaction of 1q containing a 2-naphthyl substituent proceeded to afford 3q with a decreased yield and enantioselectivity (70% yield and 77% ee). Substrates with a heteroaryl, such as 2-furanyl (3r), 3-furanyl (3s), 3-thienyl (3t), and 3-pyridinyl (3u), were well-tolerated under this catalytic condition. The 2-pyridinyl (3v) or 4-pyridinyl (3w) substituted substrate also worked well to deliver products in excellent yields, but led to an obvious decrease in enantioselectivities. The reaction between α-phenyl-substituted 1x and Ph2PH was attempted, affording the product 3x in 93% yield and 53% ee. It was found that quinoline 1y also could be hydrophosphinated with this catalyst at 60 °C to provide the product 3y in 73% yield with 54% ee. Furthermore, alkenyl pyridines were also compatible with the reaction conditions to deliver the corresponding products in good yields and enantioselectivities (3z and 3aa).
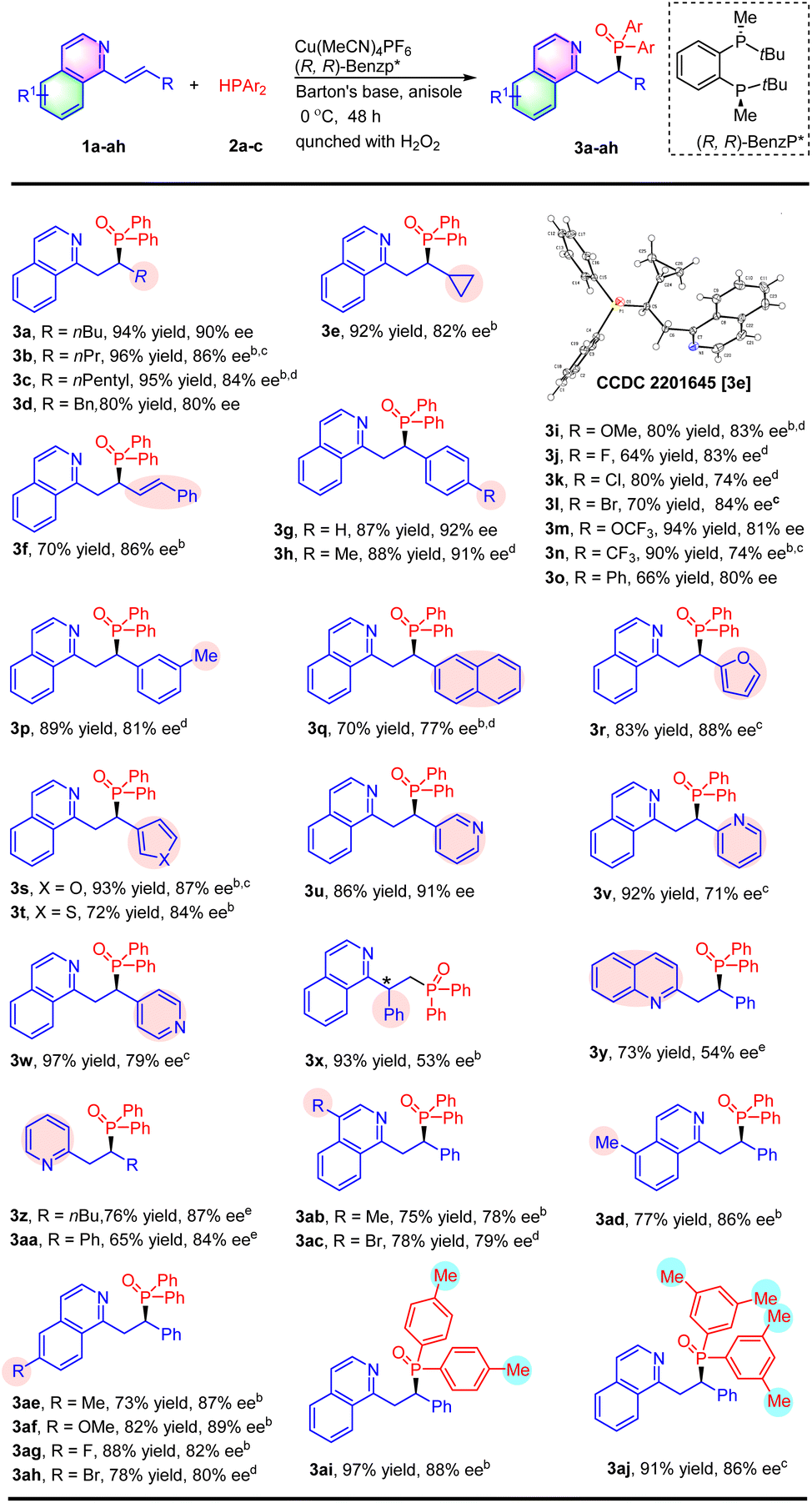
The substitute groups on isoquinolines were also evaluated (Table 2). Substrates with electron-donating groups (Me and OMe) or with electron-withdrawing groups (F and Br) at the 4-(3ab and 3ac), 5-(3ad), and 6-positions (3ae, 3af, 3ag and 3ah) of the isoquinoline ring were well-tolerated affording products in 73–93% yields with 78–89% ee. Finally, we turned our attention towards the scope of diarylphosphines 2. Hydrophosphination with (p-Me-C6H4)2PH and (3,5-Me-C6H3)2PH led to the corresponding products 3ai and 3aj with excellent yields and good enantioselectivities. However, other phosphines, such as (o-Me-C6H4)2PH, (p-MeO-C6H4)2PH, (p-F-C6H4)2PH and Cy2PH, were not suitable for the reaction and only trace expected products were detected.
Chiral bidentate P,N-ligands are one of the most important family of ligands widely used in asymmetric catalysis.13 Therefore, there is an increasing demand for the efficient synthesis of chiral bidentate P,N-ligands. However, most of the protocols for the synthesis of chiral P,N-ligands focused on the chiral resolution or required tedious multistep synthesis.14 Herein, the reduction of phosphine oxide product 3a with phenylsilane in the presence of diphenyl phosphate provided phosphine 4a in 80% yield without loss of enantiomeric purity (Scheme 2). Subsequently, we examined our P,N-ligand 4a in the Pd-catalyzed asymmetric allylic etherification of benzyl alcohol, obtaining the desired product 7 in good yield (85%) and moderate ee (57%). In addition, the product 3ah could also be transformed by Pd-catalyzed Sonogashira cross-coupling. The corresponding alkynylation product 8 was obtained in excellent yield without a decline in enantiopurity.
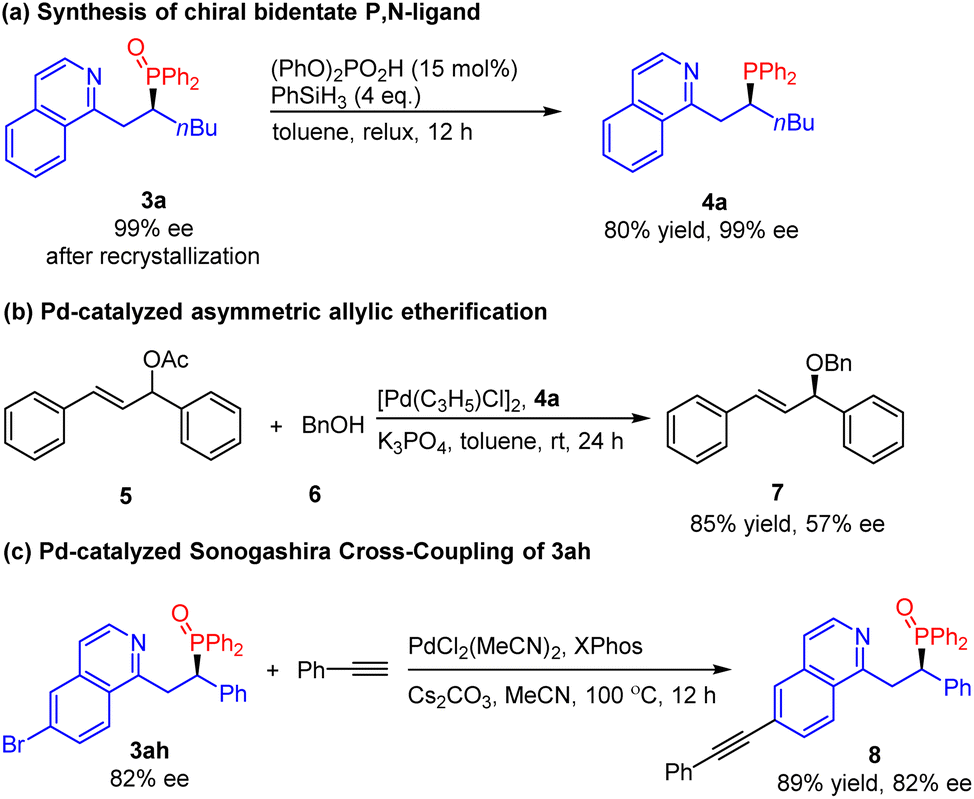 | ||
| Scheme 2 Synthesis applications. | ||
A possible mechanism for the reaction was proposed based on the literature (Scheme 3).7a,b,15 Chiral copper complex V is formed by Cu(MeCN)4PF6 and (R, R)-BenzP*. Coordination between HPPh2 and chiral copper complex V generates complex I, which undergoes deprotonation in the presence of Barton's base to produce the nucleophilic copper(I)-diphenylphosphide species II. Then, the alkene tethered with isoquinoline coordinates to complex II and affords the Cu–phosphido-alkene complex III. Next, asymmetric addition of the diarylphosphido group on copper to the alkenyl-substituted isoquinoline provides an aza-π-allylcopper intermediate IV, which is protonated to release the phosphination product 3a and regenerates copper complex V for the next catalytic cycle.
 | ||
| Scheme 3 Proposed catalytic cycle. | ||
In summary, we have established an operationally simple and highly enantioselective Cu(I)-catalyzed hydrophosphination reaction of alkenyl-substituted isoquinolines. This method afforded a direct and atom efficient access to chiral phosphines with an isoquinoline unit in high yields and enantioselectivities in most cases. Furthermore, the product was showcased as a new chiral P,N-type ligand in palladium-catalyzed asymmetric allylic etherification. Further work on the structure modification and application of these chiral phosphine products in other asymmetric reactions is underway in our laboratory.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.‡Author contributions
Dr Q. Yang performed the experiments and prepared the supplementary information. J. Zhou collected some data. Prof. J. Wang conceived and directed the project. Q. Yang and J. Wang wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank the Research Grants Council of Hong Kong (GRF 12301821) and National Natural Science Foundation of China (NSFC 21971102) for financial support. Dr Xiaoyong Chang from SUSTech is gratefully acknowledged for X-ray crystallographic analysis.Notes and references
- (a) G. P. Horsman and D. L. Zechel, Chem. Rev., 2017, 117, 5704 CrossRef CAS PubMed; (b) A. Mucha, P. Kafarski and L. Berlicki, J. Med. Chem., 2011, 54, 5955 CrossRef CAS PubMed; (c) J. P. Kahler and S. H. L. Verhelst, RSC Chem. Biol., 2021, 2, 1285 RSC; (d) C. E. Stieger, L. Franz, F. Körlin and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2021, 60, 15359 CrossRef CAS PubMed.
- For selected reviews on phosphines in asymmetric catalysis, see: (a) T. Wang, X. Han, F. Zhong, W. Yao and Y. Lu, Acc. Chem. Res., 2016, 49, 1369 CrossRef CAS PubMed; (b) H.-U. Blaser, B. Pugin, F. Spindler, E. Meja and A. Togni, Josiphos Ligands: From Discovery to Technical Applications, in Privileged Chiral Ligands and Catalysts, ed., Q.-L. Zhou, Wiley, 2011, pp. 93–136 Search PubMed; (c) P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741 CrossRef CAS PubMed; (d) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049 CrossRef CAS PubMed; (e) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed; (f) C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981 CrossRef CAS PubMed.
- (a) W. Chen, F. Spindler, B. Pugin and U. Nettekoven, Angew. Chem., Int. Ed., 2013, 52, 8652 CrossRef CAS PubMed; (b) J. P. Djukic, Cyclopalladated Compounds as Resolving Agents for Racemic Mixtures of Ligands, in Palladacycles: Synthesis, Characterization and Applications, ed. J. Dupont and M. Pfeffer, Wiley, Weinheim, 2008, pp. 123–153 Search PubMed.
- (a) J.-J. Feng, X.-F. Chen, M. Shi and W.-L. Duan, J. Am. Chem. Soc., 2010, 132, 5562 CrossRef CAS PubMed; (b) D. Du and W.-L. Duan, Chem. Commun., 2011, 47, 11101 RSC; (c) Y.-R. Chen and W.-L. Duan, Org. Lett., 2011, 13, 5824 CrossRef CAS; (d) J.-J. Feng, M. Huang, Z.-Q. Lin and W.-L. Duan, Adv. Synth. Catal., 2012, 354, 3122 CrossRef CAS; (e) J. Lu, J. Ye and W.-L. Duan, Org. Lett., 2013, 15, 5016 CrossRef CAS PubMed; (f) D. Du, Z.-Q. Lin, J.-Z. Lu, C. Li and W.-L. Duan, Asian J. Org. Chem., 2013, 2, 392 CrossRef CAS; (g) J. Lu, J. Ye and W.-L. Duan, Chem. Commun., 2014, 50, 698 RSC; (h) J. Huang, M.-X. Zhao and W.-L. Duan, Tetrahedron Lett., 2014, 55, 629 CrossRef CAS; (i) Y.-C. Song, G.-F. Dai, F. Xiao and W.-L. Duan, Tetrahedron Lett., 2016, 57, 2990 CrossRef CAS; (j) Y. Huang, S. A. Pullarkat, Y. Li and P.-H. Leung, Chem. Commun., 2010, 46, 6950 RSC; (k) Y. Huang, R. J. Chew, Y. Li, S. A. Pullarkat and P.-H. Leung, Org. Lett., 2011, 13, 5862 CrossRef CAS PubMed; (l) Y. Huang, S. A. Pullarkat, S. Teong, R. J. Chew, Y. Li and P.-H. Leung, Organometallics, 2012, 31, 4871 CrossRef CAS; (m) R. J. Chew, Y. Huang, Y. Li, S. A. Pullarkat and P.-H. Leung, Adv. Synth. Catal., 2013, 355, 1403 CrossRef CAS; (n) R. J. Chew, Y. Lu, Y.-X. Jia, B.-B. Li, E. H. Y. Wong, R. Goh, Y. Li, Y. Huang, S. A. Pullarkat and P.-H. Leung, Chem.–Eur. J., 2014, 20, 14514 CrossRef CAS PubMed; (o) R. J. Chew, K. Sepp, B.-B. Li, Y. Li, P.-C. Zhu, N. S. Tan and P.-H. Leung, Adv. Synth. Catal., 2015, 357, 3297 CrossRef CAS; (p) X.-Y. Yang, W. S. Tay, Y. Li, S. A. Pullarkat and P.-H. Leung, Organometallics, 2015, 34, 5196 CrossRef CAS; (q) X.-Y. Yang, Y.-X. Jia, W. S. Tay, Y. Li, S. A. Pullarkat and P.-H. Leung, Dalton Trans., 2016, 45, 13449 RSC; (r) R. H. X. Teo, H. J. Chen, Y. Li, S. A. Pullarkat and P.-H. Leung, Adv. Synth. Catal., 2020, 362, 2373 CrossRef CAS; (s) B. Ding, Z. Zhang, Y. Xu, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Org. Lett., 2013, 15, 5476 CrossRef CAS PubMed; (t) Y. Xu, Z. Yang, B. Ding, D. Liu, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Tetrahedron, 2015, 71, 6832 CrossRef CAS; (u) K. Gan, A. Sadeer, C. Xu, Y. Li and S. A. Pullarkat, Organometallics, 2014, 33, 5074 CrossRef CAS; (v) X.-Q. Hao, Y.-W. Zhao, J.-J. Yang, J.-L. Niu, J.-F. Gong and M.-P. Song, Organometallics, 2014, 33, 1801 CrossRef CAS; (w) X.-Q. Hao, J.-J. Huang, T. Wang, J. Lv, J.-F. Gong and M.-P. Song, J. Org. Chem., 2014, 79, 9512 CrossRef CAS PubMed.
- (a) A. D. Sadow, I. Haller, L. Fadini and A. Togni, J. Am. Chem. Soc., 2004, 126, 14704 CrossRef CAS PubMed; (b) A. D. Sadow and A. Togni, J. Am. Chem. Soc., 2005, 127, 17012 CrossRef CAS PubMed; (c) L. Huang, E. Q. Lim and M. J. Koh, Chem Catal., 2022, 2, 508 CrossRef CAS.
- I. Kovacik, D. K. Wicht, N. S. Grewal and D. S. Glueck, Organometallics, 2000, 19, 950 CrossRef CAS.
- (a) Y.-B. Li, H. Tian and L. Yin, J. Am. Chem. Soc., 2020, 142, 20098 CrossRef CAS PubMed; (b) W.-J. Yue, J.-Z. Xiao, S. Zhang and L. Yin, Angew. Chem., Int. Ed., 2020, 59, 7057 CrossRef CAS PubMed; (c) Y.-R. Chen, J.-J. Feng and W.-L. Duan, Tetrahedron Lett., 2014, 55, 595 CrossRef CAS.
- (a) J. M. Pérez, R. Postolache, M. C. Reis, E. G. Sinnema, D. Vargová, F. de Vries, E. Otten, L. Ge and S. R. Harutyunyan, J. Am. Chem. Soc., 2021, 143, 20071 CrossRef PubMed; (b) L. Ge and S. R. Harutyunyan, Chem. Sci., 2022, 13, 1307 RSC.
- (a) G. Bartoli, M. Bosco, A. Carlone, M. Locatelli, A. Mazzanti, L. Sambri and P. Melchiorre, Chem. Commun., 2007, 722 RSC; (b) A. Carlone, G. Bartoli, M. Bosco, L. Sambri and P. Melchiorre, Angew. Chem., Int. Ed., 2007, 46, 4504 CrossRef CAS PubMed; (c) I. Ibrahem, R. Rios, J. Vesely, P. Hammar, L. Eriksson, F. Himo and A. Córdova, Angew. Chem., Int. Ed., 2007, 46, 4507 CrossRef CAS PubMed; (d) I. Ibrahem, P. Hammar, J. Vesely, R. Rios, L. Eriksson and A. Córdova, Adv. Synth. Catal., 2008, 350, 1875 CrossRef CAS; (e) R. Mait, J.-L. Yan, X. Yang, B. Mondal, J. Xu, H. Chai, Z. Jin and Y. R. Chi, Angew. Chem., Int. Ed., 2021, 60, 26616 CrossRef PubMed.
- S. Das, Q. Hu, A. Kondoh and M. Terada, Angew. Chem., Int. Ed., 2021, 60, 1417 CrossRef CAS PubMed.
- (a) Z. Lu, H. Zhang, Z. Yang, N. Ding, L. Meng and J. Wang, ACS Catal., 2019, 9, 1457 CrossRef CAS; (b) L. Zhang, S.-H. Xiang, J. Wang, J. Xiao, J.-Q. Wang and B. Tan, Nat. Commun., 2019, 10, 566 CrossRef PubMed; (c) Z. Yang, X. Gu, L.-B. Han and J. Wang, Chem. Sci., 2020, 11, 7451 RSC; (d) Z. Yang and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 27288 CrossRef CAS PubMed; (e) Z. Yang, Q. Du, Y. Jiang and J. Wang, CCS Chem., 2022, 4, 1901 CrossRef CAS; (f) Y. Zhang, Y. Jiang, M. Li, Z. Huang and J. Wang, Chem. Catal., 2022, 2, 3163–3173 CrossRef CAS; (g) B. Cai, Y. Cui, J. Zhou, Y.-B. Wang, L. Yang, B. Tan and J. Wang, Angew. Chem., Int. Ed., 2022, 61 DOI:10.1002/anie.202215820.
- CCDC 2201645 [for 3e] contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/data_request/cif.‡.
- (a) G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336 CrossRef CAS PubMed; (b) M. P. Carroll and P. J. Guiry, Chem. Soc. Rev., 2014, 43, 819 RSC; (c) B. V. Rokade and P. J. Guiry, ACS Catal., 2018, 8, 624 CrossRef CAS; (d) W. A. Munzeiwa, B. Omondi and V. O. Nyamori, Beilstein J. Org. Chem., 2020, 16, 362 CrossRef CAS PubMed.
- For selected examples, see: (a) T. Hayashi, T. Mise, M. Fukushima, M. Kagotani, N. Nagashima, Y. Hamada, A. Matsumoto, S. Kawakami, M. Konishi, K. Yamamoto and M. Kumada, Bull. Chem. Soc. Jpn., 1980, 53, 1138 CrossRef CAS; (b) N. W. Alcock, J. M. Brown and D. I. Hulmes, Tetrahedron: Asymmetry, 1993, 4, 743 CrossRef CAS; (c) A. Lightfoot, P. Schnider and A. Pfaltz, Angew. Chem., Int. Ed., 1998, 37, 2897 CrossRef CAS; (d) T. Bunlaksananusorn, K. Polborn and P. Knochel, Angew. Chem., Int. Ed., 2003, 42, 3941 CrossRef CAS PubMed; (e) S. Kaiser, S. P. Smidt and A. Pfaltz, Angew. Chem., Int. Ed., 2006, 45, 5194 CrossRef PubMed; (f) Q.-B. Liu and Y.-G. Zhou, Tetrahedron Lett., 2007, 48, 2101 CrossRef CAS; (g) Z. Han, Z. Wang, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2009, 48, 5345 CrossRef CAS PubMed.
- Y.-L. Zeng, B. Chen, Y.-T. Wang, C.-Y. He, Z.-Y. Mu, J.-Y. Du, L. He, W.-D. Chu and Q.-Z. Liu, Chem. Commun., 2020, 56, 1693 RSC.
Footnotes |
| † Dedicated to Professor Guo-Qiang Lin on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2201645. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06950d |
| This journal is © The Royal Society of Chemistry 2023 |